Strategic Control of Supersaturation Thresholds for Predictive Crystal Morphology Engineering
This article provides a comprehensive guide for scientists and engineers on leveraging supersaturation control to dictate crystal morphology—a critical determinant of product performance in pharmaceuticals and materials science.
Strategic Control of Supersaturation Thresholds for Predictive Crystal Morphology Engineering
Abstract
This article provides a comprehensive guide for scientists and engineers on leveraging supersaturation control to dictate crystal morphology—a critical determinant of product performance in pharmaceuticals and materials science. It synthesizes foundational theory, modern prediction models, and advanced control strategies like continuous flow reactors and additive-mediated crystallization. The content explores practical methodologies for application, tackles common troubleshooting scenarios, and validates approaches through comparative case studies, offering a holistic framework for achieving targeted crystal size and shape with enhanced yield and quality.
The Science of Supersaturation: From Nucleation to Crystal Habit
FAQs: Core Concepts and Troubleshooting
What is supersaturation and why is it critical for crystallization? Supersaturation occurs when the concentration of a solute in a solution exceeds its equilibrium solubility. This state is metastable; it is not at equilibrium and the system can return to equilibrium by separating the excess solute from the solution, typically through crystallization [1]. It is the essential driving force behind both the initial formation of crystals (nucleation) and their subsequent growth [2].
How can I create a supersaturated solution? Common laboratory methods include:
- Cooling: A solution saturated at a high temperature often becomes supersaturated when cooled, as solubility typically decreases with temperature [1] [3].
- Evaporation: Removing solvent from a saturated solution increases the solute concentration beyond the saturation point [4].
- Antisolvent Addition: Adding a solvent in which the solute has low solubility can rapidly induce supersaturation [4].
My crystallization doesn't start. What should I do? If no crystals form after your solution has cooled, try these troubleshooting methods in order [3]:
- Scratching: Use a glass rod to scratch the inner surface of the flask. This can create microscopic particles that act as nucleation sites.
- Seeding: Introduce a tiny seed crystal of the pure compound to provide a template for growth.
- Solvent Evaporation: Return the solution to the heat source and boil off a portion of the solvent to increase concentration, then cool again.
- Cooling Bath: Lower the temperature of the cooling bath further to increase the supersaturation level.
My crystals form too quickly, and I suspect they are impure. How can I slow it down? Rapid crystallization can trap impurities within the crystal lattice. To slow the process [3]:
- Add Solvent: Use slightly more than the minimum amount of hot solvent needed for dissolution. This creates a less concentrated solution upon cooling.
- Improve Insulation: Place the crystallization flask on an insulating surface (e.g., paper towels) and cover it with a watch glass to slow the cooling rate.
- Use a Smaller Flask: If the solvent pool is very shallow, transfer to a smaller flask to reduce the surface area and slow cooling.
How does solvent choice influence crystal morphology? The solvent can significantly alter the final shape (habit) of crystals by interacting differently with various crystal faces, thereby affecting their growth rates. For example, studies on tolfenamic acid show:
- Polar solvents (e.g., ethanol) can disrupt hydrogen bonding at specific crystal faces (prismatic), leading to higher aspect ratio (needle-like) crystals [5].
- Non-polar solvents (e.g., toluene) can interact strongly with the "capping" faces of the crystal, hindering elongation and resulting in different morphologies [5].
Troubleshooting Guides
Guide 1: Optimizing Supersaturation for Improved Crystal Purity
Problem: Low product purity, inconsistent crystal size, or poor yield.
| Step | Action | Principle and Goal |
|---|---|---|
| 1 | Check feed solution quality. | Ensure the concentration, pH, and temperature are within the optimal range and free from contaminants that can act as unintended nucleation sites [6]. |
| 2 | Optimize cooling profile. | Avoid rapid, uncontrolled cooling. A slow, linear cooling rate promotes the growth of existing crystals over the formation of new nuclei, improving size and purity [7]. |
| 3 | Introduce controlled seeding. | Adding pure seed crystals at the correct supersaturation level provides designated growth sites, preventing spontaneous nucleation that leads to impure, small crystals [3] [4]. |
| 4 | Analyze the product. | Use microscopy and X-ray diffraction (XRD) to assess crystal morphology, size, and structure, comparing them to your quality targets [6]. |
Guide 2: Controlling Crystal Morphology and Size Distribution
Problem: Crystals have an undesirable shape (habit) or a wide size range, impacting downstream processing and product performance.
| Strategy | Methodology | Key Consideration |
|---|---|---|
| Solvent Selection | Crystallize the compound from different solvents of varying polarity and bonding motifs [5]. | The solvent can selectively inhibit or promote the growth of specific crystal faces by adsorbing to them, thereby changing the crystal's overall shape [5]. |
| Programmed Cooling | Use a non-linear cooling path (e.g., staggered cooling) instead of simple linear cooling [7]. | This allows the system to desaturate existing crystals at constant temperature steps, optimizing growth over nucleation and leading to more uniform crystals in a shorter time [7]. |
| Supersaturation Control | In membrane distillation crystallization, use membrane area to adjust the concentration rate [2]. | A higher supersaturation driving force favors nucleation, while lower, sustained supersaturation favors the growth of larger, more regular crystals [2]. |
Experimental Protocols for Supersaturation Control
Protocol 1: Drop Volume Ratio and Temperature (DVR/T) Optimization
This high-throughput method efficiently refines crystallization conditions for biological macromolecules without reformulating solutions [8].
Workflow Diagram
Materials and Reagents
- Protein Solution: Purified biological macromolecule.
- Cocktail Solution: Precipitant solution from initial screening that produced a "hit".
- Oil: A light, inert oil (e.g., paraffin oil) to containerize and prevent evaporation.
- Microplates: 1536-well or similar microassay plates.
- Liquid Handling System: Robotic pipetting system (optional, for high-throughput).
Procedure
- Prepare Experiment Drops: Using the same cocktail solution from the initial screen, create a matrix of experiment drops by combining the protein and cocktail solutions in different volume ratios (e.g., from 25:75 to 75:25 protein:cocktail) [8].
- Dispense Under Oil: Each experiment drop is dispensed under a layer of oil to prevent evaporation [8].
- Incubate at Multiple Temperatures: Replicate the entire plate matrix and incubate at different temperatures (e.g., 4°C, 12°C, 18°C, 23°C). Temperature is a key, underutilized variable that controls supersaturation [8].
- Analyze Outcomes: Microscopically examine all drops for outcomes (clear, precipitate, microcrystals, single crystals). The goal is to find the combination of volume ratio and temperature that produces large, single crystals of good optical quality [8].
Protocol 2: Staggered Cooling for Crystallization Time Optimization
This protocol uses a non-linear cooling path to optimize crystallisation time and crystal quality, as demonstrated in a granular model system [7].
Workflow Diagram
Materials and Reagents
- Crystallizer Setup: A temperature-controlled crystallizer vessel with accurate programming capabilities.
- Solution: A pre-filtered, saturated solution of the target compound at an elevated temperature.
Procedure
- Initial Condition: Start with the system at a high temperature where it is in a fluid, undersaturated, or slightly saturated state [7].
- Staggered Cooling Path: Instead of applying a linear cooling ramp, decrease the temperature in discrete steps:
- Step-Height: Suddenly quench the temperature by a predetermined amount (e.g., ΔT = 4-6°C). This increases supersaturation [7].
- Step-Width: Hold the system at this new, lower temperature for a defined period (e.g., 45-60 seconds). This provides time for the system to desaturate through crystal growth without generating excessive new nuclei [7].
- Repeat: Continue the sequence of step-height quenches followed by step-width holds until the final target temperature is reached.
- Optimization: The specific step-height and step-width that minimize total crystallization time and improve crystal quality must be determined experimentally for each system [7].
Supersaturation Thresholds and Growth Kinetics
Table 1: Face-Specific Crystal Growth Kinetics of Tolfenamic Acid Form I
Data obtained from growth rate measurements in ethanolic solutions, showing anisotropic growth behavior [5].
| Crystal Face (hkl) | Supersaturation (σ) | Growth Rate (μm/s) | Rate-Limiting Step |
|---|---|---|---|
| {1 0 0} (Capping) | ~0.3 | 0.044 - 0.555 | Mixed control (mass transfer and surface integration) [5]. |
| {0 1 1} (Prismatic) | ~0.3 | 0 - 0.020 | Surface integration [5]. |
Table 2: Optimized Staggered Cooling Parameters for Model System
Data from a 2D granular system showing parameters that minimize crystallization time [7].
| Cooling Step Height (ΔB in Gauss) | Minimum Crystallization Width (s) | Total Crystallization Time (s) |
|---|---|---|
| ~4.5 G | 60 | ~1000 [7] |
| ~5.8 G | 45 | Minimum time (theoretical) [7] |
The Scientist's Toolkit: Key Research Reagents and Materials
| Item | Function in Crystallization Research |
|---|---|
| Precipitants (e.g., PEG, Salts) | Primary agents used to reduce solute solubility and drive the solution into a supersaturated state [8] [4]. |
| Seeding Crystals | Small, pure crystals of the target compound used to provide a template for growth, promoting controlled crystallization in the metastable zone and improving crystal quality [3] [4]. |
| Additive Screens | Collections of various chemicals (e.g., salts, inhibitors, co-factors) used to subtly modify crystal growth kinetics and morphology when added in small quantities to a crystallization experiment [4]. |
| Liquid Metal Solvents (e.g., Ga, EGaIn) | Unconventional solvents for growing metallic crystals, allowing for dissolution and crystallization of metals at relatively low temperatures [9]. |
FAQs: Core Concepts and Troubleshooting
Q1: What is the fundamental difference between homogeneous and heterogeneous nucleation?
Homogeneous nucleation occurs spontaneously within a bulk supersaturated solution when random molecular clusters overcome a characteristic energy barrier to form stable nuclei. In contrast, heterogeneous nucleation occurs on foreign surfaces, impurities, or container walls, which act as catalysts by significantly reducing this energy barrier. Classical Nucleation Theory (CNT) quantifies this relationship through a potency factor, ( fc(\thetac) ), which scales the nucleation barrier based on the contact angle (( \thetac )) between the forming crystal and the substrate: ( \Delta G{\text{het}}^* = fc(\thetac) \Delta G_{\text{hom}}^* ) [10]. This makes heterogeneous nucleation the dominant mechanism in most experimental and industrial systems.
Q2: During membrane crystallization experiments, how can I determine if observed scaling is due to homogeneous nucleation?
Recent research indicates that membrane scaling occurs through a homogeneous nucleation mechanism when the boundary layer reaches extremely high supersaturation levels. You can distinguish this from heterogeneous nucleation by measuring induction times in both the bulk solution and at the membrane surface. Homogeneous scaling is characterized by a distinct log-linear relationship between nucleation rate and boundary layer supersaturation, which is a hallmark of CNT. Furthermore, the crystal habit of homogeneously formed scale is often distinctive from crystals formed in the bulk solution [11].
Q3: Our crystalline API product consistently shows unacceptable impurity levels. What is a systematic approach to identify the incorporation mechanism?
A structured Impurity Rejection Workflow is recommended to diagnose this issue. The five principal mechanisms of impurity incorporation are: agglomeration, surface deposition, inclusions, cocrystal formation, and solid solution formation [12]. The workflow involves a series of four experimental stages:
- Establish Baseline Knowledge: Collate essential data including product specifications, crystallization procedure, and analytical method calibration [12].
- Perform a Wash-Filtration Test: A simple wash step can remove impurities from the crystal surface. If purity improves, the mechanism is likely surface deposition or entrapped mother liquor from agglomeration.
- Conduct a Dissolution-Recrystallization Test: Dissolving the crystals and allowing them to slowly recrystallize can help identify inclusions. If the new crystals have higher purity, the original issue was likely solvent or impurity inclusions within the crystal lattice.
- Analyze the Solid-State: Techniques like X-ray diffraction and thermal analysis can identify the formation of cocrystals or solid solutions, where the impurity is incorporated directly into the crystal lattice [12].
Q4: Can Classical Nucleation Theory accurately predict nucleation on chemically heterogeneous surfaces?
Surprisingly, CNT demonstrates significant robustness even on non-uniform surfaces. Molecular dynamics simulations on checkerboard-patterned surfaces with alternating liquiphilic and liquiphobic patches show that nucleation rates retain their canonical temperature dependence as predicted by CNT. The study revealed that crystalline nuclei maintain a nearly fixed contact angle through a pinning mechanism at patch boundaries, which validates a key assumption of the theory despite surface chemical heterogeneity [10].
Q5: What is a critical operational parameter to prevent homogeneous scaling and promote controlled crystal growth in the bulk?
Research has identified a critical supersaturation threshold below which homogeneous scaling can be effectively 'switched off.' Operating below this threshold prevents the extreme supersaturation that triggers homogeneous nucleation at membrane surfaces or other interfaces. Below this threshold, crystal formation occurs solely in the bulk solution, typically yielding crystals with a preferred, uniform morphology. Temperature (T) and temperature difference (ΔT) are key control parameters to fix the boundary layer supersaturation at this desired set point [11].
Experimental Protocols
Protocol: Discriminating Nucleation Mechanisms via Induction Time Measurement
This non-invasive technique measures induction times in discrete domains to identify the nucleation pathway and establish a safe operating supersaturation [11].
- Objective: To directly relate mass and heat transfer in the boundary layer to CNT and discriminate between homogeneous and heterogeneous primary nucleation mechanisms.
- Materials:
- Membrane crystallization system with precise temperature control for both feed and permeate sides.
- Non-invasive monitoring tools (e.g., in-situ microscope, turbidity probe, focused beam reflectance measurement (FBRM)).
- Data acquisition system for temperature and process monitoring.
- Procedure:
- System Setup: Calibrate the system and set the bulk solution temperature (T) to a target value (e.g., 45°C).
- Impose Driving Force: Apply a temperature difference (ΔT) across the membrane (e.g., 15-30°C) to generate supersaturation in the boundary layer.
- Monitor Induction: Simultaneously record the induction time for the first detectable crystals in the bulk solution and for scale formation on the membrane surface.
- Systematic Variation: Repeat steps 1-3 for a matrix of T and ΔT values.
- Data Analysis: Plot the nucleation rate (inverse of induction time) against the calculated boundary layer supersaturation. A log-linear relation confirms behavior characteristic of CNT. Shorter induction times on the membrane surface at high supersaturation indicate a shift towards homogeneous nucleation leading to scaling [11].
Protocol: Seeding a Supersaturated Solution
This classic demonstration provides a clear, visual representation of triggering crystallization and is an excellent tool for understanding metastable states.
- Objective: To demonstrate the instability of a supersaturated solution and how seeding can trigger rapid, exothermic crystallization.
- Materials:
- 175 g sodium acetate trihydrate (
NaC2H3O2·3H2O) - 50 mL distilled water
- 500 mL Erlenmeyer flask
- Hot plate & 2 L beaker for water bath
- Tweezers and small seed crystals of sodium acetate trihydrate [13]
- 175 g sodium acetate trihydrate (
- Procedure:
- Prepare Solution: Combine sodium acetate trihydrate and distilled water in the flask.
- Dissolve Solute: Heat the mixture in a boiling water bath, swirling occasionally until a clear, homogeneous solution is obtained.
- Create Supersaturation: Remove the flask from the bath, cover it with an inverted beaker to prevent contamination, and allow it to cool undisturbed to room temperature (1-3 hours). This creates a metastable, supersaturated solution.
- Trigger Crystallization: Place a few seed crystals into a clean, dry beaker. Pour the supersaturated solution onto the seeds. Crystallization will begin immediately, forming a solid column and releasing heat (exothermic process) [14] [13].
- Troubleshooting: If the solution crystallizes during cooling, ensure all glassware is clean and scratch-free. Prepare multiple flasks as backups, as even minor disturbances can trigger nucleation [13].
Data Presentation
Table 1: Comparative Analysis of Nucleation Mechanisms
| Feature | Homogeneous Nucleation | Heterogeneous Nucleation |
|---|---|---|
| Definition | Spontaneous formation in the bulk solution | Catalyzed formation on a surface or impurity |
| Energy Barrier | High (( \Delta G_{\text{hom}}^* )) | Reduced (( \Delta G{\text{het}}^* = fc(\thetac) \Delta G{\text{hom}}^* )) [10] |
| Location | Bulk solution | Membrane surfaces, impurities, vessel walls [11] [10] |
| Induction Time | Generally longer at equivalent supersaturation | Shorter |
| Typical Outcome | Uncontrolled scaling, fine particles | Controlled crystal growth, preferred morphology [11] |
| Primary Control Parameter | Supersaturation level in the boundary layer [11] | Surface chemistry and topography [10] |
Table 2: Key Reagents and Materials for Nucleation Studies
| Research Reagent/Material | Function in Experiment |
|---|---|
| Sodium Acetate Trihydrate | Model compound for demonstrating supersaturation and seeding-induced crystallization [14] [13] |
| Detergents (e.g., DDM, OG) | Solubilizes membrane proteins for crystallization studies, critical for creating a homogeneous solution [15] |
| Seeding Crystals | Provides a surface to induce controlled heterogeneous nucleation and guide polymorphic form [16] |
| Anti-Solvent | Reduces API solubility to generate supersaturation and induce nucleation [16] |
| Chemically Patterned Surfaces | Substrates with defined liquiphilic/liquiphobic patches for studying the robustness of CNT on heterogeneous surfaces [10] |
Workflow Visualization
Core Theory FAQs
How do the BFDH and Attachment Energy models differ in their fundamental approach to predicting crystal morphology?
The Bravais–Friedel–Donnay–Harker (BFDH) and Attachment Energy (AE) models represent two key theoretical stages in predicting crystal morphology. Their core differences are summarized in the table below.
Table 1: Comparison of the BFDH and AE Model Approaches
| Feature | BFDH Model | Attachment Energy (AE) Model |
|---|---|---|
| Theoretical Basis | Geometric crystallography; Gibbs-Curie-Wulff principle for minimum surface energy [17] [18]. | Periodic Bond Chain (PBC) theory; energy-based analysis [17] [18]. |
| Key Predicting Parameter | Interplanar spacing ((d{hkl})); Growth rate (G{hkl} \propto 1/d_{hkl}) [18]. | Attachment Energy ((E{att})); Growth rate (R{hkl} \propto E_{att}) [18]. |
| Molecular Interactions | Not considered; purely geometric model [18]. | Explicitly considered via intermolecular forces [18]. |
| Typical Calculation Output | List of possible crystal faces and their relative growth rates based on lattice parameters and symmetry [19] [18]. | Relative growth rates of crystal faces based on the energy released upon layer attachment [20] [21] [18]. |
| Primary Limitation | Does not consider intermolecular interactions or external factors like solvents [18]. | Standard AE model predicts morphology in a vacuum; requires modification for solvents [20] [21]. |
The BFDH model is an excellent first step for identifying potential crystal faces, while the AE model provides a more physically realistic prediction for crystals grown from vapor. However, for solution-based crystallization, both models require corrections to account for solvent interactions [20] [18].
Why does my experimental crystal morphology differ from the morphology predicted by the AE model in a vacuum?
This is a common challenge, primarily because the standard AE model calculates attachment energies in a vacuum, while real-world crystallization occurs in a solution environment. The solvent significantly impacts the morphology by interacting differently with various crystal faces.
The mechanism can be understood through a corrected attachment energy model [20] [21]: [ E{att}^{solvent} = E{att} + E{int} \cdot (A{acc}/A{box}) ] where (E{att}^{solvent}) is the solvent-corrected attachment energy, (E{int}) is the interaction energy between the solvent and the crystal surface, and (A{acc}/A_{box}) is the ratio of the solvent-accessible area to the total crystal face area [21].
A solvent molecule (e.g., from acetone or water) will adsorb more strongly to a crystal face with which it has a high interaction energy ((E{int})). This strong interaction stabilizes the face, effectively lowering its growth rate ((R{hkl} \propto E_{att}^{solvent})) and making it more morphologically important in the final crystal habit [20] [21]. This explains why the dominant (020) face of HMX in vacuum predictions disappears when grown from acetone, a change correctly predicted by the solvent-corrected AE model [20].
What advanced models can predict morphology changes across different growth regimes (e.g., spiral vs. rough growth) driven by supersaturation?
Supersaturation is a key driver of growth regime transitions, and modern kinetic Monte Carlo (kMC) models are now capable of predicting the resulting morphology changes.
Table 2: Crystal Growth Regimes and Their Dependence on Supersaturation
| Growth Regime | Supersaturation Level | Growth Mechanism | Key Feature of Model |
|---|---|---|---|
| Spiral Growth | Low | Growth is driven by screw dislocations, which create self-perpetuating steps on the crystal surface [22]. | The model uses an adsorption rate that considers the lower adhesion energy barrier ((\triangle G)) at kink sites created by dislocations [22]. |
| Step Growth (2D Nucleation) | Medium | Occasional deposition onto flat surfaces creates new 2D islands that spread across the face [22]. | The increased supersaturation lowers (\triangle G) sufficiently to allow deposition on flat surfaces, not just at defects [22]. |
| Rough Growth | High | Growth units adsorb onto adatoms and isolated sites, leading to chaotic, non-layer-by-layer growth and rough surfaces [22]. | The model accounts for adsorption on adatoms and the formation of isolated islands that coalesce [22]. |
Advanced "adaptive kMC" models can seamlessly simulate transitions between these regimes by redefining the crystallization driving force to include the attachment energies of different adsorption sites (kink, adatom, edge) and the supersaturation level [22]. This allows a single model to predict morphology evolution across a wide range of operating conditions.
Troubleshooting Guides
How do I resolve the issue of "No Crystal Growth" or "Seed Crystals Dissolving"?
This problem typically indicates that the solution is not sufficiently saturated to drive crystallization.
Table 3: Troubleshooting No Crystal Growth or Dissolving Seeds
| Problem | Potential Cause | Solution | Underlying Principle |
|---|---|---|---|
| No Crystal Growth | Unsaturated solution [23]. | Continue adding solute until undissolved solid remains (saturation). Filter the warm solution before use [24] [23]. | The solution must be supersaturated for nucleation and growth to occur. |
| Slow evaporation rate [23]. | Loosely cover the container with a cloth or paper to allow solvent evaporation while keeping out dust [23]. | Evaporation increases concentration, driving the solution into a supersaturated state. | |
| Environmental vibrations [23]. | Move the setup to a quiet, undisturbed location [23]. | Vibrations can disrupt the delicate process of nucleation and ordered crystal growth. | |
| Seed Crystals Dissolve | New solution is undersaturated [23]. | Ensure the new growth solution is saturated. Let it evaporate slightly or add more solute before introducing the seed [23]. | If the solution is undersaturated, the crystal will dissolve to reach equilibrium. |
| Temperature mismatch. | Ensure the seed crystal and the new solution are at the same temperature to prevent localized dissolution [23]. | A warm solution can locally dissolve a colder seed crystal upon introduction. |
How can I control crystal shape, such as preventing needles and promoting equidimensional crystals?
Achieving a stout, equidimensional crystal habit is a common industrial goal to improve downstream processing.
Table 4: Troubleshooting and Controlling Crystal Habit
| Problem | Cause | Solution | Theoretical Insight |
|---|---|---|---|
| Needle-like/Plate-like Crystals | Anisotropic growth due to internal structure [19] [18]. | Use a tailor-made additive (e.g., polymer) that selectively binds to and inhibits the growth of the fastest-growing faces [19]. | Additives act by changing the relative growth rates ((R_{hkl})) of different faces. The additive adsorbs to specific faces, lowering their effective attachment energy [19]. |
| Solvent-specific interactions [20]. | Change the solvent. Different solvents will interact differently with crystal faces, altering the habit [20] [18]. | Solvent changes the solvent-corrected attachment energy ((E_{att}^{solvent})) for each face, modifying the growth morphology [20]. | |
| High supersaturation [22]. | Reduce supersaturation to promote slower, more orderly growth and prevent unstable, dendritic shapes [22]. | High supersaturation can lead to a transition from a spiral or step growth regime to a rough growth regime, promoting unstable morphologies [22]. | |
| Irregular Shapes | Rapid growth/cooling [23]. | Slow the cooling rate to promote growth of large, well-defined crystals [23]. | Rapid growth leads to incorporation of impurities and lattice defects, disrupting face development. |
| Impurities in solution [23]. | Use purified solute and distilled water. Filter the solution before crystallization [24] [23]. | Impurities can act as random inhibitors, disrupting the layered growth of crystal faces. |
Experimental Protocols
Protocol: Predicting and Validating Solvent-Mediated Crystal Morphology using a Modified AE Model
This protocol outlines the steps for using molecular dynamics (MD) simulations to predict crystal morphology in solution and validating it experimentally, as demonstrated for HMX [20] and Li₂CO₃ [21].
Objective: To theoretically predict the crystal morphology of a target compound in a specific solvent and confirm the prediction through experimental recrystallization.
Workflow Overview:
Materials:
- Software: Materials Studio (Accelrys) or similar MD package [20] [21].
- Force Field: COMPASS or other validated force field [21].
- Compound: Purified sample of the target compound (e.g., β-HMX, Li₂CO₃) [20] [21].
- Solvent: Analytical grade solvent (e.g., Acetone for HMX) [20].
Procedure:
Part A: Molecular Dynamics Simulation
- Unit Cell Optimization: Obtain the crystallographic unit cell of your compound from a database (e.g., ICSD). Perform a geometry optimization using a smart minimizer to optimize the cell parameters [20] [21].
- Morphology Prediction in Vacuum: Use the AE model on the optimized structure to generate a list of morphologically important (hkl) faces and predict the vacuum morphology [20] [21].
- Cleave Crystal Surfaces: Construct a periodic superstructure (e.g., 5x6 unit cells) and cleave the crystal along the dominant faces identified in the previous step (e.g., (020), (011), (100) for HMX) [20].
- Build Solvent Layer: Use the "Amorphous Cell" tool to build a solvent layer filled with solvent molecules (e.g., 500 H₂O molecules) at a target density of 1 g/cm³. Match the size of the solvent layer to the cleaved crystal surface [21].
- Construct and Optimize the Simulation System: Place two solvent layers on the crystal surface and add a vacuum slab (~50 Å) above to eliminate boundary effects. Perform a global geometry optimization on the entire system [21].
- Run Molecular Dynamics: Conduct MD simulations using an NVT ensemble (e.g., 2000 ps with a 1 fs time step at 298.15 K). Use the Andersen thermostat and standard Ewald method for electrostatic interactions [21].
- Calculate Solvent-Corrected Attachment Energy:
- Calculate the interaction energy ((E{int})) using: (E{int} = E{total} - (E{surface} + E{solvent})) [21].
- Calculate the solvent-accessible area ((A{acc})) and the total crystal face area ((A{box})) [21].
- Compute the corrected attachment energy: (E{att}^{solvent} = E{att} + E{int} \cdot (A{acc}/A{box})) [21].
- Predict Final Morphology: Calculate the relative growth rates ((R{hkl} \propto E{att}^{solvent})) and construct the predicted crystal habit for the solution environment [20] [21].
Part B: Experimental Recrystallization and Validation
- Prepare Saturated Solution: Dissolve the target compound in the chosen solvent at an elevated temperature (e.g., 60°C for HMX in acetone) to create a saturated solution [20].
- Crystallize: Slowly cool the solution to ambient temperature to induce crystallization. Alternatively, use slow evaporation at room temperature with minimal disturbance [21].
- Characterize Crystal Habit: Analyze the resulting crystals using Scanning Electron Microscopy (SEM) to observe the actual crystal morphology [19] [21].
- Compare and Analyze: Compare the experimentally observed crystal shape with the morphology predicted by the solvent-corrected AE model. The (hkl) indices of the dominant faces in the experiment should correspond to the faces with the lowest (E_{att}^{solvent}) in the simulation [20].
The Scientist's Toolkit
Table 5: Essential Research Reagents and Materials for Crystal Growth Studies
| Item | Function/Application | Example Use Case |
|---|---|---|
| Hydroxypropyl Cellulose (HPC) | Polymer additive used for crystal habit modification. Selectively adsorbs to specific crystal faces, inhibiting their growth [19]. | Used to modify the plate-like habit of Erythromycin A Dihydrate to an elongated plate-like shape, improving compaction properties [19]. |
| Monoammonium Phosphate (MAP) | Model compound for crystal growth experiments. Non-toxic, easily available, and grows clear, well-defined crystals from solution [24]. | Used in fundamental studies on crystal growth mechanics and for demonstrating the effects of additives (e.g., alum) on crystal shape (prismatic vs. needle-like) [24]. |
| Alum (Ammonium Aluminum Sulfate) | Additive used to manipulate the aspect ratio and sharpness of growing crystals [24]. | Adding 0.25-1.25 g per 100 mL water to a MAP solution changes the crystal habit from lumpy to sharp, needle-like clusters [24]. |
| Analytical Grade Solvents (e.g., Acetone, Ethanol) | Medium for solution crystallization. The choice of solvent is a critical parameter as it can significantly alter the final crystal morphology [20] [18]. | Acetone was used as a solvent to change the crystal habit of HMX, suppressing the (020) face and resulting in a morphology that matched the MD simulation [20]. |
| Distilled / Deionized Water | Purified water used to eliminate the effects of ionic impurities that can inhibit crystal growth or lead to irregular shapes [23]. | Essential for preparing clean, saturated solutions to ensure reproducible growth of high-quality crystals without contamination [23]. |
Frequently Asked Questions (FAQs)
FAQ 1: Why does my crystallization experiment sometimes produce fine needles instead of the desired polyhedral crystals? This is a classic sign of operating at an excessively high supersaturation level. High supersaturation strongly favors rapid primary nucleation, leading to a high number of crystal nuclei [2]. This depletes the available solute quickly, leaving insufficient material for the crystals to grow large and well-defined, resulting in numerous small, needle-like crystals. To correct this, you should aim to lower the initial supersaturation. Strategies include reducing the cooling rate, using a anti-solvent addition more gradually, or employing seeded crystallization to provide controlled growth sites [25].
FAQ 2: What techniques can I use to accurately determine the metastable zone width (MSZW) for my system? The metastable zone width is the region between the solubility curve and the spontaneous nucleation curve, and its determination is crucial for process control. A standard method involves using an automated reactor system (e.g., Crystal16) with transmissivity probes [25]. The process involves:
- Preparing a clear solution at an elevated temperature.
- Cooling the solution at a fixed, controlled rate while agitating.
- Recording the temperature at which a sustained decrease in transmissivity (e.g., below 50%) is observed, indicating nucleation. This temperature at a given concentration defines the metastable limit. Performing this at multiple concentrations maps out the MSZW.
FAQ 3: How can I promote crystal growth over nucleation to achieve larger crystals? The competition between nucleation and growth is directly influenced by supersaturation. To favor growth:
- Use Seeded Experiments: Introducing seed crystals provides a surface for growth without the need for spontaneous nucleation, allowing you to operate at lower supersaturations safely within the metastable zone [25].
- Control Supersaturation Rate: Implementing strategies like membrane distillation crystallization or continuous flow reactors can maintain a constant, low supersaturation, providing time for crystals to grow without generating new nuclei [2] [26].
- Extended Hold-up Time: After the initial nucleation event, maintaining the system under agitation for a longer period allows crystal growth to desaturate the solution, which in turn suppresses further nucleation and leads to larger crystal sizes [2].
FAQ 4: In a continuous flow reactor, how does supersaturation specifically lead to a morphology change from nanoplates to nanoflowers? In a continuous flow system, supersaturation is precisely controlled. Research on NiCo layered double hydroxide (LDH) synthesis has shown that increasing supersaturation triggers a shift in the dominant growth mechanism [26]. At lower supersaturations, growth occurs more uniformly, leading to well-defined 2D nanoplates. As supersaturation increases, it passes a threshold that favors a high nucleation rate. This leads to the formation of many small nuclei that aggregate and grow through a mechanism that results in complex 3D nanoflower morphologies [26].
Troubleshooting Guides
Problem: Uncontrolled Primary Nucleation Leading to Fines
Symptoms: A shower of small, needle-like crystals; broad crystal size distribution; inconsistent batch-to-batch results.
Root Cause: Operation at a supersaturation level that is too high, deep within the labile zone, which promotes homogeneous primary nucleation.
Solution Steps:
- Characterize Your System: First, determine the solubility and metastable zone width (MSZW) for your compound-solvent system using cooling curve or isothermal induction time methods [25].
- Operate in the Metastable Zone: Design your crystallization process to operate within the MSZW, not at its boundary.
- Implement Seeding: Introduce seed crystals at a supersaturation level that is high enough to support growth but low enough to prevent secondary nucleation [25].
- Fine-tune Supersaturation Control: For advanced control, consider technologies like Membrane Distillation Crystallization (MDC) or Continuous Flow Reactors (CFR), which allow for precise manipulation of supersaturation by adjusting parameters like membrane area or flow rate [2] [26].
Problem: Inconsistent Morphology Between Batch and Scale-up
Symptoms: Crystal habit (e.g., platelet, polyhedral) changes when moving from small-scale vial experiments to larger reactors.
Root Cause: Differences in mixing, heat transfer, and local supersaturation profiles between scales can shift the dominant nucleation pathway and growth kinetics.
Solution Steps:
- Decouple Kinetics: Use small-scale experiments to quantitatively estimate the primary nucleation, secondary nucleation, and growth rates as separate functions of supersaturation [25].
- Match Agitation and Supersaturation: Understand that nucleation kinetics are sensitive to agitation conditions [25]. During scale-up, ensure that the agitation intensity and supersaturation profile (e.g., cooling profile) are matched as closely as possible to the well-characterized small-scale experiment.
- Utilize a Continuous Process: Transitioning to a Continuous Flow Reactor (CFR) can inherently improve consistency. CFRs maintain constant supersaturation under pseudo-steady-state conditions, minimizing the variations that lead to morphology shifts during scale-up [26].
Data Presentation
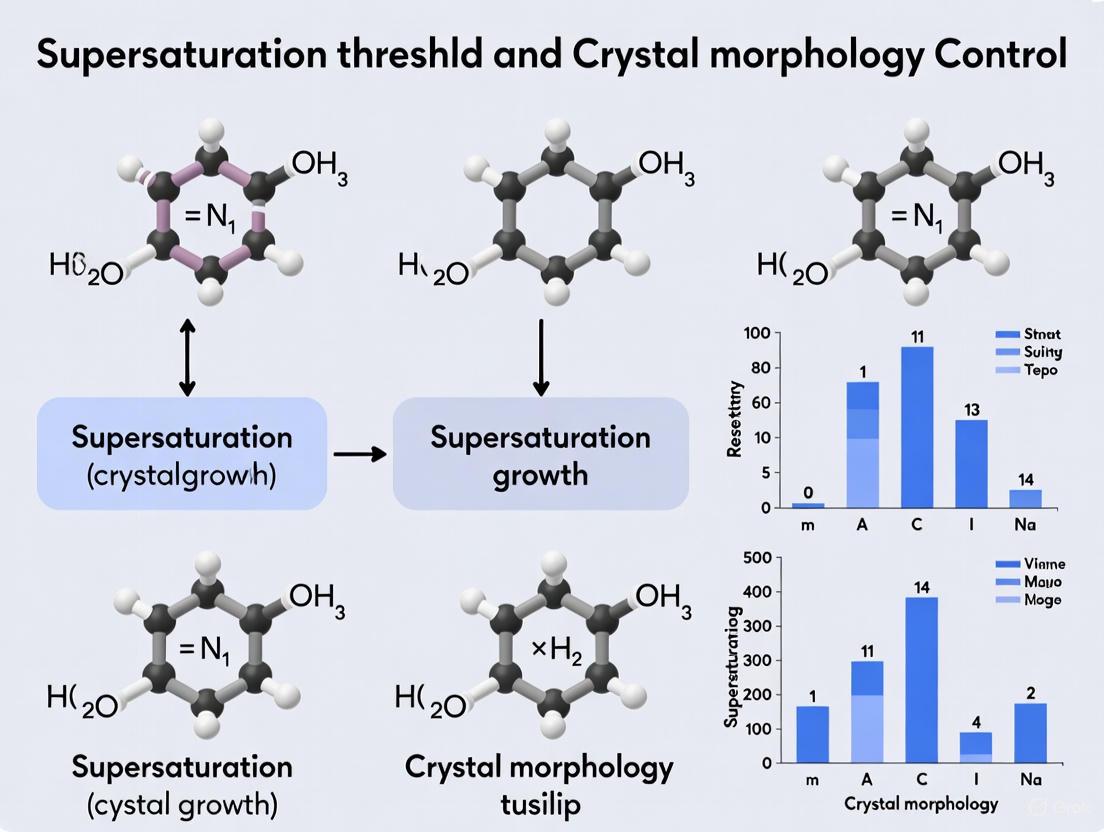
Table 1: Supersaturation Thresholds and Resulting Crystal Morphologies
| System / Material | Supersaturation (S) / Threshold | Nucleation Type Favored | Resulting Crystal Morphology |
|---|---|---|---|
| NiCo LDH [26] | Low S | Heterogeneous Nucleation | Isolated 2D Nanoplates |
| High S | Homogeneous Nucleation | 3D Nanoflowers | |
| Generic Methodology [25] | S < MSZW | Crystal Growth | Larger, Polyhedral Crystals |
| S > MSZW | Primary Nucleation | Small Needles / Fines |
Table 2: Experimental Protocols for Kinetic Analysis
| Experiment Type | Key Steps | Measured Output | Application |
|---|---|---|---|
| Isothermal Induction Time [25] | 1. Create clear solution.2. Rapidly cool to target temperature.3. Hold isothermally with agitation.4. Record time until transmissivity drop. | Distribution of induction times; estimates of primary nucleation rate (J) and growth time (tg). | Quantifies stochastic primary nucleation kinetics at different supersaturations. |
| Seeded Crystal Growth [25] | 1. Generate a clear solution at target S.2. Introduce seeds of known size.3. Monitor desupersaturation curve (e.g., via in-situ analytics). | Crystal growth rate as a function of supersaturation. | Decouples growth kinetics from nucleation; essential for low-S processes. |
| Continuous Flow Synthesis [26] | 1. Set up separate precursor feeds.2. Mix via T-junction into heated column.3. Control residence time and temperature.4. Collect product continuously. | Steady-state particle morphology and size as a function of feed concentration/flow rate. | Provides precise, constant supersaturation control for uniform morphology. |
Experimental Workflow & Pathway Visualization
Workflow for Crystal Morphology Control
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for Controlled Crystallization
| Item | Function / Application | Example from Literature |
|---|---|---|
| Hexamethylenetetramine (HMTA) | A common homogeneous precipitating agent; hydrolyzes upon heating to slowly release hydroxyl ions, enabling a more controlled pH increase and supersaturation generation [26]. | Used in the continuous flow synthesis of NiCo LDH nanoplates and nanoflowers [26]. |
| Seed Crystals | Pre-formed crystals of the target compound used to provide controlled growth sites, suppress primary nucleation, and enable operation at lower supersaturations for larger crystal growth [25]. | Essential for assessing crystal growth kinetics in α-glycine crystallization, especially at lower supersaturations [25]. |
| Metal Salt Precursors | Provide the cation source for the crystallization of inorganic or metal-organic materials. | Nickel nitrate hexahydrate and cobalt nitrate hexahydrate were used as precursors for NiCo LDH synthesis [26]. |
| Continuous Flow Reactor (CFR) | A system that maintains constant supersaturation under pseudo-steady-state conditions, enabling precise control over nucleation and growth for uniform morphology [26]. | A jacketed chromatography column with separate precursor feed lines was used to synthesize NiCo LDH with tunable morphologies [26]. |
The Role of Phase Diagrams and the Metastable Zone in Crystallization Design
Frequently Asked Questions (FAQs)
FAQ 1: What is the fundamental role of a phase diagram in crystallization design? A phase diagram is a thermodynamic map that illustrates the state of matter under varying conditions, such as concentration and temperature [27]. In crystallization design, it is used to conceptualize phase relations and identify regions where stable crystals can form. For crystallization from solution, common diagrams plot temperature against composition (T/x) or precipitant concentration against protein concentration (x/x) [27]. These diagrams help identify key zones: an undersaturated zone where the protein is completely soluble, a metastable zone where crystal growth can occur but nucleation is unlikely, and a labile or nucleation zone where spontaneous nucleation and precipitation are probable [28]. Understanding this map is the first step in rationally designing a crystallization process to target the desired outcome, whether it is for growing large, high-quality crystals or for obtaining a specific crystal form.
FAQ 2: How does the Metastable Zone Width (MSZW) influence my crystallization process? The Metastable Zone Width (MSZW) represents the range of supersaturation within which a solution remains metastable—meaning crystals will not form spontaneously—before nucleation occurs [29]. It is a critical parameter because it defines the operational window for controlled crystal growth. A wider MSZW allows for a larger range of supersaturation levels where crystal growth can proceed without the unwanted formation of new nuclei, which can lead to small crystals or showers of microcrystals [28]. The MSZW is not a fixed property; it is influenced by factors such as the cooling rate, agitation, solution volume, and vessel geometry [29]. Accurately determining the MSZW for your system is therefore essential for developing a robust crystallization procedure that avoids uncontrolled nucleation and favors the growth of large, well-ordered crystals.
FAQ 3: Why is controlling supersaturation so important, and how is it achieved? Supersaturation is the fundamental driving force for both nucleation and crystal growth [29]. The level of supersaturation directly determines the kinetics of these processes. High supersaturation favors rapid nucleation, leading to many small crystals, while moderate supersaturation within the metastable zone favors slower, controlled growth on existing crystals, resulting in larger and better-ordered specimens [30]. Supersaturation can be controlled through several methods:
- Temperature Change: Cooling a solution to decrease solubility [31].
- Antisolvent Addition: Adding a solvent in which the compound has low solubility, thereby reducing its overall solubility in the mixture [31].
- Evaporation: Removing solvent to increase concentration [30]. Advanced strategies in processes like Membrane Distillation Crystallization (MDC) use the membrane area to adjust the supersaturation rate precisely, allowing the process to be repositioned within specific regions of the metastable zone to favor either growth or nucleation as needed [30].
FAQ 4: What are common crystallization issues that phase diagrams help troubleshoot? Phase diagrams provide a framework for diagnosing and solving common crystallization problems. Issues and their phase diagram-based solutions include:
- No Crystallization: The experimental conditions may lie in the undersaturated region. The solution is to increase the concentration of the precipitant or protein to move into the metastable or labile zone [28].
- Showers of Microcrystals or Precipitate: This indicates conditions are firmly within the labile zone, where nucleation is excessive. The remedy is to lower the supersaturation by reducing precipitant or protein concentration, moving the conditions back toward the metastable zone [28] [32].
- Poor Crystal Morphology or Quality: Crystals grown at high supersaturation near the labile zone boundary are often poorly formed. Using seeding techniques to introduce crystal nuclei directly into the metastable zone allows for slow, controlled growth, typically improving crystal size and diffraction quality [28] [32].
Troubleshooting Guides
Guide 1: Addressing Poor Product Purity
Problem: Crystals contain high levels of impurities or exhibit inconsistent purity.
| Troubleshooting Step | Action & Rationale | Relevant Experimental Parameters to Check |
|---|---|---|
| 1. Check Feed Quality | Monitor and control the composition of the feed stream. Impurities in the feed can be incorporated into the crystal lattice or interfere with growth [6]. | Concentration, pH, temperature, dissolved solids. |
| 2. Optimize Operating Conditions | Adjust parameters to achieve a controlled supersaturation level that favors pure crystal growth over rapid, disordered nucleation [6]. | Temperature, cooling rate, agitation, residence time. |
| 3. Utilize Seeding | Introduce purified seed crystals into the metastable zone. This provides a defined surface for growth, reducing the supersaturation required and minimizing spontaneous nucleation that can trap impurities [28]. | Seeding rate, seed quality, timing of seed addition. |
Guide 2: Controlling Crystal Size and Morphology
Problem: Crystals are too small, have a wide size distribution, or an undesirable shape.
| Troubleshooting Step | Action & Rationale | Relevant Experimental Parameters to Check |
|---|---|---|
| 1. Map the Metastable Zone | Determine the MSZW for your system using PAT tools. This identifies the supersaturation range where growth occurs without nucleation [29]. | Cooling rates, induction times, solubility curve. |
| 2. Modulate Supersaturation | After nucleation is induced, carefully control the supersaturation rate to remain in a region of the metastable zone that favors growth. Slower desaturation of the solvent allows for larger crystal sizes [30]. | Antisolvent addition rate, evaporation rate, cooling profile. |
| 3. Segregate Crystal Phase | Use in-line filtration or controlled agitation to keep crystals suspended in the bulk solution and reduce scaling on vessel walls. This ensures consistent growth conditions and improves crystal habit [30]. | Agitation rate, crystallizer design, use of filters. |
Key Quantitative Data for Crystallization Design
The following table summarizes critical nucleation parameters for paracetamol in isopropanol, obtained through Process Analytical Technology (PAT), which are essential for modeling and controlling the crystallization process [29].
Table 1: Experimentally Determined Nucleation Parameters for a Model API (Paracetamol in Isopropanol)
| Parameter | Value | Significance in Crystallization Design |
|---|---|---|
| Nucleation Rate Constant | 10²¹ - 10²² molecules/m³·s | Quantifies the rate of formation of new crystals; crucial for predicting crystal population. |
| Gibbs Free Energy of Nucleation | 3.6 kJ/mol | Represents the energy barrier for nucleus formation; lower values favor easier nucleation. |
| Surface Energy (Interfacial Tension) | 2.6 - 8.8 mJ/m² | Reflects the energy at the crystal-solution interface; affects the critical nucleus size and nucleation rate. |
| Critical Nucleus Radius | ~10⁻³ m | The minimum size a nucleus must achieve to be stable and continue growing. |
Essential Experimental Protocols
Protocol 1: Determination of Solubility and Metastable Zone Width (MSZW) using PAT
Objective: To accurately measure the solubility curve and MSZW using in-situ Fourier Transform Infrared (FTIR) spectroscopy and Focused Beam Reflectance Measurement (FBRM) [29].
Materials:
- Crystalline compound (e.g., Paracetamol)
- Solvent (e.g., Isopropanol)
- Crystallization reactor with temperature control
- In-situ FTIR spectrometer with probe
- FBRM probe
- Crystal16 Multiple Reactor Setup (or equivalent)
Methodology:
- Sample Preparation: A known mass of the crystalline compound is added to a vial, followed by a precise mass of solvent or solvent-antisolvent mixture to create a specific solute-free antisolvent mass fraction (x~AS~) and solute concentration (C) [31].
- Solubility Determination (Clear Point Method):
- The suspension is heated at a controlled rate (e.g., 0.2 °C/min).
- The in-situ FTIR spectrometer monitors a specific IR peak (e.g., at 1516 cm⁻¹ for paracetamol). The intensity of this peak is proportional to concentration.
- The clear point temperature is recorded as the temperature at which the last crystal dissolves and the IR signal stabilizes. This temperature-concentration pair defines one point on the solubility curve [31].
- The process is repeated for multiple starting concentrations to define the full solubility curve. Temperature effects on the IR signal are corrected mathematically [29].
- MSZW Determination:
- A clear solution is cooled from above the saturation temperature at a defined cooling rate (e.g., 0.4 °C/min).
- The FBRM probe monitors the chord length count, which sharply increases when nucleation occurs.
- The temperature at which the nucleation event is detected by FBRM defines the supersolubility temperature at that cooling rate.
- The MSZW is the concentration or temperature difference between the solubility curve and the supersolubility curve at a given cooling rate [29].
Protocol 2: Protein Crystallization via Microseeding Using a Phase Diagram
Objective: To reproducibly grow high-quality protein crystals by using a phase diagram to identify the metastable zone for microseeding [28].
Materials:
- Purified protein solution
- Precipitant solutions (e.g., PEG 4000)
- Crystallization plates (e.g., sitting drop vapor diffusion plates)
- Microbatch plates (e.g., for IMPAX dispenser)
- Harvesting buffer
- Glass fiber for crushing crystals
Methodology:
- Phase Diagram Construction:
- A fine-screen microbatch experiment is set up, testing a wide matrix of protein concentrations against precipitant concentrations.
- Results are analyzed to map the phase diagram: identifying conditions that yield clear drops, showers of microcrystals, few large crystals, and amorphous precipitate [28].
- Identify Metastable Zone:
- The area of the diagram at lower precipitant and protein concentrations that remains clear but can support crystal growth is identified as the metastable zone [28].
- Seed Stock Preparation:
- A well-formed crystal from the initial screen is harvested and crushed in a harvesting buffer with a precipitant concentration slightly higher than the target growth condition.
- The resulting suspension is centrifuged at low speed (~100 g) to remove large fragments.
- The supernatant, containing the seeds, is serially diluted to create a range of seeding solutions [28].
- Seeded Crystal Growth:
- Sitting drop vapor diffusion trials are set up where the reservoir contains precipitant at the target concentration (e.g., well 16 from the diagram).
- The droplet is dispensed with a precipitant concentration marginally below the reservoir (e.g., well 12 concentration), placing it in the metastable zone even before equilibration.
- A small volume (e.g., 0.3 μL) of the diluted seed stock is added to the droplet. The optimal dilution (e.g., 10⁻³) is determined empirically.
- The plate is sealed, allowing the drop to equilibrate slowly, promoting growth on the introduced seeds without spontaneous nucleation [28].
Research Reagent Solutions
Table 2: Essential Materials for Crystallization Experiments
| Item | Function in Crystallization | Example Use Case |
|---|---|---|
| Polyethylene Glycol (PEG) | A polymer precipitant that excludes protein from solution, driving self-assembly and crystallization [28]. | Commonly used precipitant for proteins (e.g., PEG 4000). |
| Process Analytical Technology (PAT): FTIR | In-situ spectrometer for real-time concentration measurement; used for determining solubility curves [29]. | Tracking API concentration during clear point measurements. |
| Process Analytical Technology (PAT): FBRM | In-situ probe that measures chord length distribution of particles in suspension; used for detecting nucleation events [29]. | Determining the metastable zone width (MSZW) by detecting the onset of nucleation. |
| Antisolvent (e.g., Water) | A solvent added to a solution to reduce the solubility of the solute, thereby generating supersaturation [31]. | Used in antisolvent crystallization of small molecules (e.g., Lovastatin in acetone/water). |
| Crystal16 Reactor | A multiple-reactor workstation for automated temperature cycling and turbidity measurements [31]. | High-throughput determination of saturation temperatures and MSZW. |
Workflow and Relationship Visualizations
Crystallization Design Workflow
Phase Diagram Zones
Advanced Techniques for Supersaturation and Morphology Control
In the context of optimizing supersaturation thresholds for crystal morphology control, maintaining constant supersaturation is a foundational objective. Continuous Flow Reactors (CFRs) present a transformative alternative to traditional batch crystallization processes. Unlike batch reactors, where supersaturation levels fluctuate as the reaction progresses, CFRs are designed to operate at a continuous steady state [33]. This means the internal stream, temperature, reagent feed, and flow rates are all held constant, producing an unceasing flow of chemical reactant material and a continuous product output [33]. This operational stability is key to achieving uniform crystal growth, as it allows for precise control over the supersaturation level, a critical parameter that directly influences crystal size, shape, and purity. The ability to seamlessly scale from laboratory proof-of-concept to full-scale industrial production, often in half the time of batch systems, makes CFRs particularly valuable for pharmaceutical and fine chemical development [34].
Troubleshooting Guides
Problem: Inconsistent Crystal Size Distribution
- Issue: The product crystals exhibit a wide and inconsistent size distribution (CSD).
- Potential Causes & Solutions:
Cause Diagnostic Steps Solution Fluctuations in supersaturation Use in-line ATR-FTIR spectroscopy to monitor real-time solute concentration and supersaturation profiles [35]. Implement a closed-loop control system that adjusts the precursor feed rate based on real-time supersaturation data to maintain a constant set point. Inadequate mixing leading to hot spots Inspect for clogging or flow restrictions. Use flow visualization or tracer studies to identify dead zones. Switch to a reactor with enhanced passive mixing structures (e.g., heart-shaped designs) [34] or employ an actively mixed Continuous Stirred Tank Reactor (CSTR) [33]. Uncontrolled nucleation Perform offline microscopy on samples to identify excessive fines. Introduce a separate, controlled nucleation stage (e.g., a sonication loop) and ensure the main growth reactor operates below the secondary nucleation threshold.
Problem: Reactor Clogging
- Issue: Solid deposits cause blockages within the reactor tubing or modules.
- Potential Causes & Solutions:
Cause Diagnostic Steps Solution Rapid, localized supersaturation Review the selected supersaturation set point; it may be too high, leading to excessive nucleation. Reduce the supersaturation level in the main growth zone. Employ a seeding strategy with well-defined seed crystals introduced upstream [35]. Wall fouling and scale-up Visually inspect reactor internals for crust formation. Optimize surface material and geometry of the reactor. For example, Corning's Advanced-Flow reactors use corrosion-resistant glass and/or ceramic fluidic modules to minimize fouling [34]. Precipitation of byproducts Analyze the clog material to determine its composition. Modify the solvent system or introduce purification steps (e.g., in-line filters) in the reagent feed lines to remove impurities that act as nucleation sites.
Problem: Failure to Maintain Target Supersaturation
- Issue: The system cannot achieve or hold the desired supersaturation level.
- Potential Causes & Solutions:
Cause Diagnostic Steps Solution Inaccurate temperature control Calibrate all in-line temperature sensors and check for fluctuations in the heat transfer fluid. Utilize the high surface-area-to-volume ratio of CFRs for superior thermal management [36]. Ensure the reactor design provides intense heat exchange control [34]. Precursor concentration variability Audit the feed stock preparation procedure and use in-line analytics to monitor input concentrations. Automate the feeding system to ensure a consistent and precise ratio of ingredients [34]. Implement continuous mixing before the feed enters the reactor. Residence time distribution (RTD) issues Conduct a residence time distribution study using tracers. Redesign the reactor flow path to approach ideal plug flow behavior. For CSTRs, consider connecting multiple tanks in series to narrow the RTD [33].
Frequently Asked Questions (FAQs)
Q1: Why is a Continuous Flow Reactor superior to a batch reactor for controlling crystal morphology?
CFRs offer superior control over key crystallization parameters. They maintain a continuous steady state, which allows for precise and constant control over supersaturation—the primary driver of crystal growth and morphology [33]. The enhanced heat and mass transfer capabilities due to a high surface-area-to-volume ratio enable more rapid and uniform mixing and temperature control compared to batch systems [33] [36]. This minimizes localized zones of high supersaturation that lead to inconsistent nucleation and growth, thereby promoting uniform crystal morphology.
Q2: What types of flow reactors are best suited for crystallization processes?
The choice depends on the reaction and the solids handling required.
- Plug Flow Reactors (PFR): Often ideal for crystallization, as they operate with passive mixing and provide a narrow residence time distribution, ensuring all crystals experience similar growth conditions [33]. They are typically used for single-phase chemistry.
- Continuous Stirred Tank Reactors (CSTR): Employ active mixing with an internal agitator, which can be beneficial for handling slurries or preventing settling, but may have a broader residence time distribution [33].
- Packed Bed Reactors: Suitable for heterogeneous reactions, such as those involving solid catalysts, but can be prone to clogging with precipitated products [36]. Advanced reactors, like Corning's Advanced-Flow reactors, incorporate patented mixing structures and corrosion-resistant materials specifically designed for challenging chemical processes, including those with solids formation [34].
Q3: How can I experimentally determine the optimal supersaturation level for my system in a CFR?
A systematic optimization procedure is required [32].
- Initial Screening: Use a small-volume flow system to quickly test a wide range of supersaturation levels by varying parameters like concentration, temperature, and anti-solvent ratio.
- In-line Monitoring: Employ tools like ATR-FTIR spectroscopy or laser backscattering to measure supersaturation and detect nucleation events in real-time [35].
- Incremental Refinement: Systematically refine the parameters around the most promising "hits" from the screen. For example, if a condition at 25°C yielded microcrystals, run a series of experiments incrementally adjusting the temperature (e.g., 20°C, 22°C, 24°C, 26°C, 28°C) and other interdependent variables like pH and precipitant concentration [32].
- Seeding: Once a target supersaturation is identified, introduce well-characterized seed crystals to promote controlled growth over spontaneous nucleation.
Q4: What are the common pitfalls when scaling up a crystallization process from lab-scale to production-scale CFRs?
A primary pitfall is neglecting the impact of reactor geometry and mixing efficiency on scale-up. Simply increasing the flow rate and reactor volume may not preserve the same mixing dynamics and supersaturation profile. A successful scale-up involves numbering-up or using scalable reactor designs. For instance, Corning's Advanced-Flow reactors are designed to provide a seamless scale-up path by increasing the volume of ingredients fed into the system while maintaining consistent fluid dynamics and heat/mass transfer characteristics from lab to production scale [34].
Experimental Protocols & Workflows
Workflow for Establishing Constant Supersaturation
The following diagram illustrates the logical workflow and control loops involved in establishing and maintaining constant supersaturation within a Continuous Flow Reactor.
Protocol: Optimization of Crystallization Conditions via Systematic Parameter Variation
This protocol is adapted from methodologies used in macromolecular crystal growth and process optimization [32] [37].
Objective: To incrementally refine crystallization conditions in a CFR to achieve uniform crystal growth.
Materials:
- Continuous Flow Reactor system (e.g., PFR or CSTR type)
- Precision syringe or piston pumps
- In-line ATR-FTIR spectrometer and/or particle analyzer
- Thermostatted heating/cooling unit
- Data acquisition and control software
Method:
- Initial Condition Identification: From a preliminary screen, identify a condition that yields any crystalline material, even of poor quality (e.g., microcrystals or clusters). This is your "hit" [32].
- Parameter Selection: Identify the key chemical and physical parameters of the initial hit (e.g., pH, precipitant concentration, temperature, flow rate).
- Solution Matrix Preparation: Prepare a matrix of solutions where each parameter is varied incrementally around the initial value.
- Example: If the initial hit was at pH 7.0, prepare solutions at pH 6.0, 6.2, 6.4, ..., up to 8.0, keeping other parameters constant [32].
- Note: Interdependence of parameters (e.g., temperature affecting pH) must be considered.
- Sequential Flow Experimentation:
- Prime the CFR system with solvent.
- For each condition in the matrix, set the reactor temperature and the flow rates of the separate precursor streams.
- Allow the system to stabilize at the new condition, ensuring a steady state is reached before sample collection.
- Collect the product slurry and analyze crystals for size distribution, morphology, and purity.
- Data Analysis and Iteration: Analyze the results to determine the parameter set that produces the most uniform and highest-quality crystals. Use this optimal set as a new baseline for further refinement if necessary.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key components and materials essential for setting up and operating a Continuous Flow Reactor for crystallization studies.
| Item | Function & Importance |
|---|---|
| Advanced-Flow Reactor | Reactors with specialized fluidic modules (e.g., glass, silicon carbide) that provide superior heat/mass transfer and integrated mixing structures for intensified continuous processes [34]. |
| ATR-FTIR Spectrometer | A critical in-line analytical tool for real-time measurement of solute concentration and supersaturation, enabling immediate feedback and control [35]. |
| Back-Pressure Regulator (BPR) | A device that pressurizes the flow system, allowing solvents to be heated above their atmospheric boiling points and ensuring even, gas-bubble-free fluid flow through the reactor [36]. |
| Precision Feeding System | Automated pumps that provide a continuous and precise flow of reagents. Essential for maintaining a constant feed ingredient ratio, which is critical for sustaining steady-state supersaturation [34]. |
| Passive Mixing Structures | Reactor internals (e.g., heart-shaped or coiled designs) that promote thorough mixing of reagents via diffusion and fluid dynamics alone, without moving parts, ensuring uniform concentration profiles [34] [33]. |
Troubleshooting Guide: Common Experimental Challenges
This guide addresses specific issues researchers may encounter when using additives to control crystal habit.
Table 1: Troubleshooting Common Problems in Additive-Mediated Crystallization
| Problem | Possible Causes | Recommended Solutions | Underlying Mechanism |
|---|---|---|---|
| Rapid, uncontrolled crystallization | Supersaturation too high; additive concentration insufficient or ineffective. [3] | • Increase solvent volume to reduce supersaturation. [3] • Use a smaller flask to create a deeper solvent pool for slower cooling. [3] • Improve insulation during cooling (e.g., use a watch glass, insulating pad). [3] | Slower cooling and reduced supersaturation shift the kinetic balance from rapid nucleation to controlled growth, allowing additives more time to interact with crystal faces. |
| No crystal formation | Supersaturation too low; additive overly suppresses nucleation. [38] [3] | • Scratch the inside of the flask with a glass rod. [3] • Add a seed crystal. [3] • Boil off a portion of the solvent to increase concentration and cool again. [3] | Seeding provides a nucleation site, bypassing the high energy barrier of primary nucleation. Increasing supersaturation pushes the system into the metastable zone where growth can occur. |
| Poor crystal yield | Excessive solvent use; compound loss to mother liquor; additive binding too strongly. [3] | • Boil off solvent from mother liquor for a second crop crystallization. [3] • Recover crude solid via rotary evaporation and attempt a new crystallization. [3] | Reducing solvent volume increases concentration in the mother liquor, promoting further yield. A different solvent system may alter additive-solute interactions. |
| Additive appears ineffective | Incorrect additive for the crystal system; process parameters counteracting additive mechanism. [38] | • Systematically investigate additive concentration, temperature, and supersaturation using DoE. [38] • Consider molecular compatibility (e.g., H-bond donors/acceptors) between additive and crystal surface. [38] | Additive effectiveness is highly dependent on specific molecular-scale interactions (H-bonding, steric hindrance) and is modulated by process conditions. [38] |
| Unfavorable crystal morphology (needles, plates) | Anisotropic growth due to uncontrolled nucleation or specific additive action. | • Optimize supersaturation rate to control nucleation density. [2] • Use additives that selectively bind to specific crystal faces to alter the habit. • Employ in-line filtration to reduce scaling and retain crystals in the bulk for more uniform growth. [2] | Modulating supersaturation repositions the system within the metastable zone to favor growth over nucleation. Filtration segregates the crystal phase, allowing independent control over growth. [2] |
Frequently Asked Questions (FAQs)
Q1: At which stage of crystallization do additives typically exert their most significant effect? The impact stage depends on the additive and system. While many are chosen to inhibit nucleation, their primary effect may not always be at this stage. For instance, in famotidine crystallization, Polyvinylpyrrolidone (PVP) directly inhibits nucleation, decreasing the nucleation rate by orders of magnitude. [38] In contrast, for some halide perovskites, evidence shows that common additives do not predominantly impact nucleation but instead facilitate coarsening grain growth by increasing ion mobility across grain boundaries after the initial perovskite phase has formed. [39] The effect mechanism must be understood at a molecular scale for robust process design.
Q2: How do process parameters like temperature and supersaturation interact with additive performance? Process parameters and additives are interdependent. Experimental studies, such as those applying Design of Experiment (DoE) methodology, reveal that the nucleation-inhibiting effect of an additive like PVP is dependent on temperature, while increasing solute concentration (supersaturation) generally counteracts it. [38] Furthermore, controlling the supersaturation rate, for example in membrane distillation crystallisation, can reposition the system within the metastable zone to favor either crystal growth or primary nucleation, thereby working in concert with or against the additive's function. [2]
Q3: What is the molecular-scale mechanism by which polymers like PVP modify crystal habit? Based on combined experimental and theoretical investigations, the effect mechanism of polymers is often manifested through specific interactions at the molecular level. For PVP and famotidine, molecular modeling suggests that the mechanism involves hydrogen bonding and steric hindrance. [38] The polymer molecules adsorb onto specific crystal faces, thereby inhibiting the growth of those faces and leading to a modification of the final crystal habit.
Q4: How can I optimize crystallization conditions when my initial experiments yield poor-quality crystals (e.g., microcrystals, clusters)? Systematic optimization is required. First, identify the chemical (pH, precipitant concentration, ionic strength) and physical (temperature, sample volume) parameters of your initial "hit". Then, compose solutions that incrementally and systematically vary these parameters about the initial values. [32] If you have multiple hits, look for common characteristics (e.g., a specific type of precipitant) to focus your efforts. Prioritize the optimization of conditions that yield three-dimensional, polyhedral crystals over needles, plates, or clusters, which are often more disordered. [32]
Experimental Protocols for Key Methodologies
Protocol 1: Investigating Additive Effects via Design of Experiment (DoE)
This protocol outlines a systematic approach to quantify the influence of additives and process parameters, based on a published investigation of PVP in famotidine crystallization. [38]
1. Research Reagent Solutions
Table 2: Essential Materials for DoE-based Additive Investigation
| Reagent/Material | Function |
|---|---|
| Active Pharmaceutical Ingredient (API) | The target compound to be crystallized (e.g., Famotidine). [38] |
| Polymer Additive | The habit-modifying agent (e.g., Polyvinylpyrrolidone (PVP)). [38] |
| Solvent System | A suitable solvent or solvent mixture for dissolving the API and additive. |
| Camera-Aided Analytical Set-up | To visually monitor and record the crystallization process, enabling the measurement of nucleation induction times. [38] |
2. Methodology
- DoE Setup: Define the critical process parameters (CPPs) to investigate. Typically, these include additive concentration, temperature, and supersaturation. [38] Use a statistical DoE approach (e.g., a full factorial design) to structure the experiments in a way that captures both individual and interactive effects of the CPPs.
- Experimental Execution: For each experimental run in the DoE matrix, prepare solutions with the specified concentrations of API and additive. Dissolve the components completely.
- Nucleation Monitoring: Place the solution in a temperature-controlled environment and use the camera set-up to record the solution. The time from achieving a defined supersaturation until the first detectable crystals appear is recorded as the nucleation induction time. [38]
- Data Analysis: Statistically analyze the induction time data to build a model relating the CPPs to the response. Determine which factors have a significant impact on nucleation inhibition.
- Mechanistic Interpretation: The experimental nucleation rates can be analyzed according to Classical Nucleation Theory (CNT) to understand the thermodynamic and kinetic changes induced by the additive. [38] Finally, conduct molecular dynamics simulations to suggest a molecular-scale effect mechanism (e.g., H-bonding and steric hindrance). [38]
Protocol 2: Supersaturation Control for Crystal Size and Morphology
This protocol describes strategies to regulate nucleation and growth by controlling supersaturation, as applied in membrane distillation crystallisation (MDC). [2]
1. Research Reagent Solutions
Table 3: Essential Materials for Supersaturation Control Studies
| Reagent/Material | Function |
|---|---|
| Solute | The compound to be crystallized (e.g., Sodium Chloride). [2] |
| Solvent | The liquid medium (e.g., water for brine solutions). [2] |
| Membrane Crystallization Set-up | A system to control solvent removal and thereby precisely generate supersaturation. [2] |
| In-line Filter | Placed before the membrane module to retain crystals in the crystallizer and reduce membrane scaling. [2] |
2. Methodology
- Supersaturation Generation: Use the membrane area to adjust the solvent removal (concentration) rate. A larger effective membrane area increases the rate of supersaturation generation. [2]
- Induction and Hold-up: Observe the solution for the first sign of nucleation (induction). Note the supersaturation level at this point. Following induction, the system should be maintained for a controlled hold-up time.
- Scaling Mitigation: Employ an in-line filter to keep formed crystals in the bulk crystallizer and prevent them from depositing on the membrane surface (scaling). This allows for a consistent supersaturation rate to be sustained. [2]
- Outcome Analysis: A longer hold-up time, sustained by reduced scaling, allows the solvent to desaturate as the existing crystals grow. This desaturation reduces the driving force for secondary nucleation, resulting in a lower nucleation rate and ultimately larger final crystal sizes, as confirmed by population balance modeling. [2]
Visualization of Workflows and Relationships
Additive Selection and Problem Diagnosis Workflow
The following diagram outlines a logical workflow for selecting an additive and diagnosing common problems during experimental optimization.
Mechanism of Additive-Mediated Crystallization Control
This diagram illustrates the two primary mechanistic pathways—nucleation inhibition and growth modification—through which additives control crystallization, as revealed by recent studies. [38] [39]
Core Principles: Solvent and Anti-Solvent Roles in Crystallization
Crystallization from solution is a critical process for the separation, purification, and precise control of solid forms of active pharmaceutical ingredients (APIs). The selection of solvent and anti-solvent directly influences the supersaturation level, which is the fundamental driving force for nucleation and crystal growth, thereby dictating the final crystal morphology, size, purity, and polymorphic form [40] [41].
In this context, a solvent is a liquid in which the solute (e.g., an API) is highly soluble. An anti-solvent (or non-solvent) is a liquid in which the solute has very low solubility but which is miscible with the primary solvent. The addition of an anti-solvent to a solution reduces the solute's solubility, creating a supersaturated state that initiates crystallization [42]. The "solvent-engineering" method leverages this principle to achieve fast and uniform nucleation, which is crucial for producing high-quality materials in pharmaceutical and electronics applications [43] [42].
Frequently Asked Questions (FAQs) & Troubleshooting Guides
Troubleshooting Common Crystallization Problems
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| No Crystallization [3] [44] | Excessive solvent, lack of nucleation sites, insufficient supersaturation. | 1. Scratch flask interior with glass rod.2. Add a seed crystal.3. Boil off excess solvent to increase concentration and cool again.4. Use a cooling bath to lower temperature. |
| Rapid Crystallization [3] | Excessively high supersaturation. | 1. Use more hot solvent to decrease saturation level.2. Use a smaller flask to reduce surface area and slow cooling.3. Insulate the flask to slow the cooling rate. |
| Oiling Out [44] | Low melting point of compound or high solvent boiling point; impurities. | 1. Warm to re-dissolve, add more solvent, and cool very slowly.2. Ensure high purity of the starting material.3. Consider an alternative solvent system or purification method. |
| Poor Yield [3] [44] | Too much solvent used, leading to high solute loss in mother liquor. | 1. Concentrate the mother liquor (e.g., by rotary evaporation) and repeat crystallization.2. Perform a "second crop" crystallization from the concentrated mother liquor. |
| Incorporation of Impurities [3] | Crystallization occurred too quickly. | Re-dissolve the solid and follow the recommendations for slowing down crystallization. Ensure slow, gradual crystal growth. |
FAQs on Solvent and Anti-Solvent Selection
Q1: What fundamental properties should I consider when selecting an anti-solvent? The primary prerequisite is that the anti-solvent must be miscible with the process solvent but must significantly reduce the solubility of the target solute [43] [42]. Key properties to consider include:
- Hansen Solubility Parameters (HSP): These parameters (δD: dispersion forces, δP: polar interactions, δH: hydrogen bonding) help predict miscibility and solubility. An effective anti-solvent typically has HSPs significantly different from those of the solute but somewhat similar to the solvent to ensure miscibility [42].
- Boiling Point: Influences the rate of solvent/anti-solvent removal during post-processing.
- Safety and Toxicity: Especially important for pharmaceutical applications.
Q2: How can I control polymorphism through solvent selection? Solvent selection can directly influence the polymorphic outcome by affecting the molecular conformation and interaction kinetics during nucleation [41]. For example, a study on ritonavir showed that the metastable Form I crystallized from acetone, ethyl acetate, acetonitrile, and toluene, while the stable Form II was obtained from ethanol. This was attributed to solvent-dependent formation of intramolecular hydrogen bonding and different conformations of molecular groups, which either inhibited or promoted the formation of the stable form's optimal intermolecular hydrogen-bonding network [41].
Q3: What is the "Supercritical Anti-Solvent (SAS)" process and what are its advantages? The SAS process uses supercritical carbon dioxide (scCO2) as an anti-solvent. When the liquid solution is injected into scCO2, the high diffusivity of scCO2 into the liquid and its low viscosity cause extremely high and rapid supersaturation, leading to the precipitation of fine, uniform particles [45] [43]. Its advantages over conventional techniques include:
- Precise Control: Excellent control over particle size, morphology, and crystal form (e.g., nanoparticles, microparticles, amorphous solid dispersions) [45].
- Product Quality: Avoids thermal degradation as the process can be run at near-ambient temperatures.
- Reduced Solvent Residues: scCO2 is easily removed, leaving a dry product with minimal organic solvent residues [43].
- Tunability: The solvent power of scCO2 is tunable with pressure and temperature, allowing for precise process control [45].
Quantitative Data for Solvent and Anti-Solvent Selection
Hansen Solubility Parameters of Common Solvents and Anti-Solvents
Hansen Solubility Parameters (HSPs) are a key tool for predicting miscibility and solubility. Solvents with similar HSPs are typically miscible. A large difference in HSPs between the solute and the anti-solvent is desired to induce precipitation [42].
Table 1: Hansen Solubility Parameters for Common Solvents and Anti-Solvents [42]
| Solvent Name | Type | δD (MPa¹/²) | δP (MPa¹/²) | δH (MPa¹/²) |
|---|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Solvent | 18.4 | 16.4 | 10.2 |
| Dimethyl Formamide (DMF) | Solvent | 17.4 | 13.7 | 11.3 |
| N-Methyl-2-Pyrrolidone (NMP) | Solvent | 18.0 | 12.3 | 7.2 |
| Acetonitrile (ACN) | Solvent/Anti-solvent | 15.3 | 18.0 | 6.1 |
| Tetrahydrofuran (THF) | Solvent/Anti-solvent | 16.8 | 5.7 | 8.0 |
| Toluene | Anti-solvent | 18.0 | 1.4 | 2.0 |
| Chloroform | Anti-solvent | 17.8 | 3.1 | 5.7 |
| Diethyl Ether | Anti-solvent | 14.5 | 2.9 | 4.6 |
Key Properties of Common Anti-Solvents for Perovskite Processing (as a Model System)
This table illustrates how a variety of anti-solvents are used in a advanced research context, highlighting the diversity of applicable materials.
Table 2: Properties of Common Anti-Solvents in Solvent-Engineering [42]
| Anti-Solvent | Boiling Point (°C) | Dipole Moment (D) | Common Applications |
|---|---|---|---|
| Chlorobenzene | 132 | 1.6 | Widely used for MAPbI₃ perovskite films. |
| Toluene | 111 | 0.4 | Common for various perovskite compositions. |
| Diethyl Ether | 35 | 1.2 | Fast evaporation, can lead to rapid nucleation. |
| Ethyl Acetate | 77 | 1.8 | "Green" alternative with good performance. |
Experimental Protocols for Advanced Crystallization
Protocol: Continuous Supercritical Anti-Solvent (SAS) Process for Micronization
The following methodology describes the semi-continuous SAS process, a bottom-up approach for producing micro- and nanoparticles with controlled solid-state properties [45].
1. Principle: A solution of the API (and often a polymer excipient) is continuously injected into a stream of supercritical CO2. The scCO2 acts as an anti-solvent, rapidly extracting the organic solvent and causing extreme supersaturation, which leads to the precipitation of fine, dry particles [45] [43].
2. Materials & Equipment:
- High-Pressure Pump: For delivering scCO2.
- Solution Pump: For delivering the liquid solution.
- Precipitation Vessel: A high-pressure cell with temperature control.
- Nozzle: For atomizing the solution into the scCO2 (e.g., coaxial nozzle in SEDS process).
- CO2 Cylinder and Organic Solvent (e.g., DMSO, DMF, acetone).
- API and Polymer Excipient (if making composite particles).
3. Step-by-Step Procedure: 1. System Pressurization and Heating: Pump liquefied CO2 into the precipitation vessel until the desired pressure (typically above 73 bar) and temperature (above 31°C) are reached and stabilized to ensure supercritical conditions [45] [43]. 2. Solvent Equilibration: Inject pure solvent through the nozzle for a few minutes to establish steady-state composition conditions inside the vessel. 3. Solution Injection and Precipitation: Switch the flow from pure solvent to the drug (or drug/polymer) solution. The solution is atomized into the scCO2 chamber. The rapid diffusion of scCO2 into the droplets and solvent into scCO2 causes instantaneous supersaturation and particle precipitation. 4. Washing: Continue the flow of pure scCO2 for a set time to wash the precipitated particles and remove any residual solvent trapped within them. 5. Depressurization and Collection: Slowly depressurize the vessel and collect the final, dry powder from the filter located at the bottom of the vessel [45].
4. Key Parameters to Control:
- Temperature and Pressure: Directly affect the density and solvation power of scCO2.
- Solution Flow Rate and Concentration: Higher rates/concentrations can increase supersaturation, leading to smaller particles.
- Nozzle Geometry: Affects the atomization and mixing efficiency.
- Solvent Selection: Must be miscible with scCO2 [43].
Workflow Diagram: Supercritical Anti-Solvent (SAS) Process
The following diagram illustrates the semi-continuous SAS process workflow.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagents in Solvent Engineering
| Reagent | Function & Explanation |
|---|---|
| Supercritical CO₂ | Acts as a green, tunable anti-solvent. Its liquid-like density provides good solvation power, while its gas-like diffusivity and low viscosity enable highly efficient mass transfer, leading to rapid supersaturation [45] [43]. |
| Polymeric Carriers (e.g., PVP, PLGA) | Used in composite particle production via co-precipitation. They can modify drug release kinetics (immediate or prolonged), protect the API, mask taste, and improve stability [43]. |
| Dimethyl Sulfoxide (DMSO) | A common high-boiling-point, polar aprotic solvent. Often used to dissolve perovskite precursors or poorly soluble APIs, particularly when processed with supercritical CO₂ [45] [42]. |
| Hansen Solubility Parameters (HSP) | A three-dimensional parameter set (δD, δP, δH) used to predict polymer solubility, solvent miscibility, and polymer resistance. It is a crucial quantitative tool for rational solvent/anti-solvent selection instead of relying on trial and error [42]. |
| Seed Crystals | Small crystals of the pure desired polymorph. Added to a supersaturated solution to provide a nucleation site, promoting the growth of that specific polymorph and helping to control the crystallization process and avoid oiling out [3] [41]. |
Comparative Analysis of Crystallization Techniques
Conventional vs. Supercritical Anti-Solvent Crystallization
Understanding the landscape of available techniques is vital for selecting the optimal process for a given API.
Table 4: Comparison of Crystallization Techniques
| Feature | Conventional Anti-Solvent | Supercritical Anti-Solvent (SAS) | Evaporative Crystallization |
|---|---|---|---|
| Principle | Addition of liquid anti-solvent to reduce solubility [42]. | Use of scCO₂ as anti-solvent to induce rapid supersaturation [45]. | Heating to evaporate solvent, increasing concentration to saturation [40]. |
| Particle Size Control | Moderate, can be challenging for nano-range. | Excellent, capable of producing nano- to micro-particles with narrow distribution [45] [43]. | Limited, typically produces larger crystals. |
| Thermal Stress | Can be low (cooling crystallization). | Low (near-ambient temperatures). | High (requires heating). |
| Solvent Removal | Requires separate filtration and drying steps. | Integrated; product is dry after precipitation and scCO₂ washing [45]. | Integrated into the process. |
| Polymorph Control | Possible with careful control of addition rate. | High, due to rapid and uniform nucleation [45] [41]. | Dependent on cooling and evaporation rates. |
| Scalability | Well-established for large scale. | Developed from batch to continuous mode; scalable but requires high-pressure equipment [45]. | Highly scalable and common in industry [40]. |
Decision Workflow: Selecting a Crystallization Method
This diagram provides a logical pathway for selecting an appropriate crystallization method based on research goals.
Leveraging Process Analytical Technology (PAT) for Real-Time Monitoring and Control
Technical Support Center
Frequently Asked Questions (FAQs)
Q1: What is the fundamental principle of PAT in pharmaceutical development? Process Analytical Technology (PAT) is a system for designing, analyzing, and controlling manufacturing through timely measurements of critical quality attributes (CQAs) and critical process parameters (CPPs) of raw, in-process materials, and processes to ensure final product quality [46]. It enables real-time quality control based on the actual product CQAs, moving away from traditional end-product testing to building quality directly into the product [46] [47].
Q2: How does PAT support a Quality by Design (QbD) approach in crystal morphology control? PAT is the practical pathway to implementing QbD. It provides the real-time data on CPPs and CQAs necessary to establish the design space for optimal crystal morphology. By executing experiments where real-time quality predictions are made, the relationships between CPPs (e.g., supersaturation rate, temperature) and CQAs (e.g., crystal size distribution, polymorphic form) can be established, leading to true process understanding [46] [47].
Q3: What are common analytical tools used as PAT for monitoring crystallization processes? Various in-line and on-line instrumentation can be employed. Common tools include:
- Spectroscopic probes: ATR-FTIR and Raman spectroscopy for real-time concentration and polymorph monitoring.
- Focused Beam Reflectance Measurement (FBRM): For tracking particle count and size distribution in real-time.
- Process Vision Systems: For in-situ imaging of crystal morphology.
Q4: My PAT sensor readings are erratic. What are the first steps in troubleshooting?
- Perform a visual inspection of the probe window or interface for fouling, coating, or physical damage.
- Verify calibration standards and ensure they have not degraded.
- Check process conditions: Ensure the probe is properly positioned and that process conditions (e.g., temperature, stir speed) are within the sensor's operating range.
- Confirm data preprocessing steps in your multivariate model are correctly applied.
Q5: How do I establish a control strategy for a crystallization process using PAT? A control strategy is derived from product and process understanding developed via risk assessment and experimental data [47]. It involves:
- Identifying the CQAs of the final crystal form.
- Determining the CPPs and material attributes that impact these CQAs.
- Defining the functional relationship between CPPs and CQAs.
- Implementing real-time controls using PAT to adjust CPPs and keep the process within the defined design space, ultimately enabling real-time release [47].
Troubleshooting Guides
Issue 1: Inconsistent Supersaturation Control Leading to Variable Crystal Morphology
| Probable Cause | Diagnostic Steps | Corrective Action |
|---|---|---|
| Inaccurate concentration measurement | - Verify calibration of in-situ spectrometer.- Check for probe fouling.- Compare with off-line sample. | - Clean or recalibrate probe.- Implement automated drift correction algorithms. |
| Uncontrolled cooling/antisolvent addition rate | - Review controller setpoints and logs.- Check for equipment lag or overshoot. | - Implement a feedback control loop using the real-time concentration data from the PAT tool to dynamically adjust the addition rate. |
| Insufficient mixing | - Use CFD modeling or tracer studies.- Inspect impeller. | - Optimize agitator speed or design.- Consider baffle installation. |
Issue 2: PAT Model (e.g., Multivariate Calibration) Performance Drift Over Time
| Probable Cause | Diagnostic Steps | Corrective Action |
|---|---|---|
| Changes in raw material properties | - Track raw material vendor and lot changes.- Perform Principal Component Analysis (PCA) on new spectral data. | - Update model to include new material variability.- Establish stricter raw material specifications. |
| Sensor degradation or fouling | - Monitor model residuals and diagnostic statistics (e.g., Hotelling's T², Q-residual).- Perform routine sensor checks. | - Clean or replace sensor.- Implement model robustness algorithms to compensate for minor signal changes. |
| Unaccounted for process change | - Audit process parameters and equipment. | - Revisit design space and update model accordingly.- Document all process changes. |
Issue 3: Failure to Achieve Target Crystal Size Distribution (CSD)
| Probable Cause | Diagnostic Steps | Corrective Action |
|---|---|---|
| Incorrect seed loading or quality | - Use in-situ imaging (e.g., PVM) to assess seed morphology and count.- Review seed preparation protocol. | - Optimize seed recipe (size, amount, addition point).- Ensure consistent seed generation. |
| Ostwald ripening or agglomeration | - Analyze FBRM chord length distribution trends for agglomeration signatures.- Review solvent composition and impurities. | - Adjust process trajectory to minimize time in meta-stable zones where agglomeration occurs.- Modify solvent system or additive. |
| Nucleation event not properly controlled | - Monitor FBRM counts for secondary nucleation. | - Fine-tune supersaturation profile to stay within the meta-stable zone. |
Experimental Protocols for Key PAT Applications
Protocol 1: Establishing the Supersaturation Profile for a New API using ATR-FTIR
Objective: To develop a calibration model for real-time concentration monitoring and determine the meta-stable zone width (MSZW).
Materials:
- Reactors: 500 mL jacketed glass reactor with temperature control.
- PAT Tool: In-situ ATR-FTIR probe with compatible software.
- Chemicals: API, solvent(s), antisolvent (if applicable).
Methodology:
- Calibration Model Development:
- Prepare standard solutions of known API concentration across the expected process range.
- Collect spectra at multiple temperatures relevant to the process.
- Use multivariate analysis (e.g., Partial Least Squares, PLS) to build a model correlating spectral features to concentration.
MSZW Determination:
- Charge the reactor with solvent and bring to the starting temperature.
- Add API incrementally until dissolved, monitoring concentration via the PAT model.
- Cool the solution at a controlled rate while continuously monitoring concentration and temperature.
- The point where a rapid change in the spectral baseline or FBRM count is observed indicates nucleation and defines the MSZW boundary.
Data Analysis:
- Plot supersaturation (concentration relative to equilibrium) against temperature to visualize the MSZW and design an optimal cooling curve that maintains supersaturation within safe limits.
Protocol 2: Real-Time Feedback Control of Antisolvent Addition for Crystal Size Distribution (CSD) Management
Objective: To maintain a constant supersaturation level by controlling the antisolvent addition rate based on real-time concentration feedback, thereby achieving a consistent CSD.
Materials:
- Reactors: 1 L jacketed glass reactor with overhead stirring and temperature control.
- Dosing System: Programmable pump for antisolvent addition.
- PAT Tools: In-situ ATR-FTIR probe and FBRM probe.
Methodology:
- System Setup:
- Install PAT probes in the reactor, ensuring proper positioning to avoid dead zones and representative sampling.
- Connect the PAT software and dosing pump to a control platform (e.g., Process Historian, SCADA).
Controller Configuration:
- Define the target supersaturation setpoint (e.g., 1.15) based on prior MSZW studies.
- Configure a PID (Proportional-Integral-Derivative) feedback loop that adjusts the antisolvent pump rate based on the difference between the real-time supersaturation (from ATR-FTIR) and the setpoint.
Experimental Execution:
- Dissolve the API in the solvent at a defined temperature.
- Initiate the feedback controller to start the antisolvent addition.
- The system will automatically adjust the pump rate to maintain the target supersaturation as detected by the PAT tool.
- Continuously monitor the evolving CSD using the FBRM probe.
Data Collection:
- Record the real-time concentration, antisolvent addition profile, and FBRM chord length distribution.
- Compare the final CSD (via off-line analysis) with batches run without feedback control.
PAT-Enabled Workflow for Crystal Morphology Control
The Scientist's Toolkit: Essential PAT Research Reagents & Materials
The following table details key solutions and materials crucial for implementing PAT in crystallization research [46] [47].
| Item | Function in PAT Experiment |
|---|---|
| In-situ Spectroscopy Probes (ATR-FTIR, Raman) | Provides real-time, molecular-level data on solution concentration, supersaturation, and polymorphic form without the need for sampling. |
| Particle System Analyzers (FBRM, PVM) | Tracks changes in particle count, chord length distribution (FBRM), and provides direct visual images (PVM) of crystals as they form and grow. |
| Multivariate Data Analysis (MVA) Software | Interprets complex spectral and particle data from PAT tools using chemometrics to build predictive models for CQAs and enable real-time control. |
| Calibration Standards | A series of solutions with precisely known concentrations of the API, used to build the initial quantitative model for the spectroscopic probes. |
| Design of Experiments (DoE) Software | A systematic approach to planning experiments to efficiently understand the relationship and interaction between multiple CPPs and CQAs. |
| Jacketed Laboratory Reactors | Provides a controlled environment (temperature, stirring) for crystallization, often with multiple ports for PAT probe insertion. |
| Programmable Dosing Pumps | Allows for precise and automated addition of antisolvent or reagents, which can be controlled by feedback signals from the PAT system. |
Troubleshooting Guide: NiCo LDH Synthesis & Pharmaceutical Application
Low Adsorption Capacity for Pharmaceutical Compounds
Problem: Synthesized NiCo LDH shows significantly lower adsorption capacity for pharmaceutical contaminants like carprofen than literature values.
Solutions:
- Surface Functionalization: Functionalize NiCo LDH with AEAMP-TMS, a silane coupling agent containing amine and phenyl groups, to create additional binding sites. This enables dual-function adsorption through surface and interlayer interactions [48].
- pH Optimization: Conduct adsorption experiments at pH 5-8 where carprofen exists in anionic form, maximizing electrostatic attraction with the positively charged LDH layers [48].
- Structural Confirmation: Verify successful functionalization using XRD and FTIR characterization, and examine morphology and porosity through FESEM and N₂ adsorption-desorption analysis [48].
Poor Crystallinity and Uncontrolled Morphology
Problem: NiCo LDH products exhibit amorphous structure or inconsistent morphology, leading to unreliable performance.
Solutions:
- Supersaturation Control: Implement a Continuous Flow Reactor (CFR) system to maintain constant supersaturation under pseudo-steady-state conditions for uniform nucleation and growth [26] [49].
- Nucleation Management: Identify and utilize distinct thresholds for homogeneous and heterogeneous nucleation. Use seeded substrates when working below heterogeneous nucleation thresholds [26] [49].
- Parameter Optimization: Systematically adjust total concentration, metal/alkaline ratio, and control dissolved oxygen concentration to influence phase transformation kinetics from brucite-like intermediates to well-defined NiCo LDHs [26] [49].
Material Agglomeration and Low Conductivity
Problem: NiCo LDH nanosheets aggregate, reducing active surface area and compromising electrical conductivity.
Solutions:
- Composite Formation: Create heterostructures by anchoring NiCo LDH on conductive substrates like MXene through solvent-induced interfacial-confined synthesis. This improves charge transport while preventing aggregation [50].
- Morphological Engineering: Utilize structure-directing agents like potassium fluoroborate to design specific morphologies such as hollow sheets that enhance surface area and accessibility [51].
- Microwave Synthesis: Employ ultrafast microwave-assisted synthesis (210 seconds) to produce porous NiCo LDH nanospheres with intercalated ethylene glycol, creating larger interlayer spacing (approximately 8.6 Å) for improved ion transfer [52].
Inconsistent Performance Across Synthesis Batches
Problem: Significant variation in electrochemical or adsorption performance between different batches of synthesized NiCo LDH.
Solutions:
- Anion Control: Standardize anion types in nickel and cobalt salts, as anions significantly affect morphology and grain size due to different capping effects and steric hindrance during crystal growth [53].
- Precise Reactor Control: Utilize CFR systems that enable precise control over LDH morphology and size by maintaining constant supersaturation, eliminating conventional batch-to-batch variations [26] [49].
- Substrate Optimization: For electrochemical applications, deposit NiCo LDH on specially prepared substrates like double-laser-scribed Fe-LIG, which provides a highly ordered and dense framework for more consistent deposition [54].
Experimental Performance Data
Table 1: Performance Comparison of Different NiCo LDH Synthesis Methods
| Synthesis Method | Key Features | Morphology | Performance Metrics | Reference |
|---|---|---|---|---|
| AEAMPS Functionalization | Silane coupling with amine/phenyl groups | Macroporous-mesoporous structure | 742 mg/g carprofen adsorption; >92% efficiency after 8 cycles | [48] |
| Continuous Flow Reactor | Precise supersaturation control | Tunable from nanoplates to nanoflowers | Controlled phase evolution; defined nucleation thresholds | [26] [49] |
| MXene Composite | Solvent-induced interfacial confinement | 3D brush-like heterostructure | 1310 F/g specific capacitance; 92.5% retention after 10,000 cycles | [50] |
| ZIF-67 Derived LDH | Potassium fluoroborate as SDA | Hollow sheet morphology | 1171 F/g specific capacitance; 84% stability after 10,000 cycles | [51] |
| Microwave Synthesis | Ultrafast 210-second synthesis | Porous nanospheres | 2156 F/g at 1 A/g; 86.8% capacity retention at 10 A/g | [52] |
Table 2: Adsorption Mechanisms for Pharmaceutical Compounds
| Mechanism | Functional Groups Required | Optimal Conditions | Effectiveness |
|---|---|---|---|
| Electrostatic Attraction | Positively charged LDH layers | pH 5-8 for anionic pharmaceuticals | High for charged molecules |
| π-π Stacking | Aromatic rings in functional groups | Neutral pH | Enhanced with phenyl-rich functionalization |
| Hydrogen Bonding | Hydroxyl groups, amine groups | Varied depending on pharmaceutical | Moderate to high |
| Anion Exchange | Exchangeable interlayer anions | Depends on anion affinity | High for anionic contaminants |
Detailed Experimental Protocols
Protocol 1: Ultrasonic-Assisted Functionalization for Pharmaceutical Adsorption
Application: Enhanced removal of pharmaceutical contaminants like carprofen [48]
Materials:
- Ni₅₀Co₅₀-LDH precursor
- AEAMP-TMS silane coupling agent
- Carprofen model drug
- Ultrasonic reactor system
Procedure:
- Synthesize Ni₅₀Co₅₀-LDH through standard co-precipitation method
- Functionalize with AEAMP-TMS using ultrasonic-assisted reflux method
- Characterize using XRD, FTIR, and FESEM to confirm functionalization
- Perform adsorption experiments at varying pH levels (3-10)
- Determine adsorption capacity using Langmuir isotherm models
- Conduct regeneration studies with multiple cycles
Characterization:
- Confirm successful functionalization through FTIR peaks at characteristic wavelengths
- Verify maintenance of LDH structure via XRD patterns
- Measure surface area and porosity through N₂ adsorption-desorption (expected surface area: ~65.2 m²/g)
Protocol 2: Continuous Flow Synthesis for Morphology Control
Application: Precise control over NiCo LDH morphology for consistent performance [26] [49]
Materials:
- Nickel nitrate hexahydrate (Ni(NO₃)₂·6H₂O)
- Cobalt nitrate hexahydrate (Co(NO₃)₂·6H₂O)
- Hexamethylenetetramine (HMTA)
- Continuous Flow Reactor (CFR) system
- FTO glass substrates for seeding
Procedure:
- Prepare separate precursor solutions of Ni/Co nitrates and HMTA
- For low supersaturation conditions: pre-treat substrates with seed solution (100 mM Ni(NO₃)₂ and 100 mM HMTA at 90°C for 1 minute)
- Set CFR to hydrolysis temperature of 95°C
- Position substrates in CFR and pump precursor solutions through separate lines
- Mix solutions through T-junction near column entrance
- Systematically vary supersaturation levels by adjusting concentration and metal/alkaline ratio
- Collect products and characterize morphology evolution
Key Control Parameters:
- Supersaturation level (governs nucleation type)
- Metal/alkaline ratio
- Heterogeneous nucleation conditions
- Dissolved oxygen concentration
FAQs
Q1: What are the four key factors governing the transformation of NiCo mixed hydroxides to well-defined NiCo LDH? A1: The four key factors are: (1) supersaturation level, (2) metal/alkaline ratio, (3) heterogeneous nucleation conditions, and (4) dissolved oxygen concentration. These factors collectively control the phase transformation kinetics from brucite-like intermediates to defined NiCo LDHs [26] [49].
Q2: How can I quickly synthesize NiCo LDH with good electrochemical performance? A2: Use microwave-assisted synthesis, which can produce NiCo LDH with intercalated ethylene glycol in just 210 seconds. This method creates materials with large interlayer spacing (approximately 8.6 Å) and specific capacitance up to 2156 F/g at 1 A/g with excellent rate performance (86.8% capacity retention at 10 A/g) [52].
Q3: Why does my NiCo LDH aggregate, and how can I prevent it? A3: NiCo LDH naturally tends toward layer-by-layer accumulation. Prevent aggregation by: (1) creating composite structures with conductive materials like MXene, (2) using structure-directing agents like potassium fluoroborate to design hollow morphologies, or (3) employing solvent-induced interfacial confinement strategies to achieve vertical anchoring on substrates [50] [51].
Q4: What adsorption mechanisms are responsible for pharmaceutical removal by functionalized NiCo LDH? A4: Functionalized AEAMPS-LDH captures pharmaceutical compounds like carprofen through multiple mechanisms: electrostatic attraction, hydrogen bonding, π-π stacking, and interlayer anion exchange. The combination of these mechanisms enables high adsorption capacity (742 mg/g for carprofen) [48].
Research Reagent Solutions
Table 3: Essential Research Reagents for NiCo LDH Synthesis
| Reagent | Function | Application Examples | Key Considerations |
|---|---|---|---|
| AEAMP-TMS Silane | Surface functionalization with amine/phenyl groups | Pharmaceutical adsorption | Enables dual-function adsorption; enhances π-π interactions |
| Potassium Fluoroborate | Structure-directing agent | Hollow morphology creation | Concentration-dependent effects (0.015-0.035 g optimal) |
| MXene (Ti₃C₂Tₓ) | Conductive substrate | Composite electrodes | Enables 3D brush-like heterostructures; improves conductivity |
| Hexamethylenetetramine (HMTA) | Alkaline agent for hydrolysis | Controlled precipitation | Provides uniform hydroxyl release; temperature-dependent |
| Ethylene Glycol | Solvent and intercalation agent | Microwave synthesis; layer expansion | Induces highly ordered growth; expands interlayer spacing |
Experimental Workflow Visualization
Solving Practical Challenges in Crystal Size Distribution (CSD) and Purity
Addressing Growth Rate Dispersion (GRD) and Size-Dependent Growth (SDG)
Frequently Asked Questions (FAQs)
1. What is the fundamental difference between Growth Rate Dispersion (GRD) and Size-Dependent Growth (SDG)?
Growth Rate Dispersion (GRD) refers to the variation in growth rates observed among individual crystals under identical thermodynamic and hydrodynamic conditions, including supersaturation [55]. It is a stochastic phenomenon where crystals of the same size grow at different rates. In contrast, Size-Dependent Growth (SDG) describes a deterministic relationship where a crystal's growth rate is a direct function of its size; larger crystals may grow faster or slower than smaller ones based on the specific system [56]. GRD is often linked to intrinsic factors or local fluctuations, while SDG is related to the scaling of physiological or physical processes with size.
2. How do local fluctuations in supersaturation cause Growth Rate Dispersion?
Supersaturation, the primary driving force for crystallization, is subject to local fluctuations due to the Brownian motion of solute molecules. This motion affects the local number density and temperature, creating a stochastic environment [55]. Even in a well-mixed system with fixed bulk supersaturation, these microscopic fluctuations mean that individual crystals experience slightly different local supersaturation levels. This variation leads to dispersions in both nucleation and growth rates, as these processes are highly sensitive to supersaturation. A stochastic model derived from this principle can predict the resulting distributions in crystal size and shape [55].
3. Why is controlling the supersaturation profile critical for managing crystal size distribution?
Supersaturation controls the balance between nucleation and crystal growth. A high supersaturation level favors primary nucleation, which can lead to a large number of small crystals and potentially broader size distribution [2]. Conversely, maintaining a lower, controlled supersaturation level after initial nucleation favors crystal growth over the formation of new nuclei, allowing existing crystals to grow larger and more uniformly [2]. Furthermore, the rate at which supersaturation is generated (e.g., via concentration rate in membrane distillation) can affect the metastable zone width and the dominant nucleation pathway, ultimately influencing the final crystal population [2].
4. What is the relationship between size-dependent growth and the development of size variation (inequality) in a population?
The change in relative size variation (e.g., Coefficient of Variation, CV) within a growing cohort is approximately equal to the relative change in the mean per-unit-size growth rate [56]. This means that size variation will decrease, remain unchanged, or increase depending on how the growth rate scales with size. For example, if growth rate increases with size (positive size-dependence), larger individuals will grow progressively faster than smaller ones, leading to an increase in size variation over time. Conversely, if growth rate decreases with size, size variation will tend to decrease [56].
Troubleshooting Guides
Problem 1: Excessive Polydispersity in Final Crystal Product
Potential Causes and Solutions:
- Cause: Uncontrolled Supersaturation. Rapid, uncontrolled generation of supersaturation can lead to high nucleation rates and continuous secondary nucleation, resulting in crystals of various ages and sizes.
- Solution: Implement precise supersaturation control strategies. In membrane crystallization, this can be achieved by modulating the membrane area to adjust the concentration rate, thereby controlling the supersaturation profile [2]. Using in-line filtration can retain crystals in the crystallizer and reduce scaling on equipment surfaces, helping to maintain a more consistent supersaturation level and reduce secondary nucleation [2].
- Cause: Significant Growth Rate Dispersion (GRD). Intrinsic variations in crystal growth rates under identical conditions contribute to polydispersity [55].
- Solution: While difficult to eliminate entirely, the effects of GRD can be managed by extending the "hold-up time" after initial nucleation. This allows crystal growth to desaturate the solvent, which can reduce the nucleation rate and lead to larger, more uniform crystals over time [2]. Modeling the process with population balance equations that account for growth rate dispersion can also help in optimizing process parameters [55].
Problem 2: Uncontrolled Polymorphic Form or Crystal Habit
Potential Causes and Solutions:
- Cause: Non-Optimized Crystallization Conditions. Parameters such as pH, ionic strength, precipitant concentration, and temperature can strongly influence the polymorphic outcome and crystal morphology [32].
- Solution: Employ systematic optimization. After an initial "hit" is identified, compose solutions that incrementally and systematically vary parameters (e.g., pH, temperature, precipitant concentration) about the initial values [32]. The use of additives or co-formers can also strategically promote the desired polymorph or crystal habit [32] [57].
- Cause: Fluctuations in Process Parameters. Variations in temperature, concentration, or mixing can shift the system between regions of the metastable zone that favor different nucleation pathways or growth mechanisms.
- Solution: Ensure tight environmental control throughout the crystallization process. For screening and optimization, use small-volume trials to efficiently explore a wide parameter space, but be aware that scaling up to larger volumes may be necessary to obtain crystals of sufficient size and quality [32].
Problem 3: Inconsistent Results Between Batch Experiments
Potential Causes and Solutions:
- Cause: Stochastic Nature of Nucleation. The primary nucleation event is inherently stochastic, leading to variations in the initial particle population between batches.
- Solution: Move from primary to secondary nucleation pathways. Using seed crystals can ensure a more consistent starting point for crystal growth, improving batch-to-batch reproducibility. Seeding helps control the number of growing crystals and operates at lower supersaturation, favoring growth over nucleation [57].
- Cause: Inadequate Mixing or Heat Transfer. Especially during scale-up, inhomogeneities in the solution can create local "hot spots" of supersaturation.
- Solution: Improve reactor and impeller design to ensure uniform mixing, temperature, and concentration throughout the crystallizer. Consider continuous crystallization platforms, which can provide more consistent control over process parameters compared to batch systems [57].
The table below summarizes key quantitative relationships and experimental data relevant to GRD and SDG.
Table 1: Experimental Growth Rate Data and Key Parameters
| Subject / System | Observed Phenomenon | Quantitative Relationship / Value | Source / Context |
|---|---|---|---|
| KAP Crystals | Face-specific growth rates (with 0.03 mol% ethylene glycol) | ( \dot{H}1 = 0.9078S - 0.9136 ) (for face {010})( \dot{H}2 = 1.1920S - 1.1850 ) (for face {110})( \dot{H}_3 = 2.0620S - 2.0400 ) (for face {111})(Growth rates in µm/s) | [55] |
| General Cohort Growth | Change in size variation | ( \frac{dCV}{CV} \approx -\frac{d[g(w)/w]}{[g(w)/w]} )(Relative change in CV ≈ Relative change in mean per-unit-size growth rate) | [56] |
| General Growth Model | Size-dependent growth function | ( g(w) = \frac{dw}{dt} = a1w^{b1} - a2w^{b2} )(Where ( a1, a2 ) are coefficients and ( b1, b2 ) are scaling exponents for anabolism and catabolism) | [56] |
| Soybean, Sunflower, Maize | Size inequality in plant populations | Strong hierarchies in soybean & sunflower indicated by CV and skewness in shoot biomass. Weak development in maize shoot biomass, but strong inequality in reproductive output. | [58] |
Experimental Protocols
Protocol 1: Systematic Optimization of Crystallization Conditions
Objective: To refine initial crystallization "hits" to obtain crystals with improved size, morphology, and diffraction quality. This is crucial for controlling SDG and minimizing GRD in the final product [32].
Methodology:
- Parameter Identification: From the initial screening hit, identify the key parameters defining the condition (e.g., pH, precipitant type and concentration, temperature, ion concentration, additives).
- Solution Formulation: Prepare a matrix of crystallization solutions where each parameter is varied incrementally around the initial value while keeping others constant.
- Example: If the initial hit was at pH 7.0, prepare solutions at pH 6.0, 6.2, 6.4, ..., 7.6, 7.8, 8.0.
- Similarly, vary the precipitant concentration in small, systematic steps (e.g., ±5% increments).
- Crystallization Trials: Set up trials (e.g., vapor diffusion, batch) using these optimized solutions. The use of very-small-volume trials (nanoliter scale) is efficient for broad screening, but consider larger volumes (microliter to milliliter) to improve crystal size.
- Evaluation: Monitor the trials for crystal formation. Evaluate outcomes based on crystal size, morphological perfection, and when possible, analysis by X-ray diffraction to assess internal order.
Protocol 2: Investigating Growth Rate Dispersion using a Model Compound
Objective: To quantitatively measure and analyze the dispersion in growth rates of individual crystals under constant bulk conditions [55].
Methodology:
- Saturation: Prepare a saturated solution of a model compound (e.g., Potassium Acid Phthalate - KAP) in a temperature-controlled crystallizer.
- Nucleation: Induce nucleation by creating a slight supersaturation, for example, by cooling the solution by 0.5–1.0 °C.
- Seed Selection: Once a small population of crystals is obtained, isolate several well-formed, isolated seed crystals of similar initial size.
- Growth Experiment: Re-dissolve all other crystals by slightly increasing the temperature, then bring the system back to a precise, constant supersaturation level. Monitor the growth of the individual seed crystals over time using in-situ imaging tools (e.g., video microscope).
- Data Analysis: For each crystal, track its linear dimensions over time. Calculate the growth rate for specific faces (e.g., ( \dot{H}1, \dot{H}2, etc. )) for each crystal. Analyze the distribution of growth rates across the population of crystals that started at a similar size to quantify the GRD.
Experimental Workflow and Logical Diagrams
Supersaturation Control for Morphology Workflow
GRD from Supersaturation Fluctuations
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions and Materials
| Item | Function / Explanation |
|---|---|
| Precipitants (e.g., PEG, Salts) | Agents that reduce the solubility of the target molecule in solution, driving the system towards supersaturation and phase separation [32]. |
| Buffers | Solutions used to maintain the pH of the crystallization medium within a narrow, predefined range, a critical parameter for stability and polymorph control [32]. |
| Additives / Co-formers | Small molecules or ions that specifically interact with certain crystal faces to modify growth rates (habit modifiers) or form new crystal structures (co-crystals) to improve properties like solubility and stability [32] [57]. |
| Seeds | Pre-formed, small crystals of the desired polymorph used to induce secondary nucleation and control the crystal growth phase at lower, more stable supersaturation levels, improving reproducibility [57]. |
| Anti-Solvents | A solvent, miscible with the primary solvent, in which the target compound has low solubility. Its controlled addition is a method to generate supersaturation [57]. |
Managing Crystal Clustering and 'Nests' to Prevent Local Depletion and Polydispersity
Frequently Asked Questions (FAQs)
What are crystal "nests" and why are they problematic in crystallization? Crystal "nests" are clusters of several or many crystals that grow closely spaced together due to the random and uneven spatial distribution of nuclei [59]. They are problematic because crystals within a nest compete for the available solute from a shared solution volume. This leads to local depletion of the solute concentration around the nest, causing the enclosed crystals to grow more slowly and remain smaller than isolated crystals with free access to the bulk solution [59].
How does local depletion within nests affect the final Crystal Size Distribution (CSD)? Local depletion directly increases crystal polydispersity (a wide range of sizes). The separately growing crystals, which have access to a higher solute concentration, grow faster and become larger. In contrast, crystals growing within a nest, starved of solute, end up smaller [59]. This results in a broad and often unpredictable CSD, which is undesirable for most applications, particularly in pharmaceuticals where bioavailability and processability depend on a uniform crystal size [59].
Can controlling supersaturation prevent the formation of crystal nests? Yes, precise control of supersaturation is a primary strategy. By maintaining supersaturation within a specific range, you can promote growth on existing crystals while minimizing secondary nucleation, which is a common source of new nests [2]. Furthermore, advanced reactor setups like the Continuous Flow Reactor (CFR) maintain constant, pseudo-steady-state supersaturation, allowing for superior control over nucleation and growth, thereby reducing random clustering [26] [60].
What experimental strategies can segregate crystals to prevent nesting? Implementing in-line filtration is an effective strategy. This technique retains the main crystal population within the crystallizer bulk while removing the solution, preventing crystals from depositing on reactor surfaces and forming fixed nests. Segregating the crystal phase into the bulk solution allows for better control over growth conditions for all crystals uniformly, improving habit, shape, and purity [2].
Troubleshooting Guide: Crystal Nests and Local Depletion
Problem: High Polydispersity in Final Crystal Product
| Probable Cause | Diagnostic Checks | Recommended Solutions |
|---|---|---|
| Prolonged Nucleation Period | Analyze nucleation kinetics; a longer nucleation period leads to a greater initial size disparity as crystals nucleate at different times [59]. | Shorten the nucleation period; use high-supersaturation pulses for rapid, synchronous nucleation [59]. |
| Uncontrolled Secondary Nucleation | Check for excessive agitation or mechanical shock. Look for a large number of very small crystals throughout the solution. | Optimize agitation rates; introduce a controlled seeding strategy with well-sized seeds to dominate the growth process [59] [2]. |
| Formation of Crystal Nests | Visual inspection (microscopy) reveals clusters of small, closely-spaced crystals. | Use in-line filtration to keep crystals suspended in the bulk and prevent nesting on surfaces [2]. Modulate supersaturation to favor growth over new nucleation [2]. |
Problem: Slow Crystal Growth and Variable Growth Rates
| Probable Cause | Diagnostic Checks | Recommended Solutions |
|---|---|---|
| Local Depletion in Nests | Compare crystal sizes from the reactor walls or bottom (where nests form) to those in the bulk solution. Crystals in nests will be smaller [59]. | Implement strategies to break up nests, such as optimized mixing or periodic low-frequency ultrasound. |
| Growth Rate Dispersion (GRD) | Observe individual seed crystals of similar size under identical conditions; significant variations in final size suggest GRD [59]. | Ensure a consistent and uniform preparation method for seed crystals, as their history can influence GRD [59]. |
Table 1: Key Factors Influencing Crystal Nesting and Local Depletion
| Factor | Impact on Nesting & Depletion | Control Parameter | Experimental Observation |
|---|---|---|---|
| Spatial Distribution of Crystals | Random nucleation leads to uneven distribution and nest formation [59]. | Nucleation rate & supersaturation profile. | Closely spaced crystals in nests are smaller than isolated crystals [59]. |
| Supersaturation Rate | Higher rates shorten induction time but can broaden the metastable zone, favoring homogeneous nucleation and increasing nest formation [2]. | Concentration rate / Membrane area in MDC [2]. | Faster concentration raised supersaturation at induction, promoting a primary nucleation pathway [2]. |
| Hold-up Time | Longer times after nucleation allow growth to deplete solute, reducing the nucleation rate and the creation of new nests [2]. | Residence time in crystallizer; use of in-line filtration [2]. | Longer hold-up time post-induction reduced nucleation rate and resulted in larger crystal sizes [2]. |
Experimental Protocols
Protocol 1: Supersaturation Control via Continuous Flow Reactor (CFR)
Objective: To achieve precise control over supersaturation, thereby separating nucleation and growth stages to minimize nesting and produce uniform crystals [26] [60].
Methodology:
- Setup: Use a jacketed chromatography column as the reactor. Employ separate feeding lines for metal salt solution (e.g., Ni/Co nitrates) and precipitating agent (e.g., HMTA). Mix them via a T-junction immediately before the reactor entrance [26].
- Operation: Use a peristaltic pump to maintain a constant flow rate. The outer compartment of the column is connected to a circulating water bath to control temperature precisely [26].
- Seeding (for low supersaturation): For operations below the heterogeneous nucleation threshold, use pre-coated substrates with seed crystals to dominate the crystallization process [26].
- Control: By maintaining a constant flow and temperature, the system operates under pseudo-steady-state conditions, keeping supersaturation at a stable and pre-determined level. This allows for the identification of distinct thresholds for homogeneous and heterogeneous nucleation [26] [60].
Protocol 2: Mitigating Nests via In-line Filtration in Membrane Crystallization
Objective: To retain crystals in the bulk solution and prevent their deposition as nests on reactor surfaces, thereby ensuring uniform growth conditions [2].
Methodology:
- Setup: Integrate an in-line filter into the membrane distillation crystallisation (MDC) system loop.
- Operation: As the solution is concentrated and crystals form, the in-line filter prevents crystals from circulating back to the membrane module or depositing on walls.
- Outcome: Crystals are segregated and retained within the crystallizer, reducing scaling on the membrane. This sustains a consistent supersaturation rate and allows for longer, controlled crystal growth, which desaturates the solvent and suppresses further nucleation [2].
Research Reagent Solutions
Table 2: Essential Materials for Controlled Crystallization Experiments
| Reagent / Material | Function in Experiment | Application Context |
|---|---|---|
| Hexamethylenetetramine (HMTA) | A hydrolysis-based precipitating agent that uniformly releases hydroxyl ions, enabling homogeneous precipitation [26]. | Synthesis of Layered Double Hydroxides (LDHs) [26]. |
| Hydroxypropyl Methyl Cellulose (HPMC) / HPMC Acetate Succinate (HPMCAS) | Polymers acting as precipitation inhibitors (PIs). They stabilize supersaturated solutions and inhibit nucleation and crystal growth via adsorption mechanisms [61]. | Supersaturated Drug Delivery Systems (SDDS) to maintain bioavailability [61]. |
| Monoammonium Phosphate (MAP) | Model compound for fundamental crystal growth studies due to its non-toxicity and well-defined habit [24]. | Home and educational crystal growth experiments; also used in industrial and optical applications [24]. |
| Continuous Flow Reactor (CFR) | System designed to maintain constant supersaturation under pseudo-steady-state conditions, enabling precise morphology control [26] [60]. | Controlled synthesis of nanomaterials like NiCo LDH nanoplates [26] [60]. |
| In-line Filter | A physical tool to retain crystals in the bulk of the crystallizer, preventing their deposition as "nests" on internal surfaces [2]. | Membrane Distillation Crystallisation (MDC) for scaling reduction and improved CSD [2]. |
Process Visualization Diagrams
Optimizing Seeding Strategies to Bypass Uncontrollable Primary Nucleation
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: Why should I use seeding instead of relying on spontaneous nucleation? Spontaneous, or primary, nucleation is often unpredictable and occurs at high supersaturation levels, which can lead to excessive nucleation events. This results in numerous, small, and potentially imperfect crystals. Seeding introduces a pre-formed, regular crystal surface into a solution at a controlled supersaturation level. This bypasses the need for primary nucleation, decoupling the nucleation step from crystal growth and enabling the growth of larger, higher-quality crystals [62].
Q2: What is the difference between primary and secondary nucleation? Primary nucleation is the formation of new crystalline entities from a clear, supersaturated solution in the absence of existing crystals of the same compound. It can be homogeneous (in a pure solution) or heterogeneous (induced by foreign particles or surfaces) [63]. Secondary nucleation occurs specifically because of the presence of crystals of the same compound in a supersaturated suspension. This is the mechanism exploited when you add seed crystals to a solution [63].
Q3: How do I determine the right supersaturation level for seeding? The optimal supersaturation for seeding is typically within the metastable zone, a region where the solution is supersaturated but spontaneous nucleation is unlikely. The solution should be supersaturated enough to promote growth on the added seeds but not so high that it causes secondary nucleation or "breeding" of new crystals. A well-designed seeding strategy often involves identifying a threshold supersaturation for seed propagation [63]. Advanced instruments can help map the metastable zone to define these parameters precisely [63].
Q4: My seeded experiments still result in too many small crystals. What am I doing wrong? This is a common issue where secondary nucleation outcompetes controlled crystal growth. Several factors can contribute to this:
- Supersaturation is too high: The driving force for growth may also be driving excessive secondary nucleation. Try reducing the supersaturation level at the time of seeding [63].
- Agitation is too vigorous: High agitation rates can cause crystal-crystal or crystal-impeller collisions that fracture seeds and generate new nuclei. Optimizing and reducing agitation speed can mitigate this [64].
- Seed crystal size and quality: Larger seed crystals may be less prone to breeding. Ensure your seeds are of good quality and not fractured [63].
Q5: How do seed loading and size affect the final crystal product? Both factors are critical for controlling the final Particle Size Distribution (PSD):
- Seed Loading (Quantity): A higher number of seed crystals will consume the available supersaturation across more growth sites, generally leading to a larger number of smaller final crystals. Lower seed loading typically results in fewer, larger crystals [63].
- Seed Size: Research indicates that larger single seed crystals can lead to a faster observed secondary nucleation rate. Therefore, the size of the seeds you introduce can directly influence the number of new crystals that form after seeding [63].
Troubleshooting Common Experimental Issues
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| No growth on seeds | Supersaturation too low; Non-viable seeds; Seed poisoning | Re-measure solubility & increase supersaturation to within metastable zone; Prepare fresh seed stock; Ensure chemical compatibility |
| Excessive secondary nucleation | Supersaturation too high; Excessive agitation; Poor seed quality | Lower supersaturation at seeding; Reduce agitation speed; Use larger, more robust seeds [63] [64] |
| Irregular crystal habit | Incorrect solvent system; Supersaturation gradient; Impurities | Select solvent favoring desired morphology (guided by MD simulation); Improve mixing; Use purification steps [64] |
| Ostwald ripening | Final supersaturation too low; Wide PSD; High solubility | Maintain slight positive supersaturation at end of cycle; Use tightly sized seeds; Optimize temperature profile |
Experimental Protocols
Protocol 1: Streak Seeding for Rapid Screening
Streak seeding is a powerful technique for rapidly transferring microscopic seeds to pre-equilibrated drops, ideal for screening crystallization conditions [62].
Materials:
- Supersaturated protein/solution
- Source of microcrystals (seed stock)
- Animal whisker or fine glass fiber
- Stereomicroscope
- Sitting drop plates
Methodology:
- Pre-equilibration: Bring the test solution to the desired supersaturation level within the metastable zone. This is often achieved via vapor diffusion or careful titration [62].
- Seed Transfer: Under a microscope, gently touch the tip of an animal whisker to the source of microcrystals.
- Seeding: Drag the whisker through the pre-equilibrated drop. This streaks microscopic seeds into the solution [62].
- Incubation and Monitoring: Seal the drop and monitor for crystal growth. This method allows for the rapid testing of numerous conditions to find the optimal one for crystal propagation.
Protocol 2: Optimizing Seeding for Spheroidal Crystal Morphology
This detailed protocol, inspired by premium-grade crystal production research, outlines a systematic approach to achieve uniform, spherical crystals [64].
Materials:
- Solute (e.g., HATO)
- Solvent system (e.g., Formic acid-water mixture)
- Thermostatted crystallizer with agitator
- Laser diffraction particle size analyzer
- Analytical balance
Methodology:
- Solvent System Selection:
- Determine the solubility profiles of your compound in various pure and binary solvents.
- Select a solvent system that demonstrates enthalpy-driven dissolution behavior, as this can favor a more uniform crystal growth rate across different crystal planes. For instance, a formic acid–water system (e.g., 2:8 volume ratio) has been shown to minimize growth rate disparities among crystal planes, promoting spheroidal morphology [64].
Supersaturation Establishment:
- Use the van't Hoff equation to model solubility and supersaturation as a function of temperature.
- Establish a supersaturation ratio (e.g., 0.9) that is high enough to drive growth but low enough to prevent runaway nucleation. The supersaturation ratio (S) is defined as S = C / C, where C is the concentration and C is the equilibrium saturation concentration [64].
Seeding and Crystallization:
- Based on orthogonal experimental design, use optimized parameters such as:
- Cooling rate: 0.5 °C h⁻¹ (very slow to maintain control)
- Agitation speed: 500 rpm (sufficient for mixing without excessive shear) [64]
- Introduce well-characterized seeds at the determined supersaturation ratio.
- Based on orthogonal experimental design, use optimized parameters such as:
Hold-up and Growth:
- Maintain a consistent supersaturation rate to allow for a longer hold-up time after seed induction. This desaturates the solvent through controlled crystal growth, resulting in larger crystal sizes and reduced nucleation rates [2].
Data Presentation
Table 1: Impact of Seeding Parameters on Final Crystal Properties
| Supersaturation Ratio | Seed Loading (%) | Agitation (rpm) | Median Crystal Size (µm) | Crystal Habit | Notes |
|---|---|---|---|---|---|
| 1.2 | 0.1 | 500 | 120 | Needles | High secondary nucleation |
| 0.9 | 0.1 | 500 | 250 | Spheroidal | Optimal conditions [64] |
| 0.9 | 0.5 | 500 | 150 | Spheroidal | Smaller final size due to higher seed count |
| 0.9 | 0.1 | 200 | 280 | Spheroidal | Larger size, but potential for agglomeration |
| 0.7 | 0.1 | 500 | 50 | Platelets | Insufficient growth; Ostwald ripening |
Table 2: Solvent System Selection for Morphology Control
| Solvent System | Volume Ratio | Key Thermodynamic Parameter | Predicted Morphology (from MD) | Experimental Outcome |
|---|---|---|---|---|
| Formic Acid - Water | 2 : 8 | Enthalpy-driven, minimal growth rate disparity | Near-Spheroidal | Premium-grade spheroids [64] |
| Acetic Acid - Water | 2 : 8 | — | — | Improved sphericity over pure solvents |
| Ethanol - Water | 2 : 8 | — | — | Irregular, aggregated |
| Pure Water | — | High growth rate disparity | Irregular / Needles | Defective, irregular crystals [64] |
Mandatory Visualization
Seeding Strategy Workflow
Diagram 1: Seeding strategy workflow for controlled crystallization.
Supersaturation Zone Management
Diagram 2: Supersaturation zone management for seeding.
The Scientist's Toolkit
Research Reagent Solutions
| Item | Function | Application Note |
|---|---|---|
| Binary Solvent Systems (e.g., Formic acid-water) | Modulates solubility & crystal habit by reducing growth rate disparities between different crystal faces, enabling morphology control [64]. | A 2:8 volume ratio of formic acid-water was optimal for producing spheroidal HATO crystals [64]. |
| Well-Characterized Seed Crystals | Provides a regular, pre-formed surface for orderly molecule aggregation, bypassing the stochastic primary nucleation step [62]. | Seed crystal size and loading directly impact the final particle size distribution and nucleation rate [63]. |
| Animal Whiskers (for Streak Seeding) | Allows for the rapid and sterile transfer of microscopic seeds from a seed stock to a pre-equilibrated solution [62]. | Ideal for high-throughput screening of crystallization conditions and for discriminating between microcrystals and precipitate [62]. |
| In-line Filtration | Removes particulates and can be used to retain crystals within the crystallizer, reducing scaling on vessel walls [2]. | Helps maintain a consistent supersaturation rate and longer crystal hold-up time, promoting growth over nucleation [2]. |
Controlling Polymorphic Transitions and Preventing Unwanted Phase Changes
Troubleshooting Common Issues
FAQ: Why is my active pharmaceutical ingredient (API) transforming into a different polymorph during storage? This is often caused by the relative thermodynamic instability of a metastable polymorph. Even slight changes in environmental conditions, such as humidity or temperature, can provide the necessary activation energy for a transition to a more stable form. This was the case with the protease inhibitor Norvir (retonovir), where a previously unobserved polymorph precipitated in the soft gelatin capsule, leading to a product recall [65].
FAQ: My crystallization process yields inconsistent polymorphic forms. How can I improve control? Inconsistent output typically stems from uncontrolled or fluctuating nucleation conditions. Key parameters to control include the rate of supersaturation generation, the presence of specific impurities or additives that can act as templates, and the mixing dynamics. Even minor, unmonitored variations in these factors can lead to different polymorphs nucleating [65] [66].
FAQ: A promising metastable polymorph has high solubility but converts during processing. Can it be stabilized? Yes, stabilizing a metastable polymorph is a common goal to exploit advantages like higher solubility. Strategies include:
- Using custom additives or polymers that selectively inhibit the nucleation or growth of the stable form.
- Controlling the particle size, as phase stability can be size-dependent at the nanoscale.
- Utilizing molecular simulation to rationally design stabilization approaches by understanding the transition mechanism at the molecular level [65].
FAQ: What analytical techniques are best for monitoring polymorphic transitions in real-time? While standard techniques like X-ray diffraction (XRD) provide definitive structural identification, they are often not suitable for real-time monitoring. For observing transitions as they happen, methods such as Raman spectroscopy and in-situ microscopy are highly effective. Furthermore, molecular simulation offers unparalleled, time-resolved molecular-level resolution of the processes taking place, complementing experimental data [65].
Experimental Protocols for Control and Prevention
Protocol 1: Seeding to Control Polymorphic Outcome
This protocol uses intentional seeding with a specific polymorph to direct the crystallization outcome, a key tactic for optimizing the supersaturation threshold and achieving crystal morphology control.
- Objective: To ensure the consistent reproduction of a desired polymorphic form (the "seed" polymorph) from a supersaturated solution.
- Materials: API solution, seed crystals of the target polymorph (pre-characterized by techniques like XRD), standard crystallization lab equipment (vessels, stirrers, temperature control).
- Procedure:
- Generate a supersaturated solution of your API, ensuring the concentration and temperature are within the metastable zone of the desired polymorph.
- Characterize the seed crystals thoroughly to confirm their polymorphic identity and purity.
- Introduce a precise, small amount of the seed crystals into the supersaturated solution.
- Maintain controlled conditions (temperature, agitation) to allow for crystal growth exclusively on the provided seeds.
- Isolate the final product and verify the polymorphic form against the original seeds.
Protocol 2: Optimizing Crystallization Conditions via High-Throughput Screening
This methodology systematically explores a wide range of chemical conditions to identify parameters that favor the formation and stability of a specific polymorph.
- Objective: To rapidly identify chemical conditions (solvents, additives, concentrations) that lead to the formation of a target polymorph.
- Materials: API, a range of solvents and additives, high-throughput crystallization plates (e.g., 96-well plates), an automated liquid handling system (if available), and an analytical tool for polymorph identification (e.g., Raman microscopy or XRD).
- Procedure:
- Prepare a matrix of crystallization trials in the multi-well plates by varying the solvent composition, the presence and concentration of different additives, and the API concentration.
- Induce crystallization through a chosen method (e.g., cooling, solvent evaporation, anti-solvent addition).
- Allow the plates to equilibrate and crystals to form.
- Analyze each well to identify the polymorphic form of the resulting crystals.
- Map the experimental conditions to the polymorphic outcomes to define the stable operating region for your target polymorph.
Visualization of Workflows and Relationships
Polymorph Control Strategy Map
Phase Transition Mechanism Map
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details essential materials and their functions for investigating and controlling polymorphic transitions.
| Research Reagent / Material | Primary Function in Polymorph Control |
|---|---|
| Selected Seed Crystals | Provides a pre-determined crystalline template to direct nucleation and growth, ensuring the consistent reproduction of a specific polymorphic form. |
| Chemical Additives | Modifies the crystallization environment by adsorbing to specific crystal faces or altering solvent properties to either promote a desired polymorph or inhibit an unwanted one. |
| Polymer Stabilizers | Used to coat crystals and act as a physical barrier, preventing the molecular reorganization required for a solid-state polymorphic transition during storage or processing. |
| Molecular Simulators | Not a physical reagent, but a crucial tool for predicting relative polymorph stability, understanding transition mechanisms at the molecular level, and rationally designing experiments [65]. |
| High-Throughput Screening Plates | Enables the parallel setup of hundreds of crystallization experiments with varying parameters (solvents, additives) to rapidly map the polymorphic landscape of an API. |
Strategies for Fines Removal and Scaling Up from Lab to Production
Frequently Asked Questions (FAQs)
Q1: What are "fines" in a crystallization process and why is their removal important? Fines are very small, often unwanted crystals that form due to high supersaturation levels. Their removal is crucial because they can incorporate impurities, lead to poor product flowability, and cause inconsistent filtration during scale-up. Leaving fines in the crystallizer can also consume supersaturation that would otherwise support the growth of larger, more pure crystals [3] [6].
Q2: How does supersaturation control help during scale-up from lab to production? Supersaturation is the primary driver of both nucleation and crystal growth. Precise control during scale-up is essential because production-scale equipment often has different heat and mass transfer dynamics than lab setups. Effectively managing supersaturation ensures consistent Crystal Size Distribution (CSD), desired crystal morphology, and final product purity by preventing excessive primary nucleation (which creates fines) and promoting crystal growth [2] [26].
Q3: What are common signs of poor supersaturation control in a production crystallizer? Common signs include:
- Rapid Crystal Formation: The solid forms too quickly, leading to the incorporation of impurities [3].
- Excessive Fines: A high population of very small crystals in the product slurry.
- Inconsistent Crystal Size: A wide and unpredictable variation in crystal dimensions [6].
- Scaling: Unwanted deposition of solid material on the crystallizer's walls and internals, which reduces heat transfer and process efficiency [2].
Q4: What strategies can be used to remove fines from a crystallizer? Fines can be removed by applying a Fines Destruction Cycle. This involves:
- Dissolution: Introducing heat or a small amount of solvent to a segregated stream of the crystal slurry to selectively dissolve the fine crystals, which have higher solubility than larger crystals due to their greater surface area.
- Reintroduction: Returning the resulting undersaturated solution back to the main crystallizer body. This strategy reduces the total number of crystals and provides more solute material for the growth of the remaining larger crystals.
Troubleshooting Guides
Problem: Excessive Fines Formation
Fines are small, low-quality crystals that can compromise purity and handling.
| Probable Cause | Diagnostic Steps | Corrective Actions |
|---|---|---|
| Excessively high nucleation rate [2] | Measure Crystal Size Distribution (CSD). Check if supersaturation is in the metastable zone. | Optimize cooling/evaporation rates to lower supersaturation generation. Implement a controlled seeding strategy [6]. |
| Insufficient crystal growth time | Check the residence time in the crystallizer versus the growth rate. | Increase the residence time or holdup time to allow crystals to grow [2]. |
| Localized high supersaturation | Check for poor mixing or "dead zones" in the vessel. | Optimize agitator design and stirring speed to ensure uniform conditions throughout the crystallizer [6]. |
Problem: Scaling on Crystallizer Walls and Internals
Scaling is the unwanted deposition of material on equipment surfaces, hindering operation.
| Probable Cause | Diagnostic Steps | Corrective Actions |
|---|---|---|
| High wall superheat | Check temperature difference between the heating medium and the crystallizing solution. | Reduce the temperature driving force to minimize spontaneous nucleation at heat exchange surfaces. |
| Lack of crystal retention | Determine if crystals in the slurry are being deposited on surfaces instead of suspended in the bulk. | Use in-line filtration to ensure crystals are cycled within the crystallizer, providing sites for growth in the solution rather than on walls [2]. |
| Surface-induced nucleation | Inspect for scaling on rough surfaces or scratches. | Use polished surfaces where possible and ensure proper vessel finish during design [13]. |
Experimental Protocols for Supersaturation and Morphology Control
Protocol 1: Establishing a Baseline Metastable Zone Width (MSZW)
Objective: To determine the supersaturation limits where spontaneous nucleation occurs, which is critical for designing a controlled crystallization process that minimizes fines.
Materials:
- Research Reagent Solutions:
- Analyte Solution: The compound of interest dissolved in a suitable solvent.
- Antisolvent/Precipitant: A solvent in which the analyte has low solubility.
- Seeds: Small, pure crystals of the analyte for seeded experiments.
- Equipment: Lasentec FBRM (Focused Beam Reflectance Measurement) or PVM (Particle Video Microscope), jacketed crystallizer, temperature probe, agitator, syringe pump.
Methodology:
- Saturation Point: Charge the crystallizer with a known volume of solvent and add the analyte incrementally while heating and stirring until a clear, saturated solution is obtained.
- Supersaturation Generation: Cool the solution at a controlled, linear rate or add antisolvent at a controlled rate using a syringe pump.
- Nucleation Detection: Monitor the solution in real-time using FBRM. The point where the particle count dramatically increases indicates the nucleation point and the limit of the MSZW.
- Data Collection: Record the temperature and concentration at the nucleation point. Repeat at different cooling/antisolvent addition rates to understand the kinetic width of the MSZW.
Protocol 2: Seeded Crystallization to Suppress Fines
Objective: To control the crystallization process by using seed crystals, thereby consuming supersaturation through growth rather than spontaneous nucleation.
Materials:
- Research Reagent Solutions:
- Seed Crystals: A carefully sized and massed sample of pure product.
- Supersaturated Solution: A solution of the product prepared at a known supersaturation level within the metastable zone.
- Equipment: Same as Protocol 1.
Methodology:
- Solution Preparation: Create a supersaturated solution of your product, ensuring it is clear and within the metastable zone (no spontaneous nucleation).
- Seeding: Introduce a known mass and size distribution of seed crystals to the solution. The seed amount and surface area should be sufficient to consume the available supersaturation.
- Growth Phase: Carefully control the cooling or evaporation profile to maintain a low, constant supersaturation level that promotes growth on the existing seeds without generating new nuclei.
- Monitoring: Use in-line tools (FBRM/PVM) to track the increase in crystal size and ensure no "secondary nucleation" occurs.
Data Presentation: Key Crystallization Parameters
The following table summarizes quantitative data and relationships between operating parameters and crystal product attributes, as established in research.
| Operating Parameter | Impact on Supersaturation | Effect on Nucleation & Growth | Resulting Crystal Product Attribute |
|---|---|---|---|
| Cooling/Evaporation Rate [3] | Higher rate increases supersaturation generation. | Increases nucleation rate, favors primary nucleation. | Smaller Crystal Size (more fines), broader CSD, potential impurity incorporation. |
| Seed Loading & Size [3] [26] | Provides surface area to consume supersaturation. | Shifts balance from nucleation to growth. | Larger Average Size, narrower CSD, improved reproducibility. |
| Agitation Intensity [6] | Reduces localized high supersaturation. | Can abrade crystals (secondary nucleation) if too high; prevents settling if too low. | Optimum CSD; excessive agitation can create fines. |
| Hold-up Time (after induction) [2] | Allows supersaturation to be depleted by growth. | Reduces nucleation rate; promotes crystal growth. | Larger Crystal Size, improved yield. |
Process Visualization
The diagram below illustrates the logical workflow and decision points for implementing strategies that target fines removal and scaling prevention, all within the framework of supersaturation control.
Supersaturation Control for Fines and Scaling Management
Evaluating Success: Case Studies, Model Predictions, and Performance Metrics
Troubleshooting Guides and FAQs
This section addresses common challenges researchers face when developing and scaling up crystallization processes.
FAQ 1: Why does my continuous crystallization process produce crystals with a wider size distribution than my batch process, and how can I improve it?
Answer: This is a common issue arising from the fundamental difference in residence time distribution (RTD) between batch and continuous crystallizers, particularly Continuous Stirred-Tank Reactors (CSTRs). In a batch system, all crystals have nearly identical residence times. In a continuous Mixed-Suspension Mixed-Product Removal (MSMPR) crystallizer, the broad RTD means crystals spend varying amounts of time in the vessel, leading to a wider Crystal Size Distribution (CSD) [67]. To mitigate this:
- Optimize Operating Conditions: Carefully control supersaturation, temperature, and agitation to minimize unintended secondary nucleation, which generates fine crystals [68].
- Implement Fines Removal: Integrate a fines dissolution loop to continuously remove and dissolve small crystals, preventing them from reporting to the product and narrowing the CSD.
- Use Seeding in Continuous Mode: Just as in batch processes, seeding a continuous crystallizer can provide a uniform population of growth sites, dominating the nucleation process and leading to a more consistent CSD [68].
FAQ 2: How can I prevent unwanted polymorphic transformation during scale-up from batch to continuous operation?
Answer: Unwanted polymorphism often occurs due to shifts in local supersaturation or seeding efficiency. Control strategies must be adapted for continuous systems [68].
- Precise Supersaturation Control: In continuous flow crystallizers (e.g., tubular), supersaturation can be more uniformly controlled compared to large batch vessels, reducing the risk of local zones where a metastable polymorph might form [67].
- Robust Seeding Strategy: Ensure a consistent and active seed load is introduced into the continuous process. The seed form must be the most stable polymorph under process conditions.
- Advanced Process Control: Implement control strategies like Model Predictive Control (MPC) to maintain process parameters (temperature, concentration) within a tight window that exclusively favors the desired polymorph [69].
FAQ 3: My batch process yields a large mean crystal size, but my continuous process yields smaller crystals. What control strategies can help increase crystal size in continuous operation?
Answer: Smaller crystals in continuous systems are often due to high nucleation rates driven by the need to maintain a higher steady-state supersaturation to initiate and sustain crystallization [70].
- Reduce Nucleation Rate: Lower the supersaturation set point. While this may slow growth, it disproportionately reduces the nucleation rate, favoring growth over the creation of new crystals.
- Extend Hold-up Time: Research shows that maintaining crystal retention in the crystallizer for longer periods after nucleation (longer hold-up time) allows for solvent desaturation and crystal growth, resulting in larger crystals [2].
- Leverage Reinforcement Learning (RL): Novel RL-based controllers have been demonstrated to effectively track trajectories for larger mean crystal size in continuous systems by intelligently manipulating variables like jacket temperature [71].
Quantitative Data Comparison: Batch vs. Continuous Crystallization
The following table summarizes key performance indicators for different crystallization modes and control strategies, based on data from the literature.
Table 1: Benchmarking Crystallization Operational Modes and Control Strategies
| Crystallization Mode | Control Strategy | Key Performance Findings | Key Challenges |
|---|---|---|---|
| Batch [70] [68] | Uncontrolled Cooling / Evaporation | - Nucleation ceases in the presence of growing crystals [70].- Simple to implement at small scale. | - Significant batch-to-batch variability [67].- Non-uniform CSD due to cycling conditions.- Difficult to control polymorphism at scale [68]. |
| Batch [69] | Linear PI Control | - Poor output response for tracking crystallizer temperature [69].- Unsatisfactory for maintaining consistent CSD. | - Struggles with the nonlinear nature of the crystallization process. |
| Batch & Continuous [69] | Generic Model Control (GMC) | - Improves output response compared to standard PI control [69].- Better at handling process constraints. | - Performance is inferior to more advanced optimization-based controllers like MPC. |
| Batch & Continuous [69] | Model Predictive Control (MPC) | - Superior performance in setpoint tracking with minimal overshoot and fast settling time [69].- Effectively handles multi-variable constraints. | - Requires a accurate process model.- Computational cost can be high. |
| Batch & Continuous [71] | Reinforcement Learning (RL) | - Demonstrates robust control capabilities, outperforming benchmark MPC in some cases [71].- Effective at tracking temperature, supersaturation, and crystal size. | - Very high computational cost during the training phase [71]. |
| Continuous (MSMPR) [70] [67] | Steady-State Operation | - Provides consistent crystal size and morphology [67].- Higher productivity and smaller equipment footprint. | - Requires higher supersaturation to initiate crystallization [70].- Broader Crystal Size Distribution (CSD) due to Residence Time Distribution (RTD) [67]. |
| Continuous (Tubular) [67] | Plug-Flow Operation | - Narrower RTD compared to MSMPR, leading to more uniform CSD.- Excellent supersaturation control. | - Potential for fouling and clogging.- More complex to scale up. |
Experimental Protocols for Supersaturation and Morphology Control
This section provides detailed methodologies for key experiments cited in this guide.
Protocol 1: Face-Specific Crystal Growth Kinetics Measurement for Morphology Control
Objective: To quantitatively measure the growth rates of different crystal facets (e.g., {100} and {011}) of a model compound (e.g., Tolfenamic Acid) as a function of supersaturation and solvent polarity [5].
Materials:
- Single crystal seed of the desired polymorph.
- Supersaturated solution prepared in a selected solvent (e.g., ethanol).
- Temperature-controlled crystal growth cell (e.g., a glass cuvette submerged in a precision water bath) [5].
- Polarizing optical microscope with CCD camera.
- Image analysis software.
Methodology:
- Solution Preparation: Prepare a supersaturated solution of the compound in the chosen solvent at a known concentration, based on published solubility data [5].
- Seed Loading: Transfer the solution to the growth cell and introduce a single crystal seed.
- Surface Preparation: Briefly heat the cell 5-10°C above the saturation temperature to induce slight dissolution, removing surface imperfections and achieving a desired seed size (e.g., 700-1000 μm length) [5].
- Initiate Growth: Rapidly cool the cell to the target growth temperature (e.g., 20°C) to establish a fixed supersaturation (e.g., S=1.1-1.7).
- Image Acquisition: Capture time-lapse images of the growing crystal at constant intervals (e.g., every 10 minutes) [5].
- Data Analysis: Use the software to measure the displacement of specific crystal faces (e.g.,
{100}) over time. Plot the distance versus time; the slope of the initial linear region is the face-specific growth rate at that supersaturation.
Protocol 2: Implementing Supersaturation Control in Membrane Distillation Crystallization (MDC)
Objective: To regulate the competition between nucleation and crystal growth by using membrane area to modulate the rate of supersaturation generation [2].
Materials:
- Membrane distillation crystallizer setup.
- Feed solution (e.g., sodium chloride brine).
- In-line filter.
Methodology:
- System Setup: Configure the MDC system with a specific active membrane area, which directly sets the solvent removal and concentration rate.
- Induction Monitoring: Run the process and record the induction time. A shorter induction time indicates a broader Metastable Zone Width (MSZW) due to a higher supersaturation driving force, favoring homogeneous primary nucleation [2].
- Post-Induction Modulation: After nucleation is detected, use the in-line filter to retain crystals in the crystallizer bulk, reducing deposition (scaling) on the membrane.
- Growth Phase: This crystal retention allows for a consistent supersaturation rate to be maintained over a longer "hold-up time." The system desaturates as the stored crystals grow, which in turn reduces the nucleation rate [2].
- Analysis: Use population balance modeling to confirm the reduction in nucleation rate and the increase in median crystal size with longer hold-up times [2].
Workflow and Strategy Diagrams
The following diagrams illustrate the experimental workflow for crystal growth studies and the logical decision process for selecting a control strategy.
The Scientist's Toolkit: Research Reagent Solutions
This table lists key materials and computational tools used in advanced crystallization research.
Table 2: Essential Research Reagents and Tools for Crystallization Optimization
| Item Name | Function / Application in Research |
|---|---|
| Anti-Solvents [68] | A solvent in which the API has low solubility. Its controlled addition triggers supersaturation and crystallization, useful for controlling nucleation and producing fine crystals. |
| Seeds (Pre-formed Crystals) [68] | Small crystals of the desired polymorph used to promote consistent secondary nucleation and growth. Critical for controlling crystal size distribution and ensuring polymorphic purity. |
| Protic Polar Solvents (e.g., Ethanol) [5] | Solvents that can donate hydrogen bonds. Used in studies of solvent-mediated morphology, as they can disrupt specific hydrogen bonds at crystal faces, leading to higher aspect ratio (needle-like) crystals. |
| Aprotic Apolar Solvents (e.g., Toluene) [5] | Solvents that do not donate hydrogen bonds and have low polarity. Used to study morphology as they can strongly interact with aromatic moieties on capping faces, hindering crystal elongation. |
| CrystalMaker Software [72] | A visualization and modeling tool used to predict crystal morphology based on attachment energy calculations, simulate temperature effects, and model surface chemistry to rationalize growth habits. |
| CrystalGym Environment [73] | An open-source reinforcement learning (RL) environment based on Gymnasium. Used to benchmark RL algorithms for designing crystalline materials by optimizing DFT-calculated properties like band gap and bulk modulus. |
Validating Prediction Models with Experimental Data for Pharmaceutical Solids
Frequently Asked Questions (FAQs) & Troubleshooting Guides
FAQ 1: What are the fundamental crystal morphology prediction models used in pharmaceutical development?
Answer: Several established theoretical models form the foundation for predicting crystal morphology, each with specific strengths.
- BFDH Model: The Bravais-Friedel-Donnay-Harker (BFDH) model is a geometric model that predicts possible crystal faces and their relative growth rates based on the crystal's internal lattice structure and symmetry. It operates on the principle that the growth rate of a crystal face (Ghkl) is inversely proportional to its interplanar spacing (dhkl):
Ghkl ∝ 1/dhkl. This means faces with larger d-spacings (often with smaller Miller indices) grow more slowly and are more likely to appear in the final crystal habit. However, the BFDH model does not consider external factors like solvent or supersaturation [18]. - Attachment Energy (AE) Model: This model, based on the Periodic Bond Chain (PBC) theory, is more advanced and widely used. It proposes that the growth rate of a crystal face is proportional to its "attachment energy" (Eatt)—the energy released when a growth unit attaches to the crystal surface. Faces with lower attachment energies grow more slowly and become the larger, dominant faces. This model provides a more mechanistic understanding by considering intermolecular interactions [18].
- Gibbs-Curie-Wulff Principle: This is a thermodynamic principle stating that the equilibrium shape of a crystal minimizes its total surface energy. The distance from the crystal's center (Wulff point) to each face is proportional to the surface energy of that face. It describes the ideal morphology under equilibrium conditions [18].
FAQ 2: How does supersaturation specifically impact nucleation and final crystal morphology?
Answer: Supersaturation is the primary driving force for both nucleation and crystal growth, and its precise control is critical for determining final crystal properties.
- Nucleation Pathway Control: The level of supersaturation determines the nucleation pathway. At low supersaturation, heterogeneous nucleation (nucleation on surfaces like dust or vessel walls) is favored. At high supersaturation, homogeneous nucleation (spontaneous nucleation in solution) becomes dominant. Identifying the threshold between these regimes is key for reproducibility [26] [49].
- Morphological Evolution: Supersaturation directly influences the competition between nucleation and growth. Lower supersaturation favors crystal growth over new nucleation, often leading to larger, more defined crystals (e.g., isolated nanoplates). Higher supersaturation promotes rapid nucleation, resulting in a larger number of smaller crystals and can drive morphological transitions, such as the assembly of 2D nanoplates into 3D nanoflower structures [26] [2].
- Metastable Zone Width (MSZW): This is the region between the saturation curve and the spontaneous nucleation curve. Operating within the MSZW allows for growth without uncontrolled nucleation. The MSZW can be broadened by increasing the concentration rate, which raises the supersaturation at induction and favors a homogeneous primary nucleation pathway [2].
Table 1: Impact of Supersaturation on Crystallization Outcomes
| Supersaturation Level | Nucleation Pathway | Dominant Process | Expected Crystal Morphology |
|---|---|---|---|
| Low | Heterogeneous | Crystal Growth | Larger, well-defined crystals (e.g., nanoplates) |
| Moderate | Mixed | Balanced Growth & Nucleation | Uniform crystals with controlled size distribution |
| High | Homogeneous | Rapid Nucleation | Many small crystals; complex structures (e.g., nanoflowers) |
FAQ 3: What are the best experimental techniques for validating a crystal morphology prediction model?
Answer: Validating a prediction model requires experiments designed to decouple variables and provide quantitative data on growth rates and morphology under controlled conditions.
- Single-Crystal Technique for Readily Soluble Salts: This method involves studying the growth and contact nucleation of a single seed crystal in a flow system. Its key advantage is the ability to vary one operating variable (e.g., supersaturation, temperature, impact energy) while keeping all others constant. This allows for the clear identification of factors influencing secondary nucleation and crystal growth kinetics, which is difficult in a mixed-suspension crystallizer [74].
- pH-Stat / Constant-Composition Technique for Sparingly Soluble Salts: For salts like calcium carbonate, supersaturation is generated by a chemical reaction. This technique uses an autotitrator to maintain a constant pH and supersaturation by automatically adding reactants. This controlled environment allows researchers to systematically investigate the effects of factors like supersaturation, pH, ionic strength, and impurities on crystal growth and nucleation mechanisms [74].
- Continuous Flow Reactor (CFR) Synthesis: Systems like the Continuous Flow Reactor (CFR) enable precise control over supersaturation by maintaining pseudo-steady-state conditions. This is achieved by continuously feeding precursor solutions into a heated reactor. The CFR is highly effective for synthesizing materials like layered double hydroxides (LDHs) with tunable morphologies (from nanoplates to nanoflowers) by providing a constant supersaturation level, unlike batch methods where supersaturation drops rapidly [26] [49].
FAQ 4: My crystals are too small and exhibit high agglomeration. How can I troubleshoot this?
Answer: This common issue is often linked to uncontrolled nucleation and high supersaturation.
- Problem: High nucleation rate and agglomeration.
- Solution:
- Reduce Supersaturation: Decrease the cooling or evaporation rate, or reduce the concentration of anti-solvent added. This shifts the balance from nucleation toward crystal growth.
- Use a Seeding Strategy: Introduce a small number of well-defined seed crystals into the metastable solution. This provides a surface for growth and consumes supersaturation, suppressing spontaneous nucleation.
- Optimize Mixing: Improve mixing efficiency to eliminate local pockets of high supersaturation where uncontrolled nucleation occurs.
- Adjust Solvent or Additives: Consider changing the solvent system or adding specific additives or impurities that can selectively bind to crystal faces and modify growth kinetics to discourage agglomeration [18] [74].
FAQ 5: The crystal habit from my experiment does not match the model prediction. What could be the reason?
Answer: Discrepancies between model predictions and experimental results are common and highlight the models' limitations and the importance of experimental factors.
- Solvent Effects: Most basic models (like BFDH and AE) predict the morphology in a vacuum or gas phase. The interaction between the crystal surface and the solvent can significantly alter the relative growth rates of different faces. A face that grows slowly in one solvent might grow rapidly in another.
- * Presence of Impurities or Additives:* Even trace amounts of impurities or deliberately added growth modifiers can adsorb onto specific crystal faces, inhibiting their growth and radically changing the final habit.
- Inaccurate Intermolecular Force Fields: The accuracy of models like the AE model depends heavily on the force fields used to calculate interaction energies. Inaccurate force fields lead to incorrect attachment energies and flawed morphology predictions.
- Neglected Growth Mechanisms: Standard models often assume spiral growth, while 2D surface nucleation or non-classical growth pathways (like particle aggregation) may dominate under certain conditions, leading to different morphologies [18].
Experimental Protocols for Key Validation Techniques
Protocol 1: Single-Crystal Growth and Contact Nucleation Study
Objective: To quantify the effect of specific operating variables (supersaturation, impact energy) on the secondary nucleation rate of a readily soluble pharmaceutical compound.
Materials:
- Saturated solution of the compound under study.
- Single-crystal seed.
- Thermostatted flow cell apparatus with a pump.
- Microscope with camera.
- Particle counter or Coulter counter.
Methodology:
- Seed Preparation: A single seed crystal is glued to the tip of a probe and placed in the flow cell.
- Equilibration: An undersaturated solution is circulated past the seed crystal to achieve thermal and surface equilibrium.
- Supersaturation Generation: The solution temperature is lowered to create a known, controlled supersaturation.
- Induce Nucleation: A nylon rod or other implement is used to impart a controlled, reproducible impact to the seed crystal.
- Nuclei Quantification: The resulting "shower" of secondary nuclei is carried by the flowing solution to a sensor (e.g., a particle counter) to measure the nucleation rate.
- Systematic Variation: Repeat steps 3-5 while varying one parameter at a time (e.g., supersaturation, impact energy, temperature) to establish their individual effects on the secondary nucleation rate [74].
Protocol 2: Controlled Synthesis via Continuous Flow Reactor (CFR)
Objective: To synthesize crystals with precise morphology control by maintaining constant supersaturation.
Materials:
- Two precursor solutions (e.g., metal salts and alkaline precipitant).
- Dual-channel peristaltic pump.
- Jacketed chromatography column or similar heated reactor.
- Masterflex or similar chemical-resistant tubing.
- Circulating water bath.
Methodology:
- Setup: Configure the CFR system with separate feeding lines for the two precursor solutions. Use a T-junction near the entrance of the heated column for mixing.
- Temperature Control: Set the circulating water bath to the desired hydrolysis temperature (e.g., 95°C) to heat the reactor's outer jacket.
- Pump Calibration: Calibrate the peristaltic pump to ensure a precise and constant flow rate for both precursor solutions.
- Reaction Initiation: Start the pump to flow the precursors into the reactor. The solutions mix at the T-junction and react as they travel through the heated zone.
- Product Collection: Collect the reacted suspension and resulting crystals at the exit port. The steady-state conditions inside the CFR maintain a constant supersaturation, leading to uniform nucleation and growth [26] [49].
Workflow Diagrams
Crystal Morphology Validation Workflow
Supersaturation Control Logic
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Materials and Reagents for Controlled Crystallization Experiments
| Item Name | Function / Application | Key Consideration |
|---|---|---|
| Hexamethylenetetramine (HMTA) | A common hydrolyzing agent for homogeneous precipitation. It slowly decomposes in heated aqueous solutions to release hydroxyl ions, enabling a gradual and uniform increase in supersaturation. | Provides more controlled nucleation and growth compared to rapid base addition, leading to better-defined crystals [26] [49]. |
| Continuous Flow Reactor (CFR) | A system for maintaining constant supersaturation under pseudo-steady-state conditions. It consists of a pump, heated reactor, and feed lines. | Essential for decoupling nucleation and growth phases, enabling precise morphology control (e.g., from nanoplates to nanoflowers) [26]. |
| pH-Stat Autotitrator | An automated system that maintains constant pH in a crystallizing solution by titrating reactants. | Critical for studying crystallization kinetics of sparingly soluble salts, as it holds supersaturation constant, allowing isolation of variable effects [74]. |
| Seed Crystals | Well-characterized, small crystals of the target compound used to initiate growth in a metastable solution. | Seeding provides controlled nucleation sites, suppresses excessive primary nucleation, and ensures consistent crystal size and form. |
| Gold Octupole Electrode | Used to generate tunable, morphing electric field landscapes for controlling the assembly and repair of colloidal crystals. | Allows for advanced defect correction and morphology shaping via feedback control, though more common in model systems than industrial pharmaceutical production [75]. |
Crystal morphology, or habit, is a critical physical attribute of an Active Pharmaceutical Ingredient (API) that profoundly influences the efficiency of downstream pharmaceutical manufacturing processes. Defined as the external shape and appearance of a crystal, morphology is governed by the relative growth rates of different crystal faces during crystallization. It is a key factor in determining powder properties such as flowability, packability, and compactability, which directly impact the unit operations of filtration, drying, blending, and tableting [76] [19]. For instance, needle-like crystals can lead to poor filtration, slow flow, and capping during tableting, whereas more equidimensional or spherical particles are generally desired for their superior processing characteristics [77] [78]. This technical guide, framed within the context of optimizing supersaturation threshold for crystal morphology control, provides troubleshooting advice and methodologies for researchers and drug development professionals to diagnose and resolve common downstream processing issues through targeted crystal engineering.
Frequently Asked Questions (FAQs)
1. How does crystal morphology directly affect the filtration of an API slurry?
Crystal morphology impacts filtration by determining the porosity and permeability of the filter cake. Needle-like or plate-like crystals tend to form dense, impermeable cakes that trap mother liquor, resulting in prolonged filtration times and inefficient washing [77] [76]. In contrast, block-like or spherical crystals pack more uniformly, creating a porous cake structure that allows for faster liquid removal, shorter filtration cycles, and more effective removal of impurities [77].
2. Why is crystal habit a major factor in powder flowability and blending uniformity?
Powder flowability is crucial for consistent die filling during tablet production. Acicular (needle-like) and flake-like crystals have high interparticle friction and cohesion, leading to poor flowability and potential bridging in hoppers [77] [78]. This can cause significant variations in tablet weight and drug content. Spherical or equant agglomerates, with their minimal contact points and rolling ability, exhibit excellent flow properties [78]. Studies on spherical agglomerates of acebutolol hydrochloride showed a lower angle of repose and reduced cohesive stress compared to single crystals, confirming superior flowability [78].
3. Can modifying crystal habit improve the compactability of a poorly compressible API?
Yes. Crystal habit modification can significantly enhance compactability. During compression, agglomerated or spherical crystals often undergo brittle fracture at the points between primary crystallites, creating fresh, clean surfaces that form strong bonds [78]. This leads to tablets with higher tensile strength. For example, agglomerated crystals of ibuprofen produced stronger tablets than single crystals due to this fragmentation behavior [78]. Furthermore, crystal engineering of erythromycin A dihydrate using hydroxypropyl cellulose (HPC) as an additive yielded modified crystals with improved compaction properties [19].
4. What is the relationship between supersaturation control and crystal morphology?
Supersaturation is the driving force for both nucleation and crystal growth, and its level directly dictates the final crystal habit. Operating within a controlled, moderate supersaturation level in the metastable zone promotes manageable crystal growth, often leading to more uniform and desirable morphologies [77] [79]. Conversely, excessively high supersaturation, often resulting from rapid cooling, drives the solution into an unstable zone where spontaneous and rapid nucleation occurs. This typically results in fine, needle-like crystals or agglomerates with poor size control and unfavorable morphology for downstream processing [26] [79].
Troubleshooting Guides
Problem 1: Slow Filtration and Poor Cake Washing
Symptoms: Filtration cycles are excessively long; the filter cake is dense and retains high moisture content; wash solvent does not permeate the cake evenly.
Root Cause: The API crystallizes in a needle-like or thin plate-like habit, which packs densely and leaves little void space for liquid passage [77] [76].
Solutions:
- Modify the Crystallization Solvent: Switch to a solvent system that promotes more isotropic growth. For example, a study on ascorbic acid showed that crystallization from pure water produced prismatic crystals, while switching to water-isopropanol mixtures resulted in a detrimental needle-like habit [80].
- Implement a Temperature Cycling (TC) Strategy: Subject the crystal slurry to controlled heating and cooling cycles. This process, known as Ostwald ripening, dissolves fine crystals and allows their redeposition onto larger crystals, effectively rounding out sharp edges and reducing the aspect ratio (L/D) [77].
- Employ Spherical Crystallization: Use techniques like spherical agglomeration (SA) or emulsion solvent diffusion (ESD) to build large, spherical agglomerates from primary crystals. These agglomerates pack with high porosity, dramatically improving filtration efficiency [78].
Problem 2: Poor Powder Flow and Inconsistent Die Filling
Symptoms: Powder does not flow consistently from the hopper; erratic tablet weight control; observed bridging or rat-holing in powder handling equipment.
Root Cause: The crystal morphology (e.g., needles, flakes) creates high interparticle cohesion and friction [78] [76].
Solutions:
- Optimize Supersaturation Control (SSC): Use Process Analytical Technology (PAT) tools like ATR-FTIR or refractometers to maintain the concentration profile close to the solubility curve within the metastable zone. This controlled growth can reduce the L/D ratio of needle-shaped crystals like α-PABA, producing more block-like particles [77] [79].
- Induce Spherical Agglomeration: As in Problem 1, spherical crystallization is a direct solution. Spherical agglomerates have a larger particle size, a narrow size distribution, and a spherical shape, all of which contribute to excellent flowability [78].
- Introduce Ultrasound during Nucleation: Applying ultrasound at the nucleation stage generates a large number of uniform seed crystals. This leads to a more synchronized growth and a final product with a narrower crystal size distribution and improved flow properties [77].
Problem 3: Tablet Capping, Lamination, or Low Tensile Strength
Symptoms: Tablets split horizontally (capping) or vertically (lamination) after compression; tablets are mechanically weak and fail friability tests.
Root Cause: The crystal habit may lack the ability to form strong interparticle bonds upon compression. Needle-like crystals can align and entrap air, while some compact crystal forms may not bond effectively [78] [19].
Solutions:
- Use Additive-Mediated Crystallization: Introduce a pharmaceutically accepted polymer (e.g., Hydroxypropyl Cellulose - HPC) during crystallization. The additive can selectively adsorb to specific crystal faces, inhibiting their growth and modifying the overall habit to a more compact form with better compaction properties [19].
- Leverage Spherical Crystallization: Agglomerated crystals often exhibit superior compactability because they are designed to fragment under pressure, creating new binding surfaces. This fragmentation increases the number of contact points for bonding, resulting in tablets with higher tensile strength [78].
- Control the Cooling Profile: Avoid linear cooling, which can exacerbate needle growth. Instead, use a controlled cooling profile based on supersaturation to encourage more uniform crystal growth in all directions [77].
Experimental Protocols for Morphology Control
Protocol 1: Supersaturation-Controlled Cooling Crystallization
This protocol uses real-time concentration monitoring to maintain an optimal supersaturation level, promoting desirable crystal growth [77] [79].
- Objective: To produce crystals with a uniform size distribution and block-like habit by avoiding high, uncontrolled supersaturation.
- Materials:
- Jacketed crystallizer with temperature control
- PAT tool: ATR-FTIR spectrometer or Process Refractometer (e.g., Vaisala Polaris PR53AC)
- Agitator
- API and solvent system
- Method:
- Determine Solubility & Metastable Zone: Characterize the system by measuring the API's solubility and metastable zone width (MSZW) in the chosen solvent.
- Develop Calibration Model: Establish a calibration curve correlating the PAT signal (e.g., IR peak intensity or refractive index) with solution concentration and temperature.
- Execute Controlled Crystallization:
- Dissolve the API at an elevated temperature to create a saturated solution.
- Cool the solution to a temperature within the metastable zone, just above the saturation point.
- Use the PAT tool to monitor the concentration in real-time. The controller should adjust the crystallizer's temperature to maintain a constant, moderate level of supersaturation throughout the growth phase, precisely following a trajectory near the solubility curve.
- Isolate and characterize the product.
Protocol 2: Spherical Crystallization via Spherical Agglomeration (SA)
This protocol describes a method to transform fine, irregular crystals into free-flowing, compactable spherical agglomerates in a single step [78].
- Objective: To produce spherical agglomerates of an API to directly address issues of poor flow, filtration, and compactability.
- Materials:
- Good solvent (solvent in which the API is highly soluble)
- Poor solvent (solvent in which the API has low solubility; must be miscible with the good solvent)
- Bridging liquid (immiscible with the poor solvent, but wets the API crystals)
- Standard laboratory vessel with agitator
- Method:
- Dissolve the API in a minimal amount of the good solvent.
- While stirring, pour this solution into a larger volume of the poor solvent. This causes the rapid precipitation of fine API crystals.
- Add a small, calculated amount of the bridging liquid. The bridging liquid will wet the precipitated crystals and, under the correct stirring conditions, form liquid bridges between them.
- Continue agitation. The capillary forces of the liquid bridges will pull the crystals together, forming dense, spherical agglomerates.
- Isolate the agglomerates by filtration and allow the residual bridging liquid to evaporate.
Protocol 3: Habit Modification using Polymer Additives
This protocol uses a pharmaceutical polymer to selectively inhibit the growth of specific crystal faces, leading to a modified crystal habit [19].
- Objective: To change the crystal habit of an API from a needle/plate to a more equidimensional shape to improve downstream performance.
- Materials:
- API
- Crystallization solvent
- Habit modifier (e.g., Hydroxypropyl Cellulose - HPC)
- Standard crystallization setup
- Method:
- Prepare a saturated solution of the API in the chosen solvent at an elevated temperature.
- Dissolve a known concentration of the polymer additive (e.g., HPC at 0.45-4.5 wt%) in the same solvent or in an anti-solvent.
- Initiate crystallization by cooling or adding anti-solvent. The additive molecules will selectively adsorb onto specific growing crystal faces, reducing their growth rate and resulting in a habit-modified product.
- Filter, dry, and characterize the crystals for morphology and tableting properties.
Data Presentation
Table 1: Impact of Crystal Habit on Key Powder and Processing Properties
| Crystal Habit | Flowability | Packability / Bulk Density | Filtration Efficiency | Compactability / Tablet Strength |
|---|---|---|---|---|
| Needle/ Acicular | Very Poor (high cohesion, bridging) [77] | Low, highly variable [77] | Poor (forms impermeable cake) [77] [76] | Poor (capping, lamination) [77] |
| Thin Plate/ Flake | Poor | Moderate to Low | Poor (forms impermeable cake) [77] | Variable |
| Block/ Prism | Good | High | Good [77] | Good |
| Spherical Agglomerates | Excellent (low angle of repose) [78] | High (low compressibility) [78] | Excellent (high porosity cake) [78] | Excellent (fracture creates binding surfaces) [78] |
| Control Strategy | Mechanism of Action | Typical Morphological Outcome | Key Process Parameters |
|---|---|---|---|
| Supersaturation Control (SSC) [77] [79] | Maintains driving force for growth in metastable zone, preventing runaway nucleation. | Reduces aspect ratio (L/D); promotes more uniform, larger crystals. | Supersaturation setpoint, cooling profile based on solubility. |
| Spherical Crystallization [78] | Uses bridging liquid to agglomerate fine crystals into spheres via capillary forces. | Directly produces spherical agglomerates. | Good/poor solvent ratio, bridging liquid type and volume, agitation speed. |
| Additive Engineering [19] | Polymer selectively adsorbs to specific crystal faces, inhibiting their growth. | Can transform needles to plates or blocks; depends on additive and crystal structure. | Additive type, concentration, crystallization method. |
| Temperature Cycling (TC) [77] | Dissolves fines and deposits material on larger crystals (Ostwald ripening). | "Rounds out" crystals; reduces fines; narrows CSD. | Cycling amplitude and frequency. |
| Ultrasound Intensification [77] | Generates cavitation, producing a shower of uniform seed crystals. | Produces uniform seed crystals, leading to narrower Crystal Size Distribution (CSD). | Ultrasound power, duration of application. |
Workflow Visualization
The following diagram illustrates a logical workflow for troubleshooting downstream processing issues through crystal morphology optimization, integrating the strategies discussed in this guide.
The Scientist's Toolkit: Key Research Reagent Solutions
The following table lists essential materials and instruments used in the experimental protocols for crystal morphology control.
| Item / Technique | Function / Role in Morphology Control | Example from Literature |
|---|---|---|
| Process Analytical Technology (PAT) | Enables real-time monitoring and control of crystallization processes. | ATR-FTIR for concentration [77]; Refractometer for supersaturation [79]; PVM/FBRM for particle size/shape [77]. |
| Hydroxypropyl Cellulose (HPC) | A pharmaceutically accepted polymer used as a habit modifier in additive-mediated crystallization. | Selectively adsorbs to specific faces of erythromycin A dihydrate, modifying crystal habit and improving tableting properties [19]. |
| Solvent Systems (Binary Mixtures) | The choice of solvent can drastically alter the relative growth rates of crystal faces. | Water-methanol/ethanol/isopropanol mixtures used to modify the crystal habit of ascorbic acid from prisms to needles [80]. |
| Bridging Liquid | In spherical agglomeration, a liquid immiscible with the poor solvent is used to bind crystals into spheres. | A third solvent (e.g., chloroform, dichloromethane) wets the crystals and forms liquid bridges via capillary forces [78]. |
| Ultrasound Probe | An intensification technique to generate uniform secondary nucleation, creating a narrow seed distribution. | Application to α-PABA crystallization produced uniform seeds, leading to improved final product particle aspects [77]. |
Welcome to the Technical Support Center for Morphology and Bioavailability Research. This resource provides targeted troubleshooting guides and experimental protocols to help researchers overcome common challenges in controlling crystal morphology to optimize drug supersaturation, bioavailability, and product performance. The content is framed within the broader thesis that precise manipulation of supersaturation thresholds through advanced crystal morphology control represents a critical pathway for enhancing biopharmaceutical performance.
Frequently Asked Questions (FAQs)
FAQ 1: My in vitro permeability assay appears overly sensitive to excipient effects, leading to inconsistent results. How can I control this?
- Answer: Oversensitivity in cell-based permeability assays (e.g., using MDCK-wt monolayers) is a known challenge. To control this, you can employ specific experimental and statistical approaches. Experimentally, consider using a high-permeability marker like minoxidil for normalization. Statistically, using ANOVA (p < 0.01) to identify excipient effects has been shown to reduce oversensitivity compared to confidence interval (CI90) approaches, which can be more sensitive to variations. Choose the statistical method based on your goal: ANOVA for robust effect identification or CI90 for sensitive formulation comparisons [81].
FAQ 2: How can I control the crystal habit and shape of my active pharmaceutical ingredient (API) to enhance dissolution?
- Answer: Crystal habit can be modulated by controlling the crystallization environment. A demonstrated strategy involves adjusting the solution's acidity. For instance, with monoammonium phosphate, adding alum (ammonium aluminum sulfate) to the supersaturated solution progressively changes the crystal shape from thick prisms to sharp, needle-like structures. The degree of change is proportional to the amount of alum added, allowing for precise morphological control [24].
FAQ 3: What strategies can I use to regulate nucleation and favor crystal growth in my membrane crystallisation process?
- Answer: In membrane distillation crystallisation (MDC), facile supersaturation control strategies are key. You can:
- Adjust the membrane area to modify the concentration rate without altering boundary layer dynamics. A higher rate increases supersaturation, favoring a homogeneous primary nucleation pathway.
- Use in-line filtration to retain crystals in the crystalliser bulk, reducing scaling on membrane surfaces. This allows a consistent supersaturation rate to be maintained, promoting crystal growth over primary nucleation and resulting in larger crystal sizes [2].
- Answer: In membrane distillation crystallisation (MDC), facile supersaturation control strategies are key. You can:
FAQ 4: My crystals are too small for X-ray diffraction analysis. What factors should I investigate?
- Answer: Small crystal size is often due to excessive nucleation sites or mechanical disturbance. To grow larger, single crystals:
- Minimize Nucleation: Ensure your crystal growing vessel is exceptionally clean to avoid dust and particulates that act as nucleation sites.
- Reduce Mechanical Agitation: Place the crystallization setup in a quiet, low-traffic area away from vibrations (e.g., not near vacuum pumps) and avoid frequent handling.
- Optimize Solvent: Avoid solvents where your compound forms highly supersaturated solutions, as this promotes many small crystals. Choose a solvent where your compound is moderately soluble [82].
- Answer: Small crystal size is often due to excessive nucleation sites or mechanical disturbance. To grow larger, single crystals:
FAQ 5: My protein crystals crack during ligand soaking. What should I do?
- Answer: Crystal cracking typically results from mechanical strain induced by the soaking process. To mitigate this:
- Reduce the strain by soaking at a lower ligand concentration and/or for a shorter time period.
- Test if the cracking is specific to certain compounds, which may bind at crystal contacts and disrupt packing. For these, apply gentler soaking conditions.
- If available, test the soak with a known ligand to determine if active-site binding causes a conformational shift that leads to cracking [83].
- Answer: Crystal cracking typically results from mechanical strain induced by the soaking process. To mitigate this:
Troubleshooting Guides
Problem: Poor Bioavailability Due to Low Solubility and Dissolution Rate
Potential Causes and Solutions:
| Potential Cause | Diagnostic Experiments | Corrective Action & Experimental Protocols |
|---|---|---|
| Suboptimal Crystal Morphology / Habit | - Perform X-ray Powder Diffraction (XRPD) to identify polymorphic form.- Conduct dynamic vapor sorption (DVS) to assess hydformation tendency.- Measure contact angle to determine surface wettability. | - Utilize solvent engineering to target a polymorph with higher apparent solubility.- Protocol (Slow Cooling): Prepare a saturated solution of the API in a suitable solvent by heating to just below the solvent's boiling point. Transfer to a clean test tube, stopper, and place in a Dewar flask filled with hot water. Allow to cool slowly over several days [82].- Protocol (Vapor Diffusion): Dissolve the substance in a primary solvent (S1) and place it in a test tube. Place a second solvent (S2), in which the solute is less soluble, in a closed beaker. Place the test tube inside the beaker and seal it. The slow diffusion of S2 into S1 will reduce solubility and promote crystal growth [82]. |
| Insufficient Supersaturation Maintenance | - Conduct solvent-shift nucleation studies to determine the metastable zone width (MZW).- Perform pH-metric titration to assess supersaturation lifetime. | - Employ polymer-based precipitation inhibitors (e.g., HPMC, PVP) to stabilize the supersaturated state.- Protocol (Supersaturation Control in MDC): As a model for controlling nucleation, use membrane area to adjust the supersaturation rate. Combine this with in-line filtration to segregate the crystal phase in the bulk solution, which helps sustain supersaturation for longer hold-up times and favors growth over nucleation [2]. |
Problem: High Variability in In Vitro Permeability Assays
Potential Causes and Solutions:
| Potential Cause | Diagnostic Experiments | Corrective Action & Experimental Protocols |
|---|---|---|
| Oversensitivity to Excipient Effects | - Compare permeability ratios (PR = treatment/control) for your drug in the presence and absence of common excipients.- Use a high-permeability marker (e.g., minoxidil) for normalization. | - Statistically analyze the effects using ANOVA (p < 0.01) rather than confidence intervals (CI90) to reduce the identification of false-positive excipient effects [81].- If comparing two formulations specifically, the CI90 approach may be more appropriate, but ANOVA provides a more robust general assessment. |
| Inconsistent Crystal Properties between Batches | - Characterize crystal size distribution (CSD) and morphology (e.g., by microscopy) of the drug substance used in the assay. | - Strictly control the crystallization process parameters (e.g., cooling rate, agitation) to ensure consistent crystal morphology and size, which directly impact dissolution and available drug concentration [2]. |
Experimental Protocols for Key Methodologies
Protocol 1: Formulating an Inhalable Powder with Optimized Morphology and Aerodynamics
This protocol is adapted from research on developing co-spray-dried inhalable microparticles [81].
Objective: To create microparticles with enhanced physicochemical and aerodynamic properties for efficient lung deposition.
Materials:
- Active Pharmaceutical Ingredient (API) (e.g., Theophylline)
- Fine carriers (e.g., Raffinose, Leucine, Glycine)
- Solvent (e.g., Water)
- Mini spray-dryer
- Characterization equipment (e.g., Laser diffraction for particle size, Andersen Cascade Impactor for aerodynamics)
Procedure:
- Prepare Solution: Dissolve the API and your chosen carrier combination (e.g., raffinose with leucine or glycine) in the solvent.
- Spray-Drying: Process the solution using a mini spray-dryer to produce the dry powder microparticles.
- Characterization:
- Determine the Mass Median Aerodynamic Diameter (MMAD) and Fine Particle Fraction (FPF %) to evaluate aerodynamic performance. A successful formulation should ideally have an MMAD between 1-5 µm and a high FPF for deep lung deposition.
- Analyze particle size distribution, with a D [0.5] value of 3.99–5.96 µm being typical for inhalable powders.
- Perform structural analysis (e.g., XRD) to confirm an amorphous or partially amorphous structure, which is associated with solubility enhancement.
Protocol 2: Controlling Crystal Habit by Modifying Solution Conditions
This protocol is based on techniques for growing crystals with defined morphologies [24].
Objective: To systematically investigate how additives influence the crystal habit of an API.
Materials:
- API
- Solvent
- Additive (e.g., Alum)
- Beakers, stirrers, filters
- Microscopy for visualization
Procedure:
- Prepare Saturated Solution: Dissolve your API in a chosen solvent at an elevated temperature to create a saturated solution.
- Create Additive Stock Solutions: Prepare a series of solutions with increasing concentrations of the habit-modifying additive (e.g., 0, 0.25, 0.5, 0.75, 1.0 g of alum per 100 mL of solvent).
- Crystallization Setup: Add a seed crystal of your API to each solution or allow for spontaneous nucleation.
- Crystal Growth: Let the crystals grow under static, undisturbed conditions for a set period (e.g., 3-7 days).
- Analysis: Harvest the crystals and use optical or electron microscopy to characterize and compare the crystal habit (shape) across the different additive conditions.
Visualization Diagrams
Diagram 1: Morphology-Bioavailability Optimization Pathway
Diagram 2: Supersaturation Control Logic
The Scientist's Toolkit: Research Reagent Solutions
- Monoammonium Phosphate (MAP): A model compound for crystal growth studies. Its habit can be easily controlled with additives like alum, making it an excellent training tool for understanding crystallization principles [24].
- Polymers (HPMC, PVP, PVPVA): Used as precipitation inhibitors to maintain supersaturation after dissolution of an amorphous solid dispersion, thereby improving bioavailability.
- Raffinose-Amino Acid Carriers (Leucine, Glycine): Fine co-spray-dried carriers used in the development of inhalable dry powder formulations to enhance aerodynamic properties and lung deposition [81].
- Alum (Ammonium Aluminum Sulfate): A common additive used to modify crystal habit, specifically to promote the growth of needle-like or spiked crystal structures from an otherwise prismatic morphology [24].
- Cell Monolayers (e.g., MDCK, Caco-2): Standard in vitro models for assessing drug permeability. Oversensitivity to excipients in these systems requires careful experimental and statistical control [81].
Economic and Quality Trade-offs in Implementing Advanced Control Strategies
Troubleshooting Guides
Guide 1: Resolving Crystal Size and Morphology Inconsistencies
Problem: Despite a consistent supersaturation profile, final crystal batches show unacceptable variations in size distribution (CSD) and crystal habit.
Application Context: This issue is common in membrane distillation crystallisation (MDC) when the system is not optimally positioned within the metastable zone to favor growth over primary nucleation [2].
Diagnostic Procedure:
Step 1: Verify Supersaturation Control
- Check the concentration rate and induction time data. A shortened induction time and broadened metastable zone width indicate a shift towards homogeneous primary nucleation, which can lead to inconsistent crystal size [2].
- Confirm that the supersaturation control strategy can modulate the system within specific regions of the metastable zone.
Step 2: Check for Scaling and Crystal Retention
- Inspect the membrane and crystallizer for scale deposition, which reduces active surface area and disrupts supersaturation control.
- Verify the operation of the in-line filtration system. Poor crystal retention in the crystallizer leads to product loss and prevents the sustained supersaturation rates needed for growth-dominated processes [2].
Step 3: Analyze Process Data
- Use population balance models to analyze nucleation and growth rates. A high nucleation rate relative to growth rate, coupled with solvent desaturation, confirms the system is favoring nucleation.
Resolution:
- Adjust Membrane Area: Increase the effective membrane area to modify kinetics and achieve a supersaturation level that favors crystal growth.
- Optimize Hold-up Time: Increase the post-induction hold-up time. This allows crystal growth to desaturate the solvent, suppressing further nucleation and leading to larger, more uniform crystals [2].
- Ensure Proper Seeding: If using seeding, ensure seeds are added at the correct supersaturation level and are of consistent quality to guide growth.
Guide 2: Addressing Poor Economic Performance of Advanced Control Strategy
Problem: An implemented advanced control strategy (e.g., MPC) is not delivering the expected economic benefits, with high operating costs or excessive variability.
Application Context: Economic underperformance often stems from a mismatch between the controller's design (e.g., an over-aggressive benchmark like Minimum Variance Control) and realistic process constraints and costs [84].
Diagnostic Procedure:
Step 1: Performance Benchmarking
- Compare current controller performance against a more realistic Linear Quadratic Gaussian (LQG) benchmark. The LQG benchmark considers both control effort and output performance, providing a more physically meaningful assessment than MVC [84].
- Perform a step-test (with operational approval) to observe process response. If the process variable (PV) oscillation stops or reduces when the controller is in manual mode, the controller is likely over-tuned and its aggressive action is the source of instability [85].
Step 2: Stochastic Optimization Check
- Determine if the control system's economic assessment and setpoints account for process uncertainties (e.g., feed fluctuations). A deterministic approach may lead to operating points that are frequently violated in practice, eroding economic gains [84].
- Formulate the performance assessment as a stochastic optimization problem to identify an operating point that is robust to expected disturbances.
Resolution:
- Controller Re-tuning: Re-tune the controller using the LQG benchmark to find a more economically optimal balance between variability reduction and control effort [84].
- Update Economic Model: Incorporate a stochastic framework into the controller's economic optimizer to explicitly handle uncertainty, ensuring setpoints are maintained at the correct risk-adjusted levels for each variable [84].
Frequently Asked Questions (FAQs)
FAQ 1: How can I quantify the economic benefits of improving my crystallization control system?
The economic benefit is primarily realized by reducing the variability of key process variables, which allows you to safely shift the mean operating point closer to a product specification or operational constraint without increasing the risk of violation. The core calculation involves [84]:
- Estimating the potential reduction in variability (e.g., standard deviation) of your Critical Quality Attributes (CQAs) achievable with an improved controller (using benchmarks like LQG).
- Calculating how much closer to the constraint (e.g., a supersaturation limit to avoid nucleation) the process can now operate.
- Translating this shift in the operating point into an economic value, such as increased yield, reduced energy consumption, or higher throughput.
FAQ 2: What is the difference between 'voluntary isolation' and 'targeted isolation' control strategies, and how do they create a quality-economic trade-off?
These concepts, drawn from epidemic control, are analogous to process control strategies [86]:
- Voluntary Isolation (Decentralized Control): Individual units or loops react to local disturbances without coordination. This is like susceptible individuals isolating themselves. It leads to widespread, sub-optimal suppression of activity, causing a significant economic recession (high operating cost) for the process but can effectively control the "outbreak" (process variability).
- Targeted Isolation (Coordinated Control): A superior strategy that specifically identifies and isolates only the disruptive elements (the "infectious" part of the process). This allows the rest of the system to operate near optimal conditions. By resolving this coordination failure, targeted isolation avoids the sharp trade-off, suppressing variability without imposing large economic costs [86]. In process terms, this could mean using a advanced controller like MPC to pinpoint and manage a specific disruptive variable rather than slowing down the entire process.
FAQ 3: Why does my control system show oscillatory behavior even after tuning, and how can I diagnose the root cause?
Oscillations can stem from the controller itself or from external load fluctuations.
- Simple Diagnostic Test: Place the controller in manual mode. Observe the trend of the Process Variable (PV) [85].
- If the PV continues or increases its oscillation, the controller was likely helping to counteract an external oscillatory disturbance. The controller may need to be tuned more aggressively.
- If the PV oscillation stops or significantly reduces, the controller's own action is the source of the problem (over-tuning) [85].
- Further Investigation: An oscillatory load disturbance could be coming from another, poorly tuned control loop elsewhere in the system that is interacting with your loop.
Quantitative Data on Advanced Control Performance
The following table summarizes key performance indicators (KPIs) for different advanced control strategies as applied to a building energy system, illustrating typical trade-offs. While the application is building control, the relative performance characteristics are illustrative of trends in chemical process control [87].
Table 1: Benchmarking Advanced Control Strategies (Annual Simulation Data)
| Control Strategy | Energy Savings vs. Baseline | Thermal Discomfort (Constraint Violation) | Key Characteristics |
|---|---|---|---|
| Rule-Based Control (Baseline) | 0.0% | Highest | Simple, low know-how; poor adaptability |
| Black-Box MPC | 8.4% | Low | High performance; lower interpretability |
| White-Box MPC | 7.4% | Low | High interpretability; high modeling know-how |
| Gray-Box MPC | 7.2% | Low | Balance of interpretability and flexibility |
| Reinforcement Learning | 7.1% | Low | High adaptability; high computational effort |
| Approximate MPC | 4.8% | Low | Low computational effort; reduced performance |
Data adapted from evaluation of advanced controllers for building energy systems [87].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Materials and Reagents for Controlled Crystallization Experiments
| Item | Function in Experiment |
|---|---|
| Membrane Crystallizer | Provides a controlled interface for solvent removal, enabling precise supersaturation generation by adjusting membrane area and related kinetics [2]. |
| In-line Filtration Unit | Retains crystals within the crystallizer bulk, reducing scaling on equipment surfaces and enabling longer, growth-favoring hold-up times [2]. |
| Process Analytical Technology (PAT)(e.g., FBRM, PVM, NMR) | Monitors crystal size, count, and morphology in real-time, providing essential feedback for supersaturation control strategies [88]. |
| Computational Modeling Tools(echanistic & Data-Driven) | Enables resource-efficient, uncertainty-aware digital design of processes, predicting outcomes and optimizing control parameters before physical experiments [88]. |
Experimental Protocol: Supersaturation Control for Crystal Growth
Aim: To establish a robust protocol for using supersaturation control to regulate nucleation and growth, thereby optimizing crystal morphology and size distribution.
Methodology:
System Calibration:
- Characterize the metastable zone width (MZW) for your compound-solvent system under various concentration rates.
- Establish the relationship between membrane area (or other kinetic control parameter), induction time, and supersaturation at nucleation [2].
Induction Phase:
- Initiate solvent removal via the membrane to generate supersaturation.
- Monitor the solution until the induction point is detected by a sharp drop in concentration or via PAT tools.
Post-Induction Growth Phase:
- Key Step: Upon induction, modulate the supersaturation rate (e.g., by adjusting membrane area) to reposition the system within a region of the metastable zone that favors crystal growth over further primary nucleation [2].
- Activate in-line filtration to maintain crystals in the bulk solution, creating two discrete regions of supersaturation: a low level at the crystal surfaces for growth and a higher level in the bulk that is carefully controlled.
Hold-up and Harvest:
- Sustain the optimized supersaturation rate for a sufficient hold-up time. A longer, controlled hold-up time allows growth to dominate, desaturating the solvent and resulting in larger crystals [2].
- Terminate the process and harvest the product.
Workflow Diagram
Control Strategy Decision Logic
Conclusion
Mastering supersaturation control is paramount for precise crystal morphology engineering, directly impacting critical attributes from drug bioavailability to material performance. The integration of theoretical prediction models with advanced control strategies like continuous flow reactors and additive-mediation enables a shift from empirical tuning to rational design. Future progress hinges on further developing in-line monitoring and closed-loop control systems to achieve fully autonomous crystallization processes. For biomedical research, these advancements promise more effective drug formulations with tailored dissolution profiles and enhanced stability, ultimately accelerating the development of higher-quality pharmaceuticals and advanced materials.
References
- 1. Supersaturation [https://en.wikipedia.org/wiki/Supersaturation]
- 2. Facile supersaturation control strategies for regulating ... [https://www.sciencedirect.com/science/article/pii/S1383586625028497]
- 3. 3.6F: Troubleshooting [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_Lab_Techniques_(Nichols)/03%3A_Crystallization/3.06%3A_Step-by-Step_Procedures/3.6F%3A_Troubleshooting]
- 4. Optimisation – Biomolecular Crystallisation and Characterisation [https://research.csiro.au/crystal/biomolecular-crystallisation-and-characterisation/guides/c3-optimisation/]
- 5. Crystal Morphology and Associated Face-Specific Growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12593351/]
- 6. Troubleshooting Tips for Crystallization Processes [https://www.linkedin.com/advice/0/how-can-you-troubleshoot-crystallization-process-skztf]
- 7. Optimisation of the crystallisation process through ... [https://www.nature.com/articles/s41598-024-84285-4]
- 8. Efficient optimization of crystallization conditions by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2203341/]
- 9. Observing growth of metallic crystals inside liquid ... [https://www.nature.com/articles/s41467-025-66249-y]
- 10. Robustness of classical nucleation theory to chemical ... [https://arxiv.org/html/2510.01420v1]
- 11. Unifying nucleation and crystal growth mechanisms in ... [https://www.sciencedirect.com/science/article/pii/S0376738825003345]
- 12. A Structured Approach To Cope with Impurities during ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7461122/]
- 13. Crystallization from a Supersaturated Solution [https://chem.rutgers.edu/cldf-demos/1031-cldf-demo-crystallization]
- 14. Supersaturation and Crystallization [https://sciencedemonstrations.fas.harvard.edu/presentations/supersaturation-and-crystallization]
- 15. A general protocol for the crystallization of membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4075773/]
- 16. Crystallization of APIs: Methods and Challenges [https://www.bocsci.com/resources/crystallization-of-apis-methods-and-challenges.html?srsltid=AfmBOorgIZADR55e0eQ4YaVWPQ67BJeNvOwHmxs-ql9zyxHjZ6zFYjus]
- 17. Kinetic and geometric determination of the growth ... [https://www.sciencedirect.com/science/article/abs/pii/S0960897405000380]
- 18. Crystal Morphology Prediction Models and Regulating ... [https://www.mdpi.com/2073-4352/14/6/484]
- 19. Crystal Morphology Engineering of Pharmaceutical Solids [https://pmc.ncbi.nlm.nih.gov/articles/PMC2663677/]
- 20. A molecular dynamics simulation of solvent effects on the ... [https://www.sciencedirect.com/science/article/abs/pii/S0304389409014794]
- 21. Molecular dynamics simulations of solvent effects on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9049282/]
- 22. Advancing crystal growth prediction: An adaptive kMC ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250924007723]
- 23. Solutions to Common Crystal Growing Problems [https://sciencenotes.org/solutions-to-common-crystal-growing-problems/]
- 24. Growing Crystals: How to Make Beautiful Crystals at Home [https://crystalverse.com/growing-crystals/]
- 25. Rapid Assessment of Crystal Nucleation and Growth Kinetics [https://pmc.ncbi.nlm.nih.gov/articles/PMC10326855/]
- 26. Supersaturation-controlled morphology and phase evolution ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0335668]
- 27. Origin and use of crystallization phase diagrams - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4356298/]
- 28. Crystallization by Phase Diagram [https://www.douglas.co.uk/rep1.htm]
- 29. Process Analytical Technology Obtained Metastable Zone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11944372/]
- 30. Facile supersaturation control strategies for regulating ... [https://www.sciencedirect.com/science/article/abs/pii/S1383586625028497]
- 31. Phase Diagram Determination and Process Development ... [https://www.mdpi.com/2073-4352/12/8/1102]
- 32. Optimization of crystallization conditions for biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4231845/]
- 33. Continuous Flow Reactor Systems: A Guide [https://www.amt.uk/continuous-flow-reactors]
- 34. Continuous Flow Process [https://www.corning.com/worldwide/en/innovation/corning-emerging-innovations/advanced-flow-reactors/Corning-Advanced-Flow-reactors-take-continuous-flow-process-production-from-lab-to-full-scale-manufacturing.html]
- 35. Advanced control of crystallization processes [https://www.sciencedirect.com/science/article/pii/S1367578802800165]
- 36. Beginner's Guide to Flow Chemistry [https://helgroup.com/blog/beginners-guide-to-flow-chemistry/?srsltid=AfmBOopQ1CWJYGXLq8RLNXNz2xJpM6KhrLQ0T5hFXx3ku1EXBaGvugjg]
- 37. Development and Optimization of VGF-GaAs Crystal ... [https://www.mdpi.com/2073-4352/11/10/1218]
- 38. Complex investigation of the effect mechanism ... [https://www.sciencedirect.com/science/article/pii/S0378517325008312]
- 39. How crystallization additives govern halide perovskite ... [https://www.nature.com/articles/s41467-025-65484-7]
- 40. Crystallization process guide | industrial use [https://www.andritz.com/separation-en/insights/crystallizers-guide]
- 41. Influence of Solvent Selection on the Crystallizability and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11220793/]
- 42. Solvent–antisolvent interactions in metal halide perovskites [https://pubs.rsc.org/en/content/articlehtml/2023/cc/d3cc02090h]
- 43. Supercritical Antisolvent Process for Pharmaceutical ... [https://www.mdpi.com/2227-9717/8/8/938]
- 44. Problems with Recrystallisations - Chemistry Teaching Labs [https://chemtl.york.ac.uk/techniques/purification/recrystallisation/problems-with-recrystallisations]
- 45. Applications of Supercritical Anti-Solvent Process in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8067079/]
- 46. What is PAT? [https://www.bruker.com/en/products-and-solutions/process-analytical-technology/what-is-pat.html]
- 47. Process Analytical Technology Tools for Monitoring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8234957/]
- 48. Mechanistic insights using carprofen as a model drug [https://www.sciencedirect.com/science/article/pii/S0147651325015179]
- 49. Supersaturation-controlled morphology and phase evolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12571270/]
- 50. Synthesis and characterization of two-faced brush-like ... [https://www.sciencedirect.com/science/article/abs/pii/S0306261924003271]
- 51. Structure-directed synthesis of bimetallic ZIF-67 LDH ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra01889g]
- 52. Ultrafast Microwave-Assisted Synthesis of Porous NiCo ... [https://www.mdpi.com/1420-3049/29/11/2546]
- 53. Modulation of the structure and morphology of NiCo2S4 ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979725004084]
- 54. NiCo LDH deposited on novel LIG-derived substrates with ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838825023333]
- 55. Dispersions in crystal nucleation and growth rates [https://www.sciencedirect.com/science/article/abs/pii/S0009250913007835]
- 56. The effect of size-dependent growth and environmental ... [https://www.sciencedirect.com/science/article/abs/pii/S0040580906001122]
- 57. Pharmaceutical Crystallization in drug development [https://www.syrris.com/crystallization-in-drug-development/]
- 58. Size‐Dependent Growth and the Development ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4242387/]
- 59. Theoretical Analysis of the Factors Determining the Crystal ... [https://www.mdpi.com/2073-4352/15/7/653]
- 60. Supersaturation-controlled morphology and phase ... [https://pubmed.ncbi.nlm.nih.gov/41160580/]
- 61. Supersaturation-Based Drug Delivery Systems: Strategy for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9101434/]
- 62. Analytical and production seeding techniques [https://www.sciencedirect.com/science/article/abs/pii/S1046202305801458]
- 63. Understand seeding through secondary nucleation ... [https://www.crystallizationsystems.com/news/understand-seeding-through-secondary-nucleation-measurements-with-the-crystalline-instrument/]
- 64. Controlled crystallization and morphology optimization of ... [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00601e]
- 65. Polymorphic phase transitions: Macroscopic theory and ... [https://www.sciencedirect.com/science/article/abs/pii/S0169409X17301989]
- 66. formation in Crystalline -field simulations of binary... morphology phase [https://pubs.rsc.org/en/content/articlehtml/2023/tc/d3tc03047d]
- 67. Progress of Pharmaceutical Continuous Crystallization [https://www.sciencedirect.com/science/article/pii/S2095809917304277]
- 68. Crystallization of APIs: Methods and Challenges [https://www.bocsci.com/resources/crystallization-of-apis-methods-and-challenges.html?srsltid=AfmBOoqL2CEL8vzNgZ5wVZxpPzn2d6R5ePKQvcBNheA3dH_Cwjj8ICJP]
- 69. Modelling and control of crystallization process [https://www.sciencedirect.com/science/article/pii/S2405653716301166]
- 70. a comparison between batch and continuous crystallizers [https://www.sciencedirect.com/science/article/pii/0304386X7690027X]
- 71. Control of Batch and Continuous Crystallization Processes ... [https://www.sciencedirect.com/science/article/abs/pii/B9780323885065502114]
- 72. CrystalMaker: Overview [https://crystalmaker.com/crystalmaker/]
- 73. CrystalGym: A New Benchmark for Materials Discovery ... [https://arxiv.org/html/2509.23156v1]
- 74. Experimental techniques for identifying operating variables ... [https://www.sciencedirect.com/science/article/abs/pii/S1876107009000650]
- 75. Controlling colloidal crystals via morphing energy landscapes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7688337/]
- 76. The impact of crystal habit on the pharmaceutical properties of ... [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d4ce01170h]
- 77. Crystal size and morphology optimization of α-PABA [https://www.sciencedirect.com/science/article/abs/pii/S0263876222000375]
- 78. How spherical crystallization improves direct tableting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3845996/]
- 79. Cooling crystallization monitoring and control in API ... [https://www.vaisala.com/en/blog/2025-09/cooling-crystallization-monitoring-and-control-api-production-processes-ri-measurements]
- 80. Industrial importance of controlling crystal habit [https://www.crystallizationsystems.com/news/industrial-importance-of-controlling-crystal-habit/]
- 81. Drug Product Performance: Bioavailability, Relative ... [https://www.mdpi.com/journal/pharmaceutics/special_issues/M7Y0G03U6X]
- 82. Crystal Growing Guide [http://www1.udel.edu/chem/xray/Crystal_Growing_Guide.html]
- 83. Crystallography FAQ | zenfragments-copy - Zenobia Fragments [https://www.zenobiafragments.com/crystallography-faq]
- 84. Economic performance assessment of advanced process ... [https://www.sciencedirect.com/science/article/abs/pii/S0959152408001510]
- 85. Troubleshooting Problems in Control Systems Worksheet [https://control.com/worksheets/troubleshooting-problems-in-control-systems/]
- 86. Disease-economy trade-offs under alternative epidemic ... [https://www.nature.com/articles/s41467-022-30642-8]
- 87. Evaluation of advanced control strategies for building ... [https://www.sciencedirect.com/science/article/pii/S0378778822008805]
- 88. Category : 2025 [https://crystallizationsummit.com/category/2025/]