Substrate Temperature Control for Nucleation: From Fundamental Principles to Advanced Applications in Biomedical Research
This article provides a comprehensive analysis of substrate temperature as a critical parameter for controlling nucleation processes across diverse scientific fields, with a specific focus on implications for biomedical research...
Substrate Temperature Control for Nucleation: From Fundamental Principles to Advanced Applications in Biomedical Research
Abstract
This article provides a comprehensive analysis of substrate temperature as a critical parameter for controlling nucleation processes across diverse scientific fields, with a specific focus on implications for biomedical research and drug development. It explores the fundamental thermodynamic and kinetic principles governing temperature-driven nucleation, details practical methodological approaches for precise thermal control, and addresses common challenges through optimization strategies. By synthesizing insights from recent studies on protein crystallization, thin-film fabrication, and advanced materials, this review serves as a valuable resource for researchers seeking to harness nucleation control to enhance crystal quality, improve process reproducibility, and develop next-generation biotherapeutics and diagnostic technologies.
The Thermodynamic and Kinetic Foundations of Temperature-Controlled Nucleation
Theoretical Foundations of Nucleation
Classical Nucleation Theory (CNT) is the primary theoretical model used to quantitatively describe the kinetics of nucleation, which is the initial step in the spontaneous formation of a new thermodynamic phase from a metastable state [1]. The central prediction of CNT is the nucleation rate, R, which quantifies the number of nucleation events per unit volume per unit time.
The Nucleation Rate Equation
The CNT expression for the nucleation rate is: R = NS Z j exp(-ΔG^* / kBT)
Where:
- ΔG^* is the free energy barrier for the formation of a critical nucleus
- kB is the Boltzmann constant
- T is the temperature
- NS is the number of nucleation sites
- j is the rate at which molecules attach to the nucleus
- Z is the Zeldovich factor, accounting for the stability of critical clusters
The exponential term, exp(-ΔG^* / kBT), represents the probability that a fluctuation will produce a critical nucleus, making the nucleation rate exquisitely sensitive to the free energy barrier and temperature [1].
Homogeneous vs. Heterogeneous Nucleation
Homogeneous nucleation occurs within the bulk phase without preferential surfaces, while heterogeneous nucleation takes place on surfaces, impurities, or pre-existing particles. Heterogeneous nucleation is far more common because the free energy barrier is significantly reduced [1]. The reduction is described by: ΔGhet = f(θ) ΔGhom
Where f(θ) is a function of the contact angle (θ) between the nucleating phase and the substrate, ranging from 0 to 1. This relationship means that surfaces with lower contact angles (better wetting) further reduce the nucleation barrier [1].
The Critical Role of Temperature
Temperature influences nucleation kinetics through multiple mechanisms in the CNT framework, primarily by affecting the free energy barrier and molecular attachment rates.
Temperature Dependence of the Free Energy Barrier
For nucleation from a supercooled liquid, the free energy barrier has a strong temperature dependence. The free energy change per unit volume, Δgv, is approximated by: Δgv = ΔHf(Tm - T) / (VatTm)
Where:
- ΔHf is the latent heat of fusion
- Tm is the melting point temperature
- Vat is the atomic volume
This leads to expressions for the critical radius and free energy barrier that are highly temperature-dependent [1]: rc = 2σ / ΔHf × VatTm / (Tm - T) ΔG^ = 16πσ³ / [3(ΔHf)²] × [VatTm / (Tm - *T)]²
Where σ is the surface tension. These relationships show that the critical radius and energy barrier decrease as supercooling (Tm - T) increases, making nucleation more probable at lower temperatures.
Dynamic Factors and Temperature
The dynamic part of the nucleation rate, Zj, also exhibits temperature dependence. Based on the Einstein-Stokes relation: Zj ∝ T/η
Where η is viscosity. Near the melting point, Zj is approximately proportional to temperature. However, at significantly lower temperatures, the viscosity increases dramatically, causing Zj to decrease and offsetting the effect of the reduced free energy barrier [1].
Unusual Temperature Dependencies in Heterogeneous Nucleation
While homogeneous nucleation consistently shows decreasing onset saturation ratio with increasing temperature, heterogeneous nucleation can exhibit the opposite behavior. This "unusual temperature dependence" occurs when the critical cluster is more stable, on a per-molecule basis, than the bulk liquid [2]. For nucleation on silver nanoparticles, the critical saturation ratio reaches a maximum near 278 K, with positive temperature dependence (increasing saturation ratio) below this temperature and negative dependence above it [2].
Table 1: Temperature Dependence of Heterogeneous Nucleation Parameters for Water on Silver Nanoparticles [2]
| Temperature (K) | Onset Saturation Ratio (Sonset) | Critical Cluster Size (n^*) | d(lnSonset)/dT |
|---|---|---|---|
| 268.7 | 3.81 | 32 | +0.086 K⁻¹ |
| 278.2 | 4.14 | 37 | ~0 K⁻¹ |
| 288.0 | 4.02 | 31 | -0.052 K⁻¹ |
Experimental Protocols for Nucleation Research
Protocol: Isothermal Immersion Freezing with Mercuric Iodide
This protocol investigates pre-activated freezing nucleation (PFN) using mercuric iodide (HgI₂), based on the methodology of Edwards, Evans, and Zipper [3].
Research Reagent Solutions: Table 2: Essential Materials for HgI₂ Nucleation Experiments
| Item | Specification | Function |
|---|---|---|
| Mercuric Iodide (HgI₂) | 99% purity, red powder | Ice-nucleating agent exhibiting pre-activation effect |
| Distilled Water | 100 mL volume | Solvent for creating sample suspensions |
| Sterile Syringe | 0.01 cm³ capacity | Precise dispensing of uniform sample drops |
| Cold Stage | Temperature-controlled platform | Controlled cooling and warming of samples |
Procedure:
- Sample Preparation: Prepare HgI₂ suspensions by adding weighed amounts of red HgI₂ powder to distilled water to achieve concentrations of 0.02 g/mL and 0.04 g/mL [3].
- Dispensing: Draw the supernatant into a sterile syringe and dispense 121 drops of 0.01 cm³ volume onto the cold stage [3].
- Thermal Cycling:
- Cool the samples until freezing occurs at the initial nucleation temperature (Tf)
- Continue cooling below the characteristic temperature TC
- Warm the samples to a specific temperature Tw just above melting (0°C < Tw < TD)
- Repeat cycles while varying Tw [3]
- Data Collection: Record freezing temperatures for subsequent cooling cycles. Note that PFN is observed when subsequent freezing occurs at Tf^* much higher than the initial Tf (by more than 10°C) [3].
Key Findings:
- The pre-activation effect is gradually lost as the sample is heated above the melting point, with some effect still observable after heating above +5°C [3].
- PFN can be noted down to at least -6°C [3].
- The effect is interpreted in terms of surface sites, potentially through the formation of a two-dimensional ice-like monolayer on the substrate that facilitates subsequent nucleation unless destroyed by heating above TD [3].
Protocol: Investigating Temperature-Dependent Bubble Nucleation and Collapse
This protocol examines the effects of temperature on nucleation and collapse of volatile bubbles in high-density polyethylene (HDPE) melt [4].
Procedure:
- System Preparation: Adopt a HDPE/n-Hexane system in a visualization apparatus [4].
- Temperature Variation: Conduct experiments at different controlled temperatures.
- Data Measurement:
- Quantify initial bubble number, initial total bubble volume, and mean bubble diameter
- Analyze bubble diameter distribution
- Record collapse time and calculate bubble collapse rate [4]
- Model Fitting: Establish a total bubble volume collapsing model using a collapse coefficient n to describe collapse rate [4].
Key Findings:
- Initial bubble number, initial total bubble volume, and mean bubble diameter all increase with temperature [4].
- Bubble diameter distribution changes from single peak to bimodal distribution as temperature increases [4].
- Collapse time lengthens but collapse rate accelerates with increasing temperature [4].
- The maximum bubble diameter is affected by temperature mainly through changes in bubble pressure due to volatile content in the polyethylene melt [4].
Visualization of Nucleation Concepts and Workflows
Diagram 1: PFN Experimental Workflow
Diagram 2: Temperature Effect on Nucleation
Data Presentation and Analysis
Table 3: Quantitative Framework for Temperature-Dependent Nucleation Analysis
| Parameter | Symbol | Temperature Dependence | Experimental Measurement |
|---|---|---|---|
| Critical Radius | rc | ∝ 1/(Tm - T) | Indirectly via nucleation statistics |
| Free Energy Barrier | ΔG^* | ∝ 1/(Tm - T)² | Derived from nucleation rate measurements |
| Nucleation Rate | R | exp[-ΔG^/(kBT*)] | Directly measurable as events per unit time |
| Onset Saturation Ratio | Sonset | Increases or decreases with T | Measured at constant nucleation probability |
| Critical Cluster Size | n^* | Varies with T and substrate | Determined from nucleation probability slope |
Table 4: Comparison of Nucleation Types and Characteristics
| Characteristic | Homogeneous Nucleation | Heterogeneous Nucleation | Pre-activated Freezing Nucleation |
|---|---|---|---|
| Free Energy Barrier | High | Reduced by f(θ) | Further reduced by prior freezing |
| Temperature Dependence | Always negative | Can be positive or negative | Highly dependent on thermal history |
| Experimental System | Pure liquid without impurities | Liquid with immersed particles | Specific substances like HgI₂ |
| Onset Temperature | Lower | Higher | Highest (near melting point) |
| Stochastic Nature | Pure stochastic | Stochastic with site dependence | History-dependent |
Chemical potential and Gibbs Free Energy are foundational concepts in thermodynamics that describe the driving forces behind physical transformations and chemical reactions. Chemical potential (( \mu )), defined as the partial molar Gibbs free energy, represents the change in a system's free energy when particles are added or removed, serving as the potential for substance transfer. Gibbs Free Energy (( G )) combines enthalpy and entropy into a single value (( G = H - TS )) to predict process spontaneity at constant temperature and pressure. Processes proceed spontaneously in the direction of decreasing Gibbs Free Energy, reaching equilibrium when ( dG = 0 ). In nucleation research, these principles govern the initial formation of new phases from parent phases, determining critical nucleus size, nucleation rates, and the kinetic pathways of phase transformations.
The interplay between chemical potential differences and free energy barriers is crucial for understanding nucleation phenomena. The Clapeyron equation, derived from the equality of chemical potentials between coexisting phases, describes the slope of coexistence curves on phase diagrams and is essential for predicting phase stability under varying temperature and pressure conditions [5]. For homogeneous nucleation, the Gibbs free energy of formation for a nucleus involves a balance between the energy gain from creating a new volume and the energy cost of creating a new interface [6]. This framework provides the theoretical foundation for experimental control of nucleation processes through substrate temperature manipulation.
Quantitative Foundations and Theoretical Framework
The thermodynamic driving forces for nucleation can be quantitatively described through fundamental relationships between chemical potential, Gibbs free energy, and temperature. The following table summarizes key quantitative relationships essential for nucleation research:
Table 1: Fundamental Thermodynamic Relationships in Nucleation Processes
| Relationship | Mathematical Expression | Key Parameters | Application in Nucleation |
|---|---|---|---|
| Chemical Potential Equality at Phase Equilibrium | ( \mu\alpha (P, T) = \mu\beta (P, T) ) [5] | ( \mu ): chemical potential; α, β: phases | Determines phase coexistence conditions |
| Clapeyron Equation | ( \frac{dP}{dT} = \frac{\bar{S}\alpha - \bar{S}\beta}{\bar{V}\alpha - \bar{V}\beta} ) [5] | ( \bar{S} ): molar entropy; ( \bar{V} ): molar volume | Predicts phase boundary slopes on P-T diagrams |
| Gibbs Free Energy of Homogeneous Nucleation | ( \Delta G = \Delta G{\text{volume}} + \Delta G{\text{surface}} ) [6] | Volume and surface energy contributions | Determines critical nucleus size and energy barrier |
| Temperature Dependence of Nucleation Rate | ( N(T) = N0 \exp\left(-\frac{\Delta G^*}{kB T}\right) ) | ( \Delta G^* ): activation energy; ( k_B ): Boltzmann constant | Models temperature-dependent nucleation kinetics |
These quantitative relationships enable researchers to predict nucleation behavior under different experimental conditions. The Gibbs free energy of nucleation specifically includes a negative volumetric term proportional to the degree of supercooling or supersaturation, and a positive surface term proportional to the interfacial energy [6]. The maximum of this function (( \Delta G^* )) represents the nucleation barrier, with the critical nucleus size occurring at this maximum. This theoretical framework allows researchers to manipulate nucleation rates by controlling thermodynamic parameters, particularly temperature, which affects both the driving force and the kinetic prefactor.
Experimental Control of Nucleation Through Substrate Temperature
Substrate temperature serves as a critical experimental parameter for controlling nucleation processes by directly influencing the thermodynamic driving forces. Temperature affects both the chemical potential difference between phases and the thermal energy available for overcoming activation barriers. In materials science, precise temperature control enables manipulation of nucleation densities, growth modes, and ultimate material properties, as demonstrated in various experimental systems:
Table 2: Temperature-Dependent Nucleation in Experimental Systems
| Material System | Temperature Range | Observed Nucleation/Growth Effects | Reference |
|---|---|---|---|
| Zirconium Thin Films (PLD) | 300°C - 500°C | Transition from 2D layer-by-layer to 3D island growth; change in preferred crystal orientation [7] | |
| β-Ga₂O₃ on 4H-SiC (LPCVD) | ~680°C (optimized) | Suppressed vapor-phase nucleation; enhanced surface migration; improved film quality [8] | |
| CdSₓSe₁ₓ Ternary Alloys | 580°C - 690°C (gradient) | Spatial composition grading across substrate; tunable bandgap and lasing wavelengths [9] | |
| Silver Nanowires (Polyol) | 120°C - 160°C | Higher temperatures increase nucleation rates and conversion efficiency [10] | |
| Copper Oxide Sputtering | <100°C (unheated) to 1000°C | Variable phase formation (Cu₂O, Cu₄O₃, CuO) with temperature; carrier density changes [9] |
The underlying mechanism involves temperature's influence on adatom surface diffusion, which governs whether films grow in a 2D layer-by-layer fashion or transition to 3D island formation (Stranski-Krastanov growth). Computational modeling of zirconium film growth indicates that at 400°C, adatom diffusivity optimally balances crystallization and surface energy minimization, yielding the highest film quality, while at 500°C, rapid diffusivity increases cause 3D island proliferation and increased surface roughness [7]. Similarly, in Ga₂O₃ growth, optimized temperature conditions (∼680°C) suppress unwanted vapor-phase nucleation while enhancing surface migration, leading to improved film quality [8].
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Materials for Nucleation Studies
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Substrate Materials | Provides surface for heterogeneous nucleation | 4H-SiC substrates (on-axis and 4° off-axis) [8]; Silicon (100) wafers [7] |
| Precursor Sources | Supplies material for nucleus formation | Elemental Gallium (5N purity) for Ga₂O₃ growth [8]; Zirconium target (99.95%) for PLD [7]; Silver Nitrate for nanowire synthesis [10] |
| Carrier/Reactive Gases | Transport medium and reaction participant | Oxygen (O₂) for oxide formation [8] [7]; Argon (Ar) as inert carrier gas [8] [7] |
| Surface Modifiers/Etchants | Controls nucleation density and kinetics | Iron(III) chloride, Sodium chloride, Sodium bromide as etching agents in silver nanowire synthesis [10] |
| Polymeric Stabilizers | Directs anisotropic growth and prevents aggregation | Polyvinyl Pyrrolidone (PVP) of varying molecular weights (55,000-360,000) for silver nanowire growth [10] |
| Calibration Standards | Validates temperature measurement accuracy | Arizona Test Dust (ATD), Snomax for ice nucleation studies [11]; Milli-Q ultrapure water for baseline measurements [11] |
These materials enable researchers to create controlled environments for studying nucleation phenomena across different systems. The choice of substrate material significantly influences heterogeneous nucleation through lattice matching and interfacial energy considerations. Precursor purity directly affects nucleation kinetics by determining the concentration of available building blocks, while surface modifiers and polymeric stabilizers provide additional control over nucleation densities and growth morphologies.
Experimental Protocols for Nucleation Research
Protocol: Droplet Freezing Measurements for Ice Nucleating Particles (INPs)
Principle: This protocol quantifies immersion freezing initiated by atmospheric ice-nucleating particles (INPs) using a Freezing Ice Nucleation Detection Analyzer (FINDA). The method monitors the temperature-dependent frozen fraction of droplets to deduce cumulative INP number spectra [11].
Materials and Equipment:
- Freezing Ice Nucleation Detection Analyzer (FINDA-WLU or equivalent)
- 96-well PCR plates (0.2 mL capacity)
- High-precision temperature-controlled circulator (e.g., JULABO FP50-HL)
- Platinum resistance thermometers (Pt100 sensors, accuracy ±0.15°C at 0°C)
- CCD camera with adjustable zoom lens for freezing detection
- Nitrogen gas purging system
- Sample materials: Arizona Test Dust, Snomax, precipitation samples
Procedure:
- Sample Preparation: Prepare aqueous suspensions of test materials (ATD, Snomax, or environmental samples) at appropriate concentrations.
- Droplet Array Generation: Dispense uniform droplets (typically 0.2-1 μL) into 96-well PCR plate using precision pipetting or microfluidic techniques.
- Instrument Setup: Place PCR plate into the aluminum cold stage block. Ensure proper sealing with PTFE components to prevent frost formation.
- Temperature Calibration: Verify temperature measurements using four integrated Pt100 sensors sealed in thermally conductive epoxy for accurate thermal coupling.
- Experimental Run: Initiate cooling protocol with controlled rate (0.1-1.0°C/min) from 0°C to approximately -30°C while continuously monitoring droplets.
- Freezing Detection: Employ CCD camera to detect freezing events based on changes in optical properties (increased opacity or changes in reflected light).
- Data Analysis: Calculate frozen fraction (f) at each temperature. Determine INP concentration using the formula: ( N_{INP}(T) = -\frac{\ln(1-f(T))}{V} ), where V is droplet volume.
Technical Notes: The FINDA-WLU system achieves temperature uncertainty of approximately ±0.60°C, accounting for both vertical heat transfer efficiency and horizontal temperature heterogeneity. For atmospheric relevance, report results as INP concentrations per unit volume of air or mass of particulate material [11].
Protocol: Temperature-Controlled Vapor-Phase Epitaxy of β-Ga₂O₃
Principle: This protocol describes heteroepitaxial growth of β-Ga₂O₃ films on 4H-SiC substrates via low-pressure chemical vapor deposition (LPCVD), with emphasis on temperature control to suppress vapor-phase nucleation and enhance surface migration [8].
Materials and Equipment:
- Horizontal hot-wall CVD tube furnace
- 4H-SiC substrates (on-axis and 4° off-axis (0001))
- Elemental Gallium source (5N purity)
- High-purity O₂ (20 sccm) and Ar (200 sccm) gases
- RCA cleaning solutions
- Characterization tools: SEM, AFM, TEM, EDX, XPS
Procedure:
- Substrate Preparation: Clean 4H-SiC substrates using standard RCA procedure followed by Ar gas drying to remove surface contaminants.
- Reactor Setup: Place Ga metal in crucible within hot-zone of CVD reactor. Position substrates in temperature-controlled region.
- Temperature Optimization: Set substrate temperature to ∼680°C for 4° off-axis substrates. Maintain additional heating zone for gas flow preheating.
- Growth Initiation: Introduce O₂ (20 sccm) and Ar carrier gas (200 sccm) to initiate growth. Maintain low-pressure environment.
- Growth Monitoring: Observe step-flow growth morphology development, characteristic of optimized temperature conditions.
- Process Termination: Cool samples under inert atmosphere after desired growth period (typically 1-2 hours).
- Characterization: Analyze surface morphology (SEM, AFM), crystal structure (TEM), and elemental composition (EDX, XPS).
Technical Notes: The 4° off-axis substrate promotes step-flow growth by providing regular surface steps that guide adatom migration. Temperature control is critical—excessive temperature enhances vapor-phase nucleation, while insufficient temperature reduces surface migration, both degrading film quality. Optimal temperature (∼680°C) balances these factors for high-quality β-Ga₂O₃ growth [8].
Visualization of Thermodynamic Relationships and Experimental Workflows
Thermodynamic Pathways in Temperature-Controlled Nucleation
Experimental Workflow for Controlled Nucleation
Chemical potential and Gibbs Free Energy provide the fundamental thermodynamic framework for understanding and controlling nucleation processes across diverse scientific disciplines. Through precise substrate temperature control, researchers can manipulate these driving forces to achieve desired nucleation densities, growth morphologies, and material properties. The experimental protocols and quantitative relationships presented in this work establish standardized methodologies for advancing nucleation research in fields ranging from atmospheric science to materials engineering and pharmaceutical development. The integration of theoretical principles with practical experimental controls enables rational design of nucleation processes for technological applications, bridging fundamental thermodynamics with materials innovation.
Supersaturation represents the fundamental driving force for nucleation, the initial step in crystallization where solute molecules or atoms form stable clusters that can grow into crystals. The interplay between temperature and supersaturation is pivotal, as temperature directly controls the thermodynamic energy barriers and kinetic pathways of nucleation. Within the context of substrate temperature control for nucleation research, precise thermal management enables the selective activation of desired nucleation mechanisms—homogeneous, heterogeneous, classical, or non-classical—across diverse scientific domains from pharmaceutical development to advanced materials synthesis. This application note provides a consolidated framework of quantitative relationships, validated protocols, and experimental tools for exploiting temperature as a precise modulator of nucleation barriers in research and development settings.
Quantitative Relationships: Temperature-Dependent Nucleation Parameters
Table 1: Temperature-Dependent Nucleation Parameters Across Material Systems
| Material System | Temperature Range | Key Nucleation Parameter | Observed Effect on Nucleation & Growth | Reference |
|---|---|---|---|---|
| Ice Nucleation (Heterogeneous) | 230 K | Classical vs. Non-classical Pathway Flux | Co-existence of both pathways with comparable fluxes; configurational entropy stabilizes critical nucleus in non-classical pathway. | [12] |
| Ice Nucleation (Heterogeneous) | Higher than 230 K | Classical vs. Non-classical Pathway Flux | Shift towards classical pathway as potential energy contributions override configurational entropy. | [12] |
| Water Vapor on Ag Nanoparticles | ~278 K | Critical Saturation Ratio (Sonset) | Maximum critical saturation ratio observed; positive temperature dependence (increasing Sonset with T) occurs when critical cluster is more stable than bulk liquid. | [2] |
| Perovskite SCTFs (Inverse Temp. Crystallization) | Increasing Temperature | Supersaturation (ΔC) & Critical Radius (rc) | Inverse solubility increases supersaturation, reducing the critical nucleus radius and promoting uniform nucleation. | [13] |
| β-Ga2O3 CVD Growth | 680 °C (Optimized) | Vapor-Phase Nucleation vs. Surface Migration | Optimized temperature suppresses vapor-phase nucleation and enhances adatom surface migration, leading to layered growth and smooth films. | [8] |
| FeCr Alloy Decomposition | Within Miscibility Gap | Nucleation Rate (J) | Classical Nucleation Theory (CNT) valid only near solubility limit; self-consistent phase field approach required for wider temperature range. | [14] |
| Lysozyme Microcrystallization | Room Temperature | Evaporation Time (Induces Supersaturation) | Shorter evaporation time (16-20 min for 55 mg/mL) yielded high-density microcrystals via supersaturation control. | [15] |
| Poly(TFEMA) in scCO2-Toluene | 40 °C (One-Phase Region) | Nucleation Pathway | Heterogeneous surface nucleation dominates, yielding sparse, compact islands (mean diameter: 1.77 µm). | [16] |
| Poly(TFEMA) in scCO2-Toluene | 40 °C (Cloud Point) | Nucleation Pathway | Homogeneous nucleation begins, leading to agglomerated, necked spheres (mean diameter: 2.61 µm). | [16] |
| Poly(TFEMA) in scCO2-Toluene | 40 °C (Two-Phase Region) | Nucleation Pathway | Homogeneous nucleation with coalescence and solvent capture, yielding large, hollow/pitted particles (mean diameter: 2.86 µm). | [16] |
| Zirconium Thin Films (PLD) | 400 °C vs 500 °C | Adatom Surface Diffusion & Growth Mode | 400 °C: Optimal diffusivity balances crystallization energy. 500 °C: High diffusivity leads to 3D island growth (Volmer-Weber) and increased roughness. | [7] |
Underlying Mechanisms and Pathways
The quantitative data presented in Table 1 emerges from fundamental physical mechanisms linking temperature to nucleation barriers. The following diagram synthesizes these universal pathways across disparate material systems.
Temperature Modulates Nucleation Barriers: The diagram illustrates how substrate temperature controls nucleation by simultaneously influencing thermodynamic driving forces (red pathway) and kinetic parameters (green pathway). Thermally altered supersaturation and diffusion rates converge to determine the dominant nucleation mechanism and final material morphology (blue outcomes).
Thermodynamic Control
Temperature directly modulates the thermodynamic driving force for nucleation. In perovskite systems, inverse solubility behavior causes supersaturation (ΔC = C - C0) to increase with temperature, as the equilibrium solute concentration (C0) decreases [13]. This elevated supersaturation reduces the critical nucleus radius (rc ∝ γ/ΔGv) and the activation barrier (ΔG ∝ γ³/ΔGv²), facilitating nucleation at milder absolute concentrations [13]. For vapor-phase nucleation on silver nanoparticles, a positive temperature dependence (increasing onset saturation ratio with temperature) occurs when the critical cluster is more stable per molecule than the bulk liquid phase [2].
Kinetic Pathway Selection
Beyond thermodynamics, temperature governs kinetic pathways by controlling atomic/molecular mobility. Markov State Models of heterogeneous ice nucleation reveal that at deeply supercooled conditions (230 K), classical one-step and non-classical two-step pathways coexist with comparable probability, as configurational entropy from disordered ice mixtures stabilizes critical nuclei [12]. At higher temperatures, potential energy contributions prevail, shifting preference toward the classical pathway [12]. In zirconium thin film deposition, elevated substrate temperature enhances adatom surface diffusion, which controls the transition from 2D layer-by-layer growth to 3D island formation (Stranski-Krastanov or Volmer-Weber modes) [7].
Experimental Protocols
Protocol: Supersaturation-Controlled Microcrystallization for Proteins
This protocol adapts the vapor diffusion method to generate high-density microcrystals suitable for XFEL studies through precise temporal control of evaporation-induced supersaturation [15].
Research Reagent Solutions Table 2: Essential Reagents for Protein Microcrystallization
| Reagent / Material | Function | Example Specification |
|---|---|---|
| Purified Protein | Target molecule for crystallization. | Recombinant influenza virus hemagglutinin (HA), ≥95% purity. |
| Precipitant Solution | Induces supersaturation by reducing solute solubility. | 100 mM Tris-HCl (pH 8.0), 30% PEG 400, 200 mM MgCl₂. |
| Reservoir Solution | Controls vapor pressure and evaporation rate in hanging drop. | Identical to precipitant solution at higher volume. |
| 24-Well Crystallization Plate | Platform for vapor diffusion setup. | VDX plate with sealing grease. |
| Siliconized Glass Coverslips | Surface for drop dispensing; reduces heterogeneous nucleation. | 22 mm diameter, siliconized. |
| Hemocytometer | Tool for initial crystal size and density estimation. | Standard Neubauer improved. |
Procedure
- Sample Preparation: Prepare the protein solution in its storage buffer. Centrifuge at 14,000 × g for 10 minutes to remove any pre-existing aggregates.
- Drop Equilibration: For each condition, pipette 500 µL of reservoir solution into a well of the 24-well plate. Prepare a siliconized coverslip.
- Sequential Evaporation: Pipette a 1.8 µL drop of mixed protein-precipitant solution onto the coverslip. Note the start time.
- Air-dry the drop for a defined duration (td). The optimal td is protein-specific and must be determined empirically (e.g., 30 seconds to 3 minutes for HA, 16-20 minutes for lysozyme) [15].
- Quickly invert and seal the coverslip over the corresponding well. Repeat for multiple coverslips with varying td to create a time series.
- Incubation and Monitoring: Store the sealed plate at a constant temperature (e.g., 20°C). Monitor drops periodically under a light microscope for crystal formation over 6-48 hours.
- Characterization: Use a hemocytometer for initial size distribution analysis. Validate microcrystal quality using second-order nonlinear imaging of chiral crystals (SONICC) or UV fluorescence imaging [15].
Protocol: Temperature-Optimized Vapor Deposition for Thin Films
This procedure outlines the growth of high-quality β-Ga2O3 epitaxial films on 4H-SiC substrates via LPCVD, highlighting the critical role of temperature in suppressing vapor-phase nucleation and promoting step-flow growth [8].
Research Reagent Solutions Table 3: Essential Reagents for Vapor Deposition
| Reagent / Material | Function | Example Specification |
|---|---|---|
| Elemental Gallium (Ga) | High-purity Ga source. | 5N (99.999%) purity, placed in an alumina crucible. |
| Oxygen (O₂) Gas | Oxidant for Ga2O3 formation. | 5N purity, mass flow controller regulated. |
| Argon (Ar) Gas | Carrier gas. | 5N purity, mass flow controller regulated. |
| 4H-SiC Substrate | Epitaxial growth template. | 4° off-axis (0001) orientation. |
| RCA Clean Chemicals | Substrate surface preparation. | Standard SCI/SC2 solutions (NH4OH:H2O2:H2O, HCl:H2O2:H2O). |
Procedure
- Substrate Preparation: Subject 4° off-axis 4H-SiC substrates to standard RCA cleaning. Follow with an Ar gas dry to ensure complete removal of moisture and contaminants [8].
- Reactor Setup and Purging: Load the cleaned substrate and Ga source into a horizontal hot-wall CVD tube furnace. Evacuate the chamber and purge with Ar gas to establish an inert environment.
- Temperature Stabilization: Ramp the furnace temperature to the target growth temperature (optimized at 680°C for β-Ga2O3 on 4H-SiC). Maintain stability for 10 minutes before introducing the oxidant [8].
- Film Growth: Initiate film growth by introducing O2 gas at a controlled flow rate (e.g., 20 sccm) while maintaining the Ar carrier gas flow (e.g., 200 sccm). The optimal temperature suppresses premature vapor-phase nucleation in the gas stream, ensuring Ga and O species adsorb directly onto the substrate surface where they can migrate to step edges [8].
- Process Termination and Cooling: After the desired growth period, terminate the process by stopping the O2 flow. Cool the system to room temperature under continuous Ar flow.
- Characterization: Analyze film morphology by Atomic Force Microscopy (AFM) and Scanning Electron Microscopy (SEM) to confirm step-flow growth and low surface roughness. Determine crystallinity and phase purity by Transmission Electron Microscopy (TEM) and X-ray Diffraction (XRD) [8].
The Scientist's Toolkit
Table 4: Key Reagent Solutions for Nucleation Control Experiments
| Category | Item | Critical Function in Nucleation Control |
|---|---|---|
| Analytical Standards | Arizona Test Dust (ATD), Snomax | Standardized ice-nucleating particles for calibrating and validating immersion freezing measurements (e.g., in DFTs) [11]. |
| Polymer & Solvent Systems | Poly(TFEMA), scCO2, Toluene | Model system for studying thermodynamic phase-state-controlled nucleation (homogeneous vs. heterogeneous) in polymer deposition [16]. |
| Metallic Targets & Gases | Zirconium target, High-purity O2, Ar | Enables study of substrate temperature effects on adatom diffusion, growth mode, and crystallinity in PLD/CVD [8] [7]. |
| Protein Crystallization Kits | Pre-formulated Precipitant Solutions (e.g., PEGs, Salts) | Provide reproducible chemical environments for implementing supersaturation-controlled microcrystallization protocols [15]. |
| Specialized Substrates | Off-axis 4H-SiC wafers, Fluorine-doped Tin Oxide (FTO) | Crystalline templates with defined surface terraces that guide epitaxial growth and study heterogeneous nucleation kinetics [8] [16]. |
Nucleation, the initial formation of a new thermodynamic phase from a parent phase, serves as the critical first step in processes ranging from atmospheric cloud formation to pharmaceutical crystallization. The dynamics of this process can be conceptualized through two distinct frameworks: stochastic and deterministic nucleation. Stochastic nucleation treats the formation of stable nuclei as random events governed by probabilistic laws, where the exact timing and location of nucleation cannot be predicted due to the inherent randomness of molecular-scale fluctuations [17]. In contrast, deterministic approaches model nucleation as a continuous process where outcomes are precisely determined by initial conditions and system parameters, making it predictable at a macroscopic scale [14]. The choice between these frameworks depends heavily on both temporal scales and thermal conditions of the system, with temperature serving as a master variable that controls which paradigm dominates the nucleation process [14] [17].
Understanding the interplay between these frameworks is essential for researchers across disciplines. In drug development, nucleation kinetics influence polymorph selection, bioavailability, and product stability [18] [19]. In materials science, nucleation controls microstructure evolution, phase distribution, and ultimately material properties [14] [7]. This Application Note establishes the foundational principles of both stochastic and deterministic nucleation, provides quantitative comparisons, details experimental protocols, and visualizes the critical relationships governing time and temperature dependence in nucleation research.
Theoretical Foundations: Mechanisms and Temperature Dependence
Stochastic Nucleation Framework
Stochastic nucleation theory fundamentally addresses the inherent randomness of nucleation events. As an activated process, nucleation requires overcoming an energy barrier, making the precise timing of nucleation events unpredictable in individual experiments [17]. The nucleation frequency (K) in this framework is conceptualized as the product of three contributions: the number of active nucleation sites (Nactive), the frequency of attempts (fattempt), and the probability of a successful nucleation event (P_success) [17]. This relationship is expressed as:
[K = N{active} \times f{attempt} \times P_{success}]
This stochastic nature is particularly pronounced in systems dominated by primary nucleation, which includes both homogeneous and heterogeneous pathways [17]. In experimental settings, this randomness manifests as significant variability in detection times when the same crystallization process is repeated under identical conditions [18] [17]. For accurate measurement, stochastic methods typically impose a critical simplification: they assume only the first nucleus forms stochastically, with subsequent crystallization events following deterministically [17].
Deterministic Nucleation Framework
Deterministic approaches become appropriate when the system contains numerous nucleation sites or when secondary nucleation mechanisms dominate. In such cases, the collective behavior of the system becomes predictable at a macroscopic scale, averaging out molecular-scale stochasticity [14]. The Phase Field (PF) approach, particularly when employing the Cahn-Hilliard-Cook (CHC) equation, represents a powerful deterministic method that can model microstructure evolution without requiring explicit knowledge of all kinetic pathways [14].
This framework is particularly effective for modeling secondary nucleation, where existing crystals generate new nuclei through mechanisms that scale with crystal surface area or volume [17]. The deterministic nucleation rate can be described using power-law expressions:
[K{SN} = k{a} \times A_{total} \times (S-1)^b]
where (K{SN}) is the secondary nucleation frequency, (k{a}) is a rate constant, (A_{total}) is the total crystal surface area, S is supersaturation, and b is an exponent [17]. Deterministic models are implemented through population balance equations that track the evolution of crystal size distributions over time, treating nucleation as a continuous process [17].
The Master Variable: Temperature Dependence
Temperature exerts profound influence on nucleation kinetics through multiple mechanisms, regardless of the modeling framework. The following table summarizes the key quantitative relationships governing temperature-dependent nucleation across various material systems:
Table 1: Temperature Dependence in Nucleation Processes Across Material Systems
| System | Temperature Range | Key Parameter | Observed Effect | Reference |
|---|---|---|---|---|
| FeCr Alloy Decomposition | Inside miscibility gap | Nucleation rate | Complex interplay of nucleation, growth, and coarsening | [14] |
| Ice Nucleation (DFT) | 0°C to -30°C | INP concentration | Heterogeneous nucleation above -38°C; homogeneous below | [11] |
| β-Ga₂O₃ CVD Growth | Optimized at 680°C | Surface morphology | Suppressed vapor-phase nucleation and enhanced surface migration at optimal T | [8] |
| Zirconium Thin Films (PLD) | 300°C to 500°C | Crystalline orientation | Zr(100) strongest at 400°C; Zr(002) maximum at 500°C; 3D island formation at high T | [7] |
| Pharmaceutical Crystallization | Function of T | Nucleation rate | Follows Classical Nucleation Theory: (J = A \exp\left(-\frac{B}{T^3 (\ln S)^2}\right)) | [17] |
The fundamental temperature dependence in many nucleation processes is described by adaptations of the Classical Nucleation Theory (CNT), which for primary nucleation follows the general form:
[J{PN} = A{PN} \exp\left(-\frac{B_{PN}}{T^3 (\ln S)^2}\right)]
where (J{PN}) is the primary nucleation rate, (A{PN}) and (B_{PN}) are system-dependent parameters, T is temperature, and S is supersaturation [17]. This relationship highlights the dual role of temperature in nucleation kinetics: directly through thermal energy (T) and indirectly through its effect on equilibrium concentration (c*(T)) and thus supersaturation (S) [17].
Table 2: Transition Conditions Between Stochastic and Deterministic Dominance
| Factor | Stochastic Dominance | Deterministic Dominance | |
|---|---|---|---|
| Nucleation Type | Primary nucleation | Secondary nucleation | |
| System Size | Small volumes | Large volumes | |
| Nucleation Sites | Few active sites | Many active sites | |
| Supersaturation | Low | High | |
| Time Scale | Early stages | Late stages | |
| Experimental Manifestation | High variability in detection times | Reproducible crystallization profiles | [17] |
Experimental Protocols and Methodologies
Protocol 1: Droplet Freezing Measurements for Ice Nucleating Particles (INPs)
Application Notes: This protocol measures immersion freezing of INPs using a Freezing Ice Nucleation Detection Analyzer (FINDA), relevant for atmospheric science and cryopreservation research [11].
Materials and Reagents:
- Freezing Ice Nucleation Detection Analyzer (FINDA-WLU)
- 96-well PCR plate (0.2 mL)
- Milli-Q ultrapure water
- Reference materials: Arizona Test Dust (ATD), Snomax
- Precipitation or aerosol samples
- Temperature calibration standards
Procedure:
- Sample Preparation: Prepare aqueous suspensions of test particles using serial dilution in Milli-Q water.
- Droplet Generation: Pipette 50-100 µL aliquots into individual wells of the 96-well PCR plate.
- Instrument Setup: Place the PCR plate into the aluminum cold stage of FINDA-WLU. Seal with acrylic glass lid to prevent ambient air mixing.
- Temperature Programming: Cool the stage from 0.0°C to -30.0°C at a controlled rate of 0.1-1.0°C min⁻¹.
- Freezing Detection: Monitor droplet freezing using CCD camera, detecting opacity changes due to ice formation.
- Data Collection: Record freezing temperatures for each droplet alongside calibrated stage temperatures.
- Data Analysis: Calculate frozen fraction as a function of temperature. Determine INP concentration using: (n{INP}(T) = -\frac{\ln(1 - ff(T))}{V{droplet}}) where (ff(T)) is the frozen fraction at temperature T and (V_{droplet}) is droplet volume [11].
Technical Notes: Temperature uncertainty in FINDA-WLU is approximately ±0.60°C, accounting for both vertical heat transfer efficiency and horizontal temperature heterogeneity. For statistical significance, use at least 50-100 droplets per sample [11].
Protocol 2: Stochastic Nucleation Kinetics in Pharmaceutical Solutions
Application Notes: This mid-throughput approach characterizes ice nucleation kinetics of aqueous solutions in vials, essential for biopharmaceutical freezing and freeze-drying process design [18].
Materials and Reagents:
- Parallelized batch crystallizer
- Pharmaceutical vials (2-10 mL)
- Aqueous solution of active pharmaceutical ingredient (API)
- Temperature-controlled bath with precision ±0.1°C
- Visual or light-scattering detection system
Procedure:
- Solution Preparation: Prepare saturated or supersaturated solutions of the target compound in appropriate solvent.
- Sample Loading: Aseptically fill vials with equal volumes of solution.
- Thermal Equilibration: Equilibrate all vials at starting temperature above nucleation point.
- Nucleation Trigger: Apply controlled cooling ramp (0.1-1.0°C min⁻¹) or isothermal hold at supercooled temperature.
- Event Detection: Monitor for nucleation events using automated detection (turbidity, image analysis, or temperature spike).
- Data Recording: Precisely record nucleation time and temperature for each vial.
- Stochastic Analysis: For isothermal experiments, analyze detection time distribution to extract nucleation rates [18].
- Model Fitting: Apply stochastic modeling framework to compute nucleation parameters and their uncertainty [18].
Technical Notes: This protocol explicitly accounts for two variability sources: inherent stochasticity of nucleation and variability in heterogeneous nucleation sites among vials. For 100 vials, the nucleation rate can be estimated with approximately 20% precision [18].
Protocol 3: Phase Field Modeling for Microstructure Prediction
Application Notes: This computational approach predicts temperature-dependent nucleation and growth in alloys, validated against experimental measurements, using a self-consistent Phase Field approach [14].
Materials and Reagents:
- High-performance computing resources
- Phase Field modeling software
- Experimental validation data (e.g., Atom Probe Tomography)
- Material parameters (interaction energies, diffusion coefficients)
Procedure:
- Parameterization: Determine effective Hamiltonian using experimental thermodynamic data or atomic-scale simulations.
- Model Setup: Implement Cahn-Hilliard-Cook equation for conservative systems with appropriate boundary conditions.
- Saddle Point Identification: Locate index 1 saddle point of the effective Hamiltonian to define nucleation process.
- Time Integration: Simulate microstructure evolution using appropriate numerical methods.
- Nucleation Implementation: Model nucleation as either initial condition or source term based on ratio of characteristic time scales.
- Experimental Validation: Compare simulated 3D microstructures with Atom Probe Tomography measurements at different times.
- Parameter Refinement: Adjust model parameters to achieve quantitative agreement with experimental data [14].
Technical Notes: This approach circumvents limitations of Classical Nucleation Theory, which is only valid near solubility limits. It unifies modeling of nucleation, growth, and spinodal decomposition across the miscibility gap [14].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Nucleation Studies
| Reagent/Material | Function | Application Context |
|---|---|---|
| Arizona Test Dust (ATD) | Standardized ice-nucleating particle reference | Calibration and validation of ice nucleation measurements [11] |
| Snomax | Biological ice-nucleating agent containing membrane fragments of Pseudomonas syringae | Positive control for heterogeneous ice nucleation studies [11] |
| Ultra-pure FeCr Alloys | Model system for studying nucleation and growth in phase transitions | Metallurgy research on decomposition kinetics and microstructure evolution [14] |
| Elemental Gallium (5N purity) | High-purity source for Ga₂O₃ film growth | CVD growth of β-Ga₂O₃ films on SiC substrates [8] |
| 96-well PCR Plates | Micro-containers for droplet arrays | High-throughput freezing experiments in DFT and FINDA systems [11] |
| p-Aminobenzoic Acid | Model compound for crystallization kinetics | Method validation for pharmaceutical nucleation studies [17] |
| Zirconium Target (99.95%) | Source material for thin film deposition | PLD studies of temperature-dependent film morphology [7] |
Visualization: Decision Framework and Experimental Workflows
The following diagrams illustrate the critical decision pathways for selecting appropriate nucleation frameworks and the experimental workflows for characterizing nucleation kinetics.
Diagram 1: Decision framework for selecting between stochastic and deterministic nucleation approaches based on system characteristics. Systems with few nucleation sites and high detection time variability require stochastic treatment, while those with many nucleation sites and reproducible behavior suit deterministic modeling. Large systems with mixed characteristics may need hybrid approaches [17].
Diagram 2: Experimental workflows for characterizing nucleation kinetics through stochastic and deterministic pathways. The stochastic pathway uses multiple identical samples and analyzes detection time distributions, while the deterministic pathway monitors crystal size distribution evolution in a single large sample and fits population balance models [18] [17].
The dichotomy between stochastic and deterministic nucleation frameworks represents not a fundamental contradiction but a reflection of different observational scales and system complexities. Temperature serves as the master variable governing the transition between these regimes, with stochastic behavior dominating in small systems at early stages with few nucleation sites, and deterministic behavior emerging in large systems with abundant nucleation sites or secondary nucleation mechanisms [14] [17].
For researchers engaged in substrate temperature control for nucleation studies, the key insight is that method selection must align with both system characteristics and observational capabilities. Stochastic methods provide accurate primary nucleation rates when variability is properly accounted for, while deterministic methods efficiently model systems where secondary nucleation dominates [17]. Emerging approaches, such as the Phase Field method, offer unifying frameworks that can bridge these perspectives by incorporating stochastic elements into deterministic continuum models [14].
The temperature-dependent nature of nucleation kinetics underscores the critical importance of precise thermal control in experimental design. Whether studying pharmaceutical crystallization, thin film deposition, or phase transformations in alloys, recognition of the interplay between stochastic and deterministic elements enables more accurate prediction and control of nucleation processes across scientific and industrial applications.
Protein crystallization is a critical, yet often rate-limiting, step in structural biology and biopharmaceutical development [20]. A profound understanding of the phase diagram, particularly the metastable zone where nucleation is initiated, is fundamental to controlling this process. This case study examines the application of phase diagrams to enhance protein crystallization yields, with a specific focus on exploiting metastable liquid–liquid phase separation (LLPS). Framed within broader research on substrate temperature control for nucleation, we present a quantitative analysis and a detailed protocol for lysozyme crystallization, demonstrating yields exceeding 90% through precise manipulation of the metastable zone [21].
Theoretical Framework: The Phase Diagram
The phase diagram of a protein-solution system is a map that defines the thermodynamic conditions under which different phases—such as soluble, crystalline, and liquid–liquid separated—coexist or are stable [22]. For crystallization to occur, a solution must be brought into a supersaturated state where the chemical potential of the dissolved protein exceeds that of the crystalline solid [20].
The diagram below illustrates key regions and the two-step nucleation mechanism within the metastable zone.
The metastable zone, located between the solubility and the labile (or LLPS) boundary, is where nucleation is thermodynamically possible but not immediate. Within this zone, a two-step nucleation mechanism is often operative [22]: first, dense liquid protein droplets form via LLPS; second, these droplets act as precursors that lower the energy barrier for the formation of ordered crystalline nuclei [21] [22]. The width and properties of this zone are highly sensitive to solution conditions, including the type of additives present [21].
Experimental Investigation: Enhancing Yield via LLPS
Objective and Rationale
This investigation quantified the crystallization yield of Hen-Egg-White Lysozyme (HEWL) under conditions designed to exploit LLPS. The strategy employed a combination of two additives [21]:
- NaCl (0.15 M): A traditional salting-out agent that introduces attractive protein-protein interactions, inducing LLPS upon cooling.
- HEPES (0.10 M, pH 7.4): A multi-functional organic buffer that accumulates in the protein-rich liquid phase and acts as a thermodynamic stabilizer for the crystal lattice.
The protocol involved a precise temperature-cycling regimen: an initial quench below the LLPS boundary to promote crystal nucleation within the protein-rich droplets, followed by a temperature rise above the LLPS boundary to dissolve the metastable liquid phase and favor the growth of the formed nuclei [21].
Key Quantitative Findings
The following table summarizes the core quantitative data from the study, demonstrating the significant impact of additive combination and temperature profile on crystallization yield.
Table 1: Crystallization Yield Data for HEWL under Various Conditions (Protein Concentration: 50 g/L)
| Additive Combination | Ionic Strength | Key Temperature Step | Incubation Time | Crystallization Yield | Key Finding |
|---|---|---|---|---|---|
| NaCl only (0.18 M) | 0.20 M | Quench to -15°C, then to 2°C above LLPS boundary | ~60 min total | <30% | Baseline yield with standard agent [21] |
| NaCl (0.15 M) + HEPES (0.10 M) | 0.20 M | Quench to -15°C, then to 2°C above LLPS boundary | ~60 min total | >90% | >3-fold yield increase with dual additives [21] |
| NaCl (0.15 M) + HEPES (0.10 M) | 0.20 M | Varies (intersecting LLPS boundary) | Varies | Significant yield increase | Confirms LLPS boosts nucleation [21] |
A separate study highlighted the critical, yet often overlooked, parameter of solution preparation temperature. The table below shows its non-linear effect on crystallization success, underscoring the need for precise thermal control from the very first step of the protocol.
Table 2: Effect of Solution Preparation (Mixing) Temperature on Crystallization Success [23]
| Protein | Low Temp (278-283 K) | Ambient Temp (290-297 K) | High Temp (298-303 K) | Significance (P-value) |
|---|---|---|---|---|
| Lysozyme | Highest Success | Least Successful | Success Increased | 0.043 |
| Proteinase K | Highest Success | Least Successful | Success Increased | <0.001 |
| Thaumatin | Highest Success | Least Successful | Success Increased | 0.007 |
| Primary Cause | Increased supersaturation upon transfer to crystallization incubator [23] | Increased initial concentration from evaporation [23] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for LLPS-Enhanced Crystallization
| Item | Specification / Function | Role in Experiment |
|---|---|---|
| Lysozyme | Hen-Egg-White (HEWL), high purity | Model protein for crystallization studies [21]. |
| NaCl | High Purity (e.g., Molecular Biology Grade) | Salting-out agent; induces attractive protein interactions and LLPS [21]. |
| HEPES Buffer | 0.10 M, pH 7.4 | Preferentially binds to protein, stabilizes crystals, modifies phase diagram [21]. |
| Temperature-Controlled Stage/Incubator | Capable of rapid quenching and precise control from -20°C to 20°C. | To execute the precise temperature-cycling protocol critical for inducing and then clearing LLPS [21] [24]. |
| Crystallization Plates | Sitting or hanging drop vapor diffusion plates. | Standard platform for setting up crystallization trials. |
Detailed Experimental Protocol
The following diagram outlines the complete experimental workflow, from sample preparation to yield analysis, highlighting the critical temperature control points.
Step-by-Step Methodology
Step 1: Solution Preparation
- Prepare a stock solution of 50 g/L Hen-Egg-White Lysozyme (HEWL) in ultrapure water.
- Prepare a precipitant solution containing 0.15 M NaCl and 0.10 M HEPES, adjusted to pH 7.4.
- Critical Note: Control and record the ambient temperature during the mixing of protein and precipitant solutions, as this can significantly impact initial supersaturation and experimental reproducibility [23].
Step 2: Sample Setup
- Mix equal volumes (e.g., 1 µL each) of the protein and precipitant solutions directly in a sitting-drop crystallization plate.
- Seal the plate with a transparent cover to isolate the drop from the environment.
Step 3: Temperature-Quench Incubation (Nucleation Phase)
- Immediately transfer the sealed crystallization plate to a temperature-controlled stage or incubator pre-set to -15°C (T~Q~).
- Incubate the plate at this temperature for a defined period, Δt~Q~ = 30 minutes. During this time, the solution will cross the LLPS boundary, forming protein-rich micro-droplets that facilitate rapid crystal nucleation [21].
Step 4: Temperature-Rise Incubation (Crystal Growth Phase)
- After the quench period, rapidly raise the temperature of the crystallization platform to a value 2°C above the experimentally determined LLPS temperature for the specific solution condition.
- Maintain the sample at this new temperature for an additional 30 minutes. This step dissolves the metastable protein-rich liquid phase, suppressing further nucleation and providing optimal conditions for the growth of existing crystalline nuclei [21].
Step 5: Harvesting and Yield Analysis
- Carefully remove the plate from the incubator. Visually inspect the drops under a microscope for the presence of tetragonal HEWL crystals.
- To determine the crystallization yield (Y), harvest and dissolve the crystals from the drop. Measure the protein concentration in the dissolved crystal solution and the remaining mother liquor. Calculate yield as the mass of protein in the crystalline phase divided by the total initial mass of protein, expressed as a percentage. Yields of >90% are achievable with this protocol [21].
This case study demonstrates that a deep understanding of the protein crystallization phase diagram, particularly the metastable zone and LLPS, is key to developing high-yield purification protocols. The combination of a salting-out agent (NaCl) and a crystal-stabilizing agent (HEPES), coupled with a precise temperature-cycling protocol, dramatically enhanced lysozyme crystallization yield to over 90% while reducing operational time [21]. These findings provide a validated strategy and a detailed methodological framework that can be adapted for the crystallization of other therapeutically relevant proteins, directly supporting advanced research in drug development where robust and economical protein purification is paramount.
Practical Strategies for Implementing Temperature Control in Research and Development
Substrate temperature is a critical engineering parameter in thin-film deposition, directly governing adatom surface mobility, nucleation density, and crystallization kinetics. Within the framework of nucleation research, precise temperature control enables the systematic investigation of growth modes, phase evolution, and defect formation. This Application Note provides structured protocols and data for substrate temperature engineering in Plasma-Enhanced Atomic Layer Deposition (PEALD) and Pulsed Laser Deposition (PLD), two techniques with distinct thermal management requirements. The comparative analysis and standardized methodologies presented herein support reproducible experimentation and accelerated process optimization in research and development settings.
Substrate temperature systematically influences multiple thin-film properties across deposition techniques and materials. The following tables consolidate quantitative findings from recent studies.
Table 1: Effect of Substrate Temperature on Thin-Film Properties in PEALD
| Material | Temperature Range (°C) | Optimal Temperature (°C) | Key Property Enhancement | Reference |
|---|---|---|---|---|
| HfO₂ | 100 - 450 | 300 | Highest dielectric constant and breakdown field [25] | |
| Al₀.₈₈Sc₀.₁₂N | 215 - 300 | 215 (Process) | Ferroelectric switching; c-axis orientation [26] | |
| FeOx | 200 (Fixed) | 200 (Fixed) | PEALD yielded denser (~4.9 g/cm³), smoother films vs. thermal ALD [27] |
Table 2: Effect of Substrate Temperature on Thin-Film Properties in PLD and Sputtering
| Material | Deposition Technique | Temperature Range (°C) | Key Structural/Morphological Findings | Reference |
|---|---|---|---|---|
| Zirconium (Zr) | PLD | 300 - 500 | Smoothest surface at 300°C; 3D island growth & silicide formation at 500°C [7] | |
| WS₂ | RF Sputtering | 25 - 300 | Transition from nanoparticles to nanosheets at 200°C; largest crystallite size at 200°C [28] | |
| Nickel Oxide (NiO) | RF Sputtering | 100 - 300 | Increased crystallinity and bandgap shrinkage with increasing temperature [29] |
Experimental Protocols
Protocol: PEALD of HfO₂ Thin Films with Substrate Temperature Variation
This protocol details the deposition of HfO₂ films on silicon substrates to investigate the effect of temperature on crystallinity and electrical properties [25].
3.1.1 Research Reagent Solutions
- Substrate: 4-inch p-type Si wafer (1-3 Ω·m resistivity)
- Hafnium Precursor: Tetrakis(ethylemethylamino) hafnium (TEMAH), >99.9999% purity
- Oxidizing Agent: O₂/Ar plasma
- Carrier Gas: N₂, 99.999% purity
- Cleaning Agents: Deionized water, 2% diluted hydrofluoric acid (HF) solution
3.1.2 Step-by-Step Procedure
- Substrate Preparation:
- Clean Si substrates ultrasonically sequentially in deionized water (10 s), 2% diluted HF (1 min), and deionized water again (10 s).
- Dry the substrates using a stream of N₂ gas and load onto the PEALD substrate holder.
- PEALD System Setup:
- Load the TEMAH precursor into a bubbler-type canister and maintain it at 120°C.
- Heat all gas delivery lines to 130°C to prevent precursor condensation.
- Set the base pressure of the reaction chamber to 100 Pa.
- Set O₂ and Ar flow rates to 100 sccm and 50 sccm, respectively.
- Set the plasma power to 2500 W.
- Deposition Process:
- Set the substrate temperature to the desired value (e.g., 100, 200, 300, 400, or 450°C).
- Execute the following cycle sequence for the desired number of cycles (e.g., to achieve ~20 nm thickness):
- Step 1: TEMAH pulse for 1.6 s.
- Step 2: N₂ purge for 10 s.
- Step 3: O₂/Ar plasma exposure for 10 s (with RF power on for 7 s).
- Step 4: N₂ purge for 12 s.
- Post-Deposition:
- Cool the samples under N₂ atmosphere.
- Perform electrical characterization (C-V, I-V) using appropriate metal contacts.
The workflow for this protocol is summarized in the following diagram:
Protocol: PLD of Zirconium Thin Films with Substrate Temperature Variation
This protocol describes the deposition of Zr thin films to study temperature-induced morphological and crystalline phase evolution [7].
3.2.1 Research Reagent Solutions
- Target: High-purity Zirconium metal target
- Substrate: Si(100) wafer
- Laser System: KrF excimer laser (248 nm wavelength)
3.2.2 Step-by-Step Procedure
- Substrate Preparation:
- Clean Si(100) substrates using standard RCA cleaning procedures.
- Mount substrates onto the heated holder in the PLD vacuum chamber.
- PLD System Setup:
- Ensure the base pressure is in the ultra-high vacuum (UHV) range.
- Set the target-to-substrate distance according to the system geometry.
- Load the Zr target and ensure it is rotating.
- Deposition Process:
- Set the substrate temperature to the desired value (e.g., 300°C, 400°C, or 500°C).
- Set the laser parameters: 248 nm wavelength, 75 mJ energy, 10 Hz repetition rate, 1-hour deposition time.
- Initiate the laser ablation and deposition process.
- Post-Deposition & Analysis:
- Cool the samples under vacuum.
- Characterize films using XRD, SEM, and AFM to determine crystallinity, phase, and surface morphology.
The logical relationship between temperature and film properties is conceptualized below:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Substrate Temperature Experiments
| Item Name | Function / Role in Experiment | Example from Protocols |
|---|---|---|
| High-Purity Precursor | Source of the desired film material; purity is critical for reproducible film properties and growth kinetics. | TEMAH for HfO₂ PEALD [25] |
| Reactive Gas / Plasma Source | Reacts with the chemisorbed precursor monolayer to form the desired solid film. | O₂/Ar plasma for PEALD [25] |
| Carrier/Purge Gas | Transports precursor vapor and removes volatile reaction by-products from the chamber. | N₂ gas in PEALD [25] |
| Single-Crystal Substrate | Provides a well-defined, smooth, and contaminant-free surface for nucleation and growth. | Si(100) wafer for PLD [7] |
| High-Purity Sputtering/PLD Target | Source of material for physical vapor deposition techniques like PLD and sputtering. | Zirconium metal target for PLD [7] |
Substrate temperature is a fundamental parameter enabling precise control over thin-film nucleation and growth. In PEALD, temperature directly influences surface reaction rates and crystallinity, with optimal electrical properties often observed at intermediate temperatures. In PLD, temperature controls adatom mobility, dictating the transition from 2D layer growth to 3D island formation and enabling phase and orientation control. The protocols and data provided herein offer a foundational framework for designing experiments within a thesis on nucleation research, ensuring systematic and reproducible investigation of temperature-dependent phenomena in thin-film deposition.
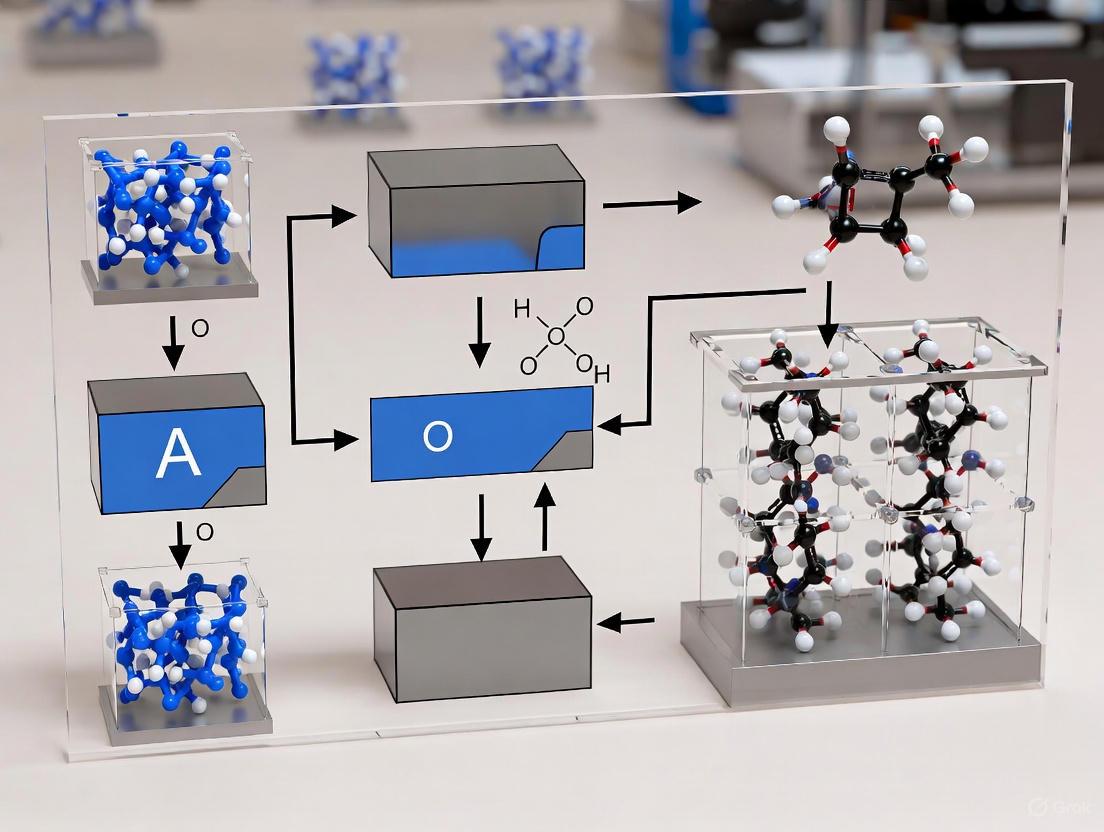
Advanced crystallization techniques are pivotal in materials science and pharmaceutical development for producing high-quality crystals with superior optoelectronic properties and purity. Inverse Temperature Crystallization (ITC) and Vapor Diffusion Crystallization represent two powerful solution-based methods that enable precise control over nucleation and growth processes. These techniques are particularly valuable for processing thermally sensitive materials, including metal halide perovskites for photovoltaics and radiation detectors, as well as organic compounds and proteins for pharmaceutical applications [30] [31] [32]. The fundamental principle underlying both methods is the achievement of a controlled supersaturated state, which is the primary driver for nucleation and subsequent crystal growth. Within a broader thesis on substrate temperature control for nucleation research, understanding how these methods modulate supersaturation—either through temperature manipulation or solvent composition changes—provides critical insights into controlling crystal quality, morphology, and phase purity.
Principle of Inverse Temperature Crystallization (ITC)
Fundamental Mechanism and Applications
Inverse Temperature Crystallization (ITC) exploits the unusual physicochemical phenomenon of retrograde solubility, where a solute's solubility decreases with increasing temperature in certain solvent systems. This behavior contradicts the typical positive solubility-temperature relationship observed for most materials. When a solution saturated at room temperature is heated, the system enters a supersaturated state without solvent evaporation, triggering spontaneous nucleation and crystal growth [31]. This method is exceptionally valuable for growing high-quality single crystals of hybrid organic-inorganic perovskites like MAPbBr₃ and MAPbI₃, which exhibit remarkable optoelectronic properties including high carrier mobility, long diffusion lengths, and low trap-state densities [30] [31].
The ITC process enables rapid crystal growth, achieving rates up to 38 mm³/h for MAPbBr₃, which is an order of magnitude faster than conventional room-temperature crystallization methods [31]. The resulting single crystals exhibit exceptional quality with trap densities as low as 10¹⁰ cm⁻³ and carrier diffusion lengths exceeding 10 micrometers for MAPbI₃, making them ideal for high-performance optoelectronic devices [31].
Experimental Protocol: ITC for MAPbX₃ Perovskite Single Crystals
Materials and Equipment:
- Lead(II) bromide (PbBr₂, ≥99.8%)
- Methylammonium bromide (MABr, ≥98%)
- Dimethylformamide (DMF) for MAPbBr₃ or γ-butyrolactone (GBL) for MAPbI₃
- Magnetic stirrer and hotplate
- Temperature-controlled oil bath or incubator
- Syringe filters (0.22 μm PTFE)
Procedure:
- Precursor Solution Preparation: Dissolve equimolar quantities of PbBr₂ and MABr in DMF (for MAPbBr₃) or GBL (for MAPbI₃) to achieve a 1.0 M concentration. Stir continuously at 50°C for 2 hours until complete dissolution [31].
Solution Filtration: Filter the precursor solution through a 0.22 μm PTFE syringe filter to remove undissolved particles and particulate impurities that can act as uncontrolled nucleation sites [31] [33].
Crystallization Initiation: Transfer the filtered solution to a sealed vial and place it in a temperature-controlled oil bath or incubator set at 80°C. Do not disturb the vial during the crystallization process.
Crystal Growth: Maintain the temperature at 80°C for 3-5 hours. Crystal nucleation typically begins within the first hour, with rapid growth occurring subsequently.
Crystal Harvesting: Carefully remove the crystals from the solution using tweezers, rinse gently with fresh solvent to remove residual solution, and air-dry.
Critical Parameters for Success:
- Temperature Control: Precise temperature control (±1°C) is essential for controlled nucleation and growth. Higher temperatures increase supersaturation, leading to faster growth but potentially more defects.
- Solvent Selection: The solvent must exhibit inverse solubility behavior with the target compound. DMF works optimally for MAPbBr₃, while GBL is preferred for MAPbI₃ [31].
- Solution Concentration: Optimal concentration is approximately 1.0 M for MAPbX₃ precursors. Higher concentrations may lead to excessive nucleation, while lower concentrations yield fewer crystals.
ITC Process Workflow
Principle of Vapor Diffusion Crystallization
Fundamental Mechanism and Applications
Vapor Diffusion Crystallization relies on the controlled equilibration of solvent composition between a droplet containing the target solute and a reservoir solution with higher osmolarity. The method operates on the principle of diffusion-controlled solvent shifting, where the vapor phase mediates the gradual change in solvent composition [34] [32]. As volatile components equilibrate between the droplet and reservoir, the droplet experiences a steady increase in supersaturation, promoting controlled nucleation and growth.
This technique is particularly valuable for growing crystals of sensitive materials, including proteins, pharmaceuticals, and hybrid perovskites, as it enables gentle, gradual approach to supersaturation. The method minimizes mechanical stress on developing crystals and allows for precise control over crystal size and quality [34] [32]. Recent advancements have expanded vapor diffusion to environmentally friendly solvent systems, such as water and isopropyl alcohol (IPA), reducing reliance on toxic solvents like DMF and DMSO while maintaining high crystal quality [34].
Experimental Protocol: Antisolvent Vapor Diffusion for CsPbBr₃ Single Crystals
Materials and Equipment:
- Cesium bromide (CsBr, ≥99.8%)
- Lead(II) bromide (PbBr₂, ≥99.8%)
- Binary solvent: DMSO/DMF (9:1 v/v)
- Antisolvent: Ethanol (≥98%)
- Crystallization plates or glass vials
- Sealed container
Procedure:
- Precursor Solution Preparation: Dissolve CsBr and PbBr₂ with a 1:1.5 molar ratio (excess PbBr₂ suppresses Cs₄PbBr₆ formation) in the DMSO/DMF binary solvent. Stir at 50°C for 2 hours until fully dissolved [35].
Solution Pretreatment (Optional): For controlled nucleation, titrate the precursor solution with ethanol until the onset of turbidity, then refilter to obtain a clear, metastable solution [35].
Experimental Setup: Place 0.5-1.0 mL of precursor solution in small open vials. Position these vials inside a larger sealed container filled with 10-20 mL of ethanol antisolvent. Ensure the vial height is less than the container height to facilitate vapor diffusion.
Crystallization: Seal the container and maintain at constant room temperature (20-25°C). Do not disturb during the crystallization process.
Crystal Growth: Crystals typically nucleate within 24-48 hours and reach centimeter sizes over 5-7 days. The slow vapor diffusion enables controlled growth with minimal defects.
Crystal Harvesting: Carefully extract crystals from the solution, rinse with DMF to remove surface impurities, and air-dry.
Critical Parameters for Success:
- Antisolvent Selection: Choose antisolvents with appropriate miscibility and vapor pressure. Ethanol offers optimal diffusion rates for CsPbBr₃ crystallization [35].
- Precursor Concentration: Optimal CsPbBr₃ precursor concentration is approximately 0.35 M in DMSO/DMF (9:1) [35].
- Container Seal: Complete sealing is essential for controlled vapor diffusion rates.
- Temperature Stability: Constant temperature prevents convective disturbances that may introduce defects.
Vapor Diffusion Process Workflow
Comparative Analysis of Crystallization Methods
Table 1: Comparative Analysis of Inverse Temperature Crystallization and Vapor Diffusion Methods
| Parameter | Inverse Temperature Crystallization (ITC) | Vapor Diffusion Crystallization |
|---|---|---|
| Fundamental Principle | Retrograde solubility with increasing temperature [31] | Diffusion-controlled solvent composition shift [34] [35] |
| Typical Growth Rate | Very fast (3-38 mm³/h) [31] | Slow to moderate (days to weeks) [35] |
| Temperature Range | Elevated temperatures (70-100°C) [31] | Room temperature or controlled (20-25°C) [35] |
| Crystal Quality | High quality, low trap density (10¹⁰ cm⁻³) [31] | High quality, minimal stress-induced defects [34] |
| Size Control | Good size control, possible shape control via vessel geometry [31] | Excellent size control, can achieve centimeter-scale crystals [35] |
| Phase Purity Challenges | Potential secondary phase formation (e.g., Cs₄PbBr₆) [35] | Better phase purity with optimized precursor ratios [35] |
| Material Systems | MAPbBr₃, MAPbI₃, CsPbBr₃ [30] [31] | CsPbBr₃, proteins, pharmaceuticals [34] [32] [35] |
| Scalability | Moderate, limited by solution volume and temperature uniformity | Good, multiple chambers possible [36] |
| Equipment Requirements | Temperature-controlled bath or incubator [31] | Sealed containers, humidity control [35] |
Table 2: Quantitative Performance Metrics for Perovskite Single Crystals Grown by Advanced Methods
| Material | Crystallization Method | Trap Density (cm⁻³) | Carrier Mobility (cm²/V·s) | Carrier Diffusion Length (μm) | Crystal Size Achievable |
|---|---|---|---|---|---|
| MAPbBr₃ | ITC [31] | 3 × 10¹⁰ | 24.0 | ~4.3 | ~10 mm (edge) |
| MAPbI₃ | ITC [31] | 1.4 × 10¹⁰ | 67.2 | ~10.0 | ~10 mm (edge) |
| CsPbBr₃ | Temperature-lowering [37] | N/A | N/A | N/A | Centimeter-scale |
| CsPbBr₃ | Antisolvent Vapor Diffusion [35] | N/A | N/A | N/A | Up to 1 cm |
| MAPbBr₃ | Solvent Vapor Diffusion [34] | N/A | N/A | N/A | Thin films |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Advanced Crystallization Methods
| Material/Reagent | Function/Application | Examples/Specific Uses |
|---|---|---|
| Dimethylformamide (DMF) | Solvent for ITC | Primary solvent for MAPbBr₃ ITC [31] |
| γ-Butyrolactone (GBL) | Solvent for ITC | Primary solvent for MAPbI₃ ITC [31] |
| Dimethyl Sulfoxide (DMSO) | Solvent component | Binary solvent with DMF for CsPbBr₃ crystallization [35] |
| Ethanol | Antisolvent for vapor diffusion | Antisolvent for CsPbBr₃ crystallization [35] |
| Methylammonium Bromide (MABr) | Perovskite precursor | Organic component for hybrid perovskites [31] |
| Cesium Bromide (CsBr) | Perovskite precursor | Inorganic component for all-inorganic perovskites [35] |
| Lead Bromide (PbBr₂) | Perovskite precursor | Metal halide component for perovskite crystals [31] [35] |
| Hydrobromic Acid (HBr) | Solvent for temperature-lowering | Growth medium for CsPbBr₃ crystals [37] |
| Water/IPA Mixture | Green solvent system | Environmentally friendly vapor diffusion crystallization [34] |
| Polyvinylpyrrolidone (PVP) | Polymer ligand | Stabilizer for perovskite nanocrystal growth [34] |
| Sealed Crystallization Chambers | Experimental setup | Controlled environment for vapor diffusion [35] |
| Temperature-Controlled Bath | Equipment | Precise temperature control for ITC [31] |
| Syringe Filters (0.22 μm) | Solution preparation | Remove particulate impurities for controlled nucleation [31] |
Inverse Temperature Crystallization and Vapor Diffusion represent sophisticated approaches to crystal engineering that enable researchers to overcome fundamental limitations of conventional crystallization techniques. ITC leverages unusual retrograde solubility behavior for rapid growth of high-quality single crystals, particularly advantageous for metal halide perovskites with exceptional optoelectronic properties. Vapor Diffusion employs controlled solvent composition shifts through the vapor phase for gentle, gradual crystallization of sensitive materials, including proteins and thermally-labile compounds. Within the context of substrate temperature control for nucleation research, these methods demonstrate how precise manipulation of thermodynamic parameters—whether temperature or chemical potential—enables unprecedented control over nucleation kinetics, crystal growth, and final material properties. The continued refinement of these advanced crystallization protocols, coupled with emerging technologies like the CrystalBreeder platform for high-throughput screening, promises to accelerate development of next-generation materials for optoelectronics, pharmaceuticals, and beyond [36].
The precise control of substrate temperature is a foundational requirement in nucleation research, directly influencing the kinetics, thermodynamics, and ultimate outcomes of phase transitions in fields ranging from materials science to pharmaceutical development [11]. Interfacial engineering offers powerful strategies to enhance thermal management at the critical interfaces where nucleation initiates. By applying functionalized coatings and incorporating nanoparticles with tailored properties, researchers can actively modulate heat transfer efficiency, minimize parasitic thermal gradients, and create surfaces with defined wettability and energy characteristics [38]. These engineered interfaces are particularly vital for the accurate quantification of ice-nucleating particles (INPs) and the study of crystallization processes, where temperature stability and homogeneity are paramount for generating reproducible and reliable data [11]. These application notes provide detailed protocols and data summaries for implementing these advanced thermal control strategies within the specific context of a research thesis on substrate temperature control for nucleation studies.
The selection of materials for interfacial engineering is critical. The following tables summarize key properties of common nanoparticles and substrate coatings used to enhance thermal control.
Table 1: Comparison of Nanoparticles for Thermal Interface Materials (TIMs)
| Nanomaterial | Intrinsic Thermal Conductivity (W/m·K) | Typical Loading in Composite (wt%) | Key Advantage | Primary Challenge |
|---|---|---|---|---|
| Nano-Copper (Cu) [39] | ~400 | 60-80 | High conductivity, cost-effective | Prone to oxidation, requires surface passivation |
| Silver (Ag) Nanowires | ~429 | 5-15 (low percolation threshold) | Forms conductive network at low loadings | High cost, potential electromigration |
| Graphene | 2000-5000 (in-plane) | 0.5-5 | Extremely high intrinsic conductivity | Anisotropic heat transfer, dispersion issues |
| Hexagonal Boron Nitride (h-BN) | ~300 (in-plane) | 10-30 | Electrically insulating, high thermal conductivity | Anisotropic, chemically inert (hard to functionalize) |
| Alumina (Al₂O₃) | ~30 | 20-50 | Electrically insulating, low cost, stable | Moderate thermal conductivity |
Table 2: Properties of Functionalized Surfaces and Coatings for Thermal Control
| Coating / Surface Treatment | Thermal Conductivity | Contact Angle (°) | Key Function in Nucleation Research | Application Method |
|---|---|---|---|---|
| Self-Assembled Monolayer (OTS) [38] | Low (thin organic layer) | ~110 (Hydrophobic) | Controls surface energy, inhibits frost formation, defines droplet contact area | Solution-based deposition |
| Polymer Matrix (e.g., PDMS) | Low (~0.2 W/m·K) | ~115 (Hydrophobic) | Provides a flexible, conformal layer for embedding nanoparticles | Spin-coating, Doctor-blading |
| Graphene Encapsulation Layer [39] | High | Tunable | Provides a conformal, oxidation-resistant barrier for metal nanoparticles | Chemical Vapor Deposition (CVD) |
| Hydrophilic Plasma Treatment | N/A | < 30 (Hydrophilic) | Increases surface energy, promotes uniform water film formation for homogeneous freezing studies | Plasma Exposure |
| Core-Shell (Cu@Ag) [39] | High (core-dependent) | N/A | Combines high conductivity of core with oxidation resistance of shell | Electroless plating, Galvanic displacement |
Experimental Protocols
Protocol: Fabrication of a Nanoparticle-Enhanced Thermal Interface Material (TIM)
Application: This protocol details the creation of a polymer nanocomposite film to be used as a thermal interface material between a Peltier cooler and a sample substrate (e.g., an aluminum cold stage) to reduce thermal contact resistance and improve temperature uniformity [11] [39].
Materials:
- Matrix: Polydimethylsiloxane (PDMS) base and curing agent (e.g., Sylgard 184).
- Nanoparticles: Surfactant-functionalized silver-coated copper nanoparticles (Ag@Cu) [39].
- Solvent: Isopropyl Alcohol (IPA).
- Equipment: Planetary centrifugal mixer, vacuum desiccator, spin coater, oven.
Procedure:
- Nanoparticle Dispersion:
- Weigh out the Ag@Cu nanoparticles to achieve a 70% by weight loading in the final composite.
- In a mixing cup, combine the nanoparticles with a volume of IPA sufficient to create a slurry. The solvent reduces viscosity for easier handling.
- Place the cup in the planetary centrifugal mixer and mix at 2000 RPM for 2 minutes.
- Transfer the mixture to a vacuum desiccator to evaporate the IPA solvent completely.
Polymer Composite Mixing:
- Add the PDMS base elastomer to the dried nanoparticles.
- Mix in the planetary centrifugal mixer at 2200 RPM for 4 minutes, pausing once to scrape down the sides. This step ensures deagglomeration and uniform distribution.
- Degas the mixture in a vacuum desiccator until no bubbles remain (approximately 30-60 minutes).
- Add the PDMS curing agent at a 10:1 base-to-agent ratio and mix again at 2000 RPM for 2 minutes. Degas briefly for 10 minutes.
Film Casting & Curing:
- Pour the composite onto the target substrate (e.g., the aluminum cold stage) or a release agent-treated surface.
- Use a spin coater or doctor blade to achieve a uniform thickness of 100-200 µm.
- Cure the film in an oven at 80°C for 2 hours.
Quality Control:
- Verify film thickness using a profilometer.
- Assess thermal performance by measuring the temperature gradient across the substrate with and without the TIM using calibrated Pt100 sensors [11].
Protocol: Application of a Superhydrophobic Coating on a Cold Stage for Droplet Immersion Freezing Studies
Application: This protocol describes the creation of a superhydrophobic surface on an aluminum cold stage to generate isolated, spherical droplets for immersion freezing experiments, minimizing surface-induced nucleation artifacts and enabling clear optical detection of freezing events [11] [38].
Materials:
- Substrate: Aluminum cold stage, cleaned and polished.
- Coating Precursor: Trichloro(octadecyl)silane (OTS).
- Solvents: Toluene, Ethanol.
- Equipment: Plasma cleaner, chemical fume hood, nitrogen gun, oven.
Procedure:
- Substrate Pre-treatment and Activation:
- Clean the aluminum stage sequentially in an ultrasonic bath with ethanol and then acetone for 15 minutes each. Dry with a stream of nitrogen.
- Place the stage in a plasma cleaner and treat with oxygen plasma for 5 minutes at high power. This step creates a dense, reactive hydroxyl (-OH) layer on the native aluminum oxide surface.
Silane Monolayer Grafting:
- Immediately after plasma treatment, prepare a 1 mM solution of OTS in anhydrous toluene within a glass container in a fume hood.
- Immerse the activated aluminum stage in the OTS solution. Seal the container and allow the reaction to proceed for 4 hours at room temperature.
Post-treatment and Curing:
- Remove the stage from the solution and rinse thoroughly with fresh toluene and then ethanol to remove any physisorbed silane molecules.
- Dry the stage with a nitrogen gun.
- Cure the coating in an oven at 120°C for 1 hour to promote the formation of stable covalent Al-O-Si bonds.
Validation:
- Measure the static water contact angle using a goniometer. A successful coating will yield a contact angle >150°.
- Test the coating's performance by pipetting an array of 1 µL Milli-Q water droplets and observing their shape and stability during a cooling ramp on the FINDA-WLU or similar apparatus [11].
Visualized Workflows and Pathways
Diagram 1: Interfacial engineering workflow for nucleation substrates.
Diagram 2: Nanoparticle thermal conductivity pathway.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Interfacial Engineering in Thermal Control
| Item Name | Function / Application | Key Characteristics |
|---|---|---|
| Platinum Resistance Thermometer (Pt100) [11] | High-precision temperature sensing and calibration of cold stages. | Accuracy of ±0.15°C at 0°C, essential for validating temperature uniformity. |
| Functionalized Nanoparticles (e.g., Ag@Cu) [39] | Filler material in Thermal Interface Materials (TIMs) to enhance thermal conductivity. | Core-shell structure provides high thermal conductivity while resisting oxidation. |
| Organosilane (e.g., OTS) [38] | Precursor for creating self-assembled monolayers (SAMs) to engineer surface wettability. | Forms a stable, hydrophobic coating on metal oxide surfaces, enabling droplet-based assays. |
| Polydimethylsiloxane (PDMS) | A flexible, biocompatible polymer matrix for creating nanocomposite TIMs. | Chemically inert, thermally stable, and easy to process with nanoparticles. |
| Polymerase Chain Reaction (PCR) Plate [11] | Serves as a multi-well sample holder for high-throughput droplet freezing assays. | Compatible with cold stages, allows parallel processing of dozens of samples. |
| Temperature-Controlled Circulator (Chiller) [11] | Provides precise and stable cooling to the cold stage or Peltier element. | Enables controlled linear cooling ramps (e.g., 0.1–1.0 °C/min) for nucleation studies. |
In-situ Growth Techniques for Single-Crystal Thin Films on Functional Layers
The integration of single-crystal thin films with functional substrates represents a frontier in advanced materials science, enabling breakthroughs in electronics, photonics, and quantum computing. This process requires precise atomic-level control over film nucleation and growth, where substrate temperature emerges as the most critical parameter governing adatom mobility, nucleation density, and ultimate crystalline quality [7]. The fundamental challenge lies in balancing the thermal energy supplied to growing surfaces: insufficient temperature results in amorphous structures with high defect densities, while excessive temperature promotes undesirable 3D island growth and interfacial reactions [8] [7].
Within this context, in-situ growth techniques offer unprecedented capability for producing epitaxial films with controlled properties. This Application Note examines two prominent vapor-phase deposition methods—Chemical Vapor Deposition (CVD) and Pulsed Laser Deposition (PLD)—framed within the broader thesis of substrate temperature control for nucleation research. We present quantitative data, detailed protocols, and visualization tools to guide researchers in optimizing these processes for specific material systems and applications.
Key Growth Techniques and Temperature-Dependent Nucleation Behavior
Chemical Vapor Deposition (CVD)
CVD involves the chemical reaction of vapor-phase precursors on a heated substrate surface, resulting in the formation of a solid thin film. The substrate temperature directly controls decomposition kinetics of precursor molecules and surface migration of adatoms.
Case Study: β-Ga₂O₃ on 4H-SiC In the growth of β-Ga₂O₃ films on 4H-SiC substrates, temperature optimization is crucial for suppressing vapor-phase nucleation and associated surface irregularities [8]. Experimental results demonstrate that a growth temperature of 680°C on a 4° off-axis (0001) substrate produces optimal film quality through step-flow growth mechanism. This specific temperature enables sufficient adatom mobility for ordered growth while minimizing premature gas-phase reactions that degrade surface morphology.
Table 1: Temperature Optimization in CVD Growth of β-Ga₂O₃ on 4H-SiC
| Growth Parameter | Optimized Condition | Impact on Nucleation & Film Quality |
|---|---|---|
| Substrate Temperature | 680°C | Balances surface migration and nucleation density |
| Substrate Off-axis Angle | 4° off-axis (0001) | Promotes step-flow growth mode |
| Ga Source | Elemental Ga (5N purity) | Reduces introduction of foreign elements |
| O₂ Flow Rate | 20 sccm | Provides sufficient oxidant without excessive gas-phase nucleation |
| Carrier Gas (Ar) Flow | 200 sccm | Ensures proper precursor delivery |
Pulsed Laser Deposition (PLD)
PLD utilizes high-power laser pulses to ablate material from a target, creating a plasma plume that deposits onto a heated substrate. The substrate temperature governs the mobility of adatoms on the substrate surface, controlling nucleation density, grain coalescence, and development of long-range crystalline order [7].
Case Study: Zirconium Thin Films on Silicon Research on zirconium thin films deposited on Si(100) substrates reveals distinct temperature-dependent growth regimes [7]:
- At 400°C, the Zr(100) plane exhibits the strongest orientation with optimal crystallinity
- At 500°C, the Zr(002) orientation dominates, but significant island formation occurs due to transition to 3D growth
- Computational modeling identifies a critical film thickness of ~1-2 nm for the transition from 2D layer-by-layer growth to 3D island formation
Table 2: Temperature-Dependent Growth Characteristics of Zirconium Thin Films via PLD
| Substrate Temperature | Crystalline Structure | Surface Morphology | Growth Mechanism |
|---|---|---|---|
| 300°C | Polycrystalline with mixed orientations | Smooth surface | Limited adatom diffusion |
| 400°C | Strong Zr(100) preferential orientation | Moderate roughness | Optimal surface migration |
| 500°C | Strong Zr(002) orientation; zirconium silicide formation | High roughness; significant island formation | 3D island growth dominated by enhanced diffusivity |
Experimental Protocols
CVD Protocol for β-Ga₂O₃ on 4H-SiC
Materials and Equipment:
- Horizontal hot-wall CVD tube furnace
- 4° off-axis (0001) 4H-SiC substrates
- Elemental Ga source (5N purity)
- High-purity O₂ (20 sccm) and Ar (200 sccm) gases
- RCA cleaning solutions
Step-by-Step Procedure:
Substrate Preparation
- Perform standard RCA cleaning on 4H-SiC substrates
- Dry substrates with Ar gas to ensure complete removal of surface contaminants
- Load substrates into CVD chamber with proper orientation
Temperature Optimization
- Ramp substrate temperature to 680°C at controlled rate of 10°C/min
- Maintain temperature stability within ±1°C during growth
- Employ additional heating zone to preheat gas flow for temperature uniformity
Film Growth
- Introduce Ar carrier gas at 200 sccm flow rate
- Initiate O₂ flow at 20 sccm once temperature stabilizes
- Maintain growth for predetermined duration (typically 60-120 minutes)
- Monitor film thickness in real-time if spectroscopic ellipsometry available
Post-growth Processing
- Cool samples gradually under Ar atmosphere (5°C/min)
- Characterize film quality using XRD, SEM, AFM, and XPS [8]
PLD Protocol for Zirconium Thin Films on Silicon
Materials and Equipment:
- KrF excimer laser (248 nm wavelength)
- High-purity zirconium target (99.95%)
- Si(100) substrates
- UHV chamber with substrate heater
- Temperature controller capable of ±0.5°C stability
Step-by-Step Procedure:
System Preparation
- Polish and clean Zr target to remove surface oxides
- Clean Si substrates using standard semiconductor cleaning protocol
- Load target and substrates into UHV chamber
- Evacuate chamber to base pressure (<1×10⁻⁶ Torr)
Temperature Calibration
- Calibrate substrate temperature using thermocouple directly bonded to substrate surface
- Account for potential temperature gradients across substrate holder
- Ramp to desired growth temperature (300-500°C) with stability of ±0.5°C
Laser Ablation and Deposition
- Set laser parameters: 248 nm, 10 ns pulse duration, 10 Hz repetition rate
- Adjust laser fluence to 75 mJ (spot size 1.65 mm²)
- Initiate ablation with substrate rotation for thickness uniformity
- Maintain deposition for 1 hour for approximately 100 nm film thickness
In-situ Monitoring and Characterization
- Monitor growth rate using quartz crystal microbalance
- Perform real-time RHEED for crystalline quality assessment (if available)
- Cool samples slowly (2-5°C/min) under vacuum to minimize thermal stress
- Characterize using XRD, SEM, AFM to correlate structure with temperature [7]
Visualization of Growth Mechanisms
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for Single-Crystal Thin Film Growth
| Material/Reagent | Specification | Function | Application Examples |
|---|---|---|---|
| 4H-SiC Substrates | 4° off-axis (0001) orientation | Provides lattice matching for epitaxial growth; enables step-flow growth mode | β-Ga₂O₃ growth for power devices [8] |
| Elemental Gallium | 5N (99.999%) purity | High-purity Ga source for oxide growth; reduces foreign element incorporation | β-Ga₂O₃ CVD growth [8] |
| Zirconium Target | 99.95% purity, polished surface | ablation source for thin film deposition | Zirconium thin films for EUV optics [7] |
| High-Purity Gases (O₂, Ar) | 99.999% purity, moisture-free | Reaction and carrier gases for CVD processes | β-Ga₂O₃ growth [8] |
| Platinum Resistance Thermometers | Pt100 sensors, ±0.15°C accuracy at 0°C | Precise temperature measurement and calibration | Temperature monitoring in deposition systems [11] |
| RCA Cleaning Solutions | Standard SC-1, SC-2 formulations | substrate surface preparation and contamination removal | Pre-deposition cleaning of Si and SiC substrates [8] [7] |
Substrate temperature control emerges as the unifying theme in optimizing in-situ growth techniques for single-crystal thin films. Through careful temperature management within material-specific windows—680°C for β-Ga₂O₃ on 4H-SiC and 400°C for Zr on Si—researchers can direct nucleation processes toward desired crystalline structures and morphologies. The protocols and data presented here provide a foundation for systematic investigation of temperature-dependent nucleation phenomena, enabling the development of next-generation electronic and optical devices requiring precise thin film architectures. Future research directions should focus on real-time monitoring of nucleation events and development of advanced temperature control systems capable of dynamic profile adjustment during growth.
In nucleation research, the precise control of process parameters is a cornerstone for achieving reproducible and meaningful results. This is particularly true for substrate temperature control, a critical variable that governs the thermodynamic and kinetic pathways of nucleation. This article details application notes and protocols for investigating heterogeneous ice nucleation, a ubiquitous process with profound implications in atmospheric science, cryobiology, and materials science. The methodologies presented herein are designed to provide researchers with a framework to systematically balance temperature, time, and the chemical environment of the substrate to decipher and optimize nucleation outcomes.
Core Principles and Quantitative Data
The transition from a liquid to a solid phase on a substrate is not governed by a single factor but by the interplay of multiple physicochemical properties. Traditional descriptors like lattice match to the crystal structure of the nucleating solid have proven insufficient for robust predictions [40]. Advanced data-driven analyses have identified a more complex set of microscopic factors that collectively determine the efficacy of a substrate as a nucleating agent [40].
The following table summarizes these key physical descriptors and their impact on the nucleation process.
Table 1: Key Physical Descriptors Governing Heterogeneous Ice Nucleation
| Descriptor | Description | Impact on Nucleation |
|---|---|---|
| Lattice Match | The alignment between the atomic or molecular structure of the substrate and the crystal lattice of the nucleating solid [40]. | A good match can lower the energy barrier for nucleation, but it is not a reliable predictor alone (e.g., AgI vs. BaF₂) [40]. |
| Local Ordering of Water | The degree of structural ordering induced in the liquid water near the substrate interface [40]. | Enhanced local ordering that resembles the ice structure promotes higher nucleation temperatures [40]. |
| Interfacial Water Density | The change in density of liquid water in the immediate vicinity of the substrate surface [40]. | A reduction in water density near the surface is correlated with improved ice-nucleating ability [40]. |
| Adsorption Energy Landscape | The spatial corrugation or variation in the energy with which water molecules bind to the substrate [40]. | A more uniform (less corrugated) energy landscape is associated with better nucleation performance [40]. |
Furthermore, the fundamental nature of the nucleation process itself must be considered. The stochastic model, which treats nucleation as a probabilistic event dependent on time and available surface area, is often required to accurately describe experimental data, particularly under isothermal conditions [41]. The number of unfrozen droplets, N_ufz, from a total, N_tot, can be modeled as:
N_ufz / N_tot = exp( -J_het * A * t )
where J_het is the heterogeneous nucleation rate coefficient (cm⁻² s⁻¹), A is the ice nucleating surface area (ISA) in a droplet, and t is time [41]. This relationship highlights the critical interplay between time (t), substrate surface properties (A), and temperature (through its direct influence on J_het).
Experimental Visualization
The following diagram illustrates the core workflow and logical relationships involved in a nucleation study, from substrate preparation to data analysis and model prediction.
Diagram Title: Nucleation Research Workflow and Key Factors
Detailed Experimental Protocols
Protocol 1: Determining Nucleation Temperature via Cooling Ramps
Objective: To establish the characteristic nucleation temperature (T_n) of a supercooled liquid in contact with a diverse set of model substrates.
Background: This protocol utilizes molecular dynamics (MD) simulations to screen a large number of substrates efficiently. The computational efficiency of coarse-grained models like mW allows for the broad screening necessary to identify robust nucleation descriptors [40].
Materials:
- Model Substrates: A library of diverse substrates (e.g., 900+ substrates including Lennard-Jones materials, OH-patterned surfaces, graphitic surfaces) [40].
- Water Model: The mW (monatomic Water) model or an equivalent computationally efficient potential [40].
- Simulation Software: A suitable MD package (e.g., LAMMPS, GROMACS) capable of implementing the chosen water model and cooling protocols.
Procedure:
- System Setup: For each substrate, construct a simulation cell with the substrate in contact with a body of supercooled liquid water.
- Equilibration: Equilibrate the entire system at a temperature above the expected freezing point to ensure a stable liquid phase.
- Cooling Ramp: Perform multiple independent MD simulation runs for each system, applying a constant cooling rate (e.g., 1 K ns⁻¹).
- Nucleation Detection: Monitor the simulation for the spontaneous formation of a critical ice nucleus. The temperature at which this occurs in each run is recorded as the nucleation temperature for that run,
T_n. - Data Collection: For each substrate, calculate the average
T_nand its standard deviation from the repeated cooling ramps. The typical uncertainty for this method is approximately ±3 K [40].
Protocol 2: Analyzing Drop-Freezing Experiments with the HUB Method
Objective: To model and extract the underlying distribution of ice nucleation temperatures from experimental drop-freezing data.
Background: Drop-freezing experiments measure the frozen fraction of droplets, f_ice(T), as temperature decreases. The HUB (heterogeneous underlying-based) method provides a robust framework for interpreting this data to obtain the differential freezing spectrum, n_m(T), which represents the density of ice-nucleating particles active at each temperature [42].
Materials:
- Drop-Freezing Apparatus: A multi-well plate or other substrate housed in a temperature-controlled stage or bath.
- Sample Preparation: A suspension containing the ice-nucleating particles of interest, prepared in a series of dilutions.
- Detection System: A method to visually or thermally detect the freezing of individual droplets.
- Analysis Software: The HUB-forward and HUB-backward Python codes [42].
Procedure:
- Experiment: Place droplets of uniform volume containing a known concentration or surface area of ice-nucleating particles on the substrate. Cool the platform gradually from above 0 °C and record the freezing temperature of each droplet.
- Data Input: Collect the fraction of frozen droplets,
f_ice(T), across the temperature range. For soluble or dispersible INs, repeat the experiment at various 10-fold dilutions [42]. - Compute Cumulative Spectrum: Calculate the cumulative freezing spectrum,
N_m(T), using the formula:N_m(T) = - (1 / X) * ln[1 - f_ice(T)]whereXis a normalization factor (e.g., mass of IN material, surface area) [42]. - HUB-backward Analysis: Use the HUB-backward code to apply a stochastic optimization method. This computes a smooth, analytical differential freezing spectrum,
n_m(T) = dN_m(T)/dT, from the experimentalN_m(T)or directly fromf_ice(T)[42]. - Interpretation: The resulting
n_m(T)spectrum reveals the underlying number of IN subpopulations and their characteristic freezing temperatures, allowing for quantitative analysis of how these depend on environmental variables.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions and Materials for Nucleation Studies
| Item | Function / Description |
|---|---|
| mW Water Model | A coarse-grained model for water that allows for high-throughput molecular dynamics screening of nucleation due to its computational efficiency [40]. |
| Lennard-Jones Substrates | A set of model substrates with tunable interaction parameters, used to systematically study the effect of substrate properties on nucleation [40]. |
| HUB Python Codes | A pair of computational tools (HUB-forward and HUB-backward) for modeling and interpreting the results of drop-freezing experiments to extract underlying nucleation temperature distributions [42]. |
| Portable Ice Nucleation Experiment (PINE) | An automated, expansion-type cloud chamber for measuring ice-nucleating particle (INP) concentrations in ambient air or laboratory samples with high temporal resolution [43]. |
| Stochastic Nucleation Rate Coefficient (J_het) | A fundamental physical parameter (in cm⁻² s⁻¹) that describes the temperature-dependent rate of nucleation per unit area of substrate, essential for time-dependent predictions [41]. |
Overcoming Challenges: Optimization Strategies for Reproducible High-Quality Outcomes
Crystallization is a pivotal process in the pharmaceutical and materials sciences, determining critical properties of the final product, from drug bioavailability to material performance. Achieving perfect crystals is often hindered by common defects such as amorphous precipitation, unwanted polymorphs, and inhomogeneous growth. These defects are intrinsically linked to the initial nucleation event and the subsequent crystal growth pathway. The phase diagram (see Figure 1) serves as the foundational map for understanding these processes, delineating zones of stability, metastability, and disorder [44]. Supersaturation (S), the driving force for crystallization, is defined as the ratio of the actual solute concentration (C) to its equilibrium solubility (C₀) [44] [13]. Operating within the metastable zone—the region between the solubility and supersolubility curves—is crucial for encouraging growth over uncontrolled nucleation. Venturing into the labile zone, with its high supersaturation, often leads to a proliferation of nucleation sites and amorphous precipitation, as the system bypasses ordered assembly for rapid, disordered aggregation [44]. This document details protocols and application notes for identifying and mitigating these defects, with a particular focus on the role of substrate temperature control in nucleation research.
Identifying and Characterizing Common Defects
A critical step in defect mitigation is the accurate identification and characterization of the resulting solid forms. Advanced analytical techniques are required to distinguish between the different states of matter that can form.
Table 1: Characterization Techniques for Common Crystallization Defects
| Defect Type | Recommended Characterization Techniques | Key Identifiers |
|---|---|---|
| Amorphous Precipitation | Small-Angle X-Ray Scattering (SAXS), Differential Scanning Calorimetry (DSC), Raman Spectroscopy, Scanning Electron Microscopy (SEM) | Broad, diffuse halos in XRPD; lack of sharp melting point in DSC; spherical, non-geometric particles under SEM [45] [46]. |
| Unwanted Polymorphs | X-Ray Powder Diffraction (XRPD), Confocal Raman Microscopy, DSC | Distinct diffraction patterns in XRPD; unique vibrational spectra in Raman; different melting enthalpies and temperatures in DSC [47] [46]. |
| Inhomogeneous Growth | Scanning Electron Microscopy (SEM), In-situ Optical Microscopy, Atomic Force Microscopy (AFM) | Wide variation in crystal size distribution (CSD); presence of fines and aggregates; irregular crystal habit [44] [45]. |
The formation pathway is often non-classical. For instance, a two-step nucleation mechanism may occur, where solute molecules first aggregate into dense, disordered clusters (a mesoscale precursor) before internally reorganizing into an ordered crystal nucleus [45]. Intercepting this pathway at the cluster stage can lead to amorphous precipitation. Furthermore, the presence of additives or specific environmental conditions can stabilize intermediate amorphous phases, as observed in the precipitation of regorafenib, where an amorphous co-precipitate with the polymer HPMCAS was identified [46].
The Scientist's Toolkit: Key Research Reagents and Materials
Controlling crystallization requires a suite of reagents and materials designed to influence nucleation and growth at the molecular level.
Table 2: Key Research Reagent Solutions for Crystallization Control
| Reagent/Material | Function & Mechanism | Example Application |
|---|---|---|
| Heteronucleants (e.g., functionalized surfaces/silica) | Provides a structured surface to lower the nucleation energy barrier and promote ordered crystal formation at lower supersaturation [44]. | Used to induce reproducible nucleation of proteins and small molecules in the metastable zone [44]. |
| Polymers (e.g., HPMCAS, Povidone) | Inhibit unwanted nucleation and stabilize amorphous phases or specific crystal faces through adsorption; can lead to amorphous co-precipitates [46]. | Formulating Amorphous Solid Dispersions (ASDs) to manage precipitation of poorly water-soluble drugs like regorafenib [46]. |
| Nucleating Agents (e.g., ZrO₂, TiO₂) | Act as catalysts for volume crystallization, forming numerous nanocrystals that template the growth of the main crystalline phase [48]. | Controlling crystallization in Lithium Aluminosilicate (LAS) glass-ceramics to achieve near-zero thermal expansion [48]. |
| Antisolvents | Rapidly decrease solute solubility during spin-coating, inducing high supersaturation and triggering instantaneous nucleation [49]. | Fabricating high-quality, pinhole-free perovskite thin films for solar cells and LEDs [49]. |
| Microreactors | Enhance micromixing and mass transfer, enabling precise control over the supersaturation profile for uniform nucleation and growth [47]. | Continuous synthesis of nano- and micro-scale crystals with optimal form and structural stability [47]. |
Experimental Protocols for Defect Mitigation
Protocol: Controlling Nucleation via Substrate Temperature
Principle: Pre-conditioning the substrate temperature alters the local chemical potential (µ) and Gibbs free energy (G) of the system. A warmer substrate accelerates solvent evaporation, rapidly driving the precursor solution to a state of high supersaturation, which promotes a high nucleation density. Conversely, a cooler substrate slows kinetics, favoring the growth of existing nuclei over the formation of new ones [49].
Materials:
- Precursor solution (e.g., perovskite solution, organic molecule in solvent).
- Hotplate with precise temperature control.
- Spin coater or other film deposition apparatus.
- Thermocouple for surface temperature verification.
Procedure:
- Substrate Pre-treatment: Clean the substrate (e.g., glass, silicon wafer) thoroughly to remove any particulate contamination.
- Temperature Setting: Set the hotplate to the target temperature (T_substrate). Typical exploration ranges are from 25°C to 70°C, depending on the solvent system [49].
- Equilibration: Place the substrate on the hotplate and allow it to thermally equilibrate for at least 10 minutes.
- Solution Deposition: Deposit a consistent volume of the precursor solution onto the pre-heated substrate.
- Film Formation: Immediately initiate the spin-coating program (or other deposition process). Ensure the substrate remains on the hotplate during this process if possible.
- Analysis: Characterize the resulting film or solid layer using SEM and XRPD to determine crystal size distribution, coverage, and polymorphism.
Application Note: This technique is highly effective for fabricating perovskite single-crystal thin films (SCTFs) where uniform, full-coverage nucleation is desired. It directly counters inhomogeneous growth by ensuring a spatially even nucleation event [49].
Protocol: Membrane Crystallization for Supersaturation Control
Principle: Membrane Distillation Crystallization (MDC) uses a hydrophobic microporous membrane to controllably remove solvent from a solution, thereby increasing solute concentration and generating supersaturation at a defined rate. This method avoids the rapid, homogeneous nucleation that leads to amorphous precipitation and instead favors controlled growth [50].
Materials:
- Membrane crystallization module with a hydrophobic membrane (e.g., PTFE, PVDF).
- Peristaltic or syringe pump.
- Heated feed solution reservoir.
- Condensate collection vessel.
- In-line particle imaging probe or turbidity meter.
Procedure:
- System Setup: Fill the feed reservoir with the solution to be crystallized. Connect the tubing to circulate the solution across the membrane module.
- Induction: Initiate solvent removal by applying a vapor pressure gradient across the membrane (e.g., by heating the feed or applying a vacuum on the permeate side).
- Supersaturation Modulation: Control the rate of concentration by adjusting the membrane area exposed to the solution, the feed temperature, or the flow rate [50].
- Nucleation Detection: Use an in-line probe to detect the first appearance of crystals, recording the induction time.
- Growth Phase: Once nucleation is detected, the system can be held at a constant supersaturation level to allow for crystal growth. Implementing in-line filtration can segregate crystals from the membrane surface, reducing scaling and promoting growth in the bulk solution [50].
- Product Harvesting: After a predetermined growth time, harvest the crystals from the bulk solution.
Application Note: MDC is exceptionally well-suited for managing the crystallization of inorganic salts from brine and for achieving high purity in biological macromolecules. By finely tuning the supersaturation rate, the operator can reposition the system within specific regions of the metastable zone to favor crystal growth over primary nucleation, resulting in larger, more uniform crystals [50] [47].
Protocol: Polymorphism Control via Thermal Engineering
Principle: The kinetics of nucleation and growth are highly temperature-dependent. By carefully controlling the thermal profile, including nucleation temperature (Tₙ) and crystallization temperature (T꜀), one can selectively favor the formation of a specific polymorph over others [48].
Materials:
- Differential Scanning Calorimeter (DSC) or thermal oven with precise programming.
- Sample pans (for DSC) or watch glasses (for oven).
- XRPD for polymorph identification.
Procedure:
- Initial Analysis: Perform a preliminary DSC scan to identify the glass transition temperature (T𝑔) and potential crystallization exotherms of the amorphous material.
- Nucleation Stage: Subject the amorphous sample to a first isothermal hold at a nucleation temperature (Tₙ) near T𝑔. As demonstrated in LAS glass-ceramics, a variation of Tₙ within T𝑔 ±15°C can significantly alter the number density of nuclei [48].
- Crystal Growth Stage: Immediately transfer the sample to a second, higher temperature (T꜀) for crystal growth. The T꜀ will influence which polymorph is the most thermodynamically stable or kinetically favored.
- Characterization: Analyze the heat-treated samples using XRPD to identify the resulting polymorphic form.
Application Note: This two-stage heat treatment is a cornerstone of producing glass-ceramics with tailored properties. For instance, in polyamide 11, the crystallization temperature was found to dictate the formation of either the α-phase or the β-mesophase, with high nucleation densities at deep supercooling favoring the latter [47] [48]. This protocol is directly applicable to controlling polymorphism in pharmaceutical compounds.
Quantitative Data and Analysis
The following table synthesizes key quantitative relationships and experimental data crucial for designing crystallization experiments that minimize defects.
Table 3: Quantitative Parameters for Crystallization Control
| Parameter | Mathematical Relation | Impact on Crystallization & Defects |
|---|---|---|
| Supersaturation (S or ΔC) | S = C/C₀ [44] or ΔC = C - C₀ [13] | High ΔC shortens induction time but broadens the Metastable Zone Width (MZW), risking amorphous precipitation. Optimal S balances nucleation and growth [44] [50]. |
| Nucleation Energy Barrier (ΔG) | ΔG = 16πγ³ / (3ν²ΔGᵥ²) [13] | A lower ΔG, achieved via heteronucleants or tailored interfaces, favors nucleation. This allows crystallization at lower S, avoiding the precipitation zone [44] [13]. |
| Critical Nucleus Radius (r_c) | r_c = 2γ / ΔGᵥ [13] | A smaller rc promotes nucleation but can lead to excessive nuclei and grain boundaries. Controlling interfacial energy (γ) is key to managing rc [13]. |
| Nucleation Rate (J) | J = A exp(-ΔG / (kBT)) [44] [13] | The nucleation rate is exponentially dependent on the energy barrier. Temperature and supersaturation are primary levers for controlling J and, consequently, crystal density [44] [13]. |
| Nucleation Temperature (Tₙ) | Experimental variable near T𝑔 [48] | In LAS systems, increasing Tₙ from 640°C to 670°C increased nuclei density, leading to a higher volume fraction of the target crystalline phase and a lower Coefficient of Thermal Expansion (CTE) [48]. |
Controlling Nucleation Density and Crystal Size Distribution
In both materials science and pharmaceutical development, the control of nucleation density and crystal size distribution is a fundamental prerequisite for obtaining materials with desired and reproducible properties. Within crystalline thin films, these parameters dictate critical characteristics such as surface roughness, electrical conductivity, and optical transparency [7] [51]. In pharmaceutical compounds, they influence the bioavailability, stability, and processability of active ingredients [52]. The substrate temperature during deposition or crystallization is one of the most influential parameters, as it directly governs the kinetic and thermodynamic forces that drive nucleation and subsequent growth [7] [53] [54]. This document outlines application notes and protocols for controlling nucleation density and crystal size distribution through precise substrate temperature management, providing a practical framework for researchers and drug development professionals.
Core Principles and the Impact of Substrate Temperature
Classical Nucleation Theory (CNT) provides the foundational framework for understanding crystal formation. It describes the formation of a stable nucleus from a supersaturated solution or vapor phase, where the free energy of formation is a balance between the volume free energy gain and the surface free energy cost [55]. The theory posits that a critical nucleus size must be exceeded for stable growth, and the rate of nucleation is exponentially dependent on the thermodynamic barrier to formation.
The substrate temperature exerts a profound and multi-faceted influence on this process, as illustrated in the diagram below:
Non-Classical Nucleation Pathways
Recent studies highlight the importance of non-classical pathways, where nucleation does not proceed directly from individual atoms or molecules to a crystalline lattice. Instead, it can occur via metastable, mesoscopic precursors such as dense-liquid droplets or amorphous aggregates [52]. This pathway is particularly relevant for organic biocrystals in pharmaceutical applications, like β-hematin (a malaria-related crystal), where nucleation is found to occur within hematin-rich clusters [52]. The activity of modifiers (e.g., antimalarial drugs) can be understood through their interactions with these precursors, either suppressing or enhancing nucleation by altering the precursor population, a mechanism that operates outside the traditional CNT framework [52].
Quantitative Data: Temperature-Dependent Nucleation and Growth
The following tables summarize key experimental findings from diverse material systems, demonstrating the quantifiable impact of substrate temperature on nucleation, crystal size, and final film properties.
Table 1: Impact of Substrate Temperature on Crystallinity and Morphology in Thin Films
| Material | Deposition Method | Temperature Range | Key Structural & Morphological Findings | Reference |
|---|---|---|---|---|
| Zirconium (Zr) | Pulsed Laser Deposition (PLD) | 300°C to 500°C | Strongest Zr(100) orientation at 400°C; transition from 2D layer-by-layer to 3D island growth at ~500°C; surface roughness increases with temperature. | [7] |
| Potassium Bromide (KBr) | Resistive Thermal Evaporation | 50°C to 250°C | Evolution from mixed polycrystalline texture to pronounced (200) orientation; smoothest surface and highest packing density at 150-200°C. | [54] |
| Cadmium Sulfide (CdS) | RF Magnetron Sputtering | 25°C to 300°C | Crystallite size increases with temperature; carrier mobility increases from 5.53 to 12.57 cm²/V·s. | [53] |
| Aluminum Nitride (AlN) | Metal-Organic Vapor Phase Epitaxy (MOVPE) | 1250°C vs. 1440°C | Constant high temperature (1440°C) yielded the best crystal quality with reduced dislocation density and smoother surface. | [56] |
Table 2: Correlating Temperature, Nucleation Density, and Crystal Size Distribution
| System / Parameter | Low Temperature Regime | Intermediate / Optimal Temperature | High Temperature Regime |
|---|---|---|---|
| Nucleation Density | High density of small, numerous nuclei due to limited adatom diffusion [7] [51]. | Balanced density; adatom mobility allows for stable nucleus formation without excessive proliferation [7]. | Lower density; high mobility leads to coalescence of islands and Oswald ripening [7] [51]. |
| Average Crystal Size | Small crystallites (e.g., ~22 nm for CdS at RT) [53]. | Optimized, larger crystallites (e.g., ~24 nm for CdS at 300°C) [53]. | Largest crystallites, but potential for abnormal grain growth and surface degradation [7]. |
| Size Distribution | Often broad due to limited surface diffusion [57]. | Can be narrowed under optimal kinetic control [54]. | Can become bimodal or broadened due to secondary nucleation and coalescence events [57]. |
| Practical Outcome | Amorphous or nanocrystalline films with high defect density [7]. | High-quality, smooth, continuous films with preferred orientation [7] [54]. | Rough, island-like morphology; potential for interfacial reactions (e.g., silicide formation) [7]. |
Experimental Protocols
Protocol: Optimizing Substrate Temperature for Thin-Film Deposition via PLD/Sputtering
This protocol is designed for the deposition of inorganic thin films (e.g., metals, semiconductors) on solid substrates.
4.1.1 Research Reagent Solutions and Essential Materials
- Substrate: Single-crystal Silicon (Si) wafers (e.g., Si(100)), 10 mm x 10 mm pieces. Function: Provides a well-defined, clean, and flat surface for heteroepitaxial or polycrystalline growth.
- Target Material: High-purity (≥99.95%) Zirconium sputtering or PLD target. Function: Source of the material to be deposited as a thin film.
- Cleaning Solvents: Analytical grade acetone, isopropanol, and methanol. Function: Removal of organic and particulate contaminants from the substrate surface prior to deposition.
- Etchant: Diluted (5%) Hydrofluoric (HF) acid. Function: Removal of the native silicon oxide layer to create a chemically pure surface. (Handle with extreme care using appropriate PPE and protocols).
- High-Purity Process Gases: Argon (for sputtering) and/or Oxygen (if depositing oxides). Function: Sputtering atmosphere and control of film stoichiometry.
4.1.2 Procedure
Substrate Preparation:
- Clean substrates ultrasonically in sequential baths of acetone, isopropanol, and methanol for 10 minutes each.
- Rinse with deionized water and dry under a stream of dry nitrogen.
- Immerse substrates in a 5% HF solution for 60 seconds to remove the native oxide layer, followed by a deionized water rinse and nitrogen dry. Load substrates into the deposition chamber immediately after drying.
System Setup and Evacuation:
- Load the target and substrates into the PLD or sputtering chamber.
- Evacuate the chamber to a base pressure of at least 2.0 x 10⁻⁵ Pa to minimize contamination [7].
Temperature Calibration and Stabilization:
- Ramp the substrate heater to the desired setpoint temperature (e.g., 300°C, 400°C, 500°C). The temperature should be monitored by a thermocouple in direct contact with or in close proximity to the substrate holder [7] [54].
- Allow the substrate to stabilize at the deposition temperature for a minimum of 2 hours to ensure thermal equilibrium across the entire substrate [54].
Deposition Process:
- Introduce high-purity process gas (if required) to the chamber, maintaining a stable dynamic pressure.
- Initiate the plasma (sputtering) or laser ablation (PLD) process. For PLD, typical parameters are a laser fluence of ~75 mJ, a repetition rate of 10 Hz, and a deposition duration of 1 hour [7].
- Maintain a constant deposition rate (e.g., 40 Å/s for thermal evaporation) as monitored by a quartz crystal microbalance [54].
Post-Deposition Cooling:
- After deposition, allow the samples to cool gradually to room temperature under vacuum or an inert atmosphere to prevent oxidation and thermal shock.
4.1.3 Validation and Analysis
- X-ray Diffraction (XRD): Perform θ–2θ scans to determine the crystalline phase and preferred orientation. Calculate crystallite size using the Scherrer equation [7] [53] [54].
- Atomic Force Microscopy (AFM): Image the film surface over a 5 µm x 5 µm area to quantify root-mean-square (RMS) roughness and observe surface morphology [7] [54].
- Scanning Electron Microscopy (SEM): Analyze surface and cross-sectional morphology to assess film continuity, island density, and thickness [7] [54].
Protocol: Controlling Nucleation in Solution-Based Crystallization
This protocol is adapted for controlling the nucleation of organic crystals, such as active pharmaceutical ingredients (APIs), from solution.
4.2.1 Research Reagent Solutions and Essential Materials
- Solute: High-purity β-Hematin (or relevant pharmaceutical compound). Function: The crystallizing species of interest.
- Solvent System: Octanol saturated with citric buffer at pH 4.8 (CBSO), or another pharmaceutically relevant solvent. Function: Dissolves the solute and mimics physiological or processing conditions [52].
- Nucleation Modifiers: Compounds such as Pyronaridine, Chloroquine, or Heme-Artesunate adduct. Function: To probe or manipulate non-classical nucleation pathways by interacting with solute precursors [52].
- Temperature-Controlled Bath/Chiller: Precision control of ±0.1°C. Function: To provide accurate and stable supersaturation control.
4.2.2 Procedure
Solution Saturation:
- Prepare a saturated solution of the solute (e.g., β-hematin) in the chosen solvent at a temperature slightly above the target crystallization temperature. Filter the solution through a 0.2 µm membrane to remove dust and pre-existing micro-crystals.
Supersaturation Generation:
- Transfer the saturated solution to a temperature-controlled crystallization vessel.
- Rapidly lower the temperature to the target supersaturation temperature to initiate nucleation. The metastable zone width (MZW) can be determined by the difference between the saturation temperature and the nucleation temperature [50].
Modifier Addition (Optional):
- To test the effect of a modifier, add it to the saturated solution at a defined concentration before inducing supersaturation. The modifier's impact on induction time and crystal number will indicate its role as a nucleation suppressor or enhancer [52].
Nucleation Monitoring:
- Use Dynamic Light Scattering (DLS) to monitor the solution in real-time. The appearance and growth of a shoulder in the autocorrelation function ( g_2(\tau) ) indicates the nucleation and growth of crystals [52].
- Alternatively, use in-situ microscopy or turbidity measurements to track the nucleation event.
4.2.3 Validation and Analysis
- Dynamic Light Scattering (DLS): Analyze the autocorrelation function to determine the nucleation induction time and estimate particle size distribution in the early stages of crystallization [52].
- Optical/Scanning Electron Microscopy: Image the final crystals to determine their habit, size, and size distribution.
- Image Analysis Software: Quantify the crystal size distribution (CSD) from microscopy images, presenting data as population density diagrams or histograms [57].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Nucleation Control Experiments
| Item | Function | Example Application |
|---|---|---|
| Patterned Sapphire Substrate (PSS) | Provides a heteroepitaxial surface with controlled topography to guide nucleation position and reduce defect density. | Growth of high-quality AlN thin films for optoelectronics [56]. |
| Trimethylaluminum (TMAl) & Ammonia (NH₃) | Metal-organic and hydride precursors for the vapor-phase deposition of nitride films via MOVPE. | Epitaxial growth of AlN layers [56]. |
| Heme-Artesunate (H-ARS) & Pyronaridine (PY) | Molecular modifiers that interact with nucleation precursors (mesoscopic clusters) in solution. | Suppressing or enhancing nucleation of β-hematin crystals for malaria research [52]. |
| Citric Buffered Solvent Octanol (CBSO) | A solvent system that mimics in-vivo lipid environments, containing ~5% water. | Studying non-classical nucleation of organic biocrystals [52]. |
Controlling nucleation density and crystal size distribution is a cornerstone of materials and pharmaceutical science. As demonstrated, substrate temperature is a master variable that directly and powerfully influences these parameters across diverse systems, from inorganic thin films to organic biocrystals. The experimental protocols provided offer a concrete starting point for systematic investigation. A deep understanding of both classical and non-classical nucleation pathways, coupled with precise control over experimental parameters like temperature, enables researchers to tailor material properties from the bottom up, paving the way for advanced technological and therapeutic applications.
Within the broader thesis on substrate temperature control for nucleation research, the precise manipulation of thermal profiles emerges as a fundamental determinant of material structure and function. This control governs atomic and molecular mobility, diffusion kinetics, and the thermodynamic driving forces that dictate nucleation pathways and ultimate material properties. Across diverse material systems—from complex proteins to inorganic metal oxides and perovskites—temperature protocols provide a powerful tool for directing self-assembly, crystallographic texture, and functional performance. The research synthesized herein demonstrates that temperature optimization is not merely a background parameter but a primary design variable that can be systematically engineered to achieve targeted material behaviors. This article provides detailed application notes and protocols for researchers, scientists, and drug development professionals seeking to exploit thermal profiles for advanced material synthesis.
Thermal Profiling for Protein Systems
Application Notes for Protein Folding and Crystallization
In protein systems, thermal profiles control the delicate balance between folding stability and aggregation, with significant implications for biopharmaceutical development. Research reveals that protein folding follows a nucleation mechanism with a distinct optimal temperature where the folding time is shortest. Computational models indicate that this temperature can be predicted by analyzing the emission and absorption rates of native residue clusters, allowing for the estimation of folding kinetics without exhaustive experimental measurements at multiple temperatures [58]. Furthermore, proteins undergo barrierless denaturation outside their stable temperature window, unfolding via spinodal decomposition when cluster emission rates exceed absorption rates across all cluster sizes. Understanding these thresholds is critical for maintaining protein stability during processing and storage.
For protein crystallization, studies on apoferritin—a protein with temperature-independent solubility—demonstrate that temperature significantly affects nucleation kinetics even when the thermodynamic driving force remains constant. The nucleation rate per active site, work for nucleus formation, and nucleation time lag all exhibit strong temperature dependence [59]. This indicates that temperature influences kinetic parameters such as molecular attachment frequencies, separate from its traditional role in controlling supersaturation. Two competing phenomena complicate temperature optimization: at higher temperatures, active sites may be consumed by competition between crystalline and amorphous phases, while lower temperatures may promote alternative crystal forms through distinct nucleation sites.
Experimental Protocol: Temperature-Dependent Protein Nucleation Kinetics
Objective: Determine the temperature dependence of nucleation kinetics for proteins with temperature-independent solubility.
Materials:
- Purified target protein (e.g., apoferritin)
- Crystallization buffers and precipulants
- Batch crystallization plates
- Temperature-controlled incubators or thermal cyclers (capable of maintaining 10-60°C ± 0.1°C)
- Automated imaging system for crystal detection
Procedure:
- Prepare identical protein solutions in batch crystallization setups across the target temperature range (e.g., 10, 20, 30, 40, 50, 60°C).
- For each temperature condition, prepare multiple replicates to account for stochastic nucleation events.
- Monitor solutions continuously for crystal appearance, recording the time of first detection for each crystallite.
- Calculate the probability of crystal detection per unit volume versus time for each temperature.
- Determine nucleation rates per active site from the probability distributions.
- Fit nucleation parameters including work for nucleus formation, nucleus size, attachment frequency, and Zeldovich factor using classical nucleation theory.
- Plot nucleation rate versus temperature to identify optimal crystallization conditions.
Troubleshooting Notes:
- For proteins susceptible to amorphous aggregation at higher temperatures, focus on the lower temperature range.
- If multiple crystal polymorphs appear, characterize their distribution as a function of temperature.
- For temperature-sensitive proteins, expand replicate numbers to improve statistical significance.
Table 1: Key Temperature-Sensitive Parameters in Protein Nucleation
| Parameter | Impact of Increased Temperature | Experimental Determination |
|---|---|---|
| Optimal Folding Temperature (Tf) | Minimizes folding time | First passage time analysis of cluster growth [58] |
| Nucleation Rate per Active Site | Typically increases to optimum then decreases | Probability of crystal detection over time [59] |
| Nucleation Time Lag | Generally decreases with temperature | Time between supersaturation creation and first crystal detection [59] |
| Thermal Denaturation Threshold | Triggered when emission > absorption rates | Stability assays via spectroscopy or calorimetry [58] |
Thermal Profiling for Metal Oxide Systems
Application Notes for Metal Oxide Thin Films
In metal oxide deposition, substrate temperature critically controls nucleation density, crystallographic texture, and electrical properties, particularly for flexible electronics and transparent conducting oxides (TCOs). Research on PEALD-grown In₂O₃ films at 100°C demonstrates that precursor-driven nucleation density—not merely film thickness—governs texture stability and carrier mobility [60]. Films deposited using the DIP3 precursor with lower nucleation density maintained preferential (222)/(400) orientation up to 80nm thickness, achieving superior resistivity of 1.1 × 10⁻³ Ω·cm. In contrast, DIP4 precursor with higher nucleation density prompted earlier random orientation and mobility degradation beyond 50nm thickness. This reveals that thermal optimization must consider precursor chemistry in conjunction with temperature, as different precursors create distinct nucleation behaviors even at identical temperatures.
For zirconium thin films deposited via pulsed laser deposition (PLD), substrate temperature dramatically influences crystalline phase evolution and surface morphology [7]. XRD analysis shows the Zr(100) plane exhibits strongest orientation at 400°C, while Zr(002) dominates at 500°C. Surface roughness increases with temperature, with the smoothest surfaces at lower temperatures and significant 3D island formation at 500°C due to transition from 2D layer-by-layer growth. Computational modeling identifies a critical film thickness of ~1-2nm for this 2D-to-3D transition, driven by adatom surface diffusion kinetics. At 400°C, adatom diffusivity optimally balances crystallization and surface energy minimization, yielding highest film quality, while 500°C causes excessive island formation.
The thermal decomposition pathway of metal carbonates to oxides involves three distinct temperature-dependent stages: dehydrogenation, decarbonization, and crystallization [61]. In situ TEM studies of La₂(CO₃)₃·8H₂O and Ce₂(CO₃)₃·8H₂O reveal concurrent compositional and structural changes during conversion, highlighting the importance of controlled thermal ramping for producing desired oxide microstructures.
Experimental Protocol: Low-Temperature PEALD of In₂O₃ Films
Objective: Deposit high-quality In₂O₃ transparent conducting films at ≤100°C with controlled crystallographic texture.
Materials:
- Plasma-enhanced ALD system
- Indium precursors (DIP3 and DIP4)
- Oxygen plasma source
- Silicon substrates (HF-cleaned, UVO-treated)
- Characterization tools: GIXRD, XPS, spectroscopic ellipsometry, Hall effect measurement system
Procedure:
- Prepare substrates with standardized surface treatment: HF dip to remove native oxide, followed by UVO cleaning to create reproducible hydroxylated surface.
- Load substrates into PEALD chamber and stabilize at 100°C.
- For DIP3-based deposition: Use precursor temperature 80°C, pulse time 0.5s, purge time 15s.
- For DIP4-based deposition: Use precursor temperature 70°C, pulse time 0.3s, purge time 20s.
- Common parameters for both: O₂ plasma power 150W, pulse time 5s, purge time 10s.
- Deposit films across target thicknesses (30, 50, 80, 100nm) by adjusting cycle count.
- Characterize films without post-annealing to assess as-deposited properties.
- Measure GPC, refractive index, crystallographic texture, carrier concentration, mobility, and resistivity.
- Compare texture stability and electrical performance versus thickness for both precursors.
Troubleshooting Notes:
- If resistivity is higher than target, optimize precursor pulse times to enhance texture stability.
- If step coverage is inadequate, adjust purge times to ensure complete precursor removal.
- For flexible substrates, verify temperature uniformity across the substrate during deposition.
Table 2: Metal Oxide Deposition Parameters and Resulting Properties
| Material System | Optimal Substrate Temperature | Key Structural Features | Resulting Functional Properties |
|---|---|---|---|
| PEALD In₂O₃ (DIP3) | 100°C | Stable (222)/(400) texture to 80nm | ρ = 1.1 × 10⁻³ Ω·cm, FoM = 1.5 × 10⁻³ Ω⁻¹ [60] |
| PEALD In₂O₃ (DIP4) | 100°C | Random orientation beyond 50nm | Higher ρ, mobility decline >50nm [60] |
| PLD Zirconium | 400°C (optimal crystallinity) | Strong Zr(100) orientation | Smoothest surface, optimal adatom diffusivity [7] |
| PLD Zirconium | 500°C | Strong Zr(002), 3D islands | Rough surface, silicide formation at interface [7] |
| Carbonate to Oxide | Multi-stage ramp | Dehydrogenation→Decarbonization→Crystallization | Controlled oxide microstructure [61] |
Thermal Profiling for Advanced Self-Assembly Systems
Application Notes for Selective Self-Assembly
In multicomponent self-assembly systems, temperature protocols enable selective retrieval of competing target structures from the same building block inventory. Research demonstrates that systems can be designed where one target structure has a lower nucleation barrier while another is globally more stable [62]. This allows selective assembly through tailored temperature protocols: a higher temperature protocol favors the structure with higher nucleation barrier, while a lower temperature protocol favors the globally stable structure. Successful design requires minimizing the number of neighboring component pairs shared between target structures to avoid chimeric aggregates, while maximizing component library sharing within this constraint.
The free-energy landscapes of competing structures differ significantly, enabling temperature-mediated pathway selection. For the model system of square (S) and plus (P) structures, the square structure can nucleate at higher temperatures where the plus structure remains unstable, allowing selective retrieval through a two-stage protocol: nucleation at higher temperature followed by growth completion at lower temperature [62]. This principle has broad applications in programmable materials and drug delivery systems where reconfigurable functionality is desired.
Experimental Protocol: Temperature-Mediated Structure Selection
Objective: Direct self-assembly toward specific target structures using temperature protocols.
Materials:
- Designed library of self-assembling components (e.g., DNA bricks, patchy particles)
- Temperature-controlled environmental chamber
- Real-time monitoring system (light scattering, microscopy)
- Analysis tools for structure identification
Procedure:
- Design two target structures with minimal shared neighbor pairs to avoid chimeras.
- Include components unique to each structure while maximizing library sharing.
- Prepare homogeneous mixture of all components at initial concentration.
- For retrieving the structure with lower nucleation barrier:
- Apply temperature T₁ just above stability threshold of competing structure
- Hold until nucleation is detected
- Gradually decrease to T₂ to complete growth
- For retrieving the globally stable structure:
- Rapidly cool to T₃ below both nucleation thresholds
- Apply thermal cycling around T₃ to promote annealing
- Monitor assembly process with real-time characterization.
- Quantify selectivity and yield of target structures.
Troubleshooting Notes:
- If chimeric aggregates form, redesign structures to further reduce shared interactions.
- If selectivity is poor, adjust temperature ramp rates to kinetically trap desired structure.
- For systems with slow kinetics, extend annealing times or introduce slight concentration gradients.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagent Solutions for Thermal Nucleation Studies
| Reagent/Material | Function in Thermal Studies | Application Examples |
|---|---|---|
| DIP3 (MeIn(Pr)₂NMe) | Indium precursor for PEALD | Lower nucleation density In₂O₃ films with stable texture [60] |
| DIP4 (InMe₃(THF)) | Alternative indium precursor | Higher nucleation density In₂O₃ films with earlier random orientation [60] |
| Arizona Test Dust (ATD) | Reference ice-nucleating particles | Validation of droplet freezing instruments [11] |
| Snomax | Biological ice-nucleating agent | Instrument calibration and comparative studies [11] |
| Apoferritin | Model protein with temperature-independent solubility | Studying pure kinetic effects on nucleation [59] |
| Metal Carbonates (La₂(CO₃)₃, Ce₂(CO₃)₃) | Solid precursor templates | Studying thermal decomposition pathways to oxides [61] |
| PCR Plates (96-well) | Microscale containment for droplets | High-throughput freezing assays [11] |
| Platinum Resistance Thermometers (Pt100) | High-accuracy temperature sensing | Temperature calibration in freezing assays [11] |
Integrated Experimental Protocols
Comprehensive Protocol: Substrate Temperature Calibration and Validation
Objective: Establish precise temperature control and calibration for nucleation studies.
Materials:
- High-accuracy temperature sensors (Pt100 recommended)
- Data acquisition system (e.g., National Instruments NI 9217)
- Temperature-controlled stage or chamber
- Calibration standards (e.g., pure water for 0°C reference)
Procedure:
- Install multiple temperature sensors at critical locations (near substrate, heat source, environment).
- Map temperature uniformity across working volume under stable conditions.
- Characterize temporal stability at setpoints relevant to experiments.
- Validate sensor readings against known phase transition temperatures (e.g., water freezing).
- Determine overall system uncertainty from combined spatial and temporal variations.
- Document calibration protocol and maintain regular recalibration schedule.
Troubleshooting Notes:
- If spatial gradients exceed requirements, modify heat transfer geometry or add active zone control.
- For rapid temperature cycling, characterize response times and overshoot.
- For microscale systems, verify that sensor measurements represent actual sample temperature.
Comprehensive Protocol: Quantitative Thermal Transport Characterization
Objective: Measure thermal transport properties in soft materials using conformable sensors.
Materials:
- Thin, conformable resistive sensors
- Data acquisition system with current source and voltmeter
- Temperature-controlled stage with variable geometry fixtures
- Reference materials with known thermal properties
Procedure:
- Fabricate or procure thin, flexible resistive sensors matching sample geometry.
- Establish sensor calibration using materials with known thermal conductivity.
- Mount sensor in conformal contact with sample material.
- Apply controlled current pulse and measure transient temperature response.
- Fit response data to thermal transport models to extract thermal conductivity.
- Validate measurements across multiple sensor sizes, measurement times, and geometries.
- For biological tissues, perform in vivo measurements where applicable.
Troubleshooting Notes:
- Ensure intimate sensor-sample contact to minimize interfacial resistance.
- For anisotropic materials, characterize directional thermal transport.
- For hydrated samples, control environmental conditions to prevent drying.
The optimization of thermal profiles for specific material systems represents a critical methodology in advanced materials synthesis and drug development. The protocols and application notes presented herein demonstrate that temperature control—from protein folding to metal oxide deposition and programmed self-assembly—provides powerful leverage over material structure and function. By implementing these detailed methodologies and leveraging the essential research tools described, scientists can systematically engineer thermal profiles to achieve targeted nucleation behaviors, advancing both fundamental understanding and practical applications in nucleation research.
The Role of Additives and Co-solvents in Modulating Temperature Effects
Within substrate temperature control research for nucleation, additives and co-solvents serve as powerful tools for directing crystallization pathways. These compounds exert their influence by modifying key interfacial energies and kinetic parameters, which in turn directly affect the critical nucleation temperature and the stochastic distribution of nucleation events. This application note provides a consolidated experimental framework for quantifying these effects, leveraging Classical Nucleation Theory (CNT) as a foundational model. The protocols and data presented herein are designed to enable researchers to systematically investigate and exploit the role of solvent additives in controlling crystallization processes for pharmaceutical and advanced material development.
Theoretical Foundation: Quantifying Additive Effects on Nucleation Kinetics
Classical Nucleation Theory provides the principal framework for relating measurable experimental parameters to the underlying thermodynamics and kinetics of nucleation. The nucleation rate, ( J ), is expressed in the Arrhenius form as shown in Equation 1, which governs the temperature-dependent appearance of stable nuclei [63].
Equation 1: Nucleation Rate $$ J = AJ \exp\left[-\frac{16\pi vm^2 \gamma^3}{3k_B^3 T^3 \ln^2 S}\right] $$
Here, ( AJ ) is the pre-exponential factor, ( \gamma ) is the solid-liquid interfacial energy, ( vm ) is the molecular volume, ( kB ) is the Boltzmann constant, ( T ) is the temperature, and ( S ) is the supersaturation ratio [63]. Additives and co-solvents primarily modulate the nucleation process by altering the interfacial energy (γ) and the pre-exponential factor (AJ ), which is related to the molecular attachment rate.
Table 1: Key CNT Parameters and the Impact of Additives
| Parameter | Physical Meaning | Influence of Additives/Co-solvents |
|---|---|---|
| Interfacial Energy (γ) | Energy required to create a new solid-liquid interface. | Can be increased or decreased depending on specific solvent-solute-additive interactions, such as adsorption at the cluster interface or changes in solvation [64]. |
| Pre-exponential Factor (A_J ) | Kinetic factor related to the rate of molecular attachment to a growing cluster. | Can be enhanced by increasing molecular diffusivity or reduced if additives impose steric or energetic barriers to attachment [64]. |
| Supersaturation (S) | Thermodynamic driving force for nucleation and growth. | Can be altered indirectly by an additive's impact on solute solubility [64]. |
Experimental Data and Comparison
A study on the model API griseofulvin (GSF) provides a quantitative example of how solvents function as co-solvents to dramatically alter nucleation kinetics. The research, involving 2960 induction time measurements, determined the following parameters [64]:
Table 2: Experimentally Determined Nucleation Kinetics of Griseofulvin in Different Solvents
| Solvent | Relative Ease of Nucleation | Interfacial Energy (γ) | Pre-exponential Factor (A_J ) | Observed Solid Form |
|---|---|---|---|---|
| Acetonitrile (ACN) | Highest | Lowest | Comparable to nBuAc | Solvate |
| n-Butyl Acetate (nBuAc) | Intermediate | Intermediate | Comparable to ACN | Solvate |
| Methanol (MeOH) | Most Difficult | Highest | Highest | Stable Form I |
This data reveals a critical nuance: while CNT suggests that higher nucleation rates are typically associated with larger pre-exponential factors, the observed ease of nucleation in GSF was primarily correlated with lower interfacial energy. The high pre-exponential factor in MeOH was insufficient to overcome its high interfacial energy, resulting in the most difficult nucleation [64]. This highlights the system-dependent nature of additive effects and the necessity for empirical measurement.
Detailed Experimental Protocols
Protocol 1: Determination of Metastable Zone Width (MSZW)
The MSZW defines the temperature range between the saturation point and the point of nucleation upon cooling and is a critical parameter for process design [63].
Procedure:
- Solution Preparation: Prepare a saturated solution of the solute in the chosen solvent or solvent-additive mixture at a known temperature, ( T_0 ). Filter the solution to remove any residual crystals.
- Equipment Setup: Load the clear solution into a jacketed crystallizer equipped with a temperature probe and a particle counter (or turbidity probe). Ensure controlled cooling via a programmable thermostat.
- Cooling and Data Logging: Cool the solution at a constant, predetermined cooling rate, ( b ). Continuously monitor and log the temperature and the transmittance/turbidity.
- Nucleation Detection: Record the temperature at which a sudden change in turbidity is detected, indicating nucleation. This is the nucleation temperature, ( Tm ). The MSZW is calculated as ( \Delta Tm = T0 - Tm ).
- Replication and Analysis: Repeat the experiment multiple times (at least 10-15 runs) to account for stochasticity. The median nucleation temperature from the cumulative distribution is used for kinetic analysis [63]. Plot ( (T0/\Delta Tm)^2 ) versus ( \ln(\Delta Tm / b) ) to determine ( \gamma ) and ( AJ ) from the slope and intercept, respectively [63].
Protocol 2: Induction Time Measurements
Induction time measurements provide nucleation kinetics at a constant supersaturation and temperature [64].
Procedure:
- Generation of Supersaturation: Prepare a clear, undersaturated solution and then rapidly create a supersaturated state. This can be achieved by:
- Method A (Cooling): Rapidly cooling a solution from a higher temperature to the target isothermal temperature.
- Method B (Anti-solvent): Rapidly adding a known volume of anti-solvent to the solution under vigorous mixing.
- Isothermal Conditioning: Maintain the supersaturated solution at a constant target temperature with controlled stirring.
- Detection and Timing: Use an in-situ probe (e.g., FBRM, PVM, or turbidity) to detect the first appearance of particles. The time elapsed from the establishment of supersaturation to this detection point is the induction time, ( t_i ).
- Replication and Analysis: Repeat the experiment numerous times at different supersaturation levels, ( S ), at a constant temperature. According to CNT, a plot of ( \ln(ti) ) versus ( 1/\ln^2 S ) should yield a straight line, from which ( \gamma ) and ( AJ ) can be derived [63].
Protocol 3: Investigating Non-Classical Pathways via Dynamic Light Scattering (DLS)
The presence of pre-nucleation clusters or mesoscale aggregates can significantly enhance nucleation rates via non-classical pathways [64].
Procedure:
- Sample Preparation: Prepare solutions in solvents of interest with and without the target additives at the desired supersaturation.
- Measurement: Load the solution into a DLS instrument's cuvette. Equilibrate at the measurement temperature for a set period.
- Data Acquisition: Perform measurements to determine the hydrodynamic diameter distribution and the count rate (particle concentration).
- Correlation with Kinetics: Compare the presence, size, and concentration of detected clusters (typically in the 10-1000 nm range) with the nucleation kinetics determined from Protocols 1 or 2. A higher concentration or larger size of mesoscale clusters is often correlated with an increased nucleation rate [64].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Nucleation Research
| Item | Function/Application | Example from Literature |
|---|---|---|
| Lewis Base Solvents (DMSO, DMF, NMP) | Co-solvents that complex with metal ions (e.g., Pb²⁺ in perovskites), influencing precursor aggregation and grain growth [65]. | Used in perovskite precursor inks to form colloidal lead complexes [65]. |
| Polymer/Protein Stabilizers (Sucrose, Trehalose) | Additives that can increase solution viscosity, modify interfacial energy, and potentially inhibit or delay nucleation. | 240 mM sucrose was used in a monoclonal antibody formulation for lyophilization [66]. |
| Surfactants (Polysorbate 20, Polysorbate 80) | Reduce surface tension, can prevent oiling out, and may act as nucleation inhibitors or modifiers. | Used at low concentrations (0.004-0.04% w/v) in protein formulations to prevent surface-induced aggregation [66]. |
| Inorganic Salt Additives (MgBr₂, LiBr) | Can tailor ion solvation structures, reduce desolvation energy, and influence interfacial energy in inorganic crystallization [67]. | MgBr₂ was used as an electrolyte additive to modify Li⁺ deposition in batteries [67]. |
| Ice Nucleating Agents | Provide heterogeneous surfaces to control and elevate ice nucleation temperature during lyophilization. | Used in "ice fog" techniques for controlled ice nucleation in freeze-drying processes [68]. |
Workflow and Conceptual Diagrams
The following diagram illustrates the logical decision-making process and key mechanisms for utilizing additives and co-solvents in temperature-controlled nucleation research.
Diagram 1: An experimental workflow for using additives to control nucleation.
The diagram below summarizes the primary molecular-level mechanisms through which additives and co-solvents influence nucleation kinetics and temperature.
Diagram 2: Molecular mechanisms of additives and co-solvents.
Nucleation, the initial formation of a new thermodynamic phase or structure, serves as the critical first step in countless processes across materials science, pharmaceuticals, and biotechnology. The control of nucleation—particularly through precise substrate temperature manipulation—directly determines key outcomes in semiconductor films, pharmaceutical crystals, and cryopreserved biologics. However, the transition from laboratory-scale nucleation protocols to industrial manufacturing presents formidable scaling challenges. These include maintaining uniform thermal profiles across larger substrates, achieving consistent nucleation densities at high throughput, and ensuring process reproducibility with different equipment geometries and volumes. This application note details specific experimental protocols and data-driven methodologies to bridge this critical gap, providing researchers with validated strategies for scaling substrate temperature control in nucleation-dependent processes.
Quantitative Data on Temperature Ramping Protocols
Table 1: Comparative Analysis of Substrate Temperature Protocols for Nucleation Control
| Protocol Name | Temperature Parameters | Target Material/System | Key Nucleation Outcomes | Scaling Considerations |
|---|---|---|---|---|
| Programmed Downward Ramping [69] | Initial: 673 K; Final: 573 K; Ramp during initial deposition | Aluminum (Metalorganic CVD) | • Highest Al(111) texturing• Highest reflectivity• Lowest resistivity• Lowest surface roughness | Requires precise control of cooling rates; thermal mass effects are significant at larger scale. |
| Single-Step High-Temperature Nucleation [70] | High-temperature pre-treatment with Group III/II reactant | III-V / II-VI Compounds (e.g., GaN) on mismatched substrates (Si, Sapphire) | • Forms nucleation layer accommodating lattice/thermal strain• Maintains crystallographic registration | Single-step process simplifies scale-up; pre-treatment reactant exposure time is critical. |
| Isothermal Immersion Freezing [41] | Constant target temperature (e.g., -20°C to -40°C) held for extended time (e.g., 30 min) | Ice in supercooled aqueous droplets | • Time-dependent freezing fraction• Described by stochastic nucleation rate coefficient (Jhet) | Total ice nucleating particle surface area per droplet is key variable; difficult to control uniformly in large volumes. |
The data summarized in Table 1 demonstrates that dynamic temperature ramping, as opposed to static isothermal holds, can yield superior material properties. The successful protocol for aluminum CVD involved a temperature decrease during the initial deposition phase, which resulted in a larger fraction of small nuclei compared to a constant lower temperature process [69]. This highlights the importance of a carefully designed thermal trajectory, not just a single target temperature.
Experimental Protocols for Scalable Nucleation Control
Protocol: Programmed Temperature Ramp for Thin-Film Deposition
This protocol is adapted from methods used in metalorganic chemical vapor deposition (MOCVD) of aluminum to achieve high nucleation density and low surface roughness [69].
- 1. Objective: To form a thin film with high nucleation density, low surface roughness, and low electrical resistivity via a programmed substrate temperature ramp.
- 2. Materials:
- Substrate (e.g., Silicon wafer)
- Precursor gases (e.g., Triisobutyl Aluminum for Al)
- MOCVD Reactor or similar deposition system
- High-purity carrier gas (e.g., N₂, H₂)
- 3. Methodology:
- 3.1. Substrate Loading and Initial Stabilization: Load the substrate into the reactor chamber. Pump down to base pressure and stabilize the initial substrate temperature at a high temperature (e.g., 673 K).
- 3.2. Initial Deposition with Ramp: Introduce the precursor gases to begin deposition. Simultaneously, initiate a controlled downward ramp of the substrate temperature from the initial high temperature (673 K).
- 3.3. Bulk Deposition at Constant Temperature: Once the temperature reaches the target lower temperature (e.g., 573 K), continue the deposition process at this constant temperature until the desired film thickness is achieved.
- 3.4. Purge and Cool: Stop the precursor flow and purge the chamber with inert gas. Cool the substrate to room temperature under vacuum or inert atmosphere before unloading.
- 4. Scaling Notes: Upon scaling, ensure the heating and cooling system can handle the thermal mass of larger or multiple substrates while maintaining the same ramp rate across the entire substrate area. The gas flow dynamics and precursor delivery must be re-optimized for the larger chamber volume.
Protocol: Single-Step High-Temperature Nucleation for Lattice-Mismatched Systems
This protocol is designed for growing high-quality epitaxial layers on lattice-mismatched substrates (e.g., GaN on silicon), a critical process in advanced semiconductor manufacturing [70].
- 1. Objective: To form a high-quality nucleation and buffer layer on a lattice-mismatched substrate in a single process step, eliminating complex multi-temperature sequences.
- 2. Materials:
- Lattice-mismatched substrate (e.g., Si, SiC, Sapphire)
- Group III reactant (e.g., Trimethylgallium, Trimethylaluminum)
- Group V reactant (e.g., Ammonia)
- Epitaxial growth system (OMVPE, MBE)
- 3. Methodology:
- 3.1. Substrate Pre-treatment: Place the substrate in the growth chamber and heat to an elevated growth temperature. Introduce only the Group III (or Group II) reactant for a defined pre-treatment period. This conditions the substrate surface.
- 3.2. Nucleation Layer Formation: Without changing the temperature, introduce the Group V (or Group VI) reactant to initiate the formation of the nucleation layer (e.g., AlxGa1-xN).
- 3.3. Buffer and Epitaxial Growth: Continue flows to grow the buffer layer (e.g., GaN) and subsequent epitaxial layers at the same temperature.
- 4. Scaling Notes: The single-step, high-temperature process is inherently less complex to scale than multi-step protocols. Focus on maintaining temperature uniformity and reactant gas distribution across the wafer. The pre-treatment time and reactant flow rates may need adjustment for larger substrate holders.
Visualization of Workflows and Relationships
Diagram 1: The scaling pathway from lab to factory.
Diagram 2: Programmed temperature ramp protocol flow.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Substrate-Mediated Nucleation Experiments
| Item | Function/Application | Example/Notes |
|---|---|---|
| Lattice-Mismatched Substrates | Provide a surface for heterogeneous nucleation of epitaxial films. | Silicon (Si), Silicon Carbide (SiC), Sapphire (Al₂O₃) [70]. |
| Organometallic Precursors | Source of metal atoms in vapor deposition of thin films and nucleation layers. | Triisobutyl Aluminum (for Al), Trimethylgallium (for Ga), Trimethylaluminum (for Al) [69] [70]. |
| Ice Nucleating Particles (INPs) | Solid substrates to initiate and control ice formation in supercooled aqueous systems. | Mineral dust (e.g., Kaolinite), organic crystals, silver iodide (AgI); potency described by nucleation rate coefficient Jhet [41]. |
| Group V/VI Reactant Gases | Source of non-metal elements during the growth of compound semiconductor nucleation layers. | Ammonia (NH₃, Group V for nitrides), Hydrogen Selenide (H₂Se, Group VI) [70]. |
| Cryoprotectant Agents | Protect cells from freezing damage during controlled-rate freezing in cryopreservation. | Dimethyl sulfoxide (DMSO), Glycerol; used in controlled-rate freezers [71]. |
Assessing Success: Analytical Techniques and Comparative Performance Metrics
This document provides detailed application notes and experimental protocols for four cornerstone characterization techniques in materials science: X-ray Diffraction (XRD), Scanning Electron Microscopy (SEM), Atomic Force Microscopy (AFM), and High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy (HAADF-STEM). The content is specifically framed within the context of a broader thesis on substrate temperature control for nucleation research, a critical parameter governing adatom mobility, phase formation, and crystallographic order during thin film growth and crystal synthesis. These methods provide the essential multi-scale structural and morphological data required to correlate synthesis conditions with resulting material properties.
Individual Methodologies and Applications
X-ray Diffraction (XRD)
Principle: XRD determines a material's crystal structure by measuring the constructive interference of a monochromatic X-ray beam diffracted by crystalline phases. The angles and intensities of the diffracted beams provide information on phase identification, lattice parameters, crystallite size, and strain.
Application in Nucleation Research: Substrate temperature ((TS)) profoundly influences phase purity and crystallinity. For instance, in the growth of BiFeO₃ films on Ti/Si, a study systematically varied (TS) from 793 K to 913 K at a fixed oxygen partial pressure. XRD analysis revealed that the film grown at 913 K exhibited the highest phase purity and crystallinity, with reflections consistent with single-phase, rhombohedrally distorted BiFeO₃. In contrast, films grown at lower temperatures showed parasitic phases. This underscores (T_S) as a critical knob for optimizing structural quality in non-epitaxial systems [72].
Table 1: Key Data from XRD Analysis of BFO Films Grown at Different Substrate Temperatures [72]
| Substrate Temperature (K) | Crystalline Phase | Phase Purity | Key Observations |
|---|---|---|---|
| 793 | Rhombohedral BiFeO₃ | Low | Presence of parasitic secondary phases |
| 853 | Rhombohedral BiFeO₃ | Medium | Reduced intensity of secondary phases |
| 913 | Rhombohedral BiFeO₃ | High | Single-phase, high crystallinity, strain-induced lattice distortion |
Experimental Protocol:
- Sample Preparation: Ensure the sample has a flat, clean surface. Powder samples should be finely ground and packed into a holder. Thin films are typically analyzed as-grown.
- Instrument Setup: Load the sample into the XRD diffractometer. Common configurations use a Cu Kα X-ray source (λ = 1.5406 Å). Set the scan parameters (e.g., 2θ range from 10° to 80°, step size of 0.02°, and counting time of 1-2 seconds per step).
- Data Collection: Execute the scan under ambient conditions unless otherwise required.
- Data Analysis:
- Phase Identification: Compare the resulting diffraction pattern with reference databases (e.g., ICDD PDF-4+).
- Crystallite Size Estimation: Use the Scherrer equation: ( τ = Kλ / (β cosθ) ), where ( τ ) is the crystallite size, ( K ) is the shape factor (~0.9), ( λ ) is the X-ray wavelength, ( β ) is the full width at half maximum (FWHM) of the peak in radians, and ( θ ) is the Bragg angle.
- Lattice Strain Analysis: Perform Williamson-Hall analysis to deconvolute size and strain contributions to peak broadening.
Scanning Electron Microscopy (SEM)
Principle: SEM generates high-resolution images of a sample's surface by scanning it with a focused beam of electrons. The interactions between the electrons and the atoms in the sample produce various signals, including secondary electrons (for topography) and backscattered electrons (for compositional contrast).
Application in Nucleation Research: In-situ high-temperature SEM enables the direct, real-time observation of phase transformation and nucleation events. A seminal study on TC21 titanium alloy used this technique to heat samples with a slow ramp rate (0.5 °C/s) to induce α to β phase transformation. The researchers directly observed that the β phase nucleates and grows at both the α/β phase boundaries and within the α/α grain boundaries. Furthermore, during the subsequent cooling (β to α transformation), α variants were seen to nucleate near the β grain boundaries and grow inwards, revealing a pronounced variant selection phenomenon driven by ( T_S ) and thermal history [73].
Experimental Protocol:
- Sample Preparation: For non-conductive samples, deposit a thin conductive coating (e.g., gold, carbon) via sputtering to prevent charging.
- Instrument Setup: Load the sample into the chamber. For in-situ heating experiments, use a specialized heating stage. Ensure the thermocouple is in good contact with the sample for accurate temperature control.
- Data Collection:
- Evacuate the chamber to high vacuum (or use variable pressure for sensitive samples).
- Set the accelerating voltage (typically 5-20 kV) and probe current.
- For in-situ studies, program the heating cycle (e.g., heat from room temperature to 1100°C at 0.5 °C/s). Continuously capture secondary electron images and/or Electron Backscatter Diffraction (EBSD) patterns at predefined temperature intervals [73].
- Data Analysis: Analyze images to track microstructural evolution, grain boundary migration, and nucleation events. EBSD data can be processed to determine crystal orientation and phase distribution.
In-situ SEM Workflow for Phase Transformation Studies
Atomic Force Microscopy (AFM)
Principle: AFM measures surface topography and other properties using a sharp probe mounted on a flexible cantilever. Deflections of the cantilever are detected optically, allowing for nanoscale resolution of surface features without the need for conductive coatings.
Application in Nucleation Research: AFM is indispensable for visualizing nucleation, early growth kinetics, and hierarchical crystal structures at the nanoscale. It has been widely used to study polymer crystallization, revealing lamellar and sub-lamellar complexities in real-time [74]. Furthermore, AFM is crucial for characterizing the quality of thin films grown at different ( TS ). In the study of BiFeO₃/Ti/Si heterostructures, AFM was used to measure surface roughness, which correlates with ( TS )-driven adatom mobility and coalescence during growth. Smooth, grain-like topographies indicate optimal growth conditions [72].
Table 2: Common AFM Modes and Their Applications in Nucleation Studies
| AFM Mode | Principle | Key Applications | Sample Considerations |
|---|---|---|---|
| Tapping Mode | Probe oscillates at resonance frequency near surface. | High-resolution imaging of soft samples (polymers, biological), minimizes damage. | Ideal for in-situ observation of crystal growth in polymer films [75]. |
| Contact Mode | Probe glides in continuous contact with surface. | High sensitivity and resolution; functionalized testing (LFM, PFM). | Can damage soft samples; used for measuring physical properties of crystals [75]. |
| PeakForce Tapping | Probe oscillates at non-resonant frequency, generating force-distance curves per tap. | Nanomechanical property mapping (modulus, adhesion) simultaneous with topography. | Crucial for measuring mechanical/electrical properties of crystals under pressure [75]. |
Experimental Protocol:
- Sample Preparation: Samples must have a surface roughness typically less than 1 μm. Minimal preparation is required; the sample should be clean and securely fixed to the sample puck.
- Probe Selection: Choose an appropriate cantilever with a specific spring constant and resonant frequency suitable for the operating mode (e.g., a stiff cantilever for contact mode, a resonant one for tapping mode).
- Instrument Setup: Mount the probe and sample. Engage the laser and align it on the cantilever. Adjust the photodetector to achieve a balanced signal.
- Engagement and Scanning:
- Approach the probe to the sample surface until engagement is achieved.
- Set the scanning parameters (scan size, rate, and setpoint).
- For in-situ studies, use a temperature-controlled stage to monitor crystal growth in real-time as a function of temperature [74].
- Data Analysis: Use the instrument's software to analyze height, phase, and amplitude images to determine surface roughness, grain size, and domain distribution.
High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy (HAADF-STEM)
Principle: In STEM, a focused electron probe is scanned across a thin sample. HAADF-STEM collects electrons scattered to high angles using an annular detector. The image intensity is approximately proportional to the square of the atomic number (Z-contrast), allowing for direct interpretation of compositional variations.
Application in Nucleation Research: While the provided search results do not contain a specific HAADF-STEM experimental dataset, its role in nucleation research is profound. It can be used to:
- Resolve the atomic-scale structure of nucleation sites at interfaces.
- Identify the initial formation of crystalline phases within an amorphous matrix.
- Characterize elemental segregation or the distribution of catalysts at nucleation sites, leveraging its Z-contrast sensitivity.
Experimental Protocol:
- Sample Preparation (Critical): Prepare electron-transparent samples (<100 nm thick) using focused ion beam (FIB) lift-out techniques or ultramicrotomy. Ensure the sample is clean and free from preparation artifacts.
- Instrument Setup: Load the sample into a (S)TEM holder. Align the microscope to achieve a parallel, monochromatic electron beam. Insert the HAADF detector.
- Data Collection:
- Switch to STEM mode.
- Set the camera length to ensure electrons are scattered onto the HAADF detector.
- Adjust the probe size, current, and convergence angle for optimal resolution and contrast.
- Acquire images and/or spectroscopic (EDS) maps simultaneously.
- Data Analysis: Analyze images for atomic column positions, interface sharpness, and compositional homogeneity. Correlate Z-contrast with EDS data for definitive elemental identification.
Integrated Workflow for Nucleation Studies
A powerful approach for nucleation research involves the sequential or correlative use of these techniques to bridge length scales from macroscopic phase identification to atomic-scale imaging. A typical workflow for investigating a new material system, such as a thin film, is outlined below.
Integrated Characterization Workflow for Nucleation Research
- Macro-scale Screening (XRD): First, use XRD to quickly screen multiple samples synthesized at different ( T_S ) to identify the temperature window for phase purity, as demonstrated in the BiFeO₃ study [72].
- Micro-scale Morphology (SEM/AFM): Select samples of interest for micro- and nano-scale analysis. Use SEM to observe grain structure, and AFM to quantitatively measure surface roughness and nanoscale features resulting from different nucleation densities and growth modes at various ( T_S ).
- Nano/Atomic-scale Analysis (HAADF-STEM): For the most promising sample(s), prepare a cross-section via FIB and perform HAADF-STEM analysis. This reveals the film-substrate interface quality, atomic-scale defects that may have acted as nucleation sites, and the elemental composition across the interface.
Research Reagent Solutions and Materials
Table 3: Essential Materials for Featured Experiments
| Material / Reagent | Function / Application | Example from Literature |
|---|---|---|
| Pure Fe and Al Plates | Model system for studying intermetallic compound (IMC) nucleation and growth kinetics at interfaces. | Used in in-situ SEM to observe IMC nucleation at 380°C and 520°C [76]. |
| Titanium-Buffered Silicon Substrate | A CMOS-compatible platform for integrating functional oxide films; Ti layer acts as adhesion layer and diffusion barrier. | Used as a substrate for growing phase-pure BiFeO₃ films at different temperatures [72]. |
| TiOF₂ & Fe₂O³ | Precursors for homogeneous and heterogeneous nucleation, respectively, in the synthesis of hollow TiO₂ nanocrystals. | Used to grow 3D hollow box TiO₂ nanostructures via different nucleation methods [77]. |
| Pulsed Laser Deposition (PLD) Target (e.g., BiFeO₃) | High-purity source material for the growth of epitaxial or polycrystalline thin films in a vacuum deposition system. | Used to deposit BFO films on Ti/Si substrates at varied temperatures [72]. |
| Hydrofluoric Acid (HF) | Etchant for removing the native SiO₂ layer from silicon wafers to ensure a clean interface for film growth. | Used in the cleaning protocol for Si substrates before BFO film deposition [72]. |
Evaluating Crystallinity, Texture, and Morphology in Temperature-Optimized Samples
Application Note
This application note provides a standardized framework for evaluating the crystallinity, texture, and morphology of samples where substrate temperature is a controlled variable. Within nucleation research, precise temperature control is a critical parameter for governing fundamental processes including self-nucleation behavior, crystalline phase evolution, and surface morphology. The protocols detailed herein are designed to establish clear structure-property relationships, enabling researchers to correlate thermal history with final material properties for applications ranging from advanced composites to pharmaceutical solids.
Key Experimental Findings
The following quantitative data, synthesized from recent investigations, summarizes the profound influence of substrate temperature and thermal history on key material characteristics.
Table 1: Influence of Temperature on Crystalline Properties in Various Material Systems
| Material System | Temperature Parameter | Key Crystallinity/Morphology Findings | Mechanical/Performance Outcome |
|---|---|---|---|
| CF/PPS Composites [78] | Secondary Melting Temp (Ts) | ↓ Ts → ↑ Crystallization Temp (Tc); Nanometer-sized crystallites | ↑ Compression, ILSS, & in-plane shear properties |
| Zirconium Thin Films [7] | Substrate Temp (300°C to 500°C) | 400°C: Strong Zr(100) orientation; 500°C: Strong Zr(002) orientation; ↑ Roughness & 3D island growth | Optimized for electronic device applications |
| YCr1-xFexO3 Perovskites [79] | Calcination Temp (1000°C vs. 1150°C) | 1150°C: ↓ Secondary phases (Y2O3, Cr2O3, Fe2O3); ↑ Crystallite size | Enhanced phase purity and crystallinity |
| Polymer MWD [80] | N/A (Intrinsic Property) | HMW: High entanglement, slow relaxation; LMW: High chain mobility; MWD drives complex textures (e.g., shish kebab) | Determines final mechanical & thermal properties |
Data Interpretation and Workflow
The following diagram outlines the core logical pathway for interpreting experimental data to optimize material properties through temperature control.
Diagram 1: Data interpretation workflow for temperature-optimized samples.
Experimental Protocols
Protocol A: Differential Scanning Calorimetry (DSC) for Self-Nucleation Behavior
This protocol is adapted from the study of carbon fiber-reinforced polyphenylene sulfide (CF/PPS) composites to investigate the self-nucleation (SN) effect and its influence on crystallization behavior and mechanical performance [78].
2.1.1. Principle The self-nucleation effect occurs when a crystallized polymer is melted, but the molecular chains in the melt do not fully revert to a random coil conformation, leaving residual self-nuclei. This dramatically increases nucleation density, reduces spherulitic size, accelerates the crystallization rate, and raises the crystallization temperature (Tc), thereby enhancing fiber/matrix interfacial interaction and mechanical properties [78].
2.1.2. Step-by-Step Procedure
- Sample Preparation: Cut a 5-10 mg sample of the composite or polymer matrix and place it in a standard aluminum DSC crucible. Ensure good thermal contact by lightly pressing the lid.
- Erase Thermal History:
- Imprint Controlled Thermal History:
- Self-Nucleation Thermal Protocol:
- Monitor Crystallization:
- Cool the sample again at a standard rate (e.g., 10 °C/min) while recording the heat flow. The crystallization temperature (Tc) and the shape of the exotherm are key metrics.
- Final Melt Scan:
- Perform a final heating scan to assess the melting behavior of the structure formed under the influence of self-nucleation.
2.1.3. Data Analysis
- Crystallization Temperature (Tc): Report the peak temperature of the crystallization exotherm from step 5. A higher Tc indicates a stronger SN effect [78].
- Melting Behavior: Analyze the final melting endotherm for changes in melting point and peak breadth, which reflect changes in crystalline perfection and lamellar thickness distribution.
Protocol B: X-ray Diffraction (XRD) with Rietveld Refinement for Phase and Texture Analysis
This protocol is essential for quantifying crystalline phase composition, preferred orientation (texture), and crystallite size, as applied in studies of perovskites and thin films [79] [7].
2.2.1. Principle XRD measures the diffraction pattern produced when X-rays interact with a crystalline material. Rietveld refinement is a powerful computational method for fitting a calculated diffraction pattern to an observed pattern, allowing for the quantitative analysis of phase fractions, lattice parameters, and crystallite size [79] [82].
2.2.2. Step-by-Step Procedure
- Sample Preparation:
- For powders: Ensure a fine, homogeneous powder and pack it into a sample holder to create a flat, level surface.
- For thin films: Mount the film as-is on the sample stage.
- Data Collection:
- Use a diffractometer with Cu Kα radiation (λ ≈ 1.54 Å).
- Scan over a relevant 2θ range (e.g., 10° to 80°) with a slow step size (e.g., 0.02°) and sufficient counting time per step to achieve good statistics.
- Rietveld Refinement:
- Use specialized software (e.g., FullProf Suite).
- Input the crystal structure models for all suspected phases (primary phase and potential secondary phases).
- Refine parameters in a sequential manner: background, scale factor, lattice parameters, peak shape, and finally atomic positions and thermal parameters.
- Continue refinement until the difference between the calculated and observed patterns is minimized, as judged by agreement factors (e.g., Rwp, χ²).
2.2.3. Data Analysis
- Phase Quantification: The refined scale factors for each phase are used to calculate their weight fractions [79].
- Crystallite Size: Calculate using the Scherrer equation based on the refined peak broadening.
- Preferred Orientation: Identify from the relative intensity changes of diffraction peaks compared to a standard powder pattern [7].
Protocol C: Atomic Force Microscopy (AFM) and Scanning Electron Microscopy (SEM) for Morphological Characterization
This protocol provides nanoscale and microscale insights into surface morphology and crystalline texture, crucial for linking structure to properties [78] [7].
2.3.1. Principle
- AFM: Uses a physical probe to scan the surface, providing topographical data with nanometer-scale resolution and enabling the visualization of nanometer-sized crystallites [78].
- SEM: Uses a focused electron beam to generate high-resolution images of the surface, revealing microstructural features like spherulites, island formations, and composite fracture surfaces.
2.3.2. Step-by-Step Procedure
- Sample Preparation:
- Ensure the sample is clean, dry, and securely mounted.
- For non-conductive samples, apply a thin conductive coating (e.g., gold sputtering) for SEM analysis.
- Imaging:
- AFM: Select an appropriate scan size. Perform scans in tapping mode to minimize sample damage. Obtain height, phase, and amplitude images.
- SEM: Place the sample in the chamber and evacuate. Select an accelerating voltage (e.g., 5-15 kV) and working distance. Capture secondary electron images at various magnifications.
- Image Analysis:
Experimental Workflow Visualization
The end-to-end process for a comprehensive characterization of temperature-optimized samples is summarized below.
Diagram 2: Comprehensive experimental workflow.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Reagents for Crystallinity and Morphology Evaluation
| Item | Function/Application | Specific Example from Literature |
|---|---|---|
| Polyphenylene Sulfide (PPS) Film | Semicrystalline polymer matrix for composite studies | 130 μm thick film, Mw ~48,200 g/mol [78] |
| Carbon Fiber Fabric | Reinforcement material to study fiber/matrix interface | Toray T300-5HS, tensile strength ~3530 MPa [78] |
| Polycrystalline Zirconium Target | Source for pulsed laser deposition of thin films | Used in PLD for EUV mirror coatings [7] |
| High-Purity Oxide Powders (Y2O3, Cr2O3, Fe2O3) | Precursors for solid-state synthesis of perovskites | Used to synthesize YCr1-xFexO3 [79] |
| Polyethylene Resins | Model polymers for thermal fractionation studies | Used in Stepwise Isothermal Segregation (SIST) & Successive Self-nucleation and Annealing (SSA) [81] |
| Supercritical CO2 | Solvent for pharmaceutical particle engineering/nanonization | Used to enhance solubility of poorly soluble drugs like Letrozole [83] |
Substrate temperature during deposition or post-deposition thermal processing is a critical parameter in materials science, directly governing nucleation, crystal growth, and the resultant electrical, optical, and stability properties of thin films and single crystals. This control is fundamental for advancing applications in electronics, photonics, and drug development. This application note provides a comparative analysis of optimized performance metrics across several material systems, details the experimental protocols for reproducible results, and outlines essential reagent solutions, all within the context of substrate temperature-induced nucleation research.
Comparative Performance Data
The following tables summarize the key performance metrics of various thin-film and crystal systems as a function of processing temperature, highlighting the optimal conditions for electrical, optical, and stability performance.
Table 1: Performance of Oxide Thin-Film Transistors (TFTs) vs. Annealing Temperature
| Material | Annealing Temperature (°C) | Field-Effect Mobility (cm²/V·s) | On/Off Current Ratio | Hysteresis Voltage (V) | Key Findings |
|---|---|---|---|---|---|
| Solution-Processed In₂O₃ [84] | 350 | Not Specified | Not Specified | 3.11 | Higher hysteresis indicates more charge trapping. |
| 450 | 4.28 | 2.15 × 10⁷ | 1.80 | Optimal balance: High mobility, excellent on/off ratio, low hysteresis. | |
| 550 | Not Specified | Not Specified | 0.92 | Lowest hysteresis, but potential degradation of other electrical properties. |
Table 2: Structural & Morphological Properties vs. Substrate/Annealing Temperature
| Material System | Process Temperature | Crystalline Phase / Orientation | Surface Roughness | Key Findings |
|---|---|---|---|---|
| Zr Thin Films on Si (PLD) [7] | 300 °C | Developing Crystallinity | Lower | Initial crystal formation. |
| 400 °C | Strong Zr(100) orientation | Moderate | Optimal crystallinity for the (100) plane. | |
| 500 °C | Strong Zr(002) orientation; Zr-silicide formation | Higher (3D islands) | Transition to 3D island growth and interfacial reaction. | |
| β-Ga₂O₃ on 4H-SiC (LPCVD) [8] | 680 °C (on 4° off-axis) | Pure β-Ga₂O₃ | Low (RMS ~0.7 nm) | Optimal condition: Suppressed vapor-phase nucleation, enhanced surface migration. |
| Crystalline Silicon Films (RTA) [85] | 750 °C | Highly Crystalline | Not Specified | Best compromise: High crystallinity without excessive dopant diffusion from film into substrate. |
Detailed Experimental Protocols
This protocol details the formation of indium oxide thin-film transistors via sol-gel and spin-coating, with a focus on the role of thermal annealing in nucleation and crystallinity.
1. Precursor Solution Preparation:
- Material: Dissolve indium nitrate hydrate (In(NO₃)₃·xH₂O, 99.999%) in 2-methoxyethanol anhydrous to create a 0.2 M solution.
- Process: Stir the solution magnetically at 350 rpm on a hot plate maintained at 75 °C for 5 hours to ensure complete dissolution and homogeneity.
2. Thermal Analysis:
- Instrument: Thermogravimetric Analysis (TGA) system.
- Method: Heat the precursor from 25 °C to 600 °C at a rate of 10 °C/min under a nitrogen atmosphere. This identifies the decomposition and crystallization temperatures, confirming that annealing above 350 °C is required for complete conversion to crystalline In₂O₃.
3. Substrate Preparation and Deposition:
- Substrates: Use quartz for optical analysis and Si/SiNₓ wafers for electrical devices.
- Cleaning: Clean substrates sequentially by ultrasonication in acetone, isopropyl alcohol, and deionized water, followed by drying with nitrogen and baking at 180 °C for 1 hour.
- Surface Treatment: Perform oxygen plasma treatment (40 W, 20 sccm O₂, 1 min) to enhance surface hydrophility.
- Spin-Coating: Filter the precursor through a 0.2 μm PTFE syringe filter. Deposit films by spin-coating at 5000 rpm for 35 seconds in a controlled environment (40-45% relative humidity).
4. Thermal Annealing for Nucleation and Crystallization:
- Process: Anneal the films in a box furnace at predefined temperatures (e.g., 350 °C, 450 °C, 550 °C) for 30 minutes in ambient air. This step is critical for solvent evaporation, precursor decomposition, and the nucleation and growth of crystalline In₂O₃.
5. Device Fabrication & Characterization:
- TFT Testing: Pattern electrodes and measure current-voltage characteristics to extract mobility, on/off ratio, and hysteresis voltage.
- Material Characterization:
- UV-Vis Spectroscopy: Determine optical transparency and bandgap.
- Atomic Force Microscopy (AFM): Analyze surface morphology and roughness.
- Raman Spectroscopy: Probe crystalline polymorphism and phase.
This protocol is essential for controlling polymorphism and particle size distribution (PSD) in pharmaceutical crystallization, a key concern for drug development.
1. Determine the Metastable Zone Width (MSZW):
- Use a tool like the Crystalline platform to generate solubility and metastability curves via transmissivity measurements. The MSZW defines the temperature and concentration window where solution is supersaturated but primary nucleation is unlikely.
2. Select Supersaturation Conditions:
- Choose operating supersaturations sufficiently close to the solubility curve to avoid spontaneous primary nucleation, ensuring that any new crystal formation is due to secondary nucleation.
3. Generate and Characterize Seed Crystals:
- Produce well-characterized single crystals (seeds) of the target compound.
- Calibrate the system's camera using polystyrene microspheres to relate the number of particles on screen (N) to the actual suspension density (Np).
4. Measure Secondary Nucleation:
- Introduce a single, characterized seed crystal into a clear, supersaturated, and agitated solution held at a constant temperature.
- Monitor the system (e.g., via in-situ imaging or transmissivity) and record the delay time and the subsequent increase in suspension density as new crystals form via secondary nucleation initiated by the seed.
5. Data Analysis:
- Correlate the secondary nucleation rate with parameters like seed crystal size and supersaturation level. This data defines the secondary nucleation threshold for process design.
The workflow for this protocol is illustrated below.
Diagram 1: Secondary nucleation study workflow.
This protocol highlights the role of substrate temperature in microstructural evolution during physical vapor deposition.
1. Substrate Preparation:
- Use Silicon (100) substrates.
- Clean substrates using a standard RCA cleaning procedure to remove organic and ionic contaminants.
2. Deposition Setup:
- Instrument: Pulsed Laser Deposition (PLD) system with a 248 nm KrF excimer laser.
- Parameters: Set laser energy to 75 mJ, repetition rate to 10 Hz, and deposition duration to 1 hour.
- Temperature: Heat the substrate to the target temperature (e.g., 300°C, 400°C, 500°C).
3. Film Growth:
- Ablate the high-purity Zr target with the laser pulses.
- The ablated material forms a plasma plume that travels and deposits on the heated substrate. The substrate temperature critically controls the adatom surface mobility, nucleation density, and growth mode (2D layer-by-layer vs. 3D island growth).
4. Post-Deposition Characterization:
- X-ray Diffraction (XRD): Determine crystalline phase and preferred orientation (e.g., Zr(100) vs. Zr(002)).
- Scanning Electron Microscopy (SEM) & Atomic Force Microscopy (AFM): Characterize surface morphology, island formation, and measure surface roughness.
- Transmission Electron Microscopy (TEM): Analyze interface quality and identify interdiffusion or reaction layers (e.g., zirconium silicide).
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Equipment for Nucleation and Crystal Growth Studies
| Item Name | Function / Application | Exemplary Specifications / Types |
|---|---|---|
| High-Temperature Chiller | Precise temperature control for droplet freezing assays and cold-stage experiments. | JULABO FP50-HL circulator used in FINDA-WLU for immersion freezing studies [11]. |
| Platinum Resistance Thermometer | Accurate temperature measurement and calibration in harsh experimental environments. | Pt100 sensors with ±0.15°C accuracy at 0°C, embedded in thermal epoxy [11]. |
| Pulsed Laser Deposition (PLD) System | Growth of high-purity, crystalline thin films from a wide range of target materials. | KrF excimer laser (248 nm) for ablating Zr targets onto heated substrates [7]. |
| The Crystalline Platform | High-throughput screening and optimization of crystallization parameters, including secondary nucleation. | In-situ visual monitoring, particle counting, and transmissivity measurements [86]. |
| Negative Liquid Crystal Mixture | Core material for studying and developing polymer-stabilized vertical alignment (PSVA) devices. | HNG 30400-200 type, used in electro-optical characteristic tuning [87]. |
| Reactive Monomer | Forming polymer networks within liquid crystal cells to stabilize alignment and improve response. | HCM 009 monomer, polymerized under UV light to create a stabilizing network [87]. |
| Vertical Alignment Agent | Inducing homeotropic (vertical) alignment of liquid crystal molecules on substrate surfaces. | DL-4018 type, applied as a thin layer to create the initial alignment [87]. |
The systematic control of substrate and annealing temperature is a powerful and universal tool for optimizing the functional properties of materials. As demonstrated across diverse systems—from solution-processed metal oxides to vapor-deposited thin films and pharmaceutical crystals—temperature directly dictates nucleation kinetics and growth mechanisms. This, in turn, determines critical performance metrics such as charge carrier mobility, optical bandgap, surface smoothness, and operational stability. The protocols and data presented herein provide a framework for researchers to reproducibly engineer material properties for targeted applications in electronics, optoelectronics, and drug development.
Within the broader thesis on advanced substrate temperature control for nucleation research, the accurate interpretation of freezing spectra and nucleation rate data is a cornerstone for validating experimental findings. This process translates raw freezing data into physically meaningful parameters that can predict ice nucleation behavior in diverse systems, from atmospheric science to pharmaceutical development. A fundamental challenge in this domain is reconciling the stochastic (probabilistic) nature of nucleation with the deterministic frameworks often used for data interpretation. This document provides detailed application notes and protocols for researchers and drug development professionals, focusing on the statistical validation of data derived from droplet freezing experiments.
Theoretical Foundation: Stochastic vs. Deterministic Frameworks
The interpretation of immersion freezing experiments is built upon two primary conceptual frameworks, each with distinct implications for statistical validation.
The Stochastic Model
The stochastic model treats ice nucleation as a random, time-dependent event, consistent with classical nucleation theory. The probability of a droplet freezing is a function of the available ice-nucleating surface area, the cooling rate or holding time, and a material-specific, temperature-dependent rate coefficient [41].
The core equation for the fraction of unfrozen droplets ((UnF)) under isothermal conditions is: $$UnF(T) = \frac{N{ufz}(T)}{N{tot}} = e^{-J_{het}(T) A t}$$ [41] where:
- (J_{het}(T)) is the heterogeneous ice nucleation rate coefficient (cm⁻² s⁻¹)
- (A) is the ice nucleating surface area (ISA) in a droplet (cm²)
- (t) is the time the droplets remain supercooled (s)
For a constant cooling rate experiment, the time dependence is implicitly accounted for in the temperature change.
The Deterministic (Active Site) Model
In contrast, the deterministic model, often described using ice nucleation active site (INAS) densities, assumes that each ice-nucleating particle (INP) contains a fixed number of specific sites that nucleate ice at characteristic temperatures, independent of time [41]. The INAS density, (ns(T)), is calculated as: $$ns(T) = \frac{- \ln(UnF(T))}{A}$$ While (n_s) is a useful empirical parameter for comparing different INP types, it is not a fundamental physical property because it inherently ignores the time-dependence of the nucleation process [41].
Key Insight for Validation: A key indicator of the underlying nucleation mechanism is the behavior of the unfrozen fraction under isothermal conditions. A straight line when plotting (\ln(UnF)) versus time confirms a single, time-dependent (J_{het}) and validates a purely stochastic process for a uniform system. Curvature in this plot suggests a distribution of ISA among droplets or multiple active sites with different nucleation energies [41].
The following tables consolidate key quantitative information and parameters essential for the statistical analysis of freezing data.
Table 1: Key Parameters for Interpreting Freezing Experiments
| Parameter | Symbol (Units) | Description | Interpretation Framework |
|---|---|---|---|
| Unfrozen Fraction | (UnF) (unitless) | Fraction of liquid droplets in a population. | Stochastic & Deterministic |
| Frozen Fraction | (FF) (unitless) | Fraction of frozen droplets in a population. | Stochastic & Deterministic |
| Nucleation Rate Coefficient | (J_{het}) (cm⁻² s⁻¹) | Rate of ice nucleation per unit surface area per time. | Stochastic |
| Ice Nucleation Active Site Density | (n_s) (cm⁻²) | Apparent density of active sites per unit surface area at temperature T. | Deterministic |
| Ice Nucleating Surface Area | (A) (cm²) | Total surface area of INPs in a droplet. | Stochastic & Deterministic |
Table 2: Instrumental Performance Metrics from a Representative Freezing Ice Nucleation Detection Analyzer (FINDA)
| Performance Characteristic | Value / Specification | Impact on Data Validation |
|---|---|---|
| Temperature Uncertainty | ±0.60 °C | Defines the confidence interval for reported nucleation temperatures [11]. |
| Temperature Range | 0.0 °C to ~ -30.0 °C | Sets the operational bounds for immersion freezing studies [11]. |
| Cooling Rate | Adjustable, 0.1 °C min⁻¹ to 1.0 °C min⁻¹ | Must be documented and consistent for valid inter-study comparisons [11]. |
| Droplet Array | 96-well PCR plate | Enables high-throughput statistical analysis of hundreds of droplets [11]. |
Experimental Protocols for Data Generation
The following protocols ensure the generation of statistically robust and validated freezing data.
Protocol: Droplet Freezing Assay for Immersion Freezing
Application: Measuring the immersion freezing efficiency of a material (e.g., an inorganic particle, a protein, or a drug excipient).
Materials:
- Freezing Ice Nucleation Detection Analyzer (FINDA-WLU) or equivalent cold-stage apparatus [11]
- 96-well PCR plates (0.2 mL volume)
- High-precision temperature sensor (e.g., Pt100, accuracy ±0.15 °C at 0 °C) [11]
- Sample suspensions in ultrapure water (e.g., Milli-Q)
- Refrigerated/heating circulator
- CCD camera for freezing detection
Procedure:
- Suspension Preparation: Prepare serial dilutions of the test material in ultrapure water. Sonicate the suspension to ensure a homogeneous distribution of particles.
- Droplet Dispensing: Using a micropipette, dispense a consistent volume (e.g., 50 µL) of the suspension into each well of a 96-well PCR plate. For statistical significance, a minimum of 50-100 droplets per sample is recommended.
- Instrument Setup: Place the PCR plate into the precooled aluminum block of the FINDA instrument. Seal the chamber with the acrylic glass lid to prevent frost formation [11].
- Temperature Calibration: Prior to experiments, perform a rigorous temperature calibration using the integrated Pt100 sensors. This step is critical for validating the reported nucleation temperatures [11].
- Experimental Run:
- Constant Cooling Rate (CCR): Program the system to cool from 0 °C to the terminal temperature (e.g., -30 °C) at a fixed, slow rate (e.g., 1 °C min⁻¹). Record the temperature at which each individual droplet freezes.
- Isothermal (ISO): Cool the stage rapidly to a target supercooled temperature (e.g., -15 °C) and hold it constant. Monitor the droplets for a defined period (e.g., 30 minutes) and record the time at which each droplet freezes [41].
- Freezing Detection: The CCD camera monitors the optical change (e.g., increased light scattering or change in shape) of each droplet upon freezing. An automated detection algorithm should be used to identify freezing events with 100% accuracy [11].
Protocol: System Validation with Reference Materials
Application: Validating the performance and calibration of the droplet freezing apparatus.
Procedure:
- Ultrapure Water Blank: Run a plate containing only Milli-Q water. The homogeneous freezing threshold should be observed near -38 °C [11]. Any freezing events at higher temperatures indicate contamination.
- Standard Reference Materials: Measure the freezing spectrum of known standards such as Arizona Test Dust (ATD) or Snomax. The resulting frozen fraction curves and derived (ns(T)) or (J{het}(T)) values must be consistent with literature data from previous studies to validate the instrument's performance [11].
Data Analysis and Statistical Workflow
The process of transforming raw freezing data into validated nucleation parameters involves a defined workflow with critical decision points.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for Freezing Experiments
| Item | Function / Application | Example / Specification |
|---|---|---|
| Reference INPs | Positive controls for system validation and inter-laboratory comparison. | Arizona Test Dust (ATD), Snomax (purified Pseudomonas syringae protein) [11]. |
| Ultrapure Water | Negative control and suspension medium. Must be particle-free. | Milli-Q water (18.2 MΩ·cm resistivity) [11]. |
| High-Precision PCR Plate | Sample holder for droplet arrays. Ensures minimal well-to-well variation. | 0.2 mL, polypropylene, low autofluorescence [11]. |
| Calibrated Temperature Sensors | Critical for accurate and traceable temperature measurement. | Pt100 platinum resistance thermometers (Accuracy: ±0.15 °C at 0 °C) [11]. |
| Microfluidic Device | Alternative to plates; generates highly uniform picoliter droplets for precise control of volume and ISA. | Used in studies requiring minimal variance in droplet volume and particle number [41]. |
Advanced Consideration: Time Dependence in Data Interpretation
A critical aspect of statistical validation is accounting for time dependence. The nucleation rate coefficient, (J{het}), is a fundamental physical parameter that is independent of the experimental method (CCR or ISO). In contrast, the ice nucleation active site density, (ns), is an empirical parameter that is only valid for the specific cooling rate at which it was measured [41]. Therefore, for predictive modeling and implementation in climate or pharmaceutical models, (J_{het}) is the superior and recommended parameter, as it can be used to predict freezing behavior under any arbitrary temperature-time history.
The curvature often observed in plots of (\ln(UnF)) versus time in isothermal experiments is not necessarily evidence against stochasticity. It can be explained by a lognormal or uniform distribution of the ice nucleating surface area (ISA) among the droplets in the population [41]. Validating a model that incorporates this ISA distribution is essential for accurately extracting the intrinsic (J_{het}) of the material.
In nucleation research, the precise control of substrate temperature is a critical determinant of experimental success, directly governing outcomes in material synthesis, pharmaceutical development, and thin-film technologies. Achieving high standards of purity, yield, and reproducibility requires robust methodologies and precise environmental control. This application note details standardized protocols and metrics for benchmarking nucleation processes, with a specific focus on substrate temperature parameters. We present quantitative data from case studies spanning ice nucleation, nanomaterial synthesis, and thin-film deposition, providing researchers with a framework for validating their systems against industry standards. The protocols emphasize temperature calibration, process characterization, and data analysis techniques essential for producing reliable, publication-quality results in both academic and industrial settings.
Quantitative Metrics and Benchmarking Data
The following tables summarize key quantitative metrics for purity, yield, and reproducibility from recent nucleation studies. These values serve as benchmarks for evaluating experimental outcomes against established industry standards.
Table 1: Purity and Crystallinity Metrics in Material Synthesis
| Material System | Characterization Technique | Key Purity/Crystallinity Metric | Reference Value | Influencing Factor |
|---|---|---|---|---|
| β-Ga₂O₃ on 4H-SiC | XPS, TEM | High elemental purity; Minimal interdiffusion | Well-ordered atomic interface with 4H-SiC [8] | Optimized growth temperature (680°C) [8] |
| Zirconium Thin Films | XRD | Strongest Zr(100) orientation | Observed at 400°C substrate temperature [7] | Substrate temperature during PLD [7] |
| Silver Nanowires | SEM, Conversion Analysis | Reaction conversion rate | ~99% conversion to nanowires [10] | Controlled reactor headspace (80% volume) [10] |
Table 2: Yield and Efficiency Metrics
| Process/System | Yield Metric | Reported Value | Optimal Conditions | Impact on Reproducibility |
|---|---|---|---|---|
| Ag Nanowire Polyol Synthesis | Conversion Rate | >99% [10] | 160°C, specific stirring, 80% reactor headspace [10] | High; Controlled parameters reduce batch variance [10] |
| CIGS Thin Film RTP | Process Throughput | Enabled by rapid temperature ramps [88] | Effective tracking of linear temperature ramps [88] | High; Dependent on precise temperature control [88] |
| Ice Nucleation (FINDA-WLU) | INP Concentration Detection | High accuracy for atmospheric samples [11] | Temperature uncertainty ±0.60°C [11] | High; Rigorous calibration ensures consistent measurement [11] |
Table 3: Reproducibility and Precision Metrics
| Experimental System | Precision/Uncertainty Metric | Value | Method for Achieving Precision |
|---|---|---|---|
| FINDA-WLU Ice Nucleation | Temperature Uncertainty | ±0.60 °C [11] | Precise temperature calibration & vertical heat transfer control [11] |
| RTP for CIGS Films | Temperature Ramp Tracking | Accurate tracking of rapid ramps [88] | Model-based observer & specialized controller with double integral term [88] |
| Multi-Lab Microbiome Study | Phenotype Consistency | Consistent plant traits across 5 labs [89] | Standardized devices (EcoFAB 2.0), protocols, and sourced materials [89] |
Detailed Experimental Protocols
Protocol: Droplet Freezing Assay for Ice Nucleating Particles (INPs)
This protocol utilizes the Freezing Ice Nucleation Detection Analyzer (FINDA-WLU) to quantify immersion freezing initiated by atmospheric ice-nucleating particles (INPs), a process critical for cloud formation and climate modeling [11].
1. Reagent and Material Setup:
- Sample: Aqueous suspension of INPs (e.g., collected from atmospheric precipitation).
- Control: Milli-Q ultrapure water for baseline freezing determination.
- Reference Materials: Arizona Test Dust (ATD) or Snomax for system validation.
- Consumables: 96-well PCR plate (0.2 mL volume per well).
- Equipment: FINDA-WLU system, comprising a temperature-controlled aluminum cold stage, refrigerated circulator, CCD camera, and Pt100 temperature sensors [11].
2. Instrument Calibration and System Preparation:
- Temperature Calibration: Perform a multi-point calibration using the four integrated Pt100 sensors. Seal sensors in PCR plate tubes with thermally conductive epoxy to ensure consistent heat transfer with sample wells. Document the vertical and horizontal temperature heterogeneity; the total system uncertainty should be within ±0.60 °C [11].
- Droplet Dispensing: Pipette a consistent volume (e.g., 1 µL) of the sample suspension or control water into individual wells of the PCR plate. Ensure no air bubbles are trapped.
- Sealing: Place the PCR plate into the cavity of the pre-cooled aluminum block. Cover the block with the acrylic glass lid, using a PTFE seal to prevent frost formation [11].
3. Experimental Execution and Data Acquisition:
- Temperature Program: Set the refrigerated circulator to a cooling rate between 0.1 °C/min and 1.0 °C/min, typically from 0.0 °C to -30.0 °C [11].
- Freezing Detection: Initiate the program and start the CCD camera. The camera monitors the reflectance of LED light from each droplet. A sudden change in reflectance indicates the phase transition from liquid to solid, recorded as the freezing event for that well [11].
- Data Logging: The customized LabVIEW program records the temperature of all four sensors and the state (frozen/unfrozen) of each well in real-time throughout the experiment.
4. Data Analysis:
- Frozen Fraction: At each temperature T, calculate the fraction f(T) of frozen droplets.
- INP Concentration: The cumulative number concentration of INPs active at temperature T, NINP(T), is calculated using the formula: *NINP(T) = - ln(1 - *f(T)) / Vdroplet* where Vdroplet* is the volume of a single droplet [11].
- Validation: Compare the freezing spectra of reference materials (ATD, Snomax) with historical data from previous studies to validate system performance [11].
Protocol: Rapid Thermal Processing (RTP) for Thin Film Selenization
This protocol outlines the use of a model-based control system for Rapid Thermal Processing (RTP) to produce high-quality CIGS thin films, with an emphasis on precise temperature ramp control [88].
1. Reagent and Material Setup:
- Substrate: Mo-coated soda lime glass with a pre-deposited Cu-In-Ga precursor film.
- Reaction Gas: H₂Se in a controlled atmosphere.
- Equipment: Pilot-scale RTP reactor with heat lamps, a pyrometer measuring the bottom surface of the sample holder, and a control system capable of running observer-based algorithms [88].
2. System and Controller Preparation:
- Observer Calibration: Conduct a process identification experiment. Use temperature-sensitive lacquers on the sample surface to obtain discrete temperature measurements. Fit the parameters of the first-principles, nonlinear observer to ensure it accurately estimates the unmeasurable film surface temperature, T(L,t), from the pyrometer-measured holder temperature, T(0,t) [88].
- Controller Tuning: Implement a specialized feedback controller (e.g., PID with a double integral term) designed to eliminate offset during linear temperature ramps. Tune the controller parameters based on a dynamic model of the system's response to heat lamp power [88].
3. Experimental Execution:
- Loading: Place the precursor sample on the graphite holder inside the RTP reactor.
- Atmosphere Control: Purge the reactor and introduce the H₂Se gas mixture at the desired concentration.
- Temperature Ramp: Execute the RTP recipe. The controller automatically adjusts the heat lamp power, u(t), to force the estimated surface temperature, T(L,t), to track a pre-defined linear ramp set point (e.g., to 550°C) [88].
- Dwell and Cool: Maintain the dwell temperature for the required reaction time before cooling.
4. Post-Processing and Analysis:
- Film Characterization: Analyze the resulting CIGS film using techniques such as SEM for morphology and XRD for crystallinity.
- Performance Validation: The control system is considered effective when it tracks the desired rapid temperature ramp accurately, with performance limited only by the physical constraints of the reactor [88].
Visualization of Experimental Workflows
Workflow for Nucleation Measurement and Control
The following diagram illustrates the logical flow and feedback mechanisms common to precise nucleation experiments, integrating elements from the featured protocols.
This workflow highlights the critical role of the feedback control loop for substrate temperature, which is essential for achieving reproducible results in nucleation research. The process begins with defining target metrics and proceeds through system setup, parameter optimization, and execution, all under the regulation of the temperature control system [11] [88].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Reagents and Materials for Nucleation Experiments
| Item | Function / Role | Example Application |
|---|---|---|
| Arizona Test Dust (ATD) | Reference ice-nucleating particle (INP) for instrument validation [11] | Calibrating and benchmarking droplet freezing assays (FINDA-WLU) [11] |
| Snomax | Organic, bacterial-based reference INP [11] | Positive control for immersion freezing measurements [11] |
| High-Purity Elemental Ga | Gallium source for CVD growth [8] | Growing β-Ga₂O₃ epilayers, minimizes foreign element introduction [8] |
| Polyvinylpyrrolidone (PVP) | Capping agent and structure-directing agent [10] | Synthesizing silver nanowires via polyol synthesis [10] |
| Ethylene Glycol | Solvent and reducing agent in polyol synthesis [10] | Reducing silver ions to form metallic nanostructures [10] |
| Temperature-Sensitive Lacquers | Provide discrete surface temperature measurements [88] | Calibrating model-based observers in RTP systems [88] |
| Pt100 Resistance Thermometers | High-accuracy temperature sensing [11] | Precise temperature monitoring and calibration in FINDA-WLU [11] |
Conclusion
Substrate temperature control emerges as a universally powerful, versatile, and indispensable tool for mastering nucleation processes across scientific disciplines. The synthesis of insights presented confirms that precise thermal management, guided by fundamental thermodynamics and tailored to specific material systems, directly dictates critical outcomes including crystal quality, defect density, and functional performance. For biomedical researchers and drug development professionals, these principles enable improved purification of biotherapeutics through crystallization, creation of high-quality protein crystals for structural biology, and development of advanced perovskite-based diagnostic devices. Future directions will likely involve increased integration of machine learning for predictive thermal protocol design, development of novel in-situ monitoring techniques for real-time optimization, and application of these refined nucleation control strategies to emerging challenges in biologics formulation and next-generation medical technologies. The continued convergence of nucleation science with biomedical engineering promises to unlock new possibilities in drug delivery, diagnostics, and therapeutic development.
References
- 1. - Wikipedia Classical nucleation theory [https://en.wikipedia.org/wiki/Classical_nucleation_theory]
- 2. Temperature Dependence in Heterogeneous Nucleation ... [https://www.nature.com/articles/s41598-017-16692-9]
- 3. Exploratory experiments on pre-activated freezing nucleation ... [https://acp.copernicus.org/articles/21/2551/2021/]
- 4. Effects of temperature on nucleation and collapse ... [https://www.sciencedirect.com/science/article/pii/S0142941820321619]
- 5. The Clapeyron equation, Gibbs phase rule, and Classical ... [https://chem.libretexts.org/Courses/New_York_University/CHEM-UA_652%3A_Thermodynamics_and_Kinetics/01%3A_Lectures/1.13%3A_The_Clapeyron_equation_Gibbs_phase_rule_and_Classical_Nucleation_Theory]
- 6. The Gibbs free energy of homogeneous nucleation [https://pubmed.ncbi.nlm.nih.gov/28915742/]
- 7. Substrate Temperature-Induced Crystalline Phase ... [https://www.mdpi.com/2079-6412/15/10/1198]
- 8. Temperature-dependent vapor-phase nucleation and ... [https://www.sciencedirect.com/science/article/abs/pii/S0042207X25006335]
- 9. Substrate Temperature - an overview [https://www.sciencedirect.com/topics/computer-science/substrate-temperature]
- 10. Quantitative understanding of nucleation and growth ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894721003090]
- 11. An improved Freezing Ice Nucleation Detection Analyzer ... [https://amt.copernicus.org/articles/18/5823/2025/]
- 12. Temperature-dependent kinetic pathways of ... [https://www.nature.com/articles/s41467-021-25267-2]
- 13. Mechanistic insights and optimization strategies for perovskite ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc08145e]
- 14. Prediction of temperature-dependent nucleation and ... [https://www.sciencedirect.com/science/article/abs/pii/S1359645425009516]
- 15. Supersaturation-controlled microcrystallization and ... [https://www.nature.com/articles/s41598-018-20899-9]
- 16. Thermodynamic Phase Control of Poly(TFEMA) Nucleation ... [https://www.preprints.org/manuscript/202510.2502]
- 17. Conceptual Validation of Stochastic and Deterministic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9896484/]
- 18. Characterizing and measuring the ice nucleation kinetics of ... [https://www.sciencedirect.com/science/article/pii/S0009250923000878]
- 19. Room-temperature X-ray fragment screening with serial ... [https://www.nature.com/articles/s41467-025-64918-6]
- 20. Computational crystallization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4909510/]
- 21. Yield of Protein Crystallization from Metastable Liquid–Liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12155984/]
- 22. Understanding protein crystallization on the basis of ... [https://www.sciencedirect.com/science/article/abs/pii/S0022024898008318]
- 23. solution preparation temperature in protein crystallization [https://www.nature.com/articles/srep07797]
- 24. An open-hardware community ice nucleation cold stage for ... [https://www.sciencedirect.com/science/article/pii/S2468067223000986]
- 25. Crystallinity Effect on Electrical Properties of PEALD –HfO2 Thin Films... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9656191/]
- 26. Ferroelectric Aluminum-Scandium Nitride by... | Research Square [https://www.researchsquare.com/article/rs-6960782/v1]
- 27. Comparative Study of Thermal and Plasma-Enhanced ... [https://www.mdpi.com/2079-4991/13/12/1858]
- 28. Effect of substrate temperature on properties of WS2 thin films [https://www.nature.com/articles/s41598-025-08916-0]
- 29. Effect of substrate temperatures on structural, optical and ... [https://www.sciencedirect.com/science/article/abs/pii/S025405842201313X]
- 30. Single-crystal perovskites for photovoltaic and high-energy ... [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d5cs00625b]
- 31. High-quality bulk hybrid perovskite single crystals within ... [https://www.nature.com/articles/ncomms8586]
- 32. (IUCr) A simple vapor-diffusion method enables protein ... [https://journals.iucr.org/paper?nj5304]
- 33. Quantifying the effect of particulate impurities on the ice ... [https://www.sciencedirect.com/science/article/pii/S0378517324013711]
- 34. Environmentally Friendly Selective Solvent Vapor Diffusion ... [https://www.sciencedirect.com/science/article/abs/pii/S0254058425011198]
- 35. Antisolvent-Assisted Growth of Centimeter-Scale CsPbBr 3 ... [https://arxiv.org/html/2511.05354v1]
- 36. Do more with less with the CrystalBreeder [https://www.crystallizationsystems.com/products/crystalbreeder/]
- 37. CsPbBr 3 Single-Crystal Growth by Temperature-Lowering ... [https://www.sciencedirect.com/science/article/abs/pii/S0969806X25009855]
- 38. Hybrid nano-interfacial engineering of special-wettable ... [https://www.sciencedirect.com/science/article/pii/S2666845925001138]
- 39. mechanisms, strategies, and innovations for oxidation resistance [https://pubs.rsc.org/en/content/articlehtml/2025/na/d5na00716j]
- 40. Predicting heterogeneous ice nucleation with a data-driven ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7509812/]
- 41. Stochastic nucleation processes and substrate abundance ... [https://www.nature.com/articles/s41612-020-0106-4]
- 42. HUB: a method to model and extract the distribution of ice ... [https://acp.copernicus.org/articles/23/5623/2023/]
- 43. ESSD - Measurement of the ice-nucleating particle concentration with... [https://essd.copernicus.org/articles/17/6165/2025/]
- 44. advances and challenges in controlling protein nucleation [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00767d]
- 45. Research on Mesoscale Nucleation and Growth Processes ... [https://www.mdpi.com/2073-4352/12/9/1234]
- 46. Precipitation from amorphous solid dispersions in ... [https://pubmed.ncbi.nlm.nih.gov/34015382/]
- 47. Nucleation and Crystal Growth: Recent Advances and ... [https://www.mdpi.com/2673-4591/56/1/22]
- 48. The effect of nucleating temperature on the crystallization ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884224001664]
- 49. Advanced Strategies to Tailor the Nucleation and Crystal ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.842924/full]
- 50. Facile supersaturation control strategies for regulating ... [https://www.sciencedirect.com/science/article/pii/S1383586625028497]
- 51. Elucidating thin film growth mechanisms for high-performance ... [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00244c]
- 52. A nonclassical pathway of β-hematin crystal nucleation ... [https://www.nature.com/articles/s42004-025-01612-0]
- 53. Effect of substrate temperature on the properties of RF ... [https://www.sciencedirect.com/science/article/pii/S2211379720300401]
- 54. Effect of Substrate Temperature on the Structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12348120/]
- 55. Robustness of classical nucleation theory to chemical ... [https://arxiv.org/html/2510.01420v1]
- 56. Enhancing the Crystal Quality of AlN Thin Films on ... [https://link.springer.com/article/10.1007/s13369-025-10248-0]
- 57. Quantitative analysis of crystal/grain sizes and their ... [https://www.sciencedirect.com/science/article/abs/pii/S0191814111001179]
- 58. Temperature effects on the nucleation mechanism of ... [https://pubmed.ncbi.nlm.nih.gov/18936853/]
- 59. Crystal nucleation from solutions of proteins with temperature ... [https://pubs.rsc.org/en/content/articlehtml/2019/ce/c8ce02016g]
- 60. Precursor-driven nucleation and texture control governing ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838825060669]
- 61. Nucleation and crystallization of metal oxides from ... [https://pubs.rsc.org/en/content/articlelanding/2025/nr/d5nr01525a]
- 62. Temperature protocols to guide selective self-assembly of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8872760/]
- 63. Comparison of the Nucleation Kinetics Obtained from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9099965/]
- 64. Rationalizing the Influence of Solvent on the Nucleation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12232334/]
- 65. How crystallization additives govern halide perovskite ... [https://www.nature.com/articles/s41467-025-65484-7]
- 66. Comparison of Techniques to Control Ice Nucleation ... [https://www.mdpi.com/2227-9717/8/11/1439]
- 67. Modulation of solvation chemistry and lithium deposition ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725022946]
- 68. Comparison of ice fog methods and monitoring ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517318309748]
- 69. Programmed substrate temperature ramping to increase nucleation density and decrease surface roughness during metalorganic chemical vapor deposition of aluminum | Journal of Materials Research | Cambridge Core [https://www.cambridge.org/core/journals/journal-of-materials-research/article/abs/programmed-substrate-temperature-ramping-to-increase-nucleation-density-and-decrease-surface-roughness-during-metalorganic-chemical-vapor-deposition-of-aluminum/AED295F4AD534C498B0D0D292B2A6C8B]
- 70. US7250360B2 - Single step, high temperature nucleation process for a lattice mismatched substrate - Google Patents [https://patents.google.com/patent/US7250360B2/en]
- 71. Freezing Technology: Control of Freezing, Thawing, and Ice Nucleation - PubMed [https://pubmed.ncbi.nlm.nih.gov/32797412/]
- 72. Substrate-temperature-driven phase stabilization and ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838825065132]
- 73. In-situ high-temperature scanning electron microscopy ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838825019474]
- 74. How atomic force microscopy has contributed to our ... [https://www.sciencedirect.com/science/article/pii/S0032386109004996]
- 75. Recent Progress on the Characterization of Polymer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12526613/]
- 76. In Situ SEM, TEM, EBSD Characterization of Nucleation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10489032/]
- 77. Structural Diversity Design, Four Nucleation Methods Growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10785669/]
- 78. Exploring the self-nucleation effect [https://www.sciencedirect.com/science/article/pii/S1359836824000428]
- 79. Structural analysis and Rietveld refinement of YCr1 ... [https://www.sciencedirect.com/science/article/pii/S2667022424001476]
- 80. The Impact of Molecular Weight Distribution on the Crystalline ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12430040/]
- 81. Differential Scanning Calorimetry – Thermal Fractionation ... [https://www.tainstruments.com/applications-notes/differential-scanning-calorimetry-thermal-fractionation-techniques-for-polymer-characterization-faster-throughput-with-the-discovery-x3-dsc/]
- 82. Rietveld Refinement on X-Ray Diffraction Patterns of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1302771/]
- 83. Computational analysis on the influence of pressure and ... [https://www.nature.com/articles/s41598-025-24019-2]
- 84. Optimizing Annealing Temperature for Enhanced Electrical ... [https://www.mdpi.com/2072-666X/16/10/1091]
- 85. Optical and electrical characterization of crystalline silicon ... [https://www.sciencedirect.com/science/article/abs/pii/S0040609016000389]
- 86. Developing seeding protocols through secondary ... [https://www.crystallizationsystems.com/news/developing-seeding-protocols-through-secondary-nucleation-measurements-on-the-crystalline/]
- 87. Tuning Electro-Optical Characteristics through Polymerization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11174515/]
- 88. Design and experimental implementation of an effective ... [https://www.sciencedirect.com/science/article/abs/pii/S0959152416300889]
- 89. Breaking the reproducibility barrier with standardized ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12416739/]