SynthNN: How Deep Learning Predicts Material Synthesizability to Accelerate Drug Discovery
This article explores SynthNN, a groundbreaking deep learning model designed to predict the synthesizability of inorganic crystalline materials—a critical challenge in materials science and drug development.
SynthNN: How Deep Learning Predicts Material Synthesizability to Accelerate Drug Discovery
Abstract
This article explores SynthNN, a groundbreaking deep learning model designed to predict the synthesizability of inorganic crystalline materials—a critical challenge in materials science and drug development. We delve into the foundational principles of synthesizability prediction, moving beyond traditional proxies like thermodynamic stability. The discussion covers SynthNN's unique methodology, which leverages positive-unlabeled learning and data from known material compositions without requiring prior chemical knowledge. For researchers and drug development professionals, we provide a comparative analysis against expert judgment and other computational methods, address common implementation challenges, and showcase its practical application in successful experimental pipelines. Finally, we examine the model's validation and its performance against newer AI approaches, concluding with its profound implications for streamlining the discovery of synthetically accessible materials and therapeutics.
The Synthesizability Challenge: Why Predicting Material Creation is Hard
Defining Synthesizability in Materials Science and Drug Development
Synthesizability is a critical concept in both materials science and drug development, referring to the feasibility of successfully creating a proposed molecule or material through chemical synthesis in a laboratory setting. It is not merely an inherent property of a substance, but a multifaceted assessment contingent on available starting materials, known reaction pathways, equipment, cost, and time [1]. The accurate prediction of synthesizability is a cornerstone for accelerating the discovery of new functional materials and therapeutic compounds, as it ensures that computationally designed candidates can be translated into physical entities for testing and application.
Defining Synthesizability Across Disciplines
The core definition of synthesizability shares common ground across fields, but the specific challenges and emphases differ, particularly between inorganic crystalline materials and organic drug-like molecules.
Inorganic Materials Science
For inorganic crystalline materials, synthesizability is defined as a material being synthetically accessible through current synthetic capabilities, regardless of whether it has been synthesized yet [2]. The primary challenge lies in the lack of well-understood reaction mechanisms compared to organic chemistry. Synthesis often depends on a complex interplay of thermodynamic and kinetic stabilization, reaction pathway selection, and selective nucleation of the target material [2] [3]. Furthermore, the decision to synthesize a material involves non-physical considerations such as reactant cost, equipment availability, and the perceived importance of the final product [2]. This makes synthesizability difficult to predict based on thermodynamic constraints alone.
Drug Development
In drug development, a molecule is considered synthesizable if a viable synthesis route of reactions from readily available starting materials to the target molecule can be found [1]. However, synthesizability is not a binary judgment but a matter of degree, heavily influenced by the stage of the drug discovery project and the resources one is willing to commit [4]. In early stages like hit-finding, the focus is on simple, tractable molecules that can be made quickly. In later stages like lead optimization, if a molecule shows high promise, chemists may engage in complex "synthetic heroics" to make it, effectively "teaching it to fly" [4]. A key emerging concept is "in-house synthesizability," which tailors the synthesizability assessment to the specific, limited collection of building blocks available in a particular laboratory, rather than assuming near-infinite commercial availability [5].
Quantitative Synthesizability Prediction: The SynthNN Model
A significant advance in computational materials science is the development of the deep learning synthesizability model, SynthNN, designed for inorganic crystalline materials.
Model Rationale and Workflow
Traditional proxies for synthesizability, such as enforcing a charge-balancing criteria, have proven inadequate, capturing only 37% of known synthesized inorganic materials [2]. SynthNN reformulates material discovery as a synthesizability classification task. It leverages the entire space of synthesized inorganic chemical compositions from the Inorganic Crystal Structure Database (ICSD) and uses a semi-supervised learning approach to learn the chemistry of synthesizability directly from the data of all experimentally realized materials [2] [6] [7].
Table 1: Key Features and Performance of the SynthNN Model
| Aspect | Description |
|---|---|
| Model Type | Deep learning classification model (SynthNN) [2] |
| Input | Chemical formulas (no structural information required) [2] |
| Core Methodology | Uses atom2vec learned atom embeddings; positive-unlabeled (PU) learning [2] |
| Key Advantage | Learns chemical principles (e.g., charge-balancing, ionicity) from data without prior knowledge [2] [7] |
| Performance vs. DFT | 7x higher precision than DFT-calculated formation energies [2] [6] |
| Performance vs. Experts | 1.5x higher precision and 100,000x faster than best human expert [2] |
Figure 1: The SynthNN prediction workflow, which transforms a chemical formula into a synthesizability classification through learned embeddings and a deep neural network [2] [6] [7].
Experimental Protocol: SynthNN Model Training and Validation
Objective: To train and validate a deep learning model (SynthNN) for predicting the synthesizability of inorganic crystalline materials from their chemical composition.
Materials and Reagents:
- Software: Python environment with deep learning libraries (e.g., TensorFlow, PyTorch).
- Training Data: Chemical formulas extracted from the Inorganic Crystal Structure Database (ICSD) [2] [8].
- Compute Resource: Standard workstation or high-performance computing node with GPU acceleration.
Procedure:
- Data Curation: Extract and clean chemical formulas of synthesized inorganic materials from the ICSD to form the set of positive (synthesized) examples [2].
- Generation of Unlabeled Data: Artificially generate a large set of chemical formulas that are not present in the ICSD. This set constitutes the unlabeled data, as these materials could be unsynthesizable or simply not yet synthesized [2].
- Model Architecture Setup: Implement a neural network using an atom embedding layer (atom2vec) to represent each chemical formula. The embedding dimensionality is a key hyperparameter [2].
- Positive-Unlabeled (PU) Training: Train the SynthNN model using a semi-supervised PU learning algorithm. This involves treating the artificially generated formulas as unlabeled data and probabilistically reweighting them according to their likelihood of being synthesizable [2] [8].
- Model Validation: Benchmark the trained model against a hold-out test set. Compare its performance against baseline methods like random guessing, charge-balancing, and predictions from human experts [2].
Synthesizability in De Novo Drug Design
In drug development, ensuring synthesizability is paramount for the practical application of generative models that design novel molecules de novo.
In-House Synthesizability Scoring
A key innovation is the development of rapidly retrainable in-house synthesizability scores. These models predict whether a molecule can be synthesized using a specific, limited inventory of building blocks available in a researcher's own laboratory [5]. This approach contrasts with traditional Computer-Aided Synthesis Planning (CASP) that assumes access to millions of commercial building blocks.
Experimental Findings: A study transferring CASP from 17.4 million commercial building blocks (Zinc) to a small laboratory setting with only ~6,000 in-house building blocks (Led3) showed a relatively modest decrease of –12% in the CASP success rate for solving synthesis routes. The primary trade-off was that routes using in-house blocks were, on average, two reaction steps longer than those using the vast commercial library [5].
Protocol: Implementing an In-House Synthesizability Workflow
Objective: To generate and experimentally validate novel, biologically active drug candidates that are synthesizable exclusively from an in-house collection of building blocks.
Materials and Reagents:
- Software: AiZynthFinder or similar CASP toolkit; QSAR modeling software; generative molecular design software (e.g., based on recurrent neural networks or variational autoencoders) [5].
- Building Blocks: Curated list of in-house available building blocks (e.g., 5,000-6,000 compounds) with associated chemical data [5].
- Laboratory Equipment: Standard synthetic chemistry apparatus for organic synthesis and purification (e.g., fume hood, glassware, rotary evaporator, HPLC). Biochemical assay kits for target protein activity evaluation [5].
Procedure:
- Workflow Setup and Synthesizability Score Training:
- Deploy a CASP tool (e.g., AiZynthFinder) configured with your in-house building block list.
- Generate a dataset of molecules and run synthesis planning to determine which are solvable with in-house blocks.
- Use this data to train a fast, random forest-based synthesizability classification model that acts as a proxy for full CASP [5].
Multi-Objective De Novo Molecular Generation:
- Use a generative model to propose new molecular structures.
- Employ a multi-objective optimization function that combines:
- A QSAR model predicting the desired biological activity (e.g., IC50 for a target protein).
- The in-house synthesizability score to prioritize molecules that can be made [5].
- Generate a library of candidate molecules ranked by the multi-objective score.
Synthesis and Experimental Validation:
- Select top-ranking candidates for further analysis.
- For each candidate, run the full CASP tool to obtain detailed, multi-step synthesis routes using only in-house blocks.
- Synthesize the candidates following the AI-suggested routes.
- Purify the compounds and experimentally evaluate their biochemical activity in assays to confirm the computational predictions [5].
Table 2: Research Reagent Solutions for In-House Drug Design
| Reagent / Resource | Function in the Workflow |
|---|---|
| In-House Building Block Collection | Provides the foundational chemical resources for all proposed synthesis routes, defining the space of in-house synthesizable molecules [5]. |
| CASP Tool (e.g., AiZynthFinder) | Performs retrosynthetic analysis to deconstruct target molecules into available building blocks and plans feasible synthetic routes [5]. |
| Generative Molecular Model | Proposes novel molecular structures that are optimized for desired properties like target activity and synthesizability [5] [1]. |
| QSAR Model | Provides a fast computational prediction of a molecule's biological activity, serving as one of the primary objectives for optimization [5]. |
Figure 2: In-house de novo drug design workflow that integrates building block availability, molecular generation, multi-objective scoring, and experimental validation [5].
The definition of synthesizability is evolving from a simplistic, binary concept to a nuanced, context-dependent one. In materials science, models like SynthNN demonstrate that synthesizability can be learned from historical data, dramatically accelerating the discovery of new inorganic crystals. In drug development, the focus is shifting towards pragmatic in-house synthesizability, which aligns computational design with practical laboratory constraints. Together, these advanced computational approaches are closing the gap between in-silico design and real-world synthesis, making the process of molecular and materials discovery more efficient and reliable.
The acceleration of materials discovery hinges on the accurate identification of synthesizable compounds. For decades, the computational materials science community has relied on two fundamental approaches for this task: the heuristic principle of charge-balancing and energy-based assessments via density functional theory (DFT). These methods serve as preliminary filters to distinguish potentially synthesizable materials from those that are not. However, within the context of developing deep learning models like SynthNN for synthesizability prediction, understanding the specific limitations of these traditional approaches becomes paramount. This document details the quantitative shortcomings and procedural constraints of charge-balancing and DFT calculations, providing a foundational rationale for the development and adoption of more advanced, data-driven synthesizability models.
Quantitative Comparison of Synthesizability Assessment Methods
The table below summarizes the key performance metrics and limitations of traditional synthesizability assessment methods compared to modern machine learning approaches.
Table 1: Performance Comparison of Synthesizability Assessment Methods
| Method | Key Principle | Reported Precision/Accuracy | Primary Limitations |
|---|---|---|---|
| Charge-Balancing | Net neutral ionic charge based on common oxidation states [2] | Only 37% of known synthesized inorganic materials are charge-balanced [2] | Overly inflexible; fails for metallic/covalent systems; poor for ionic binaries (e.g., only 23% of Cs compounds) [2] |
| DFT Formation Energy | Thermodynamic stability relative to decomposition products [2] | Captures only ~50% of synthesized inorganic materials [2] | Fails to account for kinetic stabilization and non-physical synthesis factors [2] |
| DFT (Kinetic Stability) | Absence of imaginary phonon frequencies [9] | 82.2% Accuracy [9] | Computationally expensive; materials with imaginary frequencies can be synthesized [9] |
| SynthNN (Deep Learning) | Data-driven model learning from known compositions [2] | 7x higher precision than DFT formation energy [2] | Requires large datasets; performance depends on data quality and representation |
| CSLLM (Large Language Model) | Fine-tuned LLM on crystal structure data [9] | 98.6% Accuracy [9] | Requires sophisticated text representation of crystal structures; risk of "hallucination" [9] |
Limitations of the Charge-Balancing Principle
Protocol for Charge-Balancing Assessment
Application Note: This protocol outlines the procedure for evaluating the synthesizability of an inorganic crystalline material using the charge-balancing heuristic.
Materials & Reagents:
- Chemical Formula: The stoichiometric composition of the target material.
- Oxidation State Table: A reference of common oxidation states for elements (e.g., O: -2, Alkali metals: +1).
Procedure:
- Assign Oxidation States: For each element in the chemical formula, assign its most common oxidation state.
- Calculate Total Charge: Multiply each oxidation state by its stoichiometric coefficient in the formula and sum the results to obtain the total charge.
- Assess Synthesizability: If the total charge equals zero, the material is predicted to be synthesizable. A non-zero total charge leads to a prediction of non-synthesizability.
Limitations & Data Interpretation: The critical limitation of this method is its extremely low recall. As evidenced in Table 1, this method incorrectly labels a majority of known, synthesized materials as non-synthesizable. Its performance is notably poor even for typically ionic systems like binary cesium compounds, where only 23% are charge-balanced [2]. The method fails because it cannot account for diverse bonding environments (e.g., metallic or covalent bonds) and real-world synthesis conditions that stabilize non-charge-neutral compositions [2].
Workflow and Failure Analysis of Charge-Balancing
The following diagram illustrates the charge-balancing protocol and its primary points of failure when applied to real-world material systems.
Limitations of Density Functional Theory (DFT)
Protocol for DFT-Based Synthesizability Assessment
Application Note: This protocol describes the use of DFT-calculated formation energy and energy above the convex hull to assess thermodynamic stability, a common proxy for synthesizability.
Materials & Reagents:
- Crystal Structure: An initial atomic structure of the target material (e.g., in POSCAR or CIF format).
- DFT Software: A quantum chemistry code (e.g., VASP, Quantum ESPRESSO).
- Computational Resources: High-performance computing (HPC) cluster.
- Reference Database: A database of stable phases (e.g., the Materials Project) to construct the convex hull.
Procedure:
- Structure Relaxation: Perform a full geometry optimization of the target crystal structure using DFT to find its ground state configuration.
- Formation Energy Calculation: Calculate the formation energy (ΔH~f~) of the relaxed structure relative to its constituent elements in their standard states.
- Convex Hull Construction: Compute the phase diagram for the relevant chemical system. The convex hull is defined by the set of thermodynamically stable phases with the lowest formation energies at their specific compositions.
- Energy Above Hull Calculation: Determine the energy above the convex hull (E~hull~) for the target material. This represents the energy difference between the target and the most stable combination of other phases on the convex hull at the same composition.
- Assess Synthesizability: A material is often considered potentially synthesizable if its E~hull~ is below a threshold, commonly 0 eV/atom (truly stable) or a small positive value (e.g., 10-50 meV/atom, metastable).
Limitations & Data Interpretation: While DFT is a powerful and robust electronic structure method [10], its use for synthesizability prediction has profound limitations:
- Incomplete Picture: Thermodynamic stability is a necessary but not sufficient condition for synthesizability. DFT typically overlooks finite-temperature effects, entropic factors, and kinetic barriers that govern synthetic accessibility [11].
- Metastable Materials: Many successfully synthesized materials are metastable (E~hull~ > 0) and are missed by strict hull-based filters [9].
- Functional and Basis Set Dependence: The accuracy of results is highly dependent on the choice of exchange-correlation functional and atomic orbital basis set. Outdated defaults (e.g., B3LYP/6-31G*) can yield poor results, necessitating careful selection of modern, robust method combinations [10].
- Computational Cost: Although faster than high-level wavefunction theories, DFT calculations for complex crystals remain computationally expensive, limiting high-throughput screening [2] [10].
Workflow and Failure Analysis of DFT-Based Assessment
The diagram below outlines the DFT-based assessment workflow and highlights where its fundamental approximations lead to failures in predicting real-world synthesizability.
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Computational and Data Resources for Synthesizability Research
| Item Name | Function/Application | Relevance to SynthNN Development |
|---|---|---|
| ICSD (Inorganic Crystal Structure Database) | Primary source of positive (synthesized) training data [2] [9]. | Provides the foundational dataset of experimentally realized structures for model training. |
| Materials Project / OQMD / JARVIS | Databases of calculated (including theoretical) crystal structures [9] [11]. | Source for generating negative or unlabeled training examples; benchmark for performance. |
| DFT Software (VASP, Quantum ESPRESSO) | Calculates formation energy and energy above hull for stability assessment [2]. | Provides baseline metrics for comparing and validating ML model performance. |
| Atom2Vec / Material String | Learned or engineered representation of chemical compositions or structures [2] [9]. | Converts raw chemical data into a format suitable for deep learning model input. |
| Positive-Unlabeled (PU) Learning Algorithms | Machine learning framework to learn from positive (synthesized) and unlabeled data [2] [9]. | Critical for handling the lack of definitive negative examples in materials data. |
The limitations of traditional charge-balancing and DFT-based methods are both quantitative and fundamental. Charge-balancing acts as an overly restrictive filter, while DFT's thermodynamic focus fails to capture the kinetic and pathway-dependent nature of real-world synthesis. These shortcomings, validated by low precision and accuracy metrics, create a significant bottleneck in computational materials discovery pipelines. It is this precise gap in capability that justifies the development and integration of advanced deep learning models like SynthNN. By learning directly from the full distribution of synthesized materials, SynthNN and subsequent models such as CSLLM internalize complex chemical principles beyond simple heuristics or total energy calculations, thereby offering a more reliable and effective tool for predicting material synthesizability.
A significant challenge in data-driven materials and drug discovery is the inherent bias in available data. Public databases are overwhelmingly populated with successful synthesis reports, while data on failed attempts are rarely published. This creates a "data problem" where machine learning models must learn the concept of synthesizability—whether a material or compound can be successfully synthesized—from only positive examples and artificially generated negatives. Within the context of SynthNN deep learning model research, addressing this data imbalance is crucial for developing accurate synthesizability predictors. This application note details the methodologies, protocols, and computational tools required to construct effective training datasets and models under these constrained data conditions, with applications spanning both inorganic crystalline materials and organic compound synthesis.
Dataset Construction Methods
Constructing representative datasets for synthesizability prediction requires careful consideration of data sources, labeling strategies, and augmentation techniques. The approaches vary between domains but share common principles for handling positive-unlabeled learning scenarios.
Table 1: Primary Data Sources for Synthesizability Prediction
| Data Type | Source Name | Content Description | Domain |
|---|---|---|---|
| Positive Examples | Inorganic Crystal Structure Database (ICSD) | Experimentally synthesized inorganic crystalline materials [2] | Materials Science |
| Positive Examples | ChEMBL, ZINC15 | Commercially available or synthesized molecules [12] | Drug Discovery |
| Theoretical Structures | Materials Project, OQMD, JARVIS | Computationally predicted structures [9] | Materials Science |
| Artificial Negatives | GDBChEMBL, Nonpher | Computationally generated unsynthesized molecules [12] | Drug Discovery |
| Text-Mined Synthesis Data | Literature-extracted datasets | Synthesis parameters extracted from scientific articles [13] | Cross-Domain |
Positive-Unlabeled Learning Frameworks
The core challenge in synthesizability prediction is the lack of verified negative examples. Positive-unlabeled learning provides a principled framework for this scenario, where models are trained using confirmed positive samples and "unlabeled" samples that are treated as potential negatives.
The SynthNN model addresses this through a semi-supervised approach that treats unsynthesized materials as unlabeled data and probabilistically reweights these materials according to their likelihood of being synthesizable [2]. This approach falls under the broader category of positive-unlabeled learning algorithms, which have been successfully applied to predict synthesizability across various domains:
- 2D MXenes and 3D crystals: Transductive bagging PU learning approaches have achieved over 75% and 87.9% accuracy respectively [9]
- General perovskites: Inductive PU learning with domain-specific transfer learning has demonstrated superior performance compared to tolerance factor-based approaches [13]
- Ternary oxides: Human-curated literature data enables PU learning models to predict solid-state synthesizability [13]
In the drug discovery domain, DeepSA addresses similar challenges by training on molecules labeled by retrosynthetic analysis, where compounds requiring ≤10 synthetic steps are considered easy-to-synthesize (ES) and those requiring >10 steps or failing route prediction are labeled hard-to-synthesize (HS) [12].
SynthNN Architecture and Implementation
Model Framework
The SynthNN framework implements a deep learning approach to synthesizability prediction that leverages the entire space of synthesized inorganic chemical compositions. Key architectural components include:
- Atom2Vec Representation: Chemical formulas are represented by a learned atom embedding matrix optimized alongside all other parameters of the neural network [2]
- Domain Adaptation: The model learns chemical principles of charge-balancing, chemical family relationships, and ionicity directly from data without prior chemical knowledge [2]
- Positive-Unlabeled Training: Artificially generated unsynthesized materials are treated as unlabeled data and probabilistically reweighted during training
The model reformulates material discovery as a synthesizability classification task, enabling identification of synthesizable materials with 7× higher precision than DFT-calculated formation energies and outperforming human experts by 1.5× higher precision with completion rates five orders of magnitude faster [2].
Advanced Architectures
Recent advancements have extended beyond SynthNN's composition-based approach. The Crystal Synthesis Large Language Models framework utilizes three specialized LLMs to predict synthesizability, synthetic methods, and suitable precursors for 3D crystal structures [9]. This multi-component architecture achieves state-of-the-art accuracy of 98.6% in synthesizability prediction, significantly outperforming traditional methods based on thermodynamic and kinetic stability [9].
Performance Benchmarks and Validation
Quantitative Performance Metrics
Table 2: Performance Comparison of Synthesizability Prediction Models
| Model Name | Domain | Accuracy | Precision | Key Differentiators |
|---|---|---|---|---|
| SynthNN | Inorganic Crystalline Materials | Not specified | 7× higher than DFT [2] | Composition-based; outperforms human experts |
| CSLLM | 3D Crystal Structures | 98.6% [9] | Not specified | Structure-based; suggests methods & precursors |
| DeepSA | Organic Compounds | 89.6% AUROC [12] | Not specified | SMILES-based; discriminates synthesis difficulty |
| PU Learning (Jang et al.) | Hypothetical Compounds | Not specified | Not specified | CLscore for non-synthesizable identification |
| Solid-state PU Model | Ternary Oxides | Not specified | Not specified | Human-curated literature data |
Experimental Validation Protocols
Validating synthesizability predictions requires rigorous experimental protocols to confirm model accuracy:
Protocol 1: Experimental Synthesis Verification
- Objective: Validate computationally predicted synthesizable materials through laboratory synthesis
- Materials: Predicted synthesizable compounds, precursor materials, synthesis equipment
- Procedure:
- Select high-priority candidates based on synthesizability scores (e.g., RankAvg > 0.95) [11]
- Apply retrosynthetic planning to generate viable precursor combinations
- Balance chemical reactions and compute precursor quantities
- Execute solid-state synthesis using predicted temperature parameters
- Characterize products using X-ray diffraction (XRD) for phase identification
- Validation: Compare XRD patterns with target crystal structures to confirm successful synthesis
Protocol 2: Cross-Database Benchmarking
- Objective: Evaluate model performance across multiple materials databases
- Materials: Structures from Materials Project, GNoME, Alexandria, and ICSD
- Procedure:
- Curate balanced dataset with synthesizable and non-synthesizable examples
- Calculate CLscores for all structures using pre-trained PU learning model [9]
- Set CLscore threshold (e.g., <0.1) for non-synthesizable classification
- Train model on subset and evaluate on held-out test set
- Assess generalization on structures with complexity exceeding training data
Protocol 3: Human Expert Comparison
- Objective: Benchmark model performance against domain experts
- Materials: Set of candidate materials for synthesizability assessment
- Procedure:
- Select diverse set of material compositions for evaluation
- Have domain experts assess synthesizability using traditional methods
- Run model predictions on same material set
- Compare precision, recall, and assessment time
- Statistically analyze performance differences
Computational Implementation Protocols
Data Preprocessing Workflow
Protocol 4: Training Data Preparation
- Input: Raw composition data from ICSD and generated compositions
- Processing Steps:
- Filter compositions by element count (e.g., ≤7 elements) and atom count (e.g., ≤40 atoms) [9]
- Exclude disordered structures to focus on ordered crystal structures
- Convert compositions to atom2vec representations
- Apply standardization to chemical formulas
- Split data into training/validation/test sets (typical ratio: 90/5/5)
- Output: Processed dataset ready for model training
Protocol 5: Negative Example Generation
- Input: Theoretical structures from computational databases
- Processing Steps:
- Collect theoretical structures from Materials Project, OQMD, JARVIS
- Calculate CLscores using pre-trained PU learning model [9]
- Select structures with lowest CLscores (e.g., <0.1) as non-synthesizable examples
- Balance dataset with approximately equal synthesizable and non-synthesizable examples
- Visualize dataset diversity using t-SNE for crystal systems and element distribution
- Output: Balanced dataset for synthesizability classification
Model Training and Optimization
Protocol 6: SynthNN Model Training
- Framework: Deep learning with atom embeddings
- Hyperparameters:
- Atom embedding dimension (optimized during training)
- Neural network architecture (number of layers, neurons)
- Learning rate and optimization algorithm
- Positive-unlabeled weighting parameter (N_synth)
- Batch size and training epochs
- Training Procedure:
- Initialize atom embedding matrix with random weights
- Forward pass: compute synthesizability probability
- Calculate loss with PU-weighted negative examples
- Backpropagate errors and update parameters
- Validate on held-out set and apply early stopping
- Output: Trained SynthNN model for synthesizability prediction
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Resources
| Tool/Resource | Type | Function | Example Sources |
|---|---|---|---|
| ICSD Database | Data Resource | Source of verified synthesizable inorganic materials [2] | FIZ Karlsruhe |
| Materials Project | Data Resource | Source of theoretical structures for negative examples [9] | LBNL |
| atom2vec | Algorithm | Learns optimal representation of chemical formulas [2] | Custom implementation |
| Positive-Unlabeled Learning | Framework | Handles lack of verified negative examples [2] [13] | Various implementations |
| Retrosynthetic Analysis | Software | Generates synthetic routes and identifies precursors [9] | Retro*, AiZynthFinder |
| DFT Calculations | Computational Method | Provides formation energies for stability assessment [2] | VASP, Quantum ESPRESSO |
| XRD Characterization | Experimental Method | Verifies successful synthesis of predicted materials [11] | Laboratory equipment |
| Text-Mining Pipelines | Data Extraction | Extracts synthesis information from literature [13] | Custom NLP pipelines |
Application Protocols
Materials Discovery Pipeline
Protocol 7: Integrated Synthesizability-Guided Discovery
- Objective: Identify novel synthesizable materials computationally and verify experimentally
- Workflow:
- Screen computational databases (e.g., 4.4 million structures) [11]
- Apply synthesizability score (e.g., RankAvg > 0.95) to identify candidates
- Filter by element constraints (exclude platinum group, toxic elements)
- Apply retrosynthetic planning to suggest precursors
- Predict synthesis parameters (temperature, atmosphere)
- Execute high-throughput synthesis
- Characterize products via XRD
- Expected Outcomes: Successful synthesis of 7 out of 16 targets demonstrated in recent studies [11]
Drug Discovery Implementation
Protocol 8: Compound Prioritization for Medicinal Chemistry
- Objective: Identify synthesizable drug candidates from virtually generated compounds
- Workflow:
- Generate candidate compounds using AI-based molecular generation models
- Convert structures to SMILES representation
- Apply synthesizability predictor (e.g., DeepSA, SAscore, SCScore)
- Rank compounds by synthesizability score
- Filter out compounds with synthesizability scores below threshold
- Proceed with experimental synthesis of top candidates
- Validation: Compare predicted synthesizability with actual laboratory synthesis outcomes
The "data problem" in synthesizability prediction—learning from successful syntheses and artificial negatives—represents both a challenge and opportunity in computational materials and drug discovery. The SynthNN framework and related approaches demonstrate that through careful data curation, positive-unlabeled learning strategies, and domain-adapted model architectures, it is possible to develop accurate predictors that significantly accelerate the discovery of novel materials and compounds. The protocols and methodologies outlined in this application note provide researchers with practical tools to implement these approaches in their own workflows, ultimately bridging the gap between computational prediction and experimental realization.
The discovery of novel inorganic crystalline materials is a cornerstone of technological advancement. A critical, unsolved challenge in this field is the reliable prediction of whether a hypothetical chemical composition is synthesizable—that is, synthetically accessible with current capabilities, regardless of whether its synthesis has been reported yet [2]. Traditional proxies for synthesizability, such as charge-balancing rules and density functional theory (DFT)-calculated formation energies, have proven inadequate as they fail to capture the complex and multi-factorial nature of real-world synthesis [2]. The SynthNN deep learning model represents a paradigm shift by leveraging the entire space of known inorganic compositions to directly predict synthesizability, offering a robust, data-driven solution to this complex chemical problem [2].
Core Innovation: Learning Chemistry from Data
SynthNN's foundational innovation lies in its reformulation of material discovery as a synthesizability classification task. Unlike traditional methods that rely on pre-defined chemical rules or thermodynamic calculations, SynthNN employs an atom2vec framework [2]. This approach uses a learned atom embedding matrix that is optimized alongside all other parameters of the neural network.
- Data-Driven Representation: The model learns an optimal representation of chemical formulas directly from the distribution of previously synthesized materials, without requiring prior chemical knowledge or structural information [2].
- Learned Chemical Principles: Experiments indicate that SynthNN autonomously learns fundamental chemical principles from the data, including charge-balancing, chemical family relationships, and ionicity, and utilizes these to generate predictions [2].
Quantitative Performance and Benchmarking
SynthNN's performance was rigorously benchmarked against established computational methods and human experts, demonstrating its significant advantages.
Table 1: Performance Comparison of Synthesizability Prediction Methods
| Method | Key Metric | Performance | Key Advantage |
|---|---|---|---|
| SynthNN | Precision | 7x higher than DFT-based formation energy [2] | Data-driven, learns from all known compositions |
| Charge-Balancing | Coverage of Known Materials | Only 37% of known synthesized materials are charge-balanced [2] | Chemically intuitive but inflexible |
| DFT Formation Energy | Coverage of Synthesized Materials | Captures only ~50% of synthesized inorganic crystalline materials [2] | Accounts for thermodynamics but not kinetics |
| Human Experts | Precision & Speed | 1.5x higher precision; 5 orders of magnitude faster than the best expert [2] | Scalable and consistently high-performing |
Experimental Protocol and Workflow
Implementing SynthNN involves a structured workflow from data preparation to model inference. The following protocol details the key steps.
Data Preparation and Curation
- Positive Data Source: Extract chemical formulas of synthesized inorganic crystalline materials from the Inorganic Crystal Structure Database (ICSD) [2].
- Handling Unlabeled Data: Generate a large set of artificial chemical formulas not present in the ICSD to represent unsynthesized/unsynthesizable materials. This creates a Positive-Unlabeled (PU) learning scenario [2].
- Semi-Supervised Learning: Employ a PU learning algorithm that treats unsynthesized materials as unlabeled data and probabilistically reweights them according to their likelihood of being synthesizable [2].
Model Architecture and Training
- Input Representation: Chemical formulas are fed into the model using the atom2vec framework, which learns a continuous vector representation for each element [2].
- Network Structure: A deep neural network architecture processes the atom embeddings. The specific number of layers and the dimensionality of the embeddings are treated as hyperparameters [2].
- Training Regime: The model is trained on the prepared synthesizability dataset. The ratio of artificially generated formulas to synthesized formulas (referred to as ( N_{synth} )) is a key hyperparameter optimized during training [2].
Model Inference and Screening
- Input: A candidate chemical formula.
- Processing: The model computes a synthesizability score.
- Output: A classification (synthesizable/not synthesizable) or a probability score that can be used to prioritize candidates in large-scale computational screens [2].
Figure 1: The SynthNN development and application workflow, illustrating the flow from data preparation to synthesizability prediction.
Figure 2: The core learning mechanism of SynthNN, demonstrating how chemical principles are derived directly from data.
Table 2: Essential Resources for Synthesizability Prediction Research
| Resource / Tool | Type | Primary Function in Research |
|---|---|---|
| Inorganic Crystal Structure Database (ICSD) | Database | The primary source of positive examples (synthesized materials) for model training [2]. |
| Atom2Vec | Algorithm / Framework | Learns optimal, continuous vector representations of chemical elements directly from data, forming the input layer of SynthNN [2]. |
| Positive-Unlabeled (PU) Learning | Machine Learning Paradigm | A semi-supervised approach that handles the lack of definitive negative data (unsynthesizable materials) by treating them as unlabeled examples [2]. |
| Density Functional Theory (DFT) | Computational Method | Provides thermodynamic stability metrics (e.g., formation energy) used as a baseline for comparing SynthNN's performance [2]. |
| Deep Neural Network (DNN) | Model Architecture | The core classifier that processes atom embeddings to output a synthesizability probability [2]. |
Inside SynthNN: Architecture, Training, and Real-World Deployment
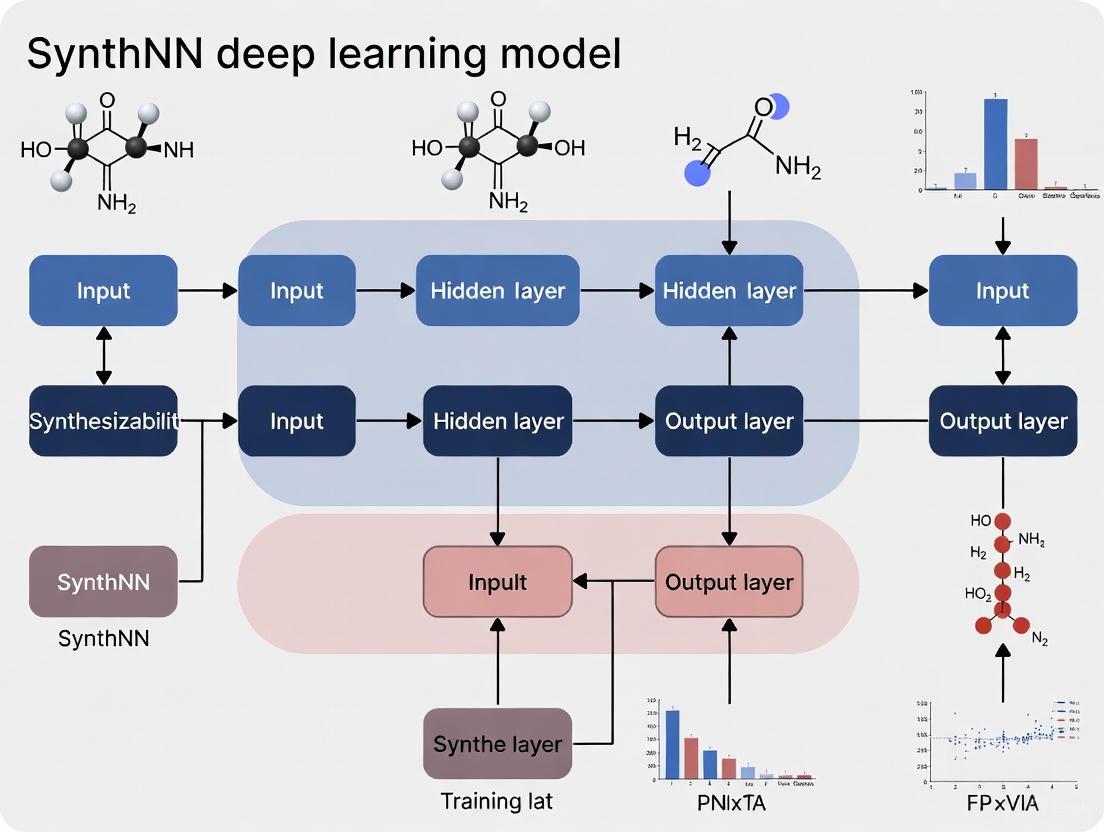
The challenge of predicting whether a hypothetical inorganic crystalline material is synthetically accessible is a fundamental bottleneck in accelerating materials discovery. Traditional computational approaches, such as density functional theory (DFT) calculations of formation energy, serve as imperfect proxies for synthesizability, while the expert judgment of solid-state chemists, though valuable, does not scale for the rapid exploration of vast chemical spaces [2]. The SynthNN deep learning model represents a paradigm shift by directly addressing the synthesizability classification task, achieving a reported 1.5× higher precision than the best human expert and completing the task five orders of magnitude faster [2]. A cornerstone of this model's architecture is its use of learned, distributed representations of atoms, a concept pioneered by the atom2vec embedding framework.
The core analogy behind atom2vec is that "if one may know a word by the company it keeps, then the same might be said of an atom" [14]. Inspired by the Word2Vec algorithm in natural language processing (NLP), atom2vec aims to derive vector representations of atoms that encapsulate their chemical nature and relationships by analyzing their co-occurrence patterns within a large database of known crystal structures [14] [15]. Within the SynthNN architecture, these embeddings are not pre-defined but are learned end-to-end. The model leverages an atom embedding matrix that is optimized alongside all other parameters of the neural network, allowing it to learn the optimal representation of chemical formulas directly from the distribution of previously synthesized materials [2]. This enables SynthNN to infer complex chemical principles such as charge-balancing, chemical family relationships, and ionicity directly from data, without prior explicit programming of these rules [2] [16].
Technical Architecture and Dataflow
The architecture integrating atom2vec principles within a synthesizability prediction model like SynthNN involves a sequential flow from chemical composition to a final synthesizability probability. The following workflow diagram delineates this process.
Diagram 1: High-level dataflow of the SynthNN model, from chemical composition to synthesizability prediction.
Input and Atom Embedding Layer
The model input is a chemical formula, represented as a set of constituent atoms. For instance, the formula "CsCl" would be decomposed into the atoms {Cs, Cl}. In the initial embedding layer, each atom in the periodic table is associated with a dense, continuous vector of a predefined dimensionality d (a model hyperparameter). This layer is implemented as a lookup table, often called an embedding matrix, where the row corresponding to an atom's index is its d-dimensional vector [2].
- Function: This layer converts an atomic symbol (a categorical value) into a numerical, differentiable representation that the neural network can process.
- Initialization: The embedding vectors are typically initialized randomly and are then updated during model training via backpropagation [14] [2].
- Learning Objective: Through training, the model adjusts these vectors so that atoms frequently found in similar chemical environments across the training database have similar vector representations. This process captures latent chemical properties [14] [15].
Compositional Representation via Pooling
A single material composition comprises multiple atoms. To create a fixed-length, composition-level representation from its constituent atom vectors, a pooling operation is applied. This step is analogous to forming a sentence representation from its constituent word vectors in NLP [14].
Common pooling strategies include:
- Sum Pooling: The vectors of all atoms in the formula are summed element-wise.
- Average Pooling: The vectors of all atoms are averaged element-wise.
For a formula like "SiO₂", the pooling layer would execute an operation such as vec(Si) + 2 * vec(O), where vec() denotes the embedding lookup. The resulting pooled vector is a single, d-dimensional representation of the entire chemical formula, which is then passed to downstream neural network layers [14] [2].
Classification Backbone
The pooled compositional representation is fed into a standard multilayer perceptron (MLP), which consists of a series of fully connected (dense) layers with non-linear activation functions (e.g., ReLU, sigmoid). This MLP acts as the classifier, learning the complex, non-linear mapping between the composed material representation and its probability of being synthesizable [2].
The final layer typically uses a sigmoid activation function to output a value between 0 and 1, interpreted as the probability that the input chemical formula is synthesizable. During training, a decision threshold (e.g., 0.5) is applied to this probability to make a binary classification, and the model's weights—including the entire embedding matrix—are updated to minimize the classification error on the training data [16].
Key Variants of Atomic Embeddings
The foundational atom2vec concept has been extended in several ways. The table below summarizes the prominent unsupervised approaches for generating distributed atomic representations.
Table 1: Comparison of Key Atomic Embedding Techniques
| Method | Core Data Source | Learning Algorithm | Key Principle |
|---|---|---|---|
| Atom2Vec [15] | Database of material compositions & structures | Matrix Factorization (SVD) | Derives atom vectors from a co-occurrence matrix of atoms and their chemical environments. |
| Mat2Vec [14] | Scientific text (abstracts from materials science literature) | Word2Vec (Skip-gram) | Learns atom representations from their context in millions of scientific abstracts. |
| SkipAtom [14] | Crystal structure graphs from materials databases | Skip-gram with Negative Sampling | Predicts neighboring atoms in a crystal structure graph to learn atom embeddings. |
The SkipAtom variant is of particular note for structural property prediction. It explicitly models a crystal structure as a graph, where atoms are nodes and bonds are edges. The unsupervised learning task is formulated to maximize the log-probability of predicting a context atom given a target atom within the same local structural environment [14]. The objective function is:
[ \frac{1}{|M|} \sum{m\in M}\sum{a\in Am}\sum{n\in N(a)}\log p(n|a) ]
Here, (M) is the set of materials, (A_m) is the set of atoms in material (m), and (N(a)) are the neighbors of atom (a) in the structure graph. The probability (p(n|a)) is typically computed using a softmax function over the inner product of the target and context atom vectors [14].
Experimental Protocol and Benchmarking
Model Training and Data Curation for SynthNN
The development of a synthesizability prediction model like SynthNN requires a specific dataset and a tailored training protocol to handle the inherent lack of confirmed negative examples.
Table 2: SynthNN Performance at Different Decision Thresholds [16]
| Decision Threshold | Precision | Recall |
|---|---|---|
| 0.10 | 0.239 | 0.859 |
| 0.20 | 0.337 | 0.783 |
| 0.30 | 0.419 | 0.721 |
| 0.40 | 0.491 | 0.658 |
| 0.50 | 0.563 | 0.604 |
| 0.60 | 0.628 | 0.545 |
| 0.70 | 0.702 | 0.483 |
| 0.80 | 0.765 | 0.404 |
| 0.90 | 0.851 | 0.294 |
Dataset Construction:
- Positive Examples: Sourced from the Inorganic Crystal Structure Database (ICSD), which contains compositions of known, synthesized crystalline inorganic materials [2] [16].
- Negative Examples: Artificially generated from a vast space of plausible but unsynthesized chemical compositions. This creates a Positive-Unlabeled (PU) learning scenario, as some "negative" examples could be synthesizable but are simply absent from the ICSD [2].
Training Protocol (PU Learning):
- The model is trained on a mixture of positive (synthesized) and artificially generated negative examples.
- A semi-supervised learning approach is employed, which treats the artificially generated materials as unlabeled data. These examples are probabilistically reweighted according to their likelihood of being synthesizable to account for the incomplete labeling [2].
- A key hyperparameter is (N_{synth}), the ratio of artificially generated formulas to synthesized formulas used during training [2].
Evaluation:
- Model performance is benchmarked against baselines like random guessing and the charge-balancing rule.
- As shown in Table 2, the choice of decision threshold on the output probability allows for a precision-recall trade-off tailored to the specific application (e.g., high-recall for broad screening vs. high-precision for candidate selection) [16].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Resources for Implementing and Experimenting with atom2vec and SynthNN
| Resource | Function/Description | Example/Reference |
|---|---|---|
| Materials Databases | Provides structured data on crystal structures and compositions for training embedding models. | Inorganic Crystal Structure Database (ICSD) [2] [16], Materials Project [17] |
| Local Environment Analysis | Algorithm for identifying coordination environments and structure motifs (e.g., octahedra, tetrahedra) in crystal structures. | Implementation in pymatgen [17] |
| Graph Construction Algorithm | Method to convert a crystal structure into a graph of atomic connections for models like SkipAtom. | Voronoi decomposition with solid angle weights [14] |
| Positive-Unlabeled Learning Algorithm | A semi-supervised learning framework to handle datasets without confirmed negative examples. | Class-weighting of unlabeled examples [2] |
| Pre-trained Models & Code | Provides a starting point for prediction and further model development. | Official SynthNN GitHub Repository [16] |
Advanced Architectural Evolutions
The principle of using learned embeddings for fundamental units has inspired architectures beyond simple compositional models. A significant advancement is the Atom-Motif Dual Graph Network (AMDNet), which incorporates higher-order building blocks into the graph representation [17].
Whereas atom-based graph networks represent crystals as graphs with atoms as nodes, AMDNet introduces structure motifs—such as SiO₄ tetrahedra or MnO₆ octahedra—as additional nodes. This creates a dual graph where motif nodes and atom nodes are connected, allowing the graph neural network to explicitly process both atomic and supra-atomic structural information. This motif-centric approach has been shown to improve the prediction of electronic properties like band gaps, demonstrating the value of embedding and combining multi-scale features [17]. The following diagram illustrates this enhanced architecture.
Diagram 2: The Atom-Motif Dual Graph Network (AMDNet) architecture, which incorporates structure motifs as explicit nodes in the graph to enhance predictive performance for electronic properties.
The discovery of novel, synthesizable materials is a fundamental driver of innovation across numerous scientific and industrial fields. However, the challenge of reliably predicting whether a hypothetical inorganic crystalline material is synthetically accessible has long hindered autonomous materials discovery. Traditional approaches, such as density-functional theory (DFT) calculations for thermodynamic stability or the enforcement of charge-balancing rules, have proven insufficient, capturing only a fraction of synthesized materials [2]. This application note details the methodology for developing a deep learning synthesizability model (SynthNN) that overcomes these limitations by integrating the Inorganic Crystal Structure Database (ICSD) with a Positive-Unlabeled (PU) Learning framework. This protocol is designed for researchers and scientists engaged in computational materials discovery and drug development, providing a robust workflow for identifying synthetically accessible candidates with high precision [2].
Core Components and Research Reagents
The experimental framework relies on several key "research reagents" – critical datasets, software tools, and algorithms. The table below catalogues these essential components.
Table 1: Key Research Reagents and Solutions
| Reagent/Solution | Type | Primary Function | Key Specifications |
|---|---|---|---|
| ICSD [18] [19] | Database | Serves as the authoritative source of positive (synthesized) material examples. | >210,000 entries; data from 1913 onwards; ~12,000 new entries annually. |
| Atom2Vec [2] | Algorithm | Generates optimal vector representations (embeddings) of chemical formulas directly from data. | Learned embedding dimensionality is a key hyperparameter. |
| PU Learning Framework [2] [20] | Machine Learning Paradigm | Enables model training using only positive (ICSD) and unlabeled (generated) examples. | Handles lack of confirmed negative data; employs semi-supervised class-weighting. |
| SynthNN Model [2] | Deep Learning Architecture | The core classifier that predicts the synthesizability of a given inorganic chemical formula. | A neural network that leverages atom embeddings and operates without structural input. |
| Artificially Generated Formulas | Dataset | Creates a pool of "unlabeled" examples, representing potentially unsynthesizable compositions. | The ratio of generated formulas to ICSD formulas ( ( N_{synth} ) ) is a critical hyperparameter. |
Workflow and Signaling Pathways
The following workflow diagram outlines the logical sequence and data flow for training the SynthNN model, from data acquisition to final model deployment.
Diagram 1: SynthNN training and deployment workflow.
Detailed Experimental Protocols
Protocol: Curation of the Positive Dataset from ICSD
Objective: To extract a high-quality set of synthesized inorganic crystalline materials to serve as positive examples for model training.
- Data Source Access: Obtain a subscription to the ICSD, available via FIZ Karlsruhe or the National Institute of Standards and Technology (NIST) [18] [19].
- Data Extraction: Download the entire database or a curated subset. The database contains over 210,000 entries, with all important crystal structure data, including unit cell parameters, space group, and complete atomic parameters [18].
- Data Parsing: Extract the chemical composition (molecular formula) for each entry. For the initial SynthNN model, structural information is not required as input [2].
- Quality Control: Rely on the ICSD's internal quality assurance processes, which involve thorough checks and continuous updates to modify, supplement, or remove duplicates [18]. The processed list of chemical formulas constitutes the set of Positive Examples.
Protocol: Generation of the Unlabeled Dataset
Objective: To create a large and diverse set of chemical formulas that represent the space of potentially unsynthesizable materials.
- Strategy: Artificially generate a vast number of plausible inorganic chemical formulas that are not present in the ICSD. The ratio of these generated formulas ((N_{synth})) to the positive ICSD formulas is a critical hyperparameter [2].
- Considerations: It is crucial to recognize that this unlabeled set is not a pure collection of negative examples. It will contain some materials that are synthesizable but have not been reported in the ICSD, or that have yet to be synthesized. This is the core challenge that PU learning is designed to address [2].
Protocol: Implementation of the PU Learning Algorithm
Objective: To train a classifier that distinguishes synthesizable materials from the unlabeled pool, accounting for the ambiguous nature of the unlabeled data.
- Feature Representation: Employ the atom2vec algorithm to convert each chemical formula into a numerical vector. The dimensionality of this learned representation is a key hyperparameter to be optimized [2].
- Model Architecture: Construct a deep neural network (SynthNN) that takes the atom2vec representations as input. The specific architecture (number of layers, nodes, activation functions) must be defined and tuned.
- PU Training Loop:
- The model is trained on the combined set of positive (ICSD) and unlabeled (generated) examples.
- Implement a semi-supervised learning approach that treats the unlabeled materials as having uncertain labels. The algorithm probabilistically reweights these examples during training according to their likelihood of being synthesizable [2].
- This process allows SynthNN to learn the underlying "chemistry of synthesizability"—such as charge-balancing principles, chemical family relationships, and ionicity—directly from the data distribution, without explicit human-defined rules [2].
Protocol: Model Benchmarking and Performance Evaluation
Objective: To quantitatively assess the performance of the trained SynthNN model against established baselines and human expertise.
- Establish Baselines: Compare SynthNN against two primary baselines:
- Random Guessing: Predictions weighted by class imbalance.
- Charge-Balancing: Predicts a material as synthesizable only if it is charge-balanced according to common oxidation states [2].
- Define Metrics: Calculate standard binary classification metrics, including Precision, Recall, and F1-score. Due to the PU learning context, the F1-score is particularly informative [2].
- Human Expert Comparison: Conduct a head-to-head discovery task where the model's candidate materials are compared against those identified by a panel of expert material scientists [2].
- Quantitative Analysis: The performance data should be summarized in a clear table for easy comparison.
Table 2: Performance Benchmarking of Synthesizability Prediction Methods
| Method | Key Principle | Precision | Relative Speed | Key Limitation |
|---|---|---|---|---|
| Charge-Balancing [2] | Net neutral ionic charge | Low (23-37% of known compounds) | Fast | Inflexible; fails for metallic/covalent materials. |
| DFT Formation Energy [2] | Thermodynamic stability | 1x (Baseline) | Slow (Calculation intensive) | Fails to account for kinetic stabilization. |
| Human Expert [2] | Specialized domain knowledge | 1x (Baseline) | 1x (Baseline) | Limited to narrow chemical domains. |
| SynthNN (PU Learning) [2] | Data-driven classification from ICSD | 7x higher than DFT; 1.5x higher than human experts | 100,000x faster than human experts | Requires a robust database like ICSD. |
The integration of the ICSD with a Positive-Unlabeled learning framework provides a powerful and efficient pipeline for predicting the synthesizability of inorganic crystalline materials. This protocol outlines a data-driven approach that surpasses traditional physical proxies and human intuition in both precision and speed. By following these application notes, researchers can implement and refine SynthNN-type models, thereby significantly enhancing the reliability of computational material screening and accelerating the discovery of novel, synthetically accessible materials.
The SynthNN model represents a significant methodological shift in predicting the synthesizability of inorganic crystalline materials by relying exclusively on chemical composition data, completely bypassing the need for atomic structural information [2]. This approach reformulates material discovery as a synthesizability classification task, leveraging the entire space of synthesized inorganic chemical compositions to generate predictions [2]. By operating solely on compositional data, SynthNN addresses a critical bottleneck in computational materials screening: the unavailability of precise crystal structures for hypothetical or yet-to-be-discovered materials. This capability is particularly valuable for high-throughput virtual screening of novel material compositions where structural details remain unknown, enabling researchers to prioritize synthetic efforts toward the most promising candidates before investing resources in structural determination or prediction.
Technical Implementation and Architecture
Atom2Vec Representation Framework
SynthNN employs a deep learning architecture based on the atom2vec framework, which represents each chemical formula through a learned atom embedding matrix that is optimized alongside all other neural network parameters [2]. This approach automatically learns optimal representations of chemical formulas directly from the distribution of previously synthesized materials without requiring pre-defined feature engineering. The dimensionality of this representation is treated as a hyperparameter determined during model development [2]. Notably, this method requires no prior chemical knowledge or assumptions about factors influencing synthesizability, as the underlying "chemistry" of synthesizability is learned entirely from the data of experimentally realized materials. The model demonstrates an ability to learn fundamental chemical principles including charge-balancing, chemical family relationships, and ionicity from composition data alone, utilizing these learned principles to generate synthesizability predictions [2].
Positive-Unlabeled Learning Methodology
A fundamental challenge in synthesizability prediction is the lack of confirmed negative examples (definitively unsynthesizable materials) in scientific literature. SynthNN addresses this through a semi-supervised positive-unlabeled (PU) learning approach that treats potentially synthesizable but unsynthesized materials as unlabeled data and probabilistically reweights them according to their likelihood of being synthesizable [2]. The training dataset is constructed from the Inorganic Crystal Structure Database (ICSD) for positive examples (confirmed synthesized materials), augmented with artificially generated unsynthesized materials. The ratio of artificially generated formulas to synthesized formulas used in training is a key model hyperparameter (N_synth) [2]. This methodology allows SynthNN to effectively learn from incomplete labeling, a common scenario in materials informatics where negative examples are rarely documented.
Comparative Performance Analysis
Quantitative Performance Metrics
The performance of SynthNN has been systematically evaluated against traditional computational methods and human experts, demonstrating significant advantages in both accuracy and efficiency.
Table 1: Performance Comparison of Synthesizability Assessment Methods
| Method | Precision | Key Advantages | Computational Requirements |
|---|---|---|---|
| SynthNN (Composition-Only) | 7× higher than DFT formation energy [2] | No structural data needed; high throughput | Computationally efficient for screening billions of candidates [2] |
| DFT Formation Energy | ~50% of synthesized materials captured [2] | Well-established physical basis | Computationally intensive; requires structural data |
| Charge-Balancing Approach | Only 37% of known compounds correctly identified [2] | Simple heuristic; no computation | Minimal computation but poor accuracy |
| Human Experts | 1.5× lower precision than SynthNN [2] | Domain knowledge application | Time-consuming; limited to specialized domains |
Advantages Over Structure-Dependent Approaches
The composition-only approach of SynthNN provides several distinct advantages over structure-dependent methods. By eliminating the requirement for atomic coordinates, space group information, and lattice parameters, SynthNN can evaluate materials for which no structural data exists, including completely novel compositions outside existing structural databases [2]. This capability is particularly valuable for exploring uncharted regions of chemical space where structural analogs are unavailable. Additionally, the computational efficiency of composition-based screening enables evaluation of billions of candidate materials, a scale impractical for structure-based methods that typically require resource-intensive density functional theory calculations [2]. In direct benchmarking against expert materials scientists, SynthNN achieved 1.5× higher precision while completing the classification task five orders of magnitude faster than the best human expert [2].
Experimental Validation Protocols
Model Training and Validation Workflow
The experimental protocol for developing and validating SynthNN follows a structured workflow to ensure robust performance evaluation and minimize overfitting.
Performance Benchmarking Protocol
To ensure meaningful evaluation, SynthNN undergoes rigorous benchmarking against multiple established methods following a standardized protocol:
- Dataset Preparation: Curate a balanced dataset containing known synthesized materials from ICSD and artificially generated unsynthesized compositions [2]
- Baseline Establishment: Implement random guessing and charge-balancing baselines for performance comparison [2]
- Expert Comparison: Conduct head-to-head material discovery comparison against domain experts with timing and precision metrics [2]
- Statistical Validation: Calculate standard performance metrics including precision, recall, and F1-score with appropriate adjustments for PU learning scenarios [2]
- Cross-Validation: Employ spatial k-fold cross-validation techniques to ensure model generalizability and avoid overfitting [11]
This comprehensive validation strategy ensures that performance claims are statistically robust and comparable across different synthesizability assessment methods.
Research Reagent Solutions
Table 2: Essential Research Materials and Computational Tools for Synthesizability Prediction
| Resource/Tool | Function | Application Context |
|---|---|---|
| Inorganic Crystal Structure Database (ICSD) | Source of positive training examples [2] | Provides confirmed synthesized materials for model training |
| Atom2Vec Framework | Composition representation learning [2] | Converts chemical formulas to optimized feature representations |
| Positive-Unlabeled Learning Algorithms | Handling of unlabeled negative examples [2] | Manages lack of confirmed unsynthesizable materials in literature |
| Deep Learning Framework (e.g., TensorFlow, PyTorch) | Neural network implementation [2] | Enables model architecture development and training |
| High-Performance Computing (HPC) Resources | Training and inference acceleration [2] | Facilitates screening of billions of candidate compositions |
Integration with Materials Discovery Workflows
Screening Pipeline Implementation
The composition-only approach of SynthNN enables seamless integration into computational materials screening pipelines, providing an efficient filter to prioritize candidates for further investigation.
Complementary Role in Multi-Stage Screening
While SynthNN operates exclusively on composition data, it serves as a critical first-pass filter in multi-stage materials discovery pipelines. Compositions flagged as highly synthesizable by SynthNN can be prioritized for subsequent computational and experimental validation, including:
- Crystal structure prediction algorithms
- Density functional theory calculations for property assessment
- Experimental synthesis planning and execution This integrated approach maximizes resource efficiency by focusing expensive computational and experimental resources on the most promising candidates identified through initial composition-based screening [2].
Limitations and Boundary Conditions
The composition-only approach of SynthNN, while broadly applicable, exhibits specific limitations that researchers should consider when implementing this methodology. The model cannot differentiate between different polymorphs of the same chemical composition, as it lacks structural information that would distinguish between alternative crystal arrangements [2]. This limitation becomes significant when synthesizability depends on specific structural features rather than overall composition. Additionally, while SynthNN learns chemical principles like charge balancing from data, its predictions remain constrained by the distribution of materials in its training dataset, potentially limiting extrapolation to completely novel chemical spaces without structural analogs in existing databases. Nevertheless, for high-throughput screening of novel compositions where structural data is unavailable, SynthNN provides an unparalleled advantage in identifying synthesizable candidates for further investigation.
The accelerating capability of computational models to design novel functional materials has starkly revealed a critical bottleneck: the profound difficulty of predicting whether a theoretically proposed material can be successfully synthesized in a laboratory. Traditional computational screening methods have heavily relied on thermodynamic stability metrics, particularly the energy above the convex hull (Ehull), as a proxy for synthesizability. However, synthesis is a complex process governed by kinetic pathways, precursor selection, and experimental conditions that extend far beyond thermodynamic equilibrium. This limitation has created a formidable barrier to the experimental realization of computationally discovered materials, necessitating a paradigm shift toward data-driven synthesizability prediction.
The development of the SynthNN deep learning model represents a significant advancement in this domain. By training directly on the distribution of known synthesized compositions from the Inorganic Crystal Structure Database (ICSD), SynthNN learns the complex chemical principles that influence synthesizability without relying on predefined rules or structural information [2]. This approach reformulates material discovery as a synthesizability classification task, enabling the identification of synthesizable materials with 7× higher precision than traditional formation energy calculations and outperforming human experts by achieving 1.5× higher precision in significantly less time [2].
Integrating synthesizability prediction early in the discovery workflow is particularly crucial for inverse design applications, where generative models produce novel material structures optimized for specific properties. Without synthesizability constraints, these generated materials often remain theoretical curiosities. This application note details protocols for embedding SynthNN and related synthesizability models into end-to-end computational workflows, bridging the gap between virtual screening and experimental realization.
Computational Frameworks for Synthesizability Prediction
Model Architectures and Performance Benchmarks
Current synthesizability prediction frameworks employ diverse architectural approaches, each with distinct advantages for integration into discovery pipelines. The table below summarizes the quantitative performance of leading models.
Table 1: Performance Comparison of Synthesizability Prediction Models
| Model Name | Input Type | Architecture | Key Performance Metric | Reference |
|---|---|---|---|---|
| SynthNN | Chemical composition | Deep learning (atom2vec) | 7× higher precision than DFT formation energy | [2] |
| CSLLM | Crystal structure | Fine-tuned Large Language Model | 98.6% accuracy, outperforms Ehull (74.1%) and phonon stability (82.2%) | [9] |
| PU Learning Model | Crystal structure | Positive-unlabeled learning | Generates CLscore for synthesizability; used to curate negative samples | [9] |
| InvDesFlow-AL | Crystal structure | Active learning-based diffusion model | Identifies 1,598,551 materials with Ehull < 50 meV/atom | [21] |
The exceptional performance of CSLLM demonstrates how large language models, when fine-tuned on comprehensive crystallographic data, can achieve unprecedented accuracy in synthesizability classification. This model utilizes a specialized text representation of crystal structures—termed "material string"—that encodes essential lattice, composition, atomic coordinate, and symmetry information in a format amenable to LLM processing [9]. This approach has shown remarkable generalization capability, maintaining 97.9% prediction accuracy even for complex structures with large unit cells that considerably exceed the complexity of its training data [9].
Workflow Integration Strategies
The integration of synthesizability prediction into computational workflows follows two principal paradigms: sequential filtering and embedded constraint. In the sequential approach, virtual screening generates candidate materials based on target properties, after which synthesizability filters (like SynthNN or CSLLM) prioritize candidates for experimental validation. This method benefits from modularity, allowing independent improvement of property prediction and synthesizability models.
In contrast, the embedded constraint approach incorporates synthesizability directly into the objective function of generative models. The InvDesFlow-AL framework exemplifies this strategy through its active learning cycle, where a generative model produces candidate structures that undergo DFT relaxation and synthesizability assessment [21]. The most promising candidates are then used to iteratively refine the generative model, gradually steering it toward regions of chemical space rich in synthesizable, high-performance materials. This tight integration has demonstrated remarkable success in inverse design tasks, notably identifying Li2AuH6 as a conventional BCS superconductor with an ultra-high transition temperature of 140 K [21].
Diagram 1: Integrated discovery workflow with synthesizability prediction. The workflow combines generative design and high-throughput screening, with synthesizability assessment acting as a critical gate before computationally intensive DFT validation.
Application Protocols for Discovery Workflows
Protocol 1: Virtual Screening with Synthesizability Filtering
This protocol details a sequential workflow for large-scale virtual screening of material databases, incorporating synthesizability as a critical filtering step.
Materials and Computational Resources:
- Starting Database: Materials Project, OQMD, JARVIS, or other crystallographic databases
- Property Prediction Models: Graph neural networks or other machine learning models for target properties
- Synthesizability Model: Pre-trained SynthNN (for composition) or CSLLM (for full structure)
- Computing Infrastructure: High-performance computing cluster for parallel screening
Procedure:
- Define Target Properties: Establish quantitative criteria for the desired material functionality (e.g., band gap range, formation energy, specific conductivity).
- Initial Database Filtering: Apply basic filters based on composition, element count, or structural complexity to reduce the search space to manageable dimensions.
- Property Prediction: Apply specialized property prediction models to identify candidates meeting target specifications. For composition-based screening without structural information, use models trained exclusively on compositional features.
- Synthesizability Assessment: Apply synthesizability classification to property-optimized candidates:
- For composition-based assessment using SynthNN:
- Input chemical formulas into the pre-trained model
- Retrieve synthesizability probability scores
- Apply threshold (typically >0.5) to identify synthesizable candidates [2]
- For structure-based assessment using CSLLM:
- Convert crystal structures to "material string" representation
- Process through fine-tuned LLM
- Classify based on output probabilities with >98% reported accuracy [9]
- For composition-based assessment using SynthNN:
- Priority Ranking: Combine property optimization and synthesizability scores to generate a ranked candidate list for experimental pursuit.
Validation: In benchmark studies, this approach identified synthesizable materials with 7× higher precision than screening based solely on DFT-calculated formation energies [2].
Protocol 2: Inverse Design with Embedded Synthesizability Constraints
This protocol implements an active learning framework where synthesizability is directly embedded into the generative process, enabling inverse design of novel, synthesizable materials.
Materials and Computational Resources:
- Generative Model: Diffusion model (e.g., InvDesFlow-AL), variational autoencoder, or other structure-generating architecture
- Property Prediction: Differentiable property predictors for guidance during generation
- Synthesizability Model: Differentiable synthesizability estimator or surrogate model
- DFT Calculator: For final validation of generated structures
Procedure:
- Model Initialization: Pre-train a generative model on a broad database of known inorganic crystals (e.g., Alex-MP-20, GNoME) to learn fundamental chemical and structural principles [21].
- Conditional Generation: Generate candidate structures conditioned on target properties through:
- Conditional diffusion processes with property-based guidance
- Latent space optimization toward property maxima
- Incorporation of synthesizability as an additional conditioning parameter
- Active Learning Cycle:
- Generate batch of candidate structures (100-1000)
- Evaluate synthesizability using CSLLM or related models
- Select top candidates for DFT validation (formation energy, Ehull, phonon stability)
- Augment training data with validated structures
- Fine-tune generative model on expanded dataset
- Iterative Refinement: Repeat the active learning cycle for multiple rounds (typically 3-10), progressively steering generation toward synthesizable, high-performance regions of chemical space.
- Experimental Prioritization: Select final candidates based on combined assessment of target properties, synthesizability scores, and thermodynamic stability metrics.
Validation: The InvDesFlow-AL implementation of this protocol successfully identified 1,598,551 materials with Ehull < 50 meV/atom after DFT structural relaxation, confirming their thermodynamic stability [21].
Essential Research Reagent Solutions
The computational tools and resources essential for implementing these workflows function as the "research reagents" of digital materials discovery. The following table details these critical components and their functions.
Table 2: Essential Computational Research Reagents for Discovery Workflows
| Reagent / Resource | Type | Function in Workflow | Access / Implementation |
|---|---|---|---|
| Pre-trained SynthNN | Deep Learning Model | Composition-based synthesizability prediction for rapid screening | Available from original publication or reimplementation [2] |
| CSLLM Framework | Fine-tuned LLM | Structure-based synthesizability classification with >98% accuracy | Custom implementation following published architecture [9] |
| ICSD Database | Data Resource | Source of confirmed synthesizable structures for training and benchmarking | Commercial license required [2] [9] |
| "Material String" Representation | Data Format | Text-based crystal structure encoding for LLM processing | Custom implementation from published specifications [9] |
| DPA-2 Interatomic Potential | Machine Learning Potential | DFT-accurate structural relaxation with reduced computational cost | Open-source packages (e.g., DeePMD-kit) [21] |
| Alex-MP-20 / GNoME Datasets | Training Data | Large-scale inorganic material datasets for model pre-training | Publicly available from respective sources [21] |
| Positive-Unlabeled (PU) Learning | Algorithmic Framework | Handling unlabeled negative samples during model training | Custom implementation from published methods [2] [9] |
Workflow Visualization and Decision Pathways
The integration of synthesizability prediction creates critical decision branches throughout the discovery pipeline. The following diagram maps these decision points and their influence on candidate progression.
Diagram 2: Decision pathway for candidate materials. Synthesizability prediction acts as a critical gate between property assessment and computationally intensive DFT validation, efficiently prioritizing experimental candidates.
The integration of synthesizability prediction models like SynthNN and CSLLM into computational discovery workflows represents a transformative advancement in materials informatics. By providing accurate assessment of synthetic accessibility directly from composition or structure, these models bridge the critical gap between theoretical prediction and experimental realization. The protocols outlined herein provide actionable frameworks for implementing synthesizability constraints in both virtual screening and inverse design paradigms.
Future developments in this domain will likely focus on several key areas: (1) integration of synthesis route prediction directly into generative models, building on the precursor identification capabilities demonstrated by CSLLM; (2) development of multi-fidelity models that incorporate both computational and experimental synthesis data; and (3) creation of unified frameworks that simultaneously optimize for target properties, synthesizability, and processability. As these capabilities mature, the integration of synthesizability prediction will evolve from a filtering step to a fundamental design constraint, ultimately enabling the direct computational design of materials that are not only high-performing but also readily realizable in the laboratory.
The acceleration of materials discovery through computational screening is often hindered by a significant bottleneck: the synthesizability of predicted crystal structures. Density functional theory (DFT) methods, while accurate for calculating zero-Kelvin formation energies, frequently identify low-energy structures that are not experimentally accessible [22] [23]. This case study details the implementation and experimental validation of a synthesizability-guided discovery pipeline, built upon the SynthNN deep learning model, which successfully bridged this gap between computational prediction and laboratory synthesis. The pipeline leveraged a combined compositional and structural synthesizability score to evaluate hypothetical materials from major databases, leading to the successful synthesis of 7 out of 16 targeted compounds in just three days [22] [23].
Computational Foundation: The SynthNN Model
The core of the predictive pipeline is SynthNN, a deep-learning classification model designed to predict the synthesizability of inorganic chemical formulas directly from composition, without requiring structural information [2] [16].
Model Architecture and Training
SynthNN was developed using a framework that leverages the entire space of synthesized inorganic chemical compositions. Its key architectural and training components are summarized below.
- Representation Learning: The model uses an
atom2vecrepresentation, which learns an optimal embedding for each element directly from the distribution of synthesized materials. This embedding matrix is optimized alongside all other parameters of the neural network, allowing the model to infer the chemical principles of synthesizability without prior chemical knowledge [2]. - Positive-Unlabeled Learning: A major challenge is the lack of confirmed "unsynthesizable" materials in literature. SynthNN addresses this with a semi-supervised Positive-Unlabeled (PU) learning approach. It is trained on positive examples from the Inorganic Crystal Structure Database (ICSD) and a large number of artificially generated "unsynthesized" compositions, which are treated as unlabeled data and probabilistically reweighted [2].
- Performance: In benchmarks, SynthNN identified synthesizable materials with 7x higher precision than DFT-calculated formation energies. In a head-to-head discovery task against 20 expert material scientists, SynthNN achieved 1.5x higher precision and completed the task five orders of magnitude faster than the best human expert [2].
Table 1: SynthNN Performance at Different Prediction Thresholds (on a dataset with a 20:1 ratio of unsynthesized:synthesized examples) [16]
| Threshold | Precision | Recall |
|---|---|---|
| 0.10 | 0.239 | 0.859 |
| 0.20 | 0.337 | 0.783 |
| 0.30 | 0.419 | 0.721 |
| 0.40 | 0.491 | 0.658 |
| 0.50 | 0.563 | 0.604 |
| 0.60 | 0.628 | 0.545 |
| 0.70 | 0.702 | 0.483 |
| 0.80 | 0.765 | 0.404 |
| 0.90 | 0.851 | 0.294 |
Advanced Synthesizability Frameworks
Complementary to the composition-based SynthNN, the field has seen the development of structure-aware models. The Crystal Synthesis Large Language Model (CSLLM) framework utilizes three specialized LLMs to predict the synthesizability of arbitrary 3D crystal structures, suggest synthetic methods, and identify suitable precursors [24]. On a test set, the Synthesizability LLM achieved a state-of-the-art accuracy of 98.6%, significantly outperforming traditional screening based on thermodynamic stability (74.1%) and kinetic stability (82.2%) [24].
Experimental Protocol: From Prediction to Synthesis
The following section provides a detailed, actionable protocol for deploying the synthesizability-guided pipeline, as validated in the featured case study.
The diagram below illustrates the complete, integrated workflow from computational screening to experimental characterization.
Protocol Steps
Step 1: Computational Screening and Synthesizability Scoring
- Input Data: Compile a list of candidate material compositions from theoretical databases such as the Materials Project (MP), GNoME, or Alexandria [22].
- Synthesizability Prediction:
- Tool: Utilize a pre-trained SynthNN model [16] or the CSLLM framework [24].
- Procedure: For SynthNN, input the chemical formulas of candidate materials. The model will output a synthesizability score between 0 and 1. A higher score indicates a higher predicted likelihood of successful synthesis.
- Thresholding: Refer to performance metrics (Table 1) to select an appropriate decision threshold based on the desired balance between precision and recall for your project. A threshold of 0.50 provides a balanced approach [16].
- Output: A ranked list of candidate materials prioritized by their synthesizability score.
Step 2: Synthesis Pathway and Precursor Prediction
- Precursor Identification: For the top-ranked candidates, use a precursor prediction model, such as the Precursor LLM within the CSLLM framework. This model specializes in identifying solid-state synthetic precursors for binary and ternary compounds with high accuracy [24].
- Pathway Confirmation: The model suggests suitable precursor combinations and may also recommend a synthetic method (e.g., solid-state vs. solution-based) [24].
Step 3: Laboratory Synthesis via Solid-State Reaction
- Weighing: Accurately weigh out the precursor powders (e.g., oxides, carbonates) according to the stoichiometry of the target compound.
- Mixing: Mechanically mix the powders using a mortar and pestle or a ball mill for a homogeneous distribution of reactants.
- Heating:
- Place the mixed powder in a high-temperature furnace inside a suitable crucible (e.g., alumina, platinum).
- Heat the sample to a target temperature (e.g., 800–1500°C, depending on the system) under an appropriate atmosphere (air, oxygen, argon, nitrogen).
- Maintain the peak temperature for a dwell time (e.g., 10-20 hours) to facilitate solid-state diffusion and crystal growth.
- Optionally, perform intermediate regrinding steps to improve reaction homogeneity.
- Cooling: Cool the sample to room temperature at a controlled rate (e.g., furnace cooling).
Step 4: Material Characterization and Validation
- Powder X-ray Diffraction (PXRD):
- Purpose: To confirm the crystal structure of the synthesized product and identify any impurity phases.
- Procedure: Grind a small amount of the synthesized pellet into a fine powder. Acquire a PXRD pattern and compare it to the simulated pattern of the target crystal structure.
- Scanning Electron Microscopy (SEM) and Energy-Dispersive X-ray Spectroscopy (EDS):
- Purpose: To analyze the material's morphology, grain size, and elemental composition, verifying the intended stoichiometry.
- Success Criteria: The synthesis is considered successful when the PXRD pattern of the product matches the target structure with minimal impurities, and EDS confirms the correct composition.
Experimental Validation and Results
The application of this pipeline to screen non-synthesized structures from the MP, GNoME, and Alexandria databases identified several hundred highly synthesizable candidates [22]. Subsequent experimental synthesis efforts targeted 16 of these candidates.
Table 2: Experimental Synthesis Outcomes [22] [23]
| Metric | Result |
|---|---|
| Total Targets Synthesized | 7 out of 16 |
| Experimental Workflow Duration | 3 days |
| Key Characterization Techniques | PXRD, SEM, EDS |
| Databases Screened | MP, GNoME, Alexandria |
The 44% success rate (7/16) within an extremely condensed timeframe of three days for the entire experimental process underscores the pipeline's practical utility and the accuracy of the synthesizability predictions in a real-world laboratory setting [22] [23]. This result highlights the pipeline's ability to counter the zero-Kelvin bias of DFT and expose omissions in existing lists of known synthesized structures [23].
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below lists key materials and reagents essential for executing the synthesizability-guided discovery pipeline.
Table 3: Key Research Reagent Solutions and Materials
| Item | Function / Purpose |
|---|---|
| Precursor Powders (e.g., metal oxides, carbonates) | High-purity starting materials for solid-state synthesis of target inorganic compounds. |
| SynthNN Model / CSLLM Framework | Deep learning models for predicting material synthesizability from composition (SynthNN) or crystal structure (CSLLM). |
| ICSD (Inorganic Crystal Structure Database) | Primary source of positive (synthesized) data for training and benchmarking synthesizability models. |
| Theoretical Databases (MP, GNoME, Alexandria) | Sources of candidate material compositions and structures for screening. |
| High-Temperature Furnace | Equipment for performing solid-state reactions at elevated temperatures (up to 1500°C+). |
| Alumina or Platinum Crucibles | Inert containers for holding powder samples during high-temperature firing. |
| PXRD Instrument | Primary tool for post-synthesis crystal structure validation and phase identification. |
This case study demonstrates that integrating deep learning-based synthesizability predictions directly into the materials discovery pipeline dramatically increases experimental efficiency and success rates. The synthesizability-guided pipeline, underpinned by models like SynthNN and CSLLM, effectively transitions materials research from high-throughput virtual screening to high-success-rate experimental validation, ensuring that computationally discovered materials are not only thermodynamically plausible but also synthetically accessible.
Overcoming Hurdles: Implementing and Optimizing Synthesizability Models
Addressing the 'Positive-Unlabeled' Data Challenge
The 'Positive-Unlabeled' (PU) data challenge is a fundamental problem in computational material science and drug discovery. It arises when researchers have a set of confirmed positive examples (e.g., synthesizable materials, successful drug-target interactions) but lack reliably confirmed negative examples; the rest of the data is merely unlabeled and may contain hidden positives. This scenario is ubiquitous in scientific research where negative results are rarely reported or cataloged. Within the context of synthesizability prediction research, the SynthNN deep learning model and related approaches directly confront this challenge to distinguish synthesizable crystalline materials from those that are not yet or cannot be synthesized [2] [25].
This application note details the core principles, methodologies, and protocols for implementing PU learning, specifically framed around the development and application of synthesizability prediction models like SynthNN. It provides researchers with structured data, visual workflows, and actionable experimental procedures to effectively address the PU learning challenge in their own work.
Core Concepts and Quantitative Comparison of PU Learning Applications
The core principle of PU learning is to learn a classification model from only positive and unlabeled data, as conventional supervised learning requires both positive and negative examples. In synthesizability prediction, the positive class (P) typically comprises experimentally verified synthesizable materials from databases like the Inorganic Crystal Structure Database (ICSD). The unlabeled set (U) contains materials with unknown synthesizability status, which is a mixture of truly synthesizable (hidden positives) and unsynthesizable materials (hidden negatives) [2] [9]. The table below summarizes the performance of various PU learning methods across different domains, demonstrating their effectiveness.
Table 1: Performance Comparison of PU Learning Applications
| Field of Application | Model/Method Name | Key Strategy | Reported Performance | Reference |
|---|---|---|---|---|
| Material Synthesizability (Composition) | SynthNN (SynthNN) | Deep learning with atom2vec embeddings | 7x higher precision than DFT formation energies; outperformed 20 human experts | [2] |
| Material Synthesizability (Structure) | Synthesizability-PU-CGCNN | Partially supervised learning with Crystal Graph Convolutional Neural Network (CGCNN) | Enables calculation of Crystal-Likeness (CL) score | [26] |
| Material Synthesizability (Structure) | CSLLM (Synthesizability LLM) | Fine-tuned Large Language Model on material strings | 98.6% accuracy | [9] |
| Material Synthesizability (Oxides) | SynCoTrain | Dual-classifier co-training (ALIGNN & SchNet) | Robust performance with high recall on test sets | [25] |
| Drug-Drug Interaction (DDI) Prediction | DDI-PULearn | Reliable Negative Sample (RNS) identification using OCSVM & KNN | Superior performance vs. 5 state-of-the-art methods | [27] |
| Drug-Target Interaction (DTI) Prediction | PUDTI | SVM-based optimization with extracted negative samples | Highest AUC on 4 datasets (Enzymes, Ion Channels, GPCRs, Nuclear Receptors) | [28] |
| Dietary Restriction Gene Prediction | Similarity-based KNN | Two-step PU learning for reliable negative selection | Significantly outperformed non-PU approach (p<0.05) | [29] |
PU Learning Workflow and the SynthNN Architecture
The general workflow for PU learning involves two key stages: the extraction of reliable negative examples from the unlabeled set, and the iterative training of a classifier using the positive and identified reliable negatives. The following diagram illustrates this generalized process, which forms the basis for many specific algorithms, including the approach used for SynthNN.
General PU Learning Workflow
SynthNN implements a specific deep-learning architecture to operationalize this workflow for synthesizability prediction. It leverages a representation learning approach to bypass the need for hand-crafted features or heuristic rules like charge-balancing, which fails to classify a majority of known synthesizable materials [2]. The following diagram details its core architecture.
SynthNN Model Architecture
Experimental Protocols
Protocol 1: Implementing a Standard PU Learning Framework for Material Classification
This protocol outlines the steps for building a PU learning model for material synthesizability prediction, based on established methodologies [2] [27] [25].
Data Preparation
- Positive Set (P): Compile a list of known synthesizable materials. The primary source is the Inorganic Crystal Structure Database (ICSD). Filter for crystalline, inorganic materials and extract their chemical compositions [2] [9].
- Unlabeled Set (U): Generate a candidate set of materials with unknown synthesizability status. This can be derived from:
- Data Representation: Convert chemical compositions into a machine-readable format. For SynthNN, this involves using an
atom2vecembedding layer that learns an optimal representation for each element directly from the data [2].
Identification of Reliable Negative Examples
- Employ a strategy to extract a preliminary set of reliable negatives (RN) from the unlabeled set (U). Common methods include:
- One-Class SVM (OCSVM): Train an OCSVM model on the positive set to describe its distribution. Materials in U that are significant outliers to this distribution are considered reliable negatives [27].
- K-Nearest Neighbors (KNN): For each material in U, compute its distance to the k-nearest neighbors in P. Materials with the largest distances are selected as reliable negatives [27] [29].
- The ratio of initial RNs to positive samples is a critical hyperparameter (often denoted as
N_synth) that requires empirical tuning [2].
- Employ a strategy to extract a preliminary set of reliable negatives (RN) from the unlabeled set (U). Common methods include:
Classifier Training and Iterative Refinement
- Train a deep neural network classifier (e.g., SynthNN) using the positive set (P) and the initial reliable negative set (RN).
- Use the trained classifier to predict labels for the remaining unlabeled data.
- From these predictions, select the most confident negative predictions and add them to the reliable negative set (RN).
- Iterate the process of training and re-labeling until a convergence criterion is met (e.g., the RN set stabilizes or a maximum number of iterations is reached) [2] [25].
Model Validation
- Hold-Out Validation: Reserve a portion of the known positive samples and a set of artificially generated "negative" samples (e.g., from the final RN set) for testing.
- Performance Metrics: Calculate precision, recall, F1-score, and accuracy on the test set. Note that precision may be underestimated as some "false positives" could be synthesizable materials that have not been synthesized yet [2].
- Benchmarking: Compare model performance against baseline methods, such as charge-balancing heuristics or formation energy thresholds from DFT calculations [2].
Protocol 2: Structure-Based Synthesizability Prediction with CGCNN
This protocol is adapted from the Synthesizability-PU-CGCNN repository for predicting the Crystal-Likeness (CL) score, a quantitative metric for synthesizability [26].
Dataset and Crystal Graph Creation
- Create a root directory (e.g.,
cif_files) containing:id_prop.csv: A two-column CSV file. The first column is a unique crystal ID, the second is1for positive (synthesizable) and0for unlabeled.atom_init.json: A JSON file containing an initialization vector for each chemical element.<ID>.cif: A CIF file for each crystal structure listed inid_prop.csv.
- Generate Crystal Graphs: Run the
generate_crystal_graph.pyscript to convert CIF files into crystal graph representations. Key parameters include cutoff radius (e.g.,--r 8Å) and maximum number of neighbors (e.g.,--n 12). The graphs will be saved as pickle files [26].
- Create a root directory (e.g.,
Model Training with PU Learning
- Execute the main training script (e.g.,
main.py). The script will:- Load the pre-generated crystal graphs.
- Create data splits for training and validation appropriate for PU learning.
- Train an ensemble of CGCNN models (e.g., 100 models via bootstrap aggregating) using the PU learning paradigm.
- The number of bootstrap samples can be controlled via a command-line option (e.g.,
--bag).
- Execute the main training script (e.g.,
Prediction and Aggregation
- After training, the model will generate synthesizability predictions (CLscores between 0 and 1) for the test-unlabeled data for each ensemble member.
- The results are aggregated (e.g., by averaging) across all models. The final output file (e.g.,
test_results_ensemble_100models.csv) contains the consensus CLscore for each candidate material, where a higher score indicates a higher predicted synthesizability [26].
The following table lists key resources required for implementing PU learning in synthesizability prediction research.
Table 2: Key Research Reagents and Computational Tools for PU Learning in Synthesizability Prediction
| Item Name | Function/Description | Example Sources / Notes |
|---|---|---|
| Inorganic Crystal Structure Database (ICSD) | Provides the canonical set of positive examples (synthesized inorganic crystalline materials) for model training. | FIZ Karlsruhe; Commercial license required [2]. |
| Materials Project (MP) Database | A primary source of unlabeled data; contains computationally derived hypothetical crystal structures whose synthesizability is unknown. | materialsproject.org; Public API [25]. |
| atom2vec / Material Compositions | A featurization method that learns optimal numerical representations of chemical elements directly from data, used in models like SynthNN. | Implementation required; alternative fixed descriptors include Magpie [2]. |
| Crystal Graph (CG) | A graph representation of a crystal structure where nodes are atoms and edges represent bonds, capturing structural information. | Generated from CIF files using scripts from repositories like Synthesizability-PU-CGCNN [26]. |
| One-Class SVM (OCSVM) | An algorithm used in the first step of PU learning to identify reliable negative samples from the unlabeled set based on deviation from the positive set distribution. | Available in scikit-learn (Python) [27]. |
| CGCNN (Crystal Graph Convolutional Neural Network) | A graph neural network architecture specifically designed for learning material properties from crystal structures. | Publicly available PyTorch implementation [26]. |
| ALIGNN & SchNet | Advanced graph neural networks used in co-training frameworks (e.g., SynCoTrain). ALIGNN incorporates bond angles, while SchNet uses continuous-filter convolutions. | ALIGNN: https://github.com/usnistgov/alignn; SchNet: SchNetPack [25]. |
Balancing Model Precision and Recall for Practical Screening
The accelerating use of computational methods has generated millions of hypothetical inorganic crystalline materials with promising functional properties. However, a significant bottleneck persists in translating these theoretical candidates into experimentally realized compounds, making accurate synthesizability prediction a critical frontier in materials science [2] [11]. Within this context, the SynthNN deep learning model emerges as a powerful framework for assessing the synthesizability of inorganic chemical compositions directly from stoichiometric data, eliminating the requirement for prior structural knowledge [2] [6]. For researchers deploying such models in practical screening scenarios, a fundamental challenge arises: the inherent trade-off between precision and recall. Optimizing this balance is not merely a statistical exercise but a practical necessity that directly determines the efficiency of experimental pipelines and the success rate of materials discovery campaigns [16].
This application note provides a structured framework for researchers aiming to implement SynthNN effectively within high-throughput screening workflows. We present quantitative performance data across operational thresholds, detailed protocols for model deployment, and visualization of strategic workflows. Furthermore, we contextualize SynthNN within the evolving landscape of synthesizability prediction, acknowledging emerging approaches like the Crystal Synthesis Large Language Models (CSLLM) framework, which has demonstrated 98.6% prediction accuracy by incorporating structural information alongside compositional data [9]. The guidance herein is designed to enable scientists to configure synthesizability filters that align precisely with their specific research objectives, whether prioritizing the confirmation of highly synthesizable candidates or conducting expansive searches across chemical space.
Performance Data and Threshold Selection
The operational performance of a synthesizability model is governed by the classification threshold applied to its output scores. Selecting this threshold allows researchers to calibrate the model's behavior along the precision-recall spectrum, making it crucial for practical screening applications. The following table synthesizes the performance metrics for the pre-trained SynthNN model across a range of decision thresholds, providing a reference for selecting an appropriate operating point based on screening goals [16].
Table 1: Performance Metrics for SynthNN Across Decision Thresholds on a Dataset with a 20:1 Ratio of Unsynthesized:Synthesized Examples [16]
| Threshold | Precision | Recall |
|---|---|---|
| 0.10 | 0.239 | 0.859 |
| 0.20 | 0.337 | 0.783 |
| 0.30 | 0.419 | 0.721 |
| 0.40 | 0.491 | 0.658 |
| 0.50 | 0.563 | 0.604 |
| 0.60 | 0.628 | 0.545 |
| 0.70 | 0.702 | 0.483 |
| 0.80 | 0.765 | 0.404 |
| 0.90 | 0.851 | 0.294 |
Interpreting the Trade-off for Screening Strategies
The data in Table 1 reveals the core trade-off: as the decision threshold increases, the model demands higher confidence to classify a material as synthesizable, resulting in higher precision but at the cost of lower recall [16]. This relationship directly informs two primary screening strategies:
- High-Precision Screening (Thresholds ≥ 0.70): This configuration is optimal when experimental resources are limited and highly expensive. A threshold of 0.70 or higher ensures that over 70% of the materials predicted to be synthesizable are likely correct, minimizing wasted effort on false leads. This approach is recommended for targeted discovery projects focused on validating a small set of highly promising candidates [16].
- High-Recall Screening (Thresholds ≤ 0.30): This strategy is suitable for exploratory phases where the primary goal is to create a comprehensive longlist of potential candidates for further computational study or preliminary experimental validation. While this generates more false positives, it minimizes the risk of missing potentially synthesizable materials (false negatives) [16].
For general-purpose screening where a balance between resource efficiency and discovery potential is desired, a moderate threshold around 0.40 to 0.50 often provides an effective compromise, offering nearly balanced precision and recall.
Integrated Synthesizability Screening Protocol
The following protocol details the steps for integrating SynthNN-based synthesizability prediction into a computational materials screening pipeline, from data preparation to final candidate selection.
Stage 1: Data Curation and Model Setup
Objective: Prepare a candidate list of material compositions and access the synthesizability model.
- Step 1.1: Candidate Compilation: Gather chemical formulas of candidate materials from computational databases (e.g., Materials Project, OQMD, GNoME). Standardize all formulas to a consistent format [11].
- Step 1.2: Model Access: Clone the official SynthNN repository from GitHub (
github.com/antoniuk1/SynthNN) to access the pre-trained model and prediction interface [16]. - Step 1.3: Environment Setup: Install required dependencies, including Python 3.x, PyTorch, NumPy, and pandas, ensuring version compatibility as specified in the repository documentation [16].
Stage 2: Threshold Calibration and Prediction
Objective: Generate synthesizability scores and apply a threshold aligned with the project's strategic goal.
- Step 2.1: Strategic Threshold Selection: Refer to Table 1 and Section 2.1 of this document to select a threshold (
T) that reflects the desired balance between precision and recall for your specific screening objective [16]. - Step 2.2: Batch Prediction: Use the
SynthNN_predict.ipynbJupyter notebook provided in the repository to process the list of candidate compositions. The model will output a synthesizability probability score for each candidate [16]. - Step 2.3: Initial Filtering: Label materials with a synthesizability score greater than the chosen threshold
Tas "predicted-synthesizable." This creates the initial filtered candidate list.
Stage 3: Prioritization and Experimental Planning
Objective: Further refine the filtered list and plan for experimental validation.
- Step 3.1: Multi-Criteria Prioritization: Rank the "predicted-synthesizable" candidates based on other relevant properties (e.g., target functional properties, calculated formation energy, cost of constituent elements) [11].
- Step 3.2: Synthesis Pathway Prediction: For top-ranked candidates, employ additional predictive tools, such as precursor suggestion models or literature mining algorithms, to propose viable synthesis routes and parameters (e.g., calcination temperature, precursor materials) [9] [11].
- Step 3.3: Final Candidate Selection: Select the final shortlist for experimental synthesis based on the combined assessment of synthesizability score, functional properties, and predicted synthesis feasibility.
Workflow Visualization
The logical relationship and data flow between the key stages of the screening protocol are visualized below.
Synthesizability Screening Workflow
The workflow begins with raw candidate materials from computational databases, which undergo curation and standardization. The core SynthNN model then processes these to generate synthesizability scores. A critical juncture is the application of a decision threshold, which is strategically chosen based on the desired precision/recall balance for the project. This filtered list is then further refined through multi-criteria prioritization and synthesis planning before yielding a final candidate list for experimental validation [2] [16] [11].
The Precision-Recall Relationship in Practice
Understanding the conceptual relationship between precision and recall is vital for interpreting model performance and making informed threshold decisions. The following diagram illustrates this fundamental trade-off.
Precision-Recall Trade-off
As visualized, a screening strategy that employs a low decision threshold will correctly identify most synthesizable materials (high recall) but will also include many non-synthesizable candidates in its predictions (low precision). Conversely, a strategy using a high threshold will produce a candidate list that is highly enriched with synthesizable materials (high precision) but will fail to identify many other viable candidates (low recall). There is no single optimal point; the choice must be dictated by the costs associated with false positives versus the opportunity costs of false negatives in a specific research context [16].
Essential Research Reagent Solutions
Successful implementation of a synthesizability-guided discovery pipeline relies on a suite of computational and data resources. The following table details key components and their functions in the research ecosystem.
Table 2: Key Resources for Synthesizability-Driven Materials Discovery
| Resource Name | Type | Primary Function in Screening |
|---|---|---|
| SynthNN [2] [16] | Deep Learning Model | Predicts synthesizability probability from chemical composition alone, enabling rapid screening before structural relaxation. |
| ICSD (Inorganic Crystal Structure Database) [2] [9] | Materials Database | Serves as the primary source of confirmed synthesizable materials for training positive-unlabeled (PU) learning models like SynthNN. |
| Materials Project [9] [11] | Computational Database | Provides a large repository of DFT-calculated hypothetical structures used as a source of candidate materials and for generating negative examples. |
| CSLLM (Crystal Synthesis LLM) [9] | Large Language Model | A state-of-the-art framework that predicts synthesizability, suggests synthetic methods, and identifies suitable precursors for crystal structures. |
| Retro-Rank-In [11] | Precursor-Suggestion Model | Generates a ranked list of viable solid-state precursors for a given target composition, bridging the gap between identification and synthesis. |
The integration of robust synthesizability filters like SynthNN into computational screening pipelines marks a significant advancement toward realistic and efficient materials discovery. By moving beyond thermodynamic stability metrics and learning directly from the distribution of known synthesized materials, these models address a critical bottleneck [2]. The ultimate effectiveness of this approach in a practical setting, however, hinges on the researcher's ability to consciously manage the precision-recall trade-off. The quantitative data, detailed protocols, and conceptual frameworks provided in this application note are designed to empower scientists to make these strategic decisions with confidence. As the field evolves with more integrated models like CSLLM [9] and automated pipelines [11], the principles of strategic threshold selection and multi-stage screening will remain foundational to translating in-silico predictions into laboratory realities.
The discovery of novel functional materials is a cornerstone of technological advancement. A critical first step in this process is identifying synthesizable materials—those that are synthetically accessible through current capabilities, regardless of whether they have been synthesized yet [2]. However, predicting synthesizability presents a significant scientific challenge. Traditional approaches, such as charge-balancing criteria or density functional theory (DFT) calculations for formation energies, often fail to accurately identify synthesizable candidates. Charge-balancing proves inflexible, incorrectly filtering out many known materials, while DFT fails to account for kinetic stabilization and non-physical considerations like cost and equipment availability that influence synthetic decisions [2]. This creates a critical bottleneck in materials discovery pipelines.
The SynthNN (Synthesizability Neural Network) deep learning model was developed to directly address this challenge [2] [16]. By reformulating material discovery as a synthesizability classification task, SynthNN leverages the entire space of known inorganic chemical compositions to make its predictions. This approach inherently forces a confrontation with a fundamental computational trade-off: the balance between the speed of screening vast chemical spaces and the depth of analysis achieved through more computationally intensive, high-fidelity methods. This article explores this trade-off within the context of SynthNN research, providing application notes and detailed protocols for researchers navigating this critical aspect of modern materials informatics and drug development, where inorganic carriers and excipients play a vital role.
SynthNN Architecture and Workflow
The SynthNN model is a deep learning classifier designed to predict the synthesizability of inorganic crystalline materials from their chemical composition alone, without requiring structural information [2]. Its architecture is built around the atom2vec framework, which represents each chemical formula through a learned atom embedding matrix that is optimized alongside all other parameters of the neural network [2]. This allows the model to learn an optimal representation of chemical formulas directly from the distribution of previously synthesized materials, without relying on pre-defined chemical descriptors or assumptions about synthesizability principles.
- Input Representation: The model takes a chemical formula as input. The atom2vec framework converts this formula into a dense vector representation that captures complex chemical relationships.
- Learning Chemistry from Data: Remarkably, without any prior chemical knowledge encoded, SynthNN learns fundamental chemical principles such as charge-balancing, chemical family relationships, and ionicity directly from the data of known material compositions [2].
- PU Learning Framework: A major innovation is the treatment of the synthesizability prediction as a Positive-Unlabeled (PU) learning problem. The model is trained on positive examples (synthesized materials from the Inorganic Crystal Structure Database, ICSD) and artificially generated unsynthesized materials, which are treated as unlabeled data and probabilistically reweighted to account for the likelihood that some may actually be synthesizable [2].
The following workflow diagram illustrates the core operational logic of SynthNN and its position within a broader materials discovery pipeline, highlighting key decision points:
Quantitative Performance: Speed vs. Accuracy Benchmarks
The performance of SynthNN must be evaluated along two primary axes: the accuracy of its predictions and the computational speed with which it makes them. These two factors are the core components of the computational trade-off. The table below summarizes key quantitative benchmarks for SynthNN and other contemporary methods, illustrating this trade-off clearly.
Table 1: Performance Comparison of Synthesizability Prediction Methods
| Method | Key Input | Reported Accuracy / Precision | Relative Speed | Primary Use Case |
|---|---|---|---|---|
| SynthNN [2] [16] | Chemical Composition | 1.5x higher precision than human experts; Precision varies with threshold (e.g., 56.3% at 0.5 threshold) [2] [16] | Five orders of magnitude faster than human experts [2] | High-throughput composition screening |
| CSLLM (Synthesizability LLM) [9] | Crystal Structure (Text Representation) | 98.6% Accuracy [9] | Fast (LLM-based), but requires structural input | High-accuracy screening when structure is known |
| DFT Formation Energy [2] | Crystal Structure | ~50% of synthesized materials captured [2] | Computationally expensive (hours/days per structure) | Depth-first analysis of thermodynamic stability |
| Charge-Balancing [2] | Chemical Composition | 37% of known synthesized materials identified [2] | Very Fast (instantaneous) | Rapid, low-accuracy pre-filtering |
| Human Expert Assessment [2] | Composition & Structure | Baseline Precision | Baseline Speed (hours/days per candidate) | Specialized, in-depth analysis |
The data in Table 1 reveals a clear spectrum. At one extreme, simple heuristics like charge-balancing are fast but lack accuracy. At the other extreme, human expertise and detailed DFT calculations provide depth but are prohibitively slow for screening large spaces. SynthNN occupies a middle ground, offering substantially improved accuracy over simple filters while maintaining a speed that enables the screening of billions of candidate compositions [2]. The CSLLM model demonstrates that even higher accuracy is achievable, but it requires crystal structure information as input, which is typically not available for truly novel, undiscovered materials [9], thus introducing a different trade-off between input requirements and predictive power.
The Precision-Recall Trade-off in SynthNN
A critical aspect of deploying SynthNN is selecting an appropriate decision threshold, which directly governs the precision-recall trade-off. The table below, derived from the official SynthNN performance data, shows how this trade-off can be managed [16].
Table 2: SynthNN Performance at Different Decision Thresholds (20:1 Unsynth:Synth Ratio)
| Threshold | Precision | Recall | Implication |
|---|---|---|---|
| 0.10 | 0.239 | 0.859 | Low precision, high recall. Casts a wide net, missing few synthesizable materials but yielding many false positives. Ideal for initial broad screening. |
| 0.50 | 0.563 | 0.604 | Balanced approach. A reasonable compromise for general-purpose discovery workflows. |
| 0.90 | 0.851 | 0.294 | High precision, low recall. Identifies a highly confident set, but misses many synthesizable materials. Best for prioritizing a shortlist for immediate experimental follow-up. |
Experimental Protocols
To ensure the reproducible application of SynthNN in research, the following detailed protocols are provided.
Protocol 1: High-Throughput Virtual Screening of Composition Space
Purpose: To rapidly identify synthesizable candidate materials from a large pool of hypothetical chemical compositions (e.g., >1 million candidates) for applications in drug development (e.g., inorganic excipient discovery) or functional materials design. Principles: This protocol prioritizes speed and scalability, accepting a moderate level of precision to quickly reduce the candidate space by orders of magnitude.
Input Data Preparation:
- Compile a list of candidate chemical formulas in a plain text file (e.g., CSV format), with one formula per row.
- Ensure formulas are written in standard notation (e.g., "Cs2TiBr6", "Fe2O3").
Model Inference with SynthNN:
- Utilize the pre-trained SynthNN model as provided in the official GitHub repository [16].
- Execute the
SynthNN_predict.ipynbJupyter notebook or an equivalent scripted interface. - Critical Parameter: Set the decision threshold to 0.10. This low threshold favors high recall, ensuring most synthesizable materials in the list are retained for the next stage, even at the cost of including many false positives [16].
Output and Triage:
- The model will output a list of compositions with synthesizability scores.
- All candidates with scores above the 0.10 threshold are considered hits from the initial screen. This list will be large but significantly smaller than the original input.
- This hit list is passed to Protocol 2 for deeper analysis.
Protocol 2: Focused High-Fidelity Synthesis Prioritization
Purpose: To create a high-confidence, shortlist of candidate materials for experimental synthesis by applying a more stringent analysis to the output of Protocol 1. Principles: This protocol prioritizes depth of analysis and precision, using slower, more resource-intensive methods to validate and rank candidates.
Input: The list of candidate materials generated from Protocol 1.
Re-Scoring with SynthNN:
- Process the candidate list through SynthNN again, this time with a decision threshold of 0.70 to 0.90 [16]. This selects only the most promising candidates based on the model's highest confidence predictions.
Structural Prediction and DFT Validation (Depth-First Analysis):
- For the high-confidence candidates from step 2, perform crystal structure prediction (e.g., using evolutionary algorithms like USPEX [6]).
- For the predicted ground-state structures, perform DFT calculations to determine:
- Note: This step is computationally intensive and should only be applied to a small number of candidates.
Final Ranking and Decision:
- Rank candidates based on a composite score incorporating the high-threshold SynthNN score, Eₕ, and the absence of imaginary phonon frequencies.
- The top-ranked candidates form the final, high-priority list for experimental synthesis efforts.
The following diagram maps these two protocols onto the computational trade-off spectrum, showing how they can be integrated into a coherent materials discovery pipeline.
Successful implementation of the aforementioned protocols requires a suite of computational tools and data resources. The table below details key components of the research toolkit for synthesizability prediction.
Table 3: Essential Resources for Synthesizability-Driven Materials Discovery
| Resource Name | Type | Function / Application | Access / Reference |
|---|---|---|---|
| SynthNN Model Code | Software | Official implementation for training and prediction; core of the high-throughput protocol. | GitHub: antoniuk1/SynthNN [16] |
| Inorganic Crystal Structure Database (ICSD) | Data | Primary source of positive examples (synthesized materials) for training and benchmarking. | Commercial License [2] [16] |
| Materials Project (MP) | Database | Source of calculated material properties and structures; can be used for generating candidates and validation. | materialsproject.org [9] |
| Vienna Ab initio Simulation Package (VASP) | Software | Industry-standard software for performing DFT calculations (e.g., for Eℎ and phonons) in the high-fidelity protocol. | Commercial License [9] |
| JARVIS | Database & Tools | Provides data and ML models for materials design; includes diverse datasets for validation. | jarvis.nist.gov [6] [9] |
| PU Learning Algorithm | Methodology | The semi-supervised learning framework crucial for handling unlabeled data in synthesizability prediction. | [2] |
The integration of SynthNN into computational materials discovery workflows provides a powerful means to navigate the inherent trade-off between speed and depth of analysis. By employing a tiered strategy—using SynthNN for initial high-speed screening of compositional space followed by high-fidelity, structure-sensitive methods for final prioritization—researchers can significantly accelerate the identification of viable synthetic targets. This approach effectively bridges the gap between massive computational searches and practical experimental synthesis, enhancing the reliability and efficiency of the discovery process for new materials and, by extension, the drug development pipelines that rely on them. The protocols and analyses provided here serve as a guide for researchers to implement this balanced strategy in their own work.
Adapting to Resource-Limited Environments and In-House Building Blocks
The process of drug discovery is inherently time-consuming and resource-intensive, often taking between six to twelve years to bring a new drug to market [30]. A significant bottleneck in this process is the synthesizability of proposed chemical compounds; molecules generated through computational models, including those from AI-driven approaches, often face major challenges in their practical synthesis [12]. This issue is acutely felt in resource-constrained environments, such as academic labs or small startups, where access to extensive compound libraries or expensive synthetic capabilities is limited. The ability to accurately predict whether a molecule can be synthesized using available in-house building blocks is therefore critical, as it can drastically reduce the time and cost associated with pursuing non-viable candidates.
Framed within the broader research on the SynthNN deep learning model, this document provides detailed application notes and protocols. SynthNN is a deep-learning classification model developed to predict the synthesizability of inorganic crystalline materials directly from their chemical formulas, without requiring structural information [2]. While originally designed for inorganic materials, the underlying principles of its data representation and classification approach offer a valuable framework that can be adapted and extended to address the synthesizability of organic drug-like molecules. By leveraging such models, researchers can prioritize compound candidates that are not only therapeutically promising but also synthetically accessible with available resources.
Model Fundamentals and Performance Benchmarks
SynthNN represents a paradigm shift in predicting material synthesizability. Instead of relying on proxy metrics like thermodynamic stability or manual expert evaluation, it learns the complex chemistry of synthesizability directly from the data of all experimentally realized materials contained in databases like the Inorganic Crystal Structure Database (ICSD) [2]. The model utilizes a semi-supervised learning approach known as Positive-Unlabeled (PU) learning. It is trained on known synthesized materials (positive examples) and a large number of artificially generated, typically unsynthesized compositions, which are treated as unlabeled data and probabilistically reweighted to account for the possibility that some might be synthesizable [2]. A key feature of SynthNN is its use of the atom2vec framework, which learns an optimal numerical representation (embedding) for each atom directly from the distribution of known chemical formulas. This allows the model to autonomously discover and utilize fundamental chemical principles such as charge-balancing, chemical family relationships, and ionicity when making its predictions [2].
Quantitative Performance Evaluation
The performance of a synthesizability prediction model is paramount for its practical application. SynthNN has been benchmarked against other methods and demonstrates superior capability. In a head-to-head comparison against 20 expert material scientists, SynthNN outperformed all experts, achieving 1.5 times higher precision and completing the task five orders of magnitude faster [2]. Furthermore, it identifies synthesizable materials with 7 times higher precision than using DFT-calculated formation energies alone [2].
For a practical deployment, the decision threshold for classifying a material as synthesizable can be adjusted based on the desired trade-off between precision and recall. The following table, derived from the official SynthNN repository, illustrates this trade-off on a dataset with a 20:1 ratio of unsynthesized to synthesized examples [16].
Table 1: SynthNN Performance at Various Decision Thresholds
| Decision Threshold | Precision | Recall |
|---|---|---|
| 0.10 | 0.239 | 0.859 |
| 0.20 | 0.337 | 0.783 |
| 0.30 | 0.419 | 0.721 |
| 0.40 | 0.491 | 0.658 |
| 0.50 | 0.563 | 0.604 |
| 0.60 | 0.628 | 0.545 |
| 0.70 | 0.702 | 0.483 |
| 0.80 | 0.765 | 0.404 |
| 0.90 | 0.851 | 0.294 |
This table is an essential tool for researchers. In a resource-constrained environment, a user might opt for a higher threshold (e.g., 0.70) to ensure that the molecules selected for synthesis have a very high probability of being synthesizable, thereby conserving precious resources, albeit at the cost of missing some viable candidates (lower recall).
Experimental Protocols for Predictions
Protocol A: Predicting Synthesizability for a Novel Composition
This protocol details the steps to use a pre-trained SynthNN model to obtain synthesizability predictions for a list of candidate chemical compositions.
Materials & Reagents:
- Hardware: A standard computer (laptop or desktop).
- Software: Python (version 3.7 or higher), Jupyter Notebook environment.
- Code & Data: The official SynthNN_predict.ipynb Jupyter Notebook from the SynthNN GitHub repository [16].
Procedure:
- Environment Setup: Install the required Python packages as specified in the SynthNN repository documentation. This typically includes core scientific computing libraries such as NumPy, Pandas, and a deep learning framework like TensorFlow or PyTorch.
- Model Acquisition: Clone the SynthNN repository and locate the
SynthNN_predict.ipynbnotebook. Ensure that the pre-trained model weights are available in the specified directory path as per the repository's instructions. - Input Preparation: Prepare a
.csvfile containing the list of target chemical formulas you wish to screen. The file should have a single column with the headercomposition, and each row should contain a single chemical formula in its standard textual representation (e.g., "SiO2", "NaCl"). - Execution: Open and run the
SynthNN_predict.ipynbnotebook in your Jupyter environment. Modify the file path within the notebook to point to your input.csvfile. - Output and Analysis: The notebook will output a file containing the original compositions alongside a
synthnn_scorefor each. This score represents the model's confidence in the synthesizability of the composition. Apply a decision threshold (see Table 1) to convert these continuous scores into binary labels (synthesizableornot synthesizable).
Troubleshooting Tip: If the model fails to load, verify the file path to the pre-trained model weights and ensure all dependencies are installed in compatible versions.
Protocol B: Fine-Tuning SynthNN with Custom Data
For researchers with specialized data, such as a curated list of molecules synthesized from a specific set of in-house building blocks, fine-tuning SynthNN can improve its predictive accuracy for that particular chemical space.
Materials & Reagents:
- Hardware: A computer with a GPU is recommended to accelerate training.
- Software: Same as Protocol A.
- Code & Data: The
train_SynthNN.ipynbJupyter Notebook from the SynthNN GitHub repository [16]. - Custom Dataset: A curated set of positive examples (successfully synthesized molecules) and a larger set of unlabeled or negative examples (molecules that failed synthesis or are theoretically generated).
Procedure:
- Data Preparation: Format your custom data into two separate lists. The positive examples file should contain chemical formulas of known, synthesizable molecules. The negative/unlabeled examples file should contain formulas of molecules considered unsynthesizable or of unknown status.
- Notebook Configuration: Open the
train_SynthNN.ipynbnotebook. Edit thepositive_example_file_pathandnegative_example_file_pathvariables to point to your custom data files. - Hyperparameter Setting: Adjust the training hyperparameters if needed. Key hyperparameters include the dimensionality of the atom embeddings, the ratio of unsynthesized to synthesized examples (
N_synth), and the learning rate. The default values provided are a good starting point [2]. - Model Training: Execute the notebook to begin the training process. The script will save the newly trained model weights in a specified directory (e.g., a 'Trained_models' folder).
- Validation: Validate the performance of your fine-tuned model on a held-out test set of molecules from your custom dataset. Use metrics such as precision and recall to compare its performance against the pre-trained model.
Note: The original SynthNN model was trained on the ICSD database, which is licensed. The provided pre-trained model and figure data in the repository allow for reproduction of results without direct ICSD access [16].
Workflow Visualization
The following diagram illustrates the integrated computational and experimental workflow for drug discovery, incorporating SynthNN as a critical filter for synthesizability.
Diagram 1: Integrated Drug Discovery Workflow with Synthesizability Filter.
The Scientist's Toolkit: Research Reagent Solutions
Successful application of these protocols relies on a combination of computational and chemical resources. The table below details key components.
Table 2: Essential Research Reagents and Resources
| Item Name | Function / Explanation | Relevance to Protocol |
|---|---|---|
| Pre-trained SynthNN Model | A deep learning model that predicts synthesizability from chemical composition, providing a baseline for predictions. | Essential for Protocol A. Serves as a starting point for Protocol B. |
| In-House Building Block Library | A curated, digitally stored list of readily available chemical precursors (e.g., from commercial suppliers or past projects). | Used to define the accessible chemical space for model fine-tuning and candidate filtering in all protocols. |
| Jupyter Notebook Environment | An interactive computing platform that enables users to combine code, visualizations, and narrative text. | The primary software environment for running both Protocol A and B. |
| ICSD / Custom Dataset | The Inorganic Crystal Structure Database (ICSD) is the original data source. A custom dataset is a lab-specific collection of synthesis outcomes. | Custom datasets are crucial for Protocol B to fine-tune the model for a specific research context. |
| Python Scientific Stack | A collection of Python libraries (e.g., NumPy, Pandas, TensorFlow/PyTorch) for data manipulation and machine learning. | Provides the computational backbone for all model operations and data handling. |
Integrating deep learning-based synthesizability predictors like SynthNN into the early stages of drug discovery represents a transformative strategy for research in resource-limited settings. By providing a computationally efficient and accurate means of prioritizing synthetically accessible compounds, these models help de-risk the discovery pipeline, saving valuable time, financial resources, and material. The protocols outlined herein offer a practical guide for researchers to implement these tools, from basic screening to custom model adaptation.
Looking forward, the field is moving towards even more integrated and lightweight approaches. The ongoing development of Tiny Machine Learning (TinyML) aims to deploy deep learning models directly on microcontrollers and mobile devices, further democratizing access to powerful AI tools [31]. Furthermore, combining synthesizability predictors with other critical property predictors, such as those for toxicity (e.g., cardiotoxicity, hepatotoxicity) [32] and bioactivity [30], into a unified screening platform will create a robust and comprehensive toolkit for the next generation of drug development professionals. This will ultimately accelerate the journey from a conceptual target to a viable, synthesizable therapeutic agent.
The acceleration of scientific discovery in fields like materials science and drug development is increasingly dependent on our ability to predict molecular behavior and synthesizability. Traditional computational methods, while valuable, often struggle with the complex, multi-factor considerations that determine whether a theoretical material can be synthesized or a novel drug candidate can be effectively produced. The emergence of Large Language Models (LLMs) and structure-aware deep learning frameworks represents a paradigm shift, moving beyond thermodynamic stability to model the intricate relationships that govern synthesis and bioactivity. This evolution is perfectly exemplified by the progression from deep learning models like SynthNN to sophisticated LLM-based frameworks such as the Crystal Synthesis LLM (CSLLM), which leverage vast chemical databases to predict synthesizability with unprecedented accuracy [2] [9]. This article details the cutting-edge applications of these models and provides standardized protocols for their implementation, empowering researchers to integrate these powerful tools into their discovery workflows.
Key Applications in Science and Drug Development
LLM-based and structure-aware models are revolutionizing discovery pipelines by providing accurate, data-driven predictions that guide experimental efforts.
Predicting Material Synthesizability
A primary challenge in materials science is bridging the gap between computationally predicted materials and those that can be experimentally realized. While traditional metrics like formation energy calculated from Density Functional Theory (DFT) are common, they often fail to accurately predict synthesizability, capturing only 50% of synthesized inorganic crystalline materials [2].
The SynthNN Model: A significant leap forward, SynthNN is a deep learning model that treats material discovery as a synthesizability classification task. It leverages the entire space of synthesized inorganic chemical compositions from databases like the Inorganic Crystal Structure Database (ICSD). Remarkably, without prior chemical knowledge, SynthNN learns fundamental chemical principles such as charge-balancing and ionicity. It has been shown to identify synthesizable materials with 7 times higher precision than DFT-calculated formation energies and, in a head-to-head discovery comparison, outperformed 20 expert material scientists, achieving 1.5 times higher precision and completing the task five orders of magnitude faster [2] [6].
The Crystal Synthesis LLM (CSLLM) Framework: Building on this concept, the CSLLM framework utilizes three specialized LLMs to address the synthesis challenge comprehensively. Fine-tuned on a massive dataset of known materials, this framework achieves a state-of-the-art 98.6% accuracy in predicting the synthesizability of arbitrary 3D crystal structures. It also exceeds 90% accuracy in classifying synthetic methods and identifying suitable solid-state precursors for binary and ternary compounds [9]. This demonstrates the powerful advantage of LLMs in integrating multiple prediction tasks into a unified, highly accurate workflow.
Accelerating de novo Drug Design
In pharmaceutical research, the "design-make-test" cycle is a major bottleneck. LLMs and structure-aware models are now enabling the de novo design of molecules with specified properties from scratch.
The DRAGONFLY framework employs deep interactome learning, combining a Graph Transformer Neural Network (GTNN) with a Chemical Language Model (CLM) to generate novel drug-like molecules. This approach leverages a drug-target interactome—a graph containing ~360,000 ligands and their targets—to design molecules based on either a ligand template or a 3D protein binding site. It accomplishes this without requiring application-specific fine-tuning, a process known as "zero-shot" learning. The generated molecules demonstrate high synthesizability (as measured by retrosynthetic accessibility score) and structural novelty, and have been prospectively validated with the successful synthesis and characterization of potent partial agonists for the PPARγ nuclear receptor [33].
Table 1: Quantitative Performance of Key Predictive Models
| Model Name | Primary Task | Key Metric | Performance | Comparison to Traditional Method |
|---|---|---|---|---|
| SynthNN [2] [6] | Synthesizability classification (composition) | Precision | 7x higher precision | DFT formation energy (Precision) |
| CSLLM Synthesizability LLM [9] | Synthesizability prediction (crystal structure) | Accuracy | 98.6% | Energy above hull ≥0.1 eV/atom (74.1% Accuracy) |
| CSLLM Precursor LLM [9] | Precursor identification | Accuracy | 80.2% Success | N/A |
| DRAGONFLY [33] | De novo drug design | Property Correlation | r ≥ 0.95 (e.g., Molecular Weight) | Outperformed fine-tuned RNNs on synthesizability, novelty, and bioactivity |
Experimental Protocols
To ensure reproducibility and facilitate adoption, this section outlines detailed protocols for key experiments cited in this field.
Protocol: Synthesizability Prediction Using the CSLLM Framework
Objective: To predict the synthesizability, suggested synthesis method, and potential precursors for a given inorganic crystalline material.
Workflow Overview:
Materials & Reagents:
- Hardware: A computer with an internet connection or local access to a GPU cluster for running large models.
- Software: Python environment, CSLLM framework (model weights and architecture), necessary libraries (e.g., PyTorch, Transformers).
- Input Data: A crystal structure file for the target material in CIF or POSCAR format.
Procedure:
- Input Preparation:
- Obtain the crystal structure of the target material. If not available, generate a reasonable candidate structure using crystal structure prediction software.
- Convert the structure into the prescribed "material string" text representation. This format efficiently encodes lattice parameters, composition, atomic coordinates, and space group symmetry, stripping redundant information found in standard CIF files [9].
Synthesizability Prediction:
- Feed the material string into the fine-tuned Synthesizability LLM.
- The LLM will return a binary classification (Synthesizable/Non-synthesizable) along with a confidence score. The model used in the cited study achieved an accuracy of 98.6% on test data [9].
- If the output is "Non-synthesizable," the protocol may be terminated, or the structure may be redesigned.
Synthesis Method Classification:
- If the material is predicted to be synthesizable, pass the material string to the Method LLM.
- This model will classify the most likely synthesis pathway, typically into categories such as solid-state reaction or solution-based synthesis [9].
Precursor Identification:
- Finally, input the material string into the Precursor LLM.
- The model will output a list of suggested chemical precursors suitable for the synthesis. The accuracy for this step in the cited CSLLM framework was 80.2% for common binary and ternary compounds [9].
Validation & Output:
- The final output is a comprehensive synthesis report. It is strongly recommended that these computational predictions be validated by cross-referencing with literature and, ultimately, through experimental synthesis.
Protocol: De Novo Drug Design with the DRAGONFLY Framework
Objective: To generate novel, synthesizable, and bioactive molecules targeting a specific protein or based on a known ligand template.
Workflow Overview:
Materials & Reagents:
- Hardware: Computer with significant computational resources (GPU recommended).
- Software: DRAGONFLY framework, molecular visualization software (e.g., PyMOL, Chimera), cheminformatics toolkit (e.g., RDKit).
- Input Data: Either a 2D molecular graph of a known ligand (e.g., as an SDF file) or the 3D coordinates of a protein binding site (e.g., as a PDB file).
Procedure:
- Input Definition:
- For ligand-based design, provide the structure of a known active molecule.
- For structure-based design, provide the 3D structure of the target protein's binding site.
Model Processing:
- The input is converted into a graph representation. For a binding site, this is a 3D graph; for a ligand, it is a 2D molecular graph.
- The graph is processed by the Graph Transformer Neural Network (GTNN), which learns a latent representation.
- This representation is passed to the Long-Short-Term Memory (LSTM) Chemical Language Model, which decodes it into a SMILES string representing a novel molecule [33].
In-silico Evaluation of Generated Molecules:
- Synthesizability: Evaluate the Retrosynthetic Accessibility Score (RAScore) to filter out molecules that are theoretically impossible or prohibitively difficult to synthesize [33].
- Novelty: Compute the structural and scaffold novelty relative to known bioactive molecules in databases like ChEMBL to ensure intellectual property potential.
- Bioactivity Prediction: Use pre-trained quantitative structure-activity relationship (QSAR) models to predict the half-maximal inhibitory concentration (pIC50) against the intended target.
- Physicochemical Properties: Ensure generated molecules adhere to drug-like criteria (e.g., molecular weight, lipophilicity). The DRAGONFLY model showed a correlation of r ≥ 0.95 between desired and generated properties [33].
Output and Prioritization:
- Rank the generated molecules based on the combined scores from Step 3.
- The output is a prioritized list of novel, synthesizable, and predicted bioactive candidates for further experimental investigation.
Successful implementation of these advanced models relies on a foundation of high-quality data and software tools.
Table 2: Key Research Reagents and Resources for LLM-Based Discovery
| Resource Name | Type | Function in Research | Relevance to Protocol |
|---|---|---|---|
| Inorganic Crystal Structure Database (ICSD) [2] [9] | Materials Database | Primary source of experimentally synthesized crystal structures used for training and benchmarking. | Serves as the ground-truth source for positive examples in synthesizability models. |
| ChEMBL Database [33] | Bioactivity Database | A manually curated database of bioactive molecules with drug-like properties, containing binding and functional assay data. | Forms the core of the drug-target interactome for training models like DRAGONFLY. |
| Materials Project (MP)/OQMD/JARVIS [9] | Materials Database | Repositories of computationally generated crystal structures and their properties. | Source of candidate structures and negative/non-synthesizable examples for training. |
| Retrosynthetic Accessibility Score (RAScore) [33] | Software Metric | A metric for assessing the feasibility of synthesizing a given molecule. | Key evaluation filter in the de novo drug design protocol (Section 3.2). |
| SMILES String [33] | Data Representation | A line notation for representing molecular structures as text, enabling LLMs to process chemical information. | The standard input/output format for chemical language models (CLMs) like the one in DRAGONFLY. |
| Material String [9] | Data Representation | A specialized text representation for crystal structures that efficiently encodes lattice, composition, and symmetry. | The required input format for the CSLLM framework in the synthesizability prediction protocol (Section 3.1). |
| Graph Transformer Neural Network (GTNN) [33] | Algorithm | A type of neural network that operates on graph-structured data, ideal for learning from molecular graphs or binding sites. | Core component of the DRAGONFLY framework for processing structural input. |
Benchmarking SynthNN: Performance Against Experts and Newer AI
The discovery of new inorganic crystalline materials is a fundamental driver of technological advancement. A critical bottleneck in this process is identifying which hypothetical materials are synthetically accessible, a challenge traditionally reliant on the expertise of solid-state chemists [2]. The SynthNN (Synthesizability Neural Network) deep learning model addresses this by reformulating material discovery as a synthesizability classification task [2] [6]. This application note details a head-to-head comparison between SynthNN and human experts, provides protocols for its application, and contextualizes its role within the evolving landscape of synthesizability prediction research.
Performance Analysis: SynthNN vs. Human Experts
In a controlled discovery task, SynthNN's performance was benchmarked against 20 expert materials scientists. The model demonstrated a fundamental shift in efficiency and precision for identifying synthesizable materials [2].
Table 1: Performance Metrics: SynthNN vs. Human Experts
| Metric | SynthNN | Best Human Expert | Improvement Factor |
|---|---|---|---|
| Precision | 1.5× higher | Baseline | 1.5× [2] |
| Task Completion Time | Minutes to hours | Weeks to months | ~5 orders of magnitude faster [2] |
| Comparative Performance | Outperformed all 20 experts | Best among human group | - [2] |
This performance is not achieved through the explicit programming of chemical rules. Experimental analyses indicate that SynthNN, trained solely on composition data from the Inorganic Crystal Structure Database (ICSD), independently learns fundamental chemical principles such as charge-balancing, chemical family relationships, and ionicity to inform its predictions [2].
Experimental Protocols & Workflows
Core SynthNN Model Protocol
The following protocol outlines the key steps in developing and applying the SynthNN model for synthesizability prediction.
Objective: To train a deep learning model that classifies inorganic chemical compositions as synthesizable or unsynthesizable, without requiring prior crystal structure information [2].
Materials & Data Sources:
- Positive Data: Chemical compositions of synthesizable, crystalline inorganic materials from the Inorganic Crystal Structure Database (ICSD) [2].
- Unlabeled Data: Artificially generated chemical compositions that represent the space of unsynthesized (and potentially unsynthesizable) materials [2].
Procedure:
- Data Preparation: Extract and curate a comprehensive set of chemical formulas from the ICSD to serve as positive (synthesizable) examples [2].
- Representation Learning: Employ the
atom2vecframework to represent each chemical formula via a learned atom embedding matrix. This model learns an optimal representation of chemical compositions directly from the data distribution [2]. - Model Training: Train a deep learning classification model (SynthNN) using a Positive-Unlabeled (PU) Learning approach. This semi-supervised method accounts for the lack of definitively negative data by treating the artificially generated materials as unlabeled and probabilistically reweighting them based on their likelihood of being synthesizable [2].
- Validation: Benchmark model performance against baseline methods, including random guessing and charge-balancing criteria, using standard classification metrics like precision and F1-score [2].
Protocol for a Modern Synthesizability-Guided Discovery Pipeline
Recent research has advanced beyond composition-only models. The following protocol describes an integrated pipeline that combines compositional and structural synthesizability scores for experimental discovery [11] [34].
Objective: To prioritize and experimentally synthesize novel, theoretically-predicted crystal structures by employing a unified synthesizability score.
Materials & Data Sources:
- Candidate Structures: Computational databases (e.g., Materials Project, GNoME, Alexandria) [11].
- Training Data: A labeled dataset where synthesizable materials have ICSD entries, and unsynthesizable materials are theoretical polymorphs from the Materials Project [34].
- Synthesis Planning Models: Precursor-suggestion (e.g., Retro-Rank-In) and calcination temperature prediction (e.g., SyntMTE) models trained on literature-mined synthesis data [11].
Procedure:
- Candidate Screening: Screen a large pool (e.g., millions) of computational crystal structures [11].
- Dual-Model Synthesizability Scoring:
- Compositional Analysis: Encode the material's composition (
x_c) using a fine-tuned transformer model (f_c) to output a compositional synthesizability score (s_c) [34]. - Structural Analysis: Encode the material's crystal structure (
x_s) using a Graph Neural Network (f_s) to output a structural synthesizability score (s_s) [34].
- Compositional Analysis: Encode the material's composition (
- Rank-Average Ensemble: Aggregate the compositional and structural scores using a rank-average ensemble (Borda fusion). This creates a robust, unified
RankAvg(i)score for each candidatei, prioritizing materials with high consensus synthesizability [11] [34]. - Precursor Selection & Reaction Planning: For high-priority candidates, use a precursor-suggestion model to generate a ranked list of viable solid-state precursors. Then, predict the required calcination temperature, balance the reaction, and compute precursor quantities [11].
- High-Throughput Experimental Validation: Execute the suggested synthesis recipes in an automated solid-state laboratory platform and characterize the products using X-ray diffraction (XRD) to verify the target structure [11] [34].
Figure 1: Integrated synthesizability prediction and discovery workflow [11] [34].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational and Experimental "Reagents"
| Item Name | Function/Brief Explanation |
|---|---|
| Inorganic Crystal Structure Database (ICSD) | A curated database of experimentally synthesized inorganic crystal structures. Serves as the primary source of "positive" data for training supervised and PU-learning models [2] [9]. |
| Materials Project / GNoME / Alexandria | Large-scale databases of DFT-computed crystal structures. Provide a pool of candidate materials for screening and a source of "theoretical" structures for training [11] [34]. |
| atom2vec / Compositional Embeddings | A framework that learns a numerical representation (embedding) for each element directly from the distribution of known materials, enabling the model to capture complex chemical relationships [2]. |
| Graph Neural Networks (GNNs) | A class of deep learning models that operate directly on the graph representation of a crystal structure (atoms as nodes, bonds as edges), capturing local coordination and structural motifs [34]. |
| Positive-Unlabeled (PU) Learning | A semi-supervised machine learning paradigm designed for situations with only positive and unlabeled data, which is ideal for synthesizability prediction where negative data is scarce [2] [25]. |
| Solid-State Precursors | High-purity, typically powdered, starting materials (e.g., metal oxides, carbonates) used in solid-state synthesis reactions to form the target crystalline material [11]. |
The Evolving Landscape of Synthesizability Prediction
The field is rapidly advancing beyond the benchmarks set by SynthNN. Newer models are demonstrating even greater accuracy and expanding their capabilities.
Table 3: Evolution of Synthesizability Prediction Models
| Model | Key Innovation | Reported Accuracy / Performance |
|---|---|---|
| SynthNN [2] | Composition-based deep learning using PU-learning. | 1.5× higher precision than best human expert; 7× higher precision than DFT formation energies. |
| CSLLM (Crystal Synthesis LLM) [9] | Uses fine-tuned Large Language Models (LLMs) with a text-based "material string" representation of crystal structures. | 98.6% accuracy in synthesizability prediction; also predicts synthetic methods and precursors with >90% accuracy. |
| SynCoTrain [25] | A dual-classifier co-training framework using two GNNs (ALIGNN & SchNet) to reduce model bias and improve generalizability in PU-learning. | Achieves high recall on internal and leave-out test sets for oxide crystals. |
The integration of structural information is critical. Research shows that a pipeline using only a compositional synthesizability model resulted in zero successful syntheses, whereas a combined composition-and-structure approach achieved a 44% experimental success rate in synthesizing target materials [34]. This highlights the complementary roles of composition (governing elemental chemistry and precursor availability) and structure (capturing local coordination and motif stability) in accurate synthesizability assessment [34].
Figure 2: Progression of synthesizability prediction paradigms.
Within computational materials science, a significant challenge persists: bridging the gap between theoretically predicted materials and those that can be experimentally synthesized. The conventional discovery cycle, often reliant on trial-and-error, can span months or even years [35]. To increase the efficiency of this process, accurate predictors of synthesizability are paramount. This application note quantitatively benchmarks a novel deep learning model, SynthNN, against two established proxies for synthesizability: the charge-balancing criterion and density functional theory (DFT)-calculated formation energies. Framed within broader thesis research on deep learning for synthesizability prediction, this analysis provides researchers with a clear, data-driven comparison of these methodologies, underscoring the performance advantages of a dedicated data-driven synthesizability model.
Quantitative Performance Comparison
The table below summarizes the key performance metrics of SynthNN, charge-balancing, and DFT-based formation energy analysis as reported in foundational literature [2] [9].
Table 1: Quantitative Benchmarking of Synthesizability Prediction Methods
| Method | Key Performance Metric | Reported Performance | Principal Advantage | Principal Limitation |
|---|---|---|---|---|
| SynthNN (Deep Learning) | Precision in identifying synthesizable materials | 7x higher precision than DFT formation energies; Outperformed 20 human experts (1.5x higher precision) [2] | Learns complex chemical principles directly from data; extremely fast screening [2] | Requires large datasets of known materials; "black box" nature can limit interpretability |
| Charge-Balancing (Heuristic) | Percentage of known synthesized materials correctly identified as synthesizable | Only 37% of known inorganic materials in ICSD are charge-balanced [2] | Computationally inexpensive; chemically intuitive [2] [35] | Inflexible; fails for metallic, covalent, and many ionic materials [2] |
| DFT Formation Energy (Thermodynamic) | Accuracy in classifying synthesizability (vs. dedicated ML models) | ~74.1% accuracy (Energy above hull ≥0.1 eV/atom) [9] | Provides foundational thermodynamic insight [2] | Fails to account for kinetic stabilization and non-thermodynamic factors; computationally expensive [2] [35] |
| CSLLM (Advanced LLM on Structure) | Accuracy on testing data | 98.6% accuracy [9] | Considers full crystal structure; suggests synthesis methods and precursors [9] | Requires known crystal structure, which is often unknown for novel materials [2] |
The data reveals that traditional proxy methods are substantially less effective than machine learning approaches for synthesizability classification. The charge-balancing criterion performs particularly poorly, failing to identify the majority of known compounds as synthesizable [2]. While DFT-based stability is a useful filter, it captures only a portion of the factors that influence real-world synthesis [2] [35]. In contrast, SynthNN leverages the entire space of synthesized inorganic compositions to achieve a significant leap in predictive precision [2]. Subsequent models like CSLLM demonstrate that even higher accuracy is achievable when structural data is available, though this is often a constraint for novel composition discovery [9].
Experimental Protocols & Workflows
Protocol for SynthNN Model Training and Benchmarking
The following protocol outlines the key experimental steps for developing and benchmarking the SynthNN model, as detailed in the original research [2] [6].
1. Data Curation: - Source: Extract positive examples of synthesizable materials from the Inorganic Crystal Structure Database (ICSD), which contains experimentally reported crystalline inorganic materials [2]. - Handling Unlabeled Data: Generate a set of artificially created chemical formulas to represent unsynthesized/unsynthesizable materials. Employ a Positive-Unlabeled (PU) learning framework to account for the possibility that some of these artificially generated materials could be synthesizable but not yet reported [2].
2. Model Architecture and Training:
- Input Representation: Utilize the atom2vec representation, which learns an optimal vector representation (embedding) for each element directly from the distribution of known chemical compositions. This bypasses the need for manual feature engineering or prior chemical knowledge [2].
- Network: Implement a deep neural network that takes the learned atom embeddings as input.
- Training Objective: Train the model as a binary classifier to distinguish between synthesizable and unsynthesizable compositions. The model learns chemical principles like charge-balancing and chemical family relationships implicitly from the data [2].
3. Benchmarking and Evaluation: - Baselines: Compare SynthNN's performance against two primary baselines: - Charge-Balancing: A material is predicted as synthesizable only if its nominal ionic charges balance to zero using common oxidation states [2]. - DFT Formation Energy: A material is predicted as synthesizable if it is thermodynamically stable (i.e., it has no decomposition products with lower energy) [2]. - Human Expert Comparison: Conduct a head-to-head discovery challenge where SynthNN and 20 expert materials scientists evaluate the synthesizability of candidate materials. Compare the precision and speed of the model against the human experts [2].
Protocol for Charge-Balancing and DFT Formation Energy Analysis
1. Charge-Balancing Workflow: - Oxidation State Assignment: Assign probable oxidation states to each element in the chemical formula based on common values from chemistry references (e.g., O = -2, Alkali metals = +1) [2]. - Calculation: Multiply the oxidation state by the stoichiometric coefficient for each element. - Decision Rule: Sum the contributions from all elements. If the total sum is zero, the material is predicted to be synthesizable; otherwise, it is not [2] [35].
2. DFT Formation Energy Workflow:
- Structure Relaxation: For a given crystal structure, perform a DFT calculation to relax the atomic coordinates and cell parameters to their ground state [36].
- Total Energy Calculation: Calculate the total energy of the relaxed compound, Etot(compound).
- Reference Phase Energies: Calculate the total energies of the most stable reference phases for each constituent element, Etot(element).
- Formation Energy (ΔH_f) Calculation: Compute the formation energy using the formula:
ΔH_f = E_tot(compound) - Σ n_i * E_tot(element_i)
where n_i is the number of atoms of element i in the compound.
- Stability Assessment: A material is considered thermodynamically stable (and thus likely synthesizable) if its formation energy is negative and it lies on the convex hull of stable phases (i.e., it has no exothermic decomposition pathway) [2] [36].
The logical flow for the comparative benchmarking study is summarized in the diagram below.
Successful development and application of synthesizability models rely on several key data, software, and computational resources.
Table 2: Key Resources for Synthesizability Prediction Research
| Resource Name | Type | Primary Function in Research | Relevance & Notes |
|---|---|---|---|
| Inorganic Crystal Structure Database (ICSD) | Data Repository | Provides a comprehensive collection of experimentally synthesized inorganic crystal structures for training positive examples in machine learning models. | The primary source of "ground truth" data for synthesizable materials [2] [9]. |
| Materials Project (MP) | Computational Database | Serves as a source of theoretically predicted, potentially unsynthesized structures for generating negative or unlabeled data samples. | Contains DFT-calculated formation energies for stability comparison [9] [11]. |
| atom2vec / Composition Descriptors | Software/Algorithm | Represents chemical compositions as numerical vectors, enabling machine learning models to process and learn from material formulas. | Learns elemental relationships directly from data, avoiding manual feature design [2]. |
| Density Functional Theory (DFT) | Computational Method | Calculates fundamental material properties, most notably formation energy, which serves as a thermodynamic proxy for synthesizability. | Computationally intensive; used as a baseline and for generating data in other databases [2] [36] [37]. |
| Positive-Unlabeled (PU) Learning | Machine Learning Framework | A semi-supervised learning technique that handles datasets where only positive labels (synthesized materials) are reliable, and negative examples are unlabeled or uncertain. | Crucial for addressing the lack of confirmed "unsynthesizable" material data [2]. |
This quantitative benchmarking study firmly establishes that dedicated deep learning models like SynthNN offer a substantial performance advantage over traditional heuristic and thermodynamic methods for predicting the synthesizability of inorganic crystalline materials. By learning directly from the full breadth of existing experimental data, SynthNN captures the complex, multi-faceted nature of solid-state synthesis that is not fully encapsulated by simple charge neutrality or formation energy alone. The integration of such data-driven synthesizability models into computational screening workflows is a critical step towards increasing the reliability and throughput of autonomous materials discovery pipelines, ensuring that predicted materials are not only thermodynamically plausible but also synthetically accessible.
The accurate prediction of material synthesizability represents a critical bottleneck in accelerating the discovery of new functional materials and drug compounds. Traditional computational screening methods, which often rely on thermodynamic or kinetic stability metrics, have proven insufficient for reliably identifying synthetically accessible structures [24]. The emergence of large language models (LLMs) offers a transformative approach by learning the complex patterns underlying successful synthesis directly from experimental data. This application note details the Crystal Synthesis Large Language Models (CSLLM) framework and places it in context against other contemporary LLM-based frameworks, such as the SynthNN deep learning model, highlighting their respective protocols, performance, and applications within drug discovery and materials science [24] [2].
Quantitative Comparison of LLM-Based Frameworks
The table below summarizes the key quantitative metrics and characteristics of CSLLM, SynthNN, and other notable LLM frameworks in drug discovery.
Table 1: Comparative Analysis of LLM-Based Frameworks for Synthesizability and Molecule Design
| Framework | Primary Application | Core Methodology | Key Performance Metrics | Input Data Format |
|---|---|---|---|---|
| CSLLM [24] | Predicting synthesizability, synthetic methods, and precursors for 3D crystal structures | Three specialized LLMs fine-tuned on a comprehensive dataset of synthesizable/non-synthesizable structures. | Synthesizability prediction accuracy: 98.6%; Method classification accuracy: >90%; Precursor prediction success: 80.2%. | Material string (text representation of crystal structure) |
| SynthNN [2] | Predicting synthesizability of inorganic crystalline materials from composition | Deep learning model using atom2vec embeddings, trained via Positive-Unlabeled (PU) learning on ICSD data. | Outperformed DFT-based formation energy screening by 7x higher precision; surpassed human expert precision by 1.5x. | Chemical composition |
| GAMES [38] | Accelerating drug discovery via molecular generation | Custom LLM fine-tuned with LoRA/QLoRA to generate valid SMILES strings. | Increased generation of valid SMILES strings; reduced invalid outputs. | SMILES strings |
| Multi-Step Retrosynthesis Framework [39] | Planning multi-step chemical synthesis routes | LLM-powered framework using molecular-similarity-based Retrieval-Augmented Generation (RAG) and iterative refinement. | Achieved 79.5% overall route validity after refinement (initial validity: 51.64%). | Target molecule (e.g., SMILES) |
| DrugAssist & MolGPT [40] | De novo drug design and molecule optimization | Transformer-based architectures conditioned on specific properties for molecular generation. | Generated bioactive HCN2 inhibitors, verified in lab settings. | SMILES strings, molecular graphs |
Detailed Experimental Protocols
Protocol for CSLLM Framework Development and Validation
Objective: To develop and validate a framework of three specialized LLMs for ultra-accurate prediction of crystal structure synthesizability, synthetic methods, and suitable precursors [24].
Materials and Reagents (Computational):
- Hardware: High-performance computing clusters.
- Software: Python, PyTorch/TensorFlow, crystallography analysis libraries.
- Data Sources: Inorganic Crystal Structure Database (ICSD), Materials Project, Computational Material Database, Open Quantum Materials Database, JARVIS database.
Methods:
- Dataset Curation:
- Positive Samples: 70,120 synthesizable crystal structures were curated from the ICSD, filtering for ordered structures with ≤40 atoms and ≤7 different elements [24].
- Negative Samples: 80,000 non-synthesizable structures were identified from a pool of 1,401,562 theoretical structures using a pre-trained PU learning model. Structures with a CLscore <0.1 were selected [24].
- The final balanced dataset of 150,120 structures was validated using t-SNE visualization to confirm coverage of diverse crystal systems and elemental compositions.
Text Representation of Crystal Structures:
- A novel "material string" representation was developed to convert crystal structures into a concise, reversible text format for efficient LLM processing.
- The format is:
Space Group | a, b, c, α, β, γ | (Element1-Site1[WyckoffPosition1,x1,y1,z1]), (Element2-Site2[WyckoffPosition2,x2,y2,z2]), ...[24]. - This representation eliminates redundant coordinate information by leveraging space group and Wyckoff position symmetry.
Model Fine-Tuning and Architecture:
- Three separate LLMs (Synthesizability LLM, Method LLM, Precursor LLM) were fine-tuned from a base pre-trained LLM.
- The fine-tuning process involved domain adaptation on the constructed dataset, aligning the models' attention mechanisms with material features critical to synthesizability to reduce hallucination [24].
Model Validation:
- Synthesizability LLM: Tested on a held-out dataset, achieving 98.6% accuracy. Its generalization was further validated on complex structures with large unit cells, achieving 97.9% accuracy [24].
- Method and Precursor LLMs: Evaluated against ground-truth data, achieving >90% accuracy in classifying solid-state or solution synthesis methods and 80.2% success in predicting precursors for binary and ternary compounds [24].
Protocol for SynthNN Model
Objective: To predict the synthesizability of inorganic crystalline materials directly from their chemical composition, without requiring structural information [2].
Methods:
- Data Sourcing and Preprocessing:
- Positive Data: Chemical formulas of synthesizable materials were extracted from the Inorganic Crystal Structure Database (ICSD).
- Unlabeled Data: A large set of artificially generated chemical formulas, not present in ICSD, served as the unlabeled (potentially non-synthesizable) class [2].
Model Training with PU Learning:
- The model, SynthNN, was built using the atom2vec framework, which learns an optimal vector representation (embedding) for each atom directly from the distribution of synthesized materials [2].
- A semi-supervised Positive-Unlabeled (PU) learning approach was employed. This method probabilistically reweights the unlabeled examples during training according to their likelihood of being synthesizable, rather than treating them as definitive negative samples [2].
Validation and Benchmarking:
- Model performance was benchmarked against traditional methods like charge-balancing and DFT-calculated formation energy.
- SynthNN was also evaluated in a head-to-head comparison against 20 expert material scientists, outperforming all in both precision and speed [2].
Table 2: Key Computational Tools and Data for LLM-Driven Discovery
| Item Name | Function/Application | Specifications/Examples | ||
|---|---|---|---|---|
| Material String [24] | A concise text representation for encoding crystal structure information (space group, lattice parameters, atomic coordinates) for LLM input. | Format: `SP | a, b, c, α, β, γ | (AS1-WS1[WP1...))` |
| SMILES Strings [40] [38] | A standardized notation system representing molecular structures as short text strings, enabling LLMs to process and generate chemical compounds. | Used by GAMES, MolGPT, DrugAssist, and other frameworks for molecular generation. | ||
| ICSD Database [24] [2] | A critical source of experimentally synthesized and characterized crystal structures, used as positive training data for synthesizability models. | Contains over 70,000 ordered crystal structures used in CSLLM and SynthNN development. | ||
| PU Learning [2] | A semi-supervised machine learning technique critical for training synthesizability models, where only positive (synthesized) data is definitive, and negative data is unlabeled. | Used by SynthNN to handle artificially generated unsynthesized materials. | ||
| LoRA / QLoRA [38] | Parameter-efficient fine-tuning techniques that dramatically reduce computational cost and hardware requirements for adapting large LLMs to specialized scientific domains. | Used by the GAMES LLM for efficient fine-tuning on SMILES strings. | ||
| RAG (Retrosynthesis) [39] | Retrieval-Augmented Generation enhances LLMs for retrosynthesis by retrieving relevant reaction examples from a database to guide the planning of valid synthetic routes. | Molecular-similarity-based RAG improved reaction round-trip validity from 24.42% to 51.64%. |
Comparative Workflow and Decision Pathways
The following diagram illustrates the logical relationships and decision pathways for selecting an appropriate LLM framework based on the research objective, highlighting the distinct approaches of CSLLM and SynthNN.
Within the broader research on the SynthNN deep learning model for synthesizability prediction, a critical evaluation of its accuracy and generalization capabilities, particularly on complex crystal structures, is paramount for adoption in real-world materials discovery and drug development pipelines. Accurately predicting whether a theoretical inorganic crystalline material can be successfully synthesized bridges the gap between computational screening and experimental realization [41]. While early synthesizability models like SynthNN demonstrated the feasibility of this task, subsequent research has significantly advanced the state-of-the-art, achieving remarkable accuracy and robustness on structurally complex compounds. This Application Note summarizes quantitative performance benchmarks against newer models, provides detailed experimental protocols for validation, and offers essential tools for researchers to implement these assessments.
Performance Benchmarking
The table below summarizes the performance of various synthesizability prediction models, highlighting the evolution in accuracy and capability.
Table 1: Performance Comparison of Synthesizability Prediction Models
| Model Name | Input Type | Reported Accuracy | Key Performance Metrics | Handles Complex Structures? |
|---|---|---|---|---|
| SynthNN [2] | Composition | Outperformed human experts (1.5x higher precision) | Precision: Up to 70.2% (at high threshold); Recall: Up to 85.9% (at low threshold) [16] | Limited data on very complex cells |
| Crystal Synthesis LLM (CSLLM) [9] | Crystal Structure (Text) | 98.6% (Overall) | 97.9% accuracy on complex structures with large unit cells [9] | Yes, demonstrated explicitly |
| Teacher-Student DNN (TSDNN) [8] | Crystal Structure | 92.9% (True Positive Rate) | Improved baseline PU learning true positive rate from 87.9% to 92.9% [8] | Not explicitly tested |
| Synthesizability-Guided Pipeline [11] | Composition & Structure | Experimental Success: 7/16 Targets | Successfully synthesized 7 of 16 computationally proposed targets [11] | Implied by experimental success |
The pursuit of higher accuracy has led to diverse approaches. The Crystal Synthesis Large Language Model (CSLLM) framework represents a significant leap, achieving 98.6% accuracy by leveraging a fine-tuned large language model on a comprehensive dataset of 150,120 crystal structures [9]. Crucially, its generalization was tested on structures with "complexity considerably exceeding that of the training data," where it maintained a 97.9% accuracy [9]. Other models, like the Teacher-Student Dual Neural Network (TSDNN), focus on efficient learning from limited data, increasing the true positive rate for synthesizability prediction to 92.9% while using 98% fewer parameters than a previous benchmark [8].
Experimental Protocols for Validation
Protocol: Benchmarking Against Thermodynamic and Kinetic Stability Metrics
This protocol outlines a head-to-head comparison between a data-driven synthesizability model and traditional physics-based stability metrics.
Table 2: Essential Reagents for Synthesizability Validation
| Research Reagent / Resource | Function / Explanation |
|---|---|
| Inorganic Crystal Structure Database (ICSD) | Provides a curated set of experimentally synthesized, and therefore synthesizable, materials as positive examples for model training and testing [2] [9]. |
| Materials Project (MP) Database | A source of DFT-calculated structures, many of which are theoretical and can be used as a source of negative or unlabeled examples in a Positive-Unlabeled (PU) learning framework [11] [8] [41]. |
| Pre-Trained Positive-Unlabeled (PU) Model | Used to generate a crystallikeness score (CLscore) to programmatically identify likely non-synthesizable structures from large databases for creating balanced test sets [9]. |
| Density Functional Theory (DFT) Code | Used to calculate formation energy and energy above the convex hull (Ehull), which are traditional thermodynamic proxies for synthesizability used as performance baselines [41]. |
| Phonon Spectrum Analysis Software | Computes phonon frequencies to assess kinetic stability, another common baseline for judging synthesizability potential [9]. |
Dataset Curation:
- Positive Examples: Select a benchmark set of confirmed synthesizable crystal structures from the ICSD. The CSLLM study used 70,120 structures, ensuring they are ordered and within a manageable size (e.g., ≤40 atoms, ≤7 elements) [9].
- Negative Examples: Construct a reliable set of non-synthesizable structures. This can be done by applying a pre-trained PU learning model (e.g., from Jang et al. [9]) to large theoretical databases (MP, OQMD, AFLOW) and selecting structures with the lowest CLscore (e.g., <0.1) [9]. The CSLLM study selected 80,000 such structures to create a balanced dataset [9].
- Complexity Stratification: Partition the test set based on structural complexity metrics, such as the number of atoms in the unit cell or the number of unique elements, to specifically evaluate performance on complex structures.
Baseline Calculation:
- Calculate the formation energy and energy above the convex hull (Ehull) for all test structures using DFT. A typical threshold for stability is Ehull ≤ 0.1 eV/atom [9].
- Compute the lowest phonon frequency to check for kinetic instability. A threshold like ≥ -0.1 THz can be used to define kinetically stable structures [9].
Model Evaluation:
- Run the synthesizability prediction model (e.g., SynthNN, CSLLM) on the curated test set.
- Compare the model's accuracy, precision, and recall against the predictions made by the thermodynamic (Ehull) and kinetic (phonon) stability criteria.
Diagram 1: Benchmark Validation Workflow (82 characters)
Protocol: Experimental Validation of Model Predictions
The most rigorous test of a synthesizability model is the successful synthesis of its predictions. The following protocol is adapted from a synthesizability-guided pipeline that achieved a 44% success rate (7/16 targets) [11].
Candidate Screening and Prioritization:
- Apply the synthesizability model to a large pool of theoretical structures (e.g., from GNoME, Materials Project). The referenced study screened 4.4 million structures [11].
- Prioritize candidates using a high synthesizability score threshold (e.g., a rank-average > 0.95) [11].
- Apply practical filters, such as excluding compounds with platinoid group elements, non-oxides, or toxic compounds [11].
Retrosynthetic Planning:
High-Throughput Synthesis and Characterization:
- Execute the synthesis reactions in an automated high-throughput laboratory platform.
- Characterize the resulting products using techniques like X-ray Diffraction (XRD) to verify if the crystal structure of the synthesized product matches the predicted target [11].
Diagram 2: Experimental Validation Protocol (73 characters)
Implementation and Technical Toolkit
For researchers seeking to implement a synthesizability prediction pipeline, particularly for handling complex structures, the following technical details are critical.
Table 3: Advanced Model Architectures for Complex Structures
| Model Architecture | Description | Advantage for Complex Structures |
|---|---|---|
| Composition & Structure Ensemble [11] | Combines a compositional transformer (MTEncoder) with a structural Graph Neural Network (GNN), aggregating their scores via rank-average. | Captures both elemental chemistry and long-range structural motifs, providing a more holistic assessment. |
| Large Language Model (LLM) [9] | Utilizes a fine-tuned LLM on a "material string" text representation that integrates essential crystal information (lattice, composition, coordinates, symmetry). | Leverages the vast knowledge and pattern recognition capabilities of LLMs, showing exceptional generalization. |
| Teacher-Student Dual Network (TSDNN) [8] | A semi-supervised model where a teacher network generates pseudo-labels for unlabeled data to train a student network. | Effectively exploits large amounts of unlabeled data, overcoming the scarcity of confirmed negative examples. |
Key Technical Considerations
- Data Representation for Complex Structures: The CSLLM framework introduced an efficient text representation for crystal structures to fine-tune LLMs, moving beyond redundant CIF or POSCAR formats. This representation includes essential information on lattice, composition, atomic coordinates, and symmetry without redundancy, which is crucial for processing large, complex unit cells efficiently [9].
- Handling the "Negative Data" Problem: A fundamental challenge is the lack of confirmed non-synthesizable materials. The field widely adopts Positive-Unlabeled (PU) learning algorithms [2] [8]. In this setup, known synthesized materials are "positives," while a vast set of theoretical structures are treated as "unlabeled." Models are then trained to probabilistically identify the most likely negatives from the unlabeled set [2].
The accurate prediction of a material's synthesizability—the likelihood that it can be successfully synthesized in a laboratory—is a critical bottleneck in computational materials discovery. While models like SynthNN can screen millions of candidate compositions, their true value is only realized when their predictions are validated through actual synthesis experiments [2] [11]. This document provides detailed application notes and protocols for the experimental validation of materials identified by synthesizability prediction models such as SynthNN, serving as a practical guide for researchers aiming to bridge the gap between computational prediction and experimental realization.
Performance Benchmarks of Synthesizability Models
Before designing validation experiments, it is essential to understand the performance capabilities of existing synthesizability prediction models. The following table summarizes key quantitative metrics for several state-of-the-art models.
Table 1: Performance comparison of synthesizability prediction models
| Model Name | Input Type | Reported Accuracy | Key Performance Metrics | Primary Data Source |
|---|---|---|---|---|
| SynthNN [2] | Chemical composition | Not explicitly stated (outperforms human experts by 1.5x in precision) | 7x higher precision than DFT formation energies; various precision/recall values at different thresholds (see Table 2) | ICSD |
| CSLLM [9] | Crystal structure (via text representation) | 98.6% | Outperforms thermodynamic (74.1%) and kinetic (82.2%) stability methods | ICSD & theoretical databases |
| Structure-Based PU Learning [9] | Crystal structure | 87.9% | Uses Positive-Unlabeled (PU) learning | ICSD & theoretical databases |
| Teacher-Student Network [9] | Crystal structure | 92.9% | Improved accuracy over basic PU learning | ICSD & theoretical databases |
SynthNN specifically provides a precision-recall profile that can guide the selection of an appropriate decision threshold for experimental campaigns, balancing the risk of false positives against the chance of missing viable candidates.
Table 2: SynthNN performance at different prediction thresholds on a dataset with a 20:1 ratio of unsynthesized to synthesized examples [16]
| Decision Threshold | Precision | Recall |
|---|---|---|
| 0.10 | 0.239 | 0.859 |
| 0.50 | 0.563 | 0.604 |
| 0.90 | 0.851 | 0.294 |
Experimental Validation Workflow
The following diagram outlines the integrated computational-experimental workflow for validating model-predicted materials, from initial candidate selection to final characterization.
Figure 1: Integrated workflow for the validation of model-predicted materials, combining computational screening with experimental synthesis.
Detailed Experimental Protocols
Computational Candidate Selection Protocol
Objective: To identify the most promising synthesizable candidates from a large pool of theoretical structures using a tiered filtering approach.
Materials & Data Sources:
- Primary Input: A database of theoretical crystal structures (e.g., from Materials Project, GNoME, or Alexandria) [11].
- Software: Access to pre-trained SynthNN model [16] or CSLLM framework [9].
Procedure:
- Composition-Based Screening:
- Input the list of candidate compositions into SynthNN.
- Obtain synthesizability scores for each candidate.
- Apply a precision-oriented threshold (e.g., 0.80, corresponding to ~76.5% precision) to create a shortlist [16].
- Structure-Based Refinement:
- For the shortlisted compositions, obtain their corresponding crystal structures.
- Apply a structure-based synthesizability model (e.g., CSLLM [9] or a CLscore filter [11]) to further prioritize candidates. The CSLLM model achieves 98.6% accuracy in predicting synthesizability from structure [9].
- The output of this stage is a final list of high-priority targets for experimental synthesis.
Synthesis Planning Protocol
Objective: To predict viable solid-state synthesis routes and parameters for the selected candidates.
Procedure:
- Precursor Identification:
- Use a precursor-suggestion model such as Retro-Rank-In [11].
- Input the target composition and crystal structure.
- The model outputs a ranked list of potential solid-state precursor compounds.
Reaction Parameter Prediction:
- Use a synthesis parameter prediction model such as SyntMTE [11].
- Input the target material and selected precursors.
- The model predicts key parameters, most notably the calcination temperature required to form the target phase.
Reaction Balancing:
- Manually balance the chemical reaction based on the selected precursors and expected products.
- Calculate the precise stoichiometric masses of each precursor required.
Solid-State Synthesis Execution Protocol
Objective: To experimentally synthesize the target material based on the computational predictions.
Materials:
- Precursor powders (typically metal oxides, carbonates, or other salts), high-purity (>99.9%).
- Mortar and pestle (agate or alumina).
- High-temperature furnace (tube or box).
- Alumina crucibles.
- Glove box (for air-sensitive compounds).
Procedure:
- Precursor Preparation: Weigh out the precursor powders according to the calculated stoichiometric masses.
- Mixing: Mechanically mix and grind the powders in a mortar and pestle for 30-45 minutes to ensure homogeneity. For air-sensitive materials, perform this step in a glove box under an inert atmosphere.
- Pelletization (Optional): Press the mixed powder into a pellet using a hydraulic press. This step improves solid-solid contact and reaction kinetics.
- Calcination: Place the sample in an alumina crucible and heat it in a furnace at the predicted temperature (e.g., from SyntMTE). Use a heating rate of 5-10°C per minute. Hold at the target temperature for 6-12 hours.
- Regrinding and Annealing: After the initial calcination, allow the sample to cool to room temperature. Regrind the powder thoroughly to expose fresh surfaces, then subject it to a second heating cycle (annealing). This process may be repeated multiple times to improve phase purity.
- Final Cooling: After the final annealing step, cool the sample to room temperature, either slowly (e.g., 2-5°C per minute) or by quenching, as appropriate for the material.
Material Characterization Protocol
Objective: To verify the successful synthesis of the target material and assess its phase purity.
Primary Technique: Powder X-ray Diffraction (XRD)
Procedure:
- Data Collection: Grind a small portion of the final synthesized product into a fine powder. Load it into an XRD sample holder and collect a diffraction pattern using Cu Kα radiation.
- Phase Identification: Compare the collected XRD pattern to the reference pattern of the target crystal structure (e.g., from the Materials Project).
- Successful Synthesis: A match between the experimental and reference patterns, including the presence of all major peaks, confirms the successful synthesis of the target phase [11].
- Failed Synthesis: The appearance of peaks corresponding to unreacted precursors or unexpected secondary phases indicates a failed or incomplete reaction.
Supplementary Techniques:
- Scanning Electron Microscopy (SEM) and Energy-Dispersive X-ray Spectroscopy (EDX): Use these to analyze the sample's morphology and elemental composition, confirming the expected stoichiometry and revealing the particle size and distribution.
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table details the key materials, equipment, and software required for the validation pipeline.
Table 3: Essential research reagents, materials, and software for the validation pipeline
| Item Name | Specification / Example | Primary Function in Protocol |
|---|---|---|
| Precursor Oxides/Carbonates | High-purity (>99.9%) powders, e.g., MgO, TiO₂, La₂O₃ | Reactants for solid-state synthesis of target inorganic materials. |
| Alumina Crucibles | High-temperature resistant ceramic containers | Holding powder samples during high-temperature calcination and annealing steps. |
| Tube/Box Furnace | Capable of reaching 1500°C+ with programmable temperature profiles | Providing the high-temperature environment required for solid-state reactions. |
| Mortar and Pestle | Agate or alumina material to avoid contamination | Grinding and homogenizing precursor powders before and during reactions. |
| Hydraulic Press | Uniaxial press, 5-10 tons capacity | Compressing mixed powders into pellets to improve interparticle contact. |
| X-ray Diffractometer (XRD) | Powder XRD system with Cu Kα source | Determining the crystal structure and phase purity of the synthesized product. |
| SynthNN Model | Pre-trained model from official repository [16] | Providing initial synthesizability score based on chemical composition alone. |
| CSLLM Framework | Fine-tuned Large Language Model for crystals [9] | Predicting synthesizability from crystal structure with high (98.6%) accuracy. |
| Precursor Prediction Model | E.g., Retro-Rank-In [11] | Suggesting viable solid-state precursor compounds for a target material. |
Case Study & Concluding Remarks
A recent large-scale validation of a synthesizability-guided pipeline, which integrated signals from both composition and structure, screened over 4.4 million theoretical structures [11]. This process identified 24 highly synthesizable candidates. Subsequent experimental synthesis and characterization of 16 of these targets resulted in the successful synthesis of 7 materials that matched the target structure, including one completely novel phase and one previously unreported compound [11]. This success rate of ~44% (7 out of 16) demonstrates a significantly higher efficiency compared to traditional, unguided exploration and provides strong practical validation for the use of machine learning models in de-risking experimental synthesis campaigns. This document's protocols are designed to empower research groups to achieve similar success in validating and discovering new materials predicted by next-generation synthesizability models.
Conclusion
SynthNN represents a paradigm shift in predicting material synthesizability, demonstrating that deep learning can not only match but surpass human expertise and traditional computational methods in both speed and precision. Its ability to learn fundamental chemical principles like charge-balancing and ionicity directly from data opens new avenues for reliable computational material discovery. The successful experimental synthesis of candidates identified through synthesizability-guided pipelines validates its practical utility. Looking forward, the integration of SynthNN and its next-generation counterparts, such as CSLLM, into automated discovery platforms promises to dramatically accelerate the identification of novel, synthesizable materials. For biomedical and clinical research, this translates directly into a faster and more reliable path from computational design to the synthesis of new drug candidates, functional biomaterials, and therapeutic agents, ultimately shortening the timeline for bringing new treatments to patients.
References
- 1. Fake it until you make it? Generative de novo design and ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X2300132X]
- 2. Predicting the synthesizability of crystalline inorganic ... [https://www.nature.com/articles/s41524-023-01114-4]
- 3. Predicting synthesizability - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6760004/]
- 4. On synthesizability - by Huafeng Xu [https://medium.com/@huafeng/on-synthesizability-4aae0372125c]
- 5. Generate what you can make: achieving in-house ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00910-4]
- 6. Predicting the synthesizability of crystalline inorganic ... [https://www.osti.gov/pages/biblio/1996869]
- 7. Predicting the synthesizability of crystalline inorganic ... [https://ui.adsabs.harvard.edu/abs/2023npjCM...9..155A/abstract]
- 8. Materials synthesizability and stability prediction using a semi ... [https://pubs.rsc.org/en/content/articlehtml/2023/dd/d2dd00098a]
- 9. Accurate prediction of synthesizability and precursors of 3D ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12264133/]
- 10. Best‐Practice DFT Protocols for Basic Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9826355/]
- 11. A Synthesizability-Guided Pipeline for Materials Discovery [https://arxiv.org/html/2511.01790v1]
- 12. DeepSA: a deep-learning driven predictor of compound ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00771-3]
- 13. Solid-state synthesizability predictions using positive ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00065c]
- 14. Distributed representations of atoms and materials for ... [https://www.nature.com/articles/s41524-022-00729-3]
- 15. Atom2Vec: learning atoms for materials discovery [https://arxiv.org/abs/1807.05617]
- 16. antoniuk1/SynthNN [https://github.com/antoniuk1/SynthNN]
- 17. Structure motif–centric learning framework for inorganic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8059928/]
- 18. ICSD: Home [https://icsd.products.fiz-karlsruhe.de/]
- 19. NIST Inorganic Crystal Structure Database (ICSD) - National ... [https://icsd.nist.gov/]
- 20. Why Positive-Unlabeled Learning? [https://www.geeksforgeeks.org/data-science/why-positive-unlabeled-learning/]
- 21. InvDesFlow-AL: active learning-based workflow for inverse ... [https://www.nature.com/articles/s41524-025-01830-z]
- 22. A Synthesizability-Guided Pipeline for Materials Discovery [https://arxiv.org/abs/2511.01790]
- 23. A Synthesizability-Guided Pipeline for Materials Discovery [https://openreview.net/forum?id=W6i4ivbAy5]
- 24. Accurate prediction of synthesizability and precursors of 3D ... [https://www.nature.com/articles/s41467-025-61778-y]
- 25. SynCoTrain: a dual classifier PU-learning framework for ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00394b]
- 26. kaist-amsg/Synthesizability-PU-CGCNN [https://github.com/kaist-amsg/Synthesizability-PU-CGCNN]
- 27. DDI-PULearn: a positive-unlabeled learning method for large ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3214-6]
- 28. Screening drug-target interactions with positive-unlabeled ... [https://www.nature.com/articles/s41598-017-08079-7]
- 29. Positive-Unlabelled learning for identifying new candidate ... [https://www.sciencedirect.com/science/article/pii/S0010482524010849]
- 30. Deep Learning-Assisted Compound Bioactivity Estimation ... [https://www.sciencedirect.com/science/article/pii/S111086652400121X]
- 31. Lightweight Deep Learning for Resource-Constrained ... [https://arxiv.org/abs/2404.07236]
- 32. A deep learning based multi-model approach for predicting ... [https://pubmed.ncbi.nlm.nih.gov/38702021/]
- 33. Prospective de novo drug design with deep interactome ... [https://www.nature.com/articles/s41467-024-47613-w]
- 34. [Literature Review] A Synthesizability-Guided Pipeline for ... [https://www.themoonlight.io/en/review/a-synthesizability-guided-pipeline-for-materials-discovery]
- 35. How to accelerate the inorganic materials synthesis [https://pmc.ncbi.nlm.nih.gov/articles/PMC11960098/]
- 36. High-throughput calculations of charged point defect ... [https://www.nature.com/articles/s41524-023-01015-6]
- 37. Density functional theory methods applied to ... [https://pubs.rsc.org/en/content/articlehtml/2024/cp/d4cp00266k]
- 38. SwRI develops chemistry LLM called GAMES for faster ... [https://www.swri.org/newsroom/press-releases/swri-develops-chemistry-llm-called-games-faster-drug-discovery]
- 39. How Well Can LLMs Synthesize Molecules? An LLM ... [https://openreview.net/forum?id=b89OyrljJD]
- 40. Integrating large language models (LLMs) with traditional ... [https://www.cognizant.com/nl/en/insights/blog/articles/large-language-models-with-traditional-drug-discovery-methods]
- 41. Predicting Synthesizability using Machine Learning on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9996807/]