Solid-State vs. Sol-Gel Synthesis: A Strategic Guide to Mastering Particle Size Control for Pharmaceutical and Material Scientists
This article provides a comprehensive comparative analysis of solid-state and sol-gel synthesis methods, with a focused lens on achieving precise particle size control—a critical parameter in pharmaceutical development and advanced...
Solid-State vs. Sol-Gel Synthesis: A Strategic Guide to Mastering Particle Size Control for Pharmaceutical and Material Scientists
Abstract
This article provides a comprehensive comparative analysis of solid-state and sol-gel synthesis methods, with a focused lens on achieving precise particle size control—a critical parameter in pharmaceutical development and advanced material science. Tailored for researchers and drug development professionals, the content explores the foundational principles of each technique, details their methodological applications for nanomaterial fabrication, addresses common scaling and homogeneity challenges with modern optimization strategies, and establishes a framework for the rigorous validation and selection of the appropriate method based on target particle characteristics and application requirements. The synthesis is supported by the latest research, including insights into microwave-assisted sol-gel processes and mechanochemical solid-state methods.
Understanding the Core Principles: How Solid-State and Sol-Gel Chemistry Govern Particle Formation
The controlled synthesis of functional particles is a cornerstone of modern materials science, with profound implications for industries ranging from pharmaceuticals to energy storage. Among the myriad of fabrication techniques, solid-state and sol-gel methods represent two fundamentally different paradigms for particle engineering. Solid-state synthesis, one of the oldest and most straightforward methods, involves direct reaction between solid precursors at high temperatures, making it particularly suitable for large-scale industrial production of ceramic oxides. In contrast, the sol-gel method is a solution-based chemical process that enables molecular-level mixing of precursors, typically yielding materials with superior homogeneity, purity, and compositional control at significantly lower processing temperatures.
The selection between these methodologies profoundly impacts critical particle characteristics including crystallite size, morphological uniformity, phase purity, and ultimately, functional performance in applications such as battery electrodes, catalysts, and drug delivery systems. This guide provides a comprehensive, objective comparison of these two foundational synthesis approaches, supported by recent experimental data and detailed protocols to inform researchers and development professionals in their methodological selections.
Comparative Analysis: Fundamental Principles and Outcome Differences
The following table summarizes the core characteristics and typical outcomes of solid-state and sol-gel synthesis methods, highlighting their distinct advantages and limitations.
Table 1: Fundamental Comparison Between Solid-State and Sol-Gel Synthesis Methods
| Aspect | Solid-State Synthesis | Sol-Gel Synthesis |
|---|---|---|
| Basic Principle | Direct reaction between solid precursors through diffusion at high temperatures | Transition from a liquid "sol" to a solid "gel" network via hydrolysis and polycondensation |
| Typical Temperature Range | High (800–1100°C) [1] [2] | Low to Moderate (Room Temperature to 400°C) [3] [4] |
| Primary Driving Force | Thermal energy overcoming diffusion barriers | Chemical reaction kinetics and supersaturation |
| Particle Size Control | Limited, often results in larger, aggregated particles | Excellent, can yield nanoparticles with narrow size distribution [5] |
| Mixing Homogeneity | Limited, requires intensive mechanical milling | Atomic-level, excellent homogeneity [2] [4] |
| Phase Purity | Can suffer from incomplete reaction and secondary phases [2] | High phase purity achievable at lower temperatures [2] |
| Product Morphology | Irregular, often aggregated particles | Can be finely tuned to produce spheres, films, or monoliths |
| Specific Surface Area | Generally low | High, especially with surfactant templating [5] [4] |
| Scalability | Excellent for industrial-scale production | More complex for large-scale production |
| Key Advantage | Simplicity, cost-effectiveness, scalability | Superior control over composition, structure, and texture at nanoscale |
Quantitative Performance Data from Recent Studies
Recent experimental studies directly comparing materials synthesized via both methods provide tangible evidence of their performance differences. The data below illustrate how the choice of synthesis method influences key physical and electrochemical properties.
Table 2: Experimental Data Comparison for Specific Material Systems
| Material Synthesized | Synthesis Method | Key Outcome Metrics | Experimental Details/Conditions |
|---|---|---|---|
| ZnFe₂O₄ (for battery applications) [3] | Sol-Gel | Improved ionic conductivity; more uniform particle size | Precursors: ZnCl₂, FeCl₃; pH ~10.5; Final firing: various temperatures |
| Solid-State | Lower ionic conductivity; broader particle size distribution | Precursors: ZnO, Fe₂O₃; Mechanochemical activation: 30 min at 1380 rpm; Final firing: various temperatures | |
| BiFeO₃ (multiferroic) [2] | Sol-Gel | Grain size: ~0.5x SS; Higher dielectric constant; Stoichiometric Bi:Fe | Precursors: Bismuth & Iron Nitrates; Calcination: ~600°C |
| Solid-State | Grain size: ~1-2 μm; Lower dielectric constant; Bi-deficient common | Precursors: Bi₂O₃, Fe₂O₃; Calcination: 800-820°C | |
| BaTiO₃ (for MLCCs) [1] | Low-Pressure Solid-State | Particle Size: 90-160 nm; High tetragonality (c/a = 1.0095) | Precursors: BaCO₃, TiO₂; Pressure: 0.01 MPa; Temperature: 800-900°C |
| P2-Na₀.₇₉Li₀.₁₁Ni₀.₂₁Mn₀.₆₇O₂ (Cathode) [6] | Solid-State | Suppressed O²⁻/(O₂)ⁿ⁻ redox; Better cycling stability; Higher crystallinity | Precursors: Na₂CO₃, LiOH, NiO, MnO₂; Calcination: 900°C for 15 h |
| Sol-Gel | Anionic redox activity at >4.0 V; Lower cycling stability | Precursors: Acetates/Nitrates; Calcination: 900°C | |
| NiO-Fe₂O₃-SiO₂/Al₂O₃ (Catalyst) [4] | Optimized Sol-Gel | Particle Size: 44 nm; Specific Surface Area: 134.79 m²/g | Precursors: Metal nitrates, TEOS; Heat treatment: 400°C; Heating rate: 5°C/min |
Detailed Experimental Protocols
Representative Solid-State Synthesis Protocol
The following protocol for synthesizing ZnFe₂O₄ is adapted from a comparative study and exemplifies the classic solid-state approach [3].
- Step 1: Precursor Preparation: Weigh stoichiometric amounts of iron(III) oxide (Fe₂O₃, 99.50%) and zinc oxide (ZnO, 99.50%).
- Step 2: Homogenization and Mechanochemical Activation: Place the precursor mixture in an agate mortar for preliminary grinding. Subsequently, transfer the mixture to a planetary ball mill (e.g., "Aktivator-2 SL") equipped with zirconium oxide-lined drums and balls.
- Step 3: Milling: Process the mixture at a high speed (e.g., 1380 rpm) for a set duration (e.g., 30 minutes) to achieve mechanochemical activation, which increases reactivity by reducing particle size and creating fresh surfaces.
- Step 4: Thermal Treatment: Transfer the activated powder to a high-temperature furnace (e.g., a muffle furnace). Heat the sample at a controlled rate (e.g., 10°C/min) to the final synthesis temperature (determined by thermal analysis, often between 800-1000°C) and hold for several hours to ensure complete reaction.
- Step 5: Product Characterization: The final product is typically characterized by X-ray diffraction (XRD) for phase identification, scanning electron microscopy (SEM) for morphology, and impedance spectroscopy for electrophysical properties.
Representative Sol-Gel Synthesis Protocol
This protocol for synthesizing ZnFe₂O₄ and other oxide materials highlights the solution-based nature of the method [3] [4].
- Step 1: Precursor Solution Preparation: Dissolve metal-containing precursors, such as zinc chloride (ZnCl₂, 96.00%) and iron(III) chloride (FeCl₃, 98.00%), in deionized water in their required molar ratio under vigorous stirring.
- Step 2: Precipitation/Gelation: Slowly add a precipitating agent, typically sodium hydroxide (NaOH, 98.00%) solution, to the mixed metal solution. The addition is controlled to reach a specific pH (e.g., pH 10.5, just before the dissolution point of zinc hydroxide). This step results in the formation of a colloidal suspension (sol) or a gel.
- Step 3: Ageing and Washing: Allow the gel to age for 30-60 minutes with continuous hydrodynamic processing. Subsequently, filter the product under vacuum using a Büchner funnel and wash thoroughly with deionized water to remove residual ions and mother liquor.
- Step 4: Drying and Thermal Treatment: Dry the washed precursor at room temperature or in an oven. The final thermal treatment (e.g., in a muffle furnace) is performed at a significantly lower temperature (e.g., 400-700°C) compared to the solid-state method to crystallize the target phase.
- Step 5: Product Characterization: Similar to the solid-state product, the final material is characterized by XRD, SEM, and other techniques like nitrogen sorption for surface area analysis.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following reagents are fundamental to executing solid-state and sol-gel syntheses in a research setting.
Table 3: Key Reagent Solutions for Synthesis Methods
| Reagent/Solution | Primary Function | Example in Protocol |
|---|---|---|
| Metal Oxides/Carbonates | Primary precursors for solid-state reactions | Fe₂O₃, ZnO, BaCO₃, TiO₂ [3] [1] |
| Metal Salts (Chlorides, Nitrates) | Soluble precursors for sol-gel synthesis | ZnCl₂, FeCl₃, metal nitrates [3] [4] |
| Alkoxides (e.g., TEOS) | Network former in sol-gel; source of Si, Al, etc. | Tetraethyl orthosilicate (TEOS) for silica matrix [5] [4] |
| Precipitating Agents (e.g., NaOH, NH₄OH) | Controls pH to induce hydrolysis/condensation or precipitation | NaOH solution, Ammonia solution [3] [5] |
| Surfactants (e.g., CTAB, F127) | Template for mesoporous structures in sol-gel | Cetyltrimethylammonium bromide (CTAB), Pluronic F127 [5] |
| Chelating Agents (e.g., Ammonia) | Controls precipitation kinetics in co-precipitation | Ammonia solution for hydroxide co-precipitation [7] |
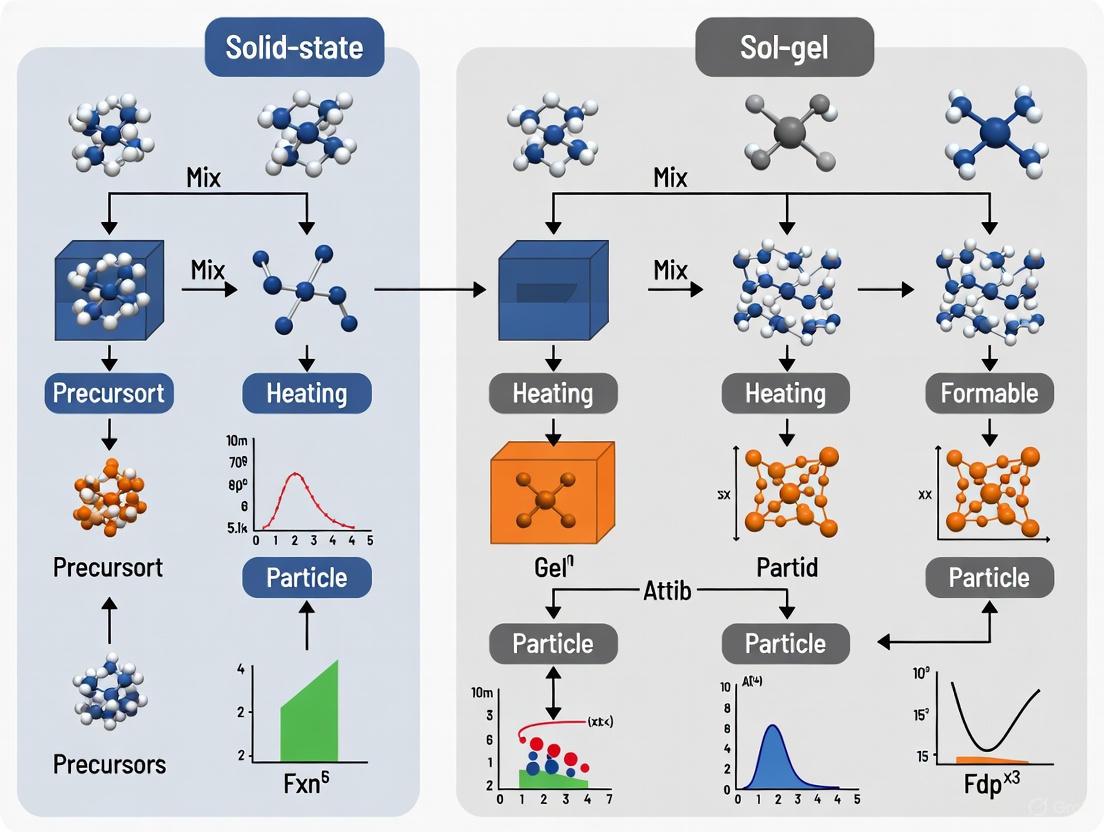
Synthesis Workflow and Particle Formation Pathways
The diagrams below illustrate the fundamental procedural workflows and underlying particle formation mechanisms for both synthesis methods, highlighting their distinct pathways.
Solid-State Synthesis Workflow
Sol-Gel Synthesis Workflow
The synthesis of advanced inorganic materials, crucial for applications ranging from electroceramics to pharmaceutical development, often hinges on the precise control of particle size, morphology, and phase purity. The choice of synthesis method fundamentally dictates these characteristics by governing the underlying chemical pathways. This guide provides a comparative analysis of two predominant synthesis strategies: the high-temperature solid-state method and the solution-based sol-gel method, with a specific focus on their mechanistic pathways. The solid-state route relies on atomic diffusion across particle boundaries at elevated temperatures, leading to particle coarsening. In contrast, the sol-gel pathway involves molecular-level reactions in a liquid medium, offering superior control over particle nucleation and growth at lower temperatures. Understanding the intrinsic relationship between each method's mechanism and the final particle properties is essential for researchers to select the optimal synthesis protocol for their specific material and application.
Comparative Mechanistic Pathways: Solid-State vs. Sol-Gel Synthesis
The fundamental difference between solid-state and sol-gel synthesis lies in the initial state of the reactants and the mechanisms that drive the formation of the final product. The journey from raw precursors to a final crystalline material follows two distinct paths, as illustrated below.
The diagram above summarizes the two core mechanistic pathways. The solid-state pathway is characterized by:
- Diffusion-Limited Kinetics: The reaction is initiated at points of contact between solid precursor particles. The rate-limiting step is the solid-state diffusion of ions across these interfaces, a process that requires high thermal energy to overcome significant activation barriers [8].
- Particle Coarsening: At the high temperatures required for diffusion (often >1000°C for oxides), rapid grain growth occurs, typically resulting in coarse, micron-sized particles with broad size distributions and irregular morphologies [3] [9].
In contrast, the sol-gel pathway is defined by:
- Molecular-Level Homogeneity: The process begins with precursors dissolved in a liquid solvent, ensuring mixing at the atomic or molecular level. This homogeneity is maintained through the hydrolysis and condensation reactions that form an amorphous gel network [5] [10].
- Nucleation-Controlled Growth: During subsequent thermal treatment, nucleation of the crystalline phase occurs uniformly throughout the amorphous matrix at a lower temperature than in solid-state reactions. This separation of the nucleation and growth stages allows for finer control over the final particle size and morphology [2] [10].
Experimental Data Comparison: Structural and Performance Outcomes
The distinct mechanistic pathways of solid-state and sol-gel synthesis lead to measurable differences in the properties of the resulting materials. The following table summarizes quantitative and qualitative outcomes from comparative studies on various metal oxides.
Table 1: Comparative experimental data from solid-state and sol-gel synthesized materials.
| Material Synthesized | Synthesis Method | Calcination Temperature & Time | Average Particle Size | Surface Area (BET) | Key Findings |
|---|---|---|---|---|---|
| ZnFe₂O₄ [3] | Solid-State + Mechanochemical | High Temperature (Not Specified) | Larger, broader distribution | Not Reported | Electrophysical properties highly dependent on firing temperature and method. |
| Sol-Gel + Solid-State Finish | Varied Temperature Regimes | Finer, more controlled | Not Reported | ||
| BiFeO₃ [2] | Solid-State Reaction | ~800°C | ~1 μm (Coarse) | Not Reported | Contained secondary Bi₂Fe₄O₉ impurity phase; coarse grains. |
| Sol-Gel | Lower Temperature | ~0.5 μm (Fine) | Not Reported | Near phase-pure; grain size reduced by half; higher dielectric constant. | |
| MgAl₂O₄ [10] | Sol-Gel | 700°C | Not Reported | 188 m²/g | Surface area decreases with increasing calcination temperature. |
| Sol-Gel | 800°C | Not Reported | ~144 m²/g (est. from trend) | Spherical morphology; agglomeration at higher temperatures. | |
| Sol-Gel | 900°C | Not Reported | 94 m²/g | ||
| LLZO Solid Electrolyte [9] | Solid-State Reaction | 900-1200°C for 12-36 hrs | 1-10 μm | Low | Simple and scalable, but requires high temperatures and long times; prone to Li loss. |
| Sol-Gel | 700-900°C | 100-500 nm | High | High phase-purity and homogeneity; lower processing temperatures. |
The data demonstrates a consistent trend: the sol-gel method enables the formation of finer particles with higher surface areas due to its molecular-level mixing and lower required calcination temperatures. For instance, in the synthesis of BiFeO₃, the sol-gel route produced particles half the size of those from the solid-state method and achieved a superior phase purity, which directly enhanced its dielectric properties [2]. Similarly, the high surface area of sol-gel derived MgAl₂O₄ is a critical factor for its effectiveness as a catalyst support [10].
Detailed Experimental Protocols
To replicate the results discussed, below are generalized protocols for the solid-state and sol-gel methods, synthesized from the reviewed literature.
- Precursor Preparation: Weigh stoichiometric amounts of high-purity solid precursors (e.g., Bi₂O₃ and Fe₂O₃ for BiFeO₃).
- Mechanochemical Mixing: Transfer the powder mixture to a milling apparatus (e.g., a planetary ball mill). Use grinding media (e.g., zirconia balls) and a liquid milling aid (e.g., ethanol) to prevent excessive heat. Mill for several hours (e.g., 4 hours) to achieve homogenization.
- Calcination: Place the homogenized powder in a high-temperature stable crucible (e.g., alumina). Insert into a muffle furnace and calcine in air at a high temperature (e.g., 800°C) for a prolonged period (e.g., 2 hours) using a defined heating rate (e.g., 5-10°C/min).
- Intermediate Grinding: After the first calcination, allow the sample to cool to room temperature. Transfer to a mortar and pestle and grind thoroughly to break up agglomerates and expose fresh particle surfaces for further reaction.
- Second Calcination (Optional): For incomplete reactions, a second calcination step at the same or a higher temperature may be required to improve phase purity.
- Final Processing: The resulting powder is ground one final time to achieve the desired consistency for characterization or application.
- Precursor Solution Preparation: Dissolve metal-containing precursors in a solvent. For MgAl₂O₄, this may involve a magnesium salt (e.g., nitrate) and an aluminum salt (e.g., nitrate) in distilled water. For metal alkoxides, organic solvents like ethanol are used.
- Complexation/Hydrolysis: Add a complexing agent (e.g., citric acid) to the solution in a defined molar ratio to the metal ions. Stir vigorously until a clear solution is obtained. Subsequently, slowly add a hydrolysis agent (e.g., water for alkoxides) under continuous stirring.
- Gelation: Continue stirring until the solution gradually increases in viscosity and transforms into a wet gel. This can be facilitated by mild heating (e.g., 80°C).
- Ageing and Drying: Allow the gel to age for several hours to strengthen the network. Then, dry the gel in an oven at elevated temperatures (e.g., 120°C) to remove the solvent and form a dry, porous xerogel.
- Calcination: Place the xerogel in a furnace and heat to a target temperature (e.g., 700-900°C) with a controlled heating ramp. This step decomposes the organic components and crystallizes the desired oxide phase.
- Post-Synthesis Processing: The resulting fine powder may be lightly ground to break up soft agglomerates.
The Scientist's Toolkit: Essential Research Reagents and Equipment
The following table lists key materials, reagents, and instruments essential for conducting and characterizing solid-state and sol-gel syntheses, based on the methodologies described in the search results.
Table 2: Key research reagents and equipment for solid-state and sol-gel syntheses.
| Category | Item | Specific Examples | Function in Synthesis |
|---|---|---|---|
| Precursors | Metal Oxides | Fe₂O₃, ZnO, Bi₂O₃, La₂O₃ [3] [2] | Solid-state starting materials for cation sources. |
| Metal Salts/Alkoxides | Chlorides (ZnCl₂), Nitrates (Fe(NO₃)₃), Tetraethyl orthosilicate (TEOS) [3] [5] | Soluble precursors for sol-gel synthesis. | |
| Reagents | Fuel/Complexing Agents | Citric Acid, Cetyltrimethylammonium bromide (CTAB) [5] [10] | Chelates metal ions in sol-gel for homogeneity; CTAB acts as a surfactant template for pores. |
| Solvents | Deionized Water, Ethanol [3] [5] | Liquid medium for dissolution and reaction in sol-gel. | |
| Equipment | Milling/Homogenization | Planetary Ball Mill, Agate Mortar & Pestle [3] [2] | Homogenizes solid precursors or breaks down agglomerates. |
| Thermal Processing | Muffle Furnace, Tube Furnace [3] [11] | Provides controlled high-temperature environment for calcination and sintering. | |
| Synthesis Automation | Open-Source Liquid Handling Platforms [5] | Enables high-throughput, reproducible sol-gel synthesis for parameter screening. | |
| Characterization | Structural Analysis | X-Ray Diffractometer (XRD) [3] [2] | Determines crystal structure, phase purity, and crystallite size. |
| Thermal Analysis | Thermogravimetric Analysis (TGA), Differential Thermal Analysis (DTA) [3] [11] | Analyzes mass changes and thermal events during precursor decomposition and phase formation. | |
| Surface Area/Porosity | BET Surface Area Analyzer [10] | Measures specific surface area and pore characteristics. | |
| Morphology Imaging | Scanning Electron Microscope (SEM), Transmission Electron Microscope (TEM) [10] | Visualizes particle size, morphology, and agglomeration. |
The choice between solid-state and sol-gel synthesis is a fundamental decision that dictates the microstructure and properties of inorganic materials. The solid-state pathway, driven by high-temperature diffusion, is a robust and scalable method but often yields coarse particles with a higher risk of impurity phases. The sol-gel pathway, governed by molecular-level reactions in solution, provides superior control over particle size, homogeneity, and phase purity at lower temperatures, albeit often with more complex chemistry and potentially higher precursor costs. The decision matrix for a researcher is clear: for applications demanding high purity, nanoscale particles, and large surface areas (e.g., advanced catalysts, specialized ceramics), the sol-gel method is objectively superior. For applications where cost and scalability are paramount and coarse powders are acceptable, the solid-state method remains a viable and effective choice. This mechanistic understanding empowers scientists and engineers to rationally select and optimize synthesis routes for targeted material performance.
The precise control over particle size, morphology, and composition is a fundamental objective in materials science, directly influencing the performance of products in fields ranging from pharmaceuticals to energy storage. Within this context, two synthesis methodologies—solid-state reaction and sol-gel processing—offer distinctly different pathways from raw materials to final product. The solid-state method relies on high-temperature diffusion and reaction of solid precursors to form crystalline powders, whereas the sol-gel technique employs solution-phase chemistry at mild temperatures to build inorganic networks through sequential hydrolysis and condensation reactions of molecular precursors [12] [13].
This guide provides an objective comparison of these methods, focusing specifically on their capabilities and limitations for controlling particle size and structural properties. Supported by experimental data and detailed protocols, it serves as a decision-making framework for researchers selecting synthesis routes for specific application requirements.
Fundamental Principles and Reaction Mechanisms
The Sol-Gel Process: Solution-Phase Network Formation
The sol-gel process is a wet-chemical technique for producing solid materials from small molecules. The process involves the conversion of monomers in solution into a colloidal solution (sol) that acts as the precursor for an integrated network (or gel) of either discrete particles or network polymers [12]. Typical precursors are metal alkoxides or metal chlorides.
The reaction sequence occurs in several distinct stages, as illustrated in the workflow below:
The chemical foundation of sol-gel processing involves two principal reactions:
Hydrolysis: Metal alkoxides (M-OR) react with water, replacing alkoxide groups (OR) with hydroxyl groups (OH):
Si(OR)₄ + H₂O → HO-Si(OR)₃ + R-OH[12]Condensation: Hydrolyzed species link together through the formation of M-O-M bonds, liberating water or alcohol:
(OR)₃-Si-OH + HO-Si-(OR)₃ → [(OR)₃Si-O-Si(OR)₃] + H₂O[12]
The kinetics and thermodynamics of these reactions are strongly influenced by catalyst type (acid or base), pH, temperature, water-to-precursor ratio, and precursor reactivity [12] [14]. Acid-catalyzed conditions typically produce more linear, polymeric networks, while base-catalyzed conditions favor the formation of discrete, colloidal particles that can self-assemble into ordered structures [12].
Solid-State Reactions: High-Temperature Diffusion Chemistry
In contrast, solid-state synthesis involves mechanically mixing solid precursors (typically oxides, carbonates, or hydroxides) followed by high-temperature treatment (often >1000°C) to facilitate diffusion and reaction between solid phases [13] [15]. This method is fundamentally governed by nucleation and growth kinetics, ionic diffusion rates, and phase transformation dynamics at elevated temperatures. The process typically yields crystalline, micron-sized powders, with particle size controlled primarily by calcination temperature, duration, mechanical milling intensity, and starting precursor morphology [3] [13].
Table 1: Fundamental Characteristics of Synthesis Methods
| Characteristic | Sol-Gel Process | Solid-State Reaction |
|---|---|---|
| Reaction Medium | Liquid solution (water, alcohol) [12] [16] | Solid-solid interface [13] |
| Typical Temperature | Room temperature to ~100°C [12] [16] | 800°C to 1500°C [3] [13] |
| Primary Driving Force | Hydrolysis and condensation kinetics [12] | Solid-state diffusion and nucleation [13] |
| Reaction Homogeneity | Molecular-level mixing [12] | Limited by powder mixing efficiency [13] |
| Fundamental Rate Controls | pH, catalyst, concentration, temperature [12] [14] | Temperature, time, precursor morphology, mixing [3] [13] |
Experimental Protocols for Comparative Studies
Sol-Gel Synthesis of ZnFe₂O₄ Nanoparticles
Objective: To synthesize zinc ferrite (ZnFe₂O₄) nanoparticles with controlled size and composition [3].
Materials and Reagents:
- Zinc chloride (ZnCl₂, 96% purity) and iron(III) chloride (FeCl₃, 98% purity) as metal precursors [3]
- Sodium hydroxide (NaOH, 98% purity) as precipitation agent [3]
- Deionized water as solvent [3]
Procedure:
- Precursor Preparation: Dissolve zinc chloride and iron(III) chloride in deionized water at a molar ratio of 1:2 (Zn:Fe) under intensive stirring [3].
- Precipitation: Slowly add sodium hydroxide solution to the metal chloride mixture at room temperature until reaching pH 10.5 [3].
- Hydrodynamic Processing: Maintain the suspension under continuous stirring for 30-60 minutes to facilitate reaction completion [3].
- Filtration and Washing: Filter the resulting precipitate under vacuum using a Büchner funnel and wash with deionized water to remove residual salts and reaction by-products [3].
- Drying: Air-dry the filtered precursor at room temperature [3].
- Thermal Treatment: Calcine the dried precursor in a muffle furnace at temperatures ranging from 600°C to 1000°C for 2-4 hours to crystallize the ZnFe₂O₄ spinel structure [3].
Solid-State Synthesis of ZnFe₂O₄
Objective: To synthesize crystalline ZnFe₂O₄ using traditional ceramic processing [3].
Materials and Reagents:
- Iron(III) oxide (Fe₂O₃, 99.5% purity) and zinc oxide (ZnO, 99.5% purity) as starting materials [3]
Procedure:
- Weighing and Mixing: Weigh Fe₂O₃ and ZnO in a 1:1 molar ratio and mix manually in an agate mortar [3].
- Mechanochemical Activation: Transfer the mixture to a planetary ball mill equipped with zirconium oxide vessels and balls. Process at 1380 rpm for 30 minutes to achieve homogenization and particle size reduction [3].
- Pelletization: Optionally, press the homogenized powder into pellets to enhance interparticle contact [3].
- High-Temperature Calcination: Heat the mixture in a muffle furnace at temperatures between 1000°C and 1300°C for 10-24 hours with a heating rate of 10°C/min to facilitate solid-state diffusion and reaction [3].
- Post-Synthesis Processing: Gently grind the sintered product to obtain a fine powder [3].
Comparative Performance Analysis: Structural and Morphological Properties
Particle Size and Surface Area Control
Table 2: Particle Size and Surface Area Comparison
| Synthesis Parameter | Sol-Gel Method | Solid-State Method | Experimental Basis |
|---|---|---|---|
| Typical Particle Size Range | 5-100 nm [3] [17] | 0.5-10 μm [3] [13] | ZnFe₂O₄, Sr₈MgEu(PO₄)₇ studies [3] [13] |
| Specific Surface Area | High (50-800 m²/g) [12] [18] | Low (1-10 m²/g) [13] | Aerogels, ferrites, phosphors [12] [18] [13] |
| Primary Size Control Mechanisms | pH, catalyst, concentration, solvent [12] [14] | Milling time, temperature, duration [3] [13] | Multiple material systems [12] [3] [13] |
| Crystallite Size | 10-50 nm [3] [13] | 100-500 nm [3] [13] | XRD analysis of multiple systems [3] [13] |
| Size Uniformity | Narrow distribution possible [12] [14] | Broad distribution typical [13] | Comparative studies [12] [13] [14] |
Structural and Compositional Properties
Table 3: Structural and Compositional Characteristics
| Property | Sol-Gel Method | Solid-State Method | Experimental Basis |
|---|---|---|---|
| Crystallinity | Requires thermal treatment [3] | High crystallinity directly achieved [13] | ZnFe₂O₄, phosphor studies [3] [13] |
| Compositional Homogeneity | Excellent (molecular mixing) [12] [3] | Inhomogeneities common [13] | Elemental mapping, spectroscopy [3] [13] |
| Phase Purity | Can contain amorphous phases [3] | High phase purity achievable [13] | XRD analysis [3] [13] |
| Dopant Incorporation | Uniform at molecular level [12] [17] | Segregation common [13] | Spectroscopic studies [13] [17] |
| Densification Temperature | Lower (reduced by 200-400°C) [12] | Higher (near melting point) [13] | Comparative sintering studies [12] [13] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Reagents for Sol-Gel and Solid-State Syntheses
| Reagent Category | Specific Examples | Function | Application Context |
|---|---|---|---|
| Alkoxide Precursors | Tetraethyl orthosilicate (TEOS), Titanium isopropoxide, Methyltrimethoxysilane (MTMS) [12] [18] | Molecular source of metal oxide network [12] | Sol-gel synthesis of SiO₂, TiO₂, PMSQ [12] [18] |
| Inorganic Salts | Metal chlorides (ZnCl₂, FeCl₃), nitrates [3] [19] | Cost-effective alternative precursor [3] | Sol-gel synthesis of ferrites, doped oxides [3] [19] |
| Oxide Precursors | ZnO, Fe₂O₃, SrCO₃ [3] [13] | Solid starting materials for ceramic reaction [3] [13] | Solid-state synthesis of ceramics, phosphors [3] [13] |
| Catalysts | HCl, acetic acid, ammonia, urea [12] [18] | Control hydrolysis/condensation rates [12] [18] | Acid/base-catalyzed sol-gel processes [12] [18] |
| Solvents | Ethanol, methanol, deionized water [12] [16] | Reaction medium for precursor dissolution [12] | All sol-gel processes [12] [16] |
| Surfactants/Structure Directors | Cetyl-trimethyl-ammonium chloride (CTAC) [18] | Control pore size, prevent phase separation [18] | Mesoporous materials, aerogels [18] |
The comparative analysis presented in this guide demonstrates that sol-gel and solid-state methods offer complementary advantages for particle size control and material synthesis. The experimental data reveal that the sol-gel process provides superior control over nanoscale dimensions, with typical particle sizes in the 5-100 nm range, high specific surface areas, and excellent compositional homogeneity due to molecular-level mixing in solution [3] [13]. These characteristics make it particularly suitable for applications requiring high surface-to-volume ratios, such as catalysis, drug delivery, and functional coatings [12] [16] [14].
Conversely, solid-state reactions excel in producing highly crystalline, micron-sized powders (0.5-10 μm) with excellent phase purity, making them ideal for applications where crystallinity and thermal stability are paramount, such as in structural ceramics, phosphors, and battery electrode materials [3] [13]. The choice between these methods ultimately depends on the specific material requirements, with sol-gel processing offering precision at the nanoscale and solid-state reactions providing robustness at the microscale.
For researchers pursuing particle size control, hybrid approaches that combine elements of both methods are increasingly emerging. For instance, sol-gel synthesis with solid-state finishing or mechanochemical activation of sol-gel precursors can potentially leverage the advantages of both techniques [3]. As material requirements continue to evolve toward more complex nanostructures with precisely engineered interfaces, understanding these fundamental synthesis paradigms remains essential for advancing materials design across scientific and industrial applications.
The precise control of particle size during synthesis is a cornerstone of advanced materials science, directly influencing the properties and performance of materials in applications ranging from ceramics and catalysts to batteries. The choice of synthesis method fundamentally determines the levers available for this control. This guide provides a detailed, experimental data-driven comparison of two predominant synthesis techniques: the solid-state method and the sol-gel method. The analysis is framed around three key parameters—precursor reactivity, temperature, and mixing homogeneity—and their collective impact on the final particle size and material characteristics.
Synthesis Methods at a Glance
The foundational principles of the two methods dictate their inherent capabilities for particle size control.
dot Synthesis Method Comparison { graph [bgcolor="#F1F3F4" labelloc="t" fontcolor="#202124" fontsize="16"] node [shape=rectangle style=filled fillcolor="#FFFFFF" color="#5F6368" fontcolor="#202124"] edge [color="#4285F4"]
}
- Solid-State Method: A direct reaction between solid precursor powders (typically oxides or carbonates) through high-temperature calcination. Mixing is achieved mechanically (e.g., ball milling), which limits homogeneity and often results in larger, micron-sized particles [3] [20].
- Sol-Gel Method: A wet-chemical technique involving the transition of a solution ("sol") into a solid "gel" network. Precursors are mixed at a molecular level in a liquid medium, allowing for exceptional homogeneity and the formation of nanometer-sized particles [3] [21] [20].
Comparative Analysis of Key Parameters
The following tables summarize how each synthesis method leverages the three key parameters, with supporting experimental data.
Table 1: Influence of Synthesis Parameters on Particle Size
| Parameter | Solid-State Method | Sol-Gel Method |
|---|---|---|
| Precursor Reactivity & Homogeneity | Limited by solid-state diffusion; mechanical mixing (ball milling) can reduce particle size but cannot achieve atomic-scale homogeneity [21] [22]. | Molecular-level mixing in solution ensures high homogeneity from the outset, directly leading to uniform nucleation and finer particles [3] [21]. |
| Temperature Profile | Requires high calcination temperatures (often >900°C) to facilitate solid-state diffusion, promoting particle coarsening and grain growth [20]. | Calcination occurs at significantly lower temperatures (e.g., 450-700°C), suppressing excessive grain growth and preserving fine, nanoscale structures [23] [20]. |
| Resulting Particle Characteristics | Larger particle size (microns), broader size distribution, and denser morphology [20]. | Smaller particle size (nanometers), narrower size distribution, and often porous morphology [23] [20]. |
Table 2: Experimental Data from Comparative Studies
| Material | Synthesis Method | Key Synthesis Conditions | Resulting Particle Size | Key Findings & Performance | Source |
|---|---|---|---|---|---|
| Li₂ZnTi₃O₈ (LZTO) Ceramics | Solid-State | Calcination: 900°C; Sintering: 1150°C | Not specified (larger grains) | Q×f value: 56,874 GHzAchieved dense structure only at high sintering temperatures. | [20] |
| Sol-Gel | Calcination: 700°C; Sintering: 900°C | 40 - 100 nm | Q×f value: 60,579 GHzSuperior dielectric properties at a 250°C lower sintering temperature. | [20] | |
| ZnFe₂O₄ | Solid-State | Mechanochemical activation & thermal treatment | Not specified | Properties highly dependent on extended milling and high-temperature treatment. | [3] |
| Sol-Gel | Co-precipitation & low-temperature firing | Not specified | Improved electrophysical properties as a prospective battery cathode material. | [3] | |
| ZrV₂O₇ | Solid-State | Multiple calcination cycles (700°C) & extended milling | Impurities present without optimized milling | Can achieve high purity, but requires "extended milling time and repeated calcination cycles." | [21] |
| Sol-Gel | - | Homogeneous, phase-pure | Enabled a "‘near-atomic’ level of mixing," producing homogeneous, phase-pure material. | [21] | |
| Ca/TiO₂-ZrO₂ Nano-catalyst | Sol-Gel | Optimized calcination: 487°C | 80 - 110 nm | High surface area (276.5 m²/g) and 97.6% conversion in esterification. | [23] |
Detailed Experimental Protocols
This protocol highlights the high-temperature requirements of the solid-state method.
- Precursors: High-purity Li₂CO₃, ZnCO₃, and TiO₂ powders.
- Milling: The precursor powders are mixed and ball-milled in a nylon jar with zirconia balls and deionized water for 24 hours.
- Calcination: The mixed powders are dried and then calcined at 900°C for 4 hours to form the desired crystalline phase.
- Sintering: The calcined powder is pressed into pellets and sintered at temperatures ranging from 1050°C to 1200°C to achieve a dense ceramic structure.
This protocol demonstrates the optimization of sol-gel parameters for nanoscale particle control.
- Precursor Mixing: Titanium and zirconium precursors are mixed in a liquid solution. A complexing agent (e.g., citric acid) is used to control reactivity.
- Complexation: The solution is maintained at a specific gelling temperature (e.g., 72°C) for a defined complex time (e.g., 2.65 hours) to form a homogeneous metal-citrate complex.
- Gel Formation: The solvent evaporates, leading to the formation of a solid gel.
- Calcination: The gel is calcined at a optimized, relatively low temperature (e.g., 487°C) to form the final crystalline nano-catalyst with high surface area.
The Scientist's Toolkit: Essential Research Reagents
dot Research Reagent Flow { graph [bgcolor="#F1F3F4" labelloc="t" fontcolor="#202124" fontsize="16"] node [shape=rectangle style=filled fillcolor="#FFFFFF" color="#5F6368" fontcolor="#202124"] edge [color="#4285F4"]
}
Table 3: Key Reagents and Their Functions in Synthesis
| Reagent Category | Example | Primary Function in Synthesis |
|---|---|---|
| Solid-State Precursors | Metal Oxides (e.g., Fe₂O₃, ZnO, ZrO₂) [3] [21] | Source of metal cations; react at high temperatures via solid-state diffusion. |
| Metal Carbonates (e.g., Li₂CO₃, ZnCO₃) [20] | Act as precursors, often releasing CO₂ during calcination to form the desired oxide. | |
| Sol-Gel Precursors | Metal Alkoxides (e.g., Tetraethyl Orthosilicate - TEOS) [24] [5] | Undergo hydrolysis and condensation to form the metal-oxide network. |
| Metal Salts (e.g., Chlorides, Nitrates) [3] [23] | Soluble alternative to alkoxides; participate in co-precipitation or complexation. | |
| Sol-Gel Modifiers | Catalysts (e.g., HCl, NH₄OH) [24] | Control the rates of hydrolysis and condensation, influencing gel structure and porosity. |
| Surfactants (e.g., CTAB, Pluronic F127) [5] | Template the formation of mesopores, providing control over pore size and surface area. | |
| Complexing Agents (e.g., Citric Acid) [23] | Chelate metal ions, improving homogeneity and preventing premature precipitation. |
The experimental data unequivocally demonstrates that the sol-gel method offers superior control over particle size, enabling the consistent production of nanomaterials with narrow size distributions. This advantage stems from its core principles: molecular-level precursor mixing (homogeneity) and lower processing temperatures. In contrast, the solid-state method, while industrially robust for larger volumes, is inherently limited by solid-state diffusion and high-temperature coarsening, typically resulting in larger, micron-sized particles.
The choice between methods is a trade-off. For applications demanding high specific surface area, tailored porosity, and nanoscale effects—such as advanced catalysis, drug delivery, and certain functional ceramics—sol-gel is the definitive choice. For applications where extreme purity and high density are the primary goals and particle size is less critical, solid-state synthesis remains a viable and effective path.
Practical Synthesis Strategies: Tailoring Solid-State and Sol-Gel Protocols for Targeted Particle Sizes
The fabrication of advanced functional materials hinges on the precise control over composition, crystal structure, and particle morphology. Among the plethora of synthesis techniques available, solid-state synthesis and sol-gel methods represent two fundamentally different philosophical approaches. Solid-state reactions, particularly those enhanced by mechanochemical activation, rely on the physical mixing and high-temperature diffusion of solid precursors to form new compounds [2] [25]. In contrast, the sol-gel process is a wet-chemical technique where a solution (sol) of small molecules evolves into a network structure (gel) encompassing a liquid phase, allowing for molecular-level mixing at low temperatures [26] [12]. This guide objectively compares these two dominant methodologies, focusing on their mechanistic principles, experimental protocols, and resulting material properties, to equip researchers with the data necessary for informed synthetic planning.
Fundamental Principles and Comparative Mechanisms
The Solid-State Synthesis Workflow
Solid-state synthesis, in its conventional form, involves the direct reaction of solid precursors through repeated high-energy ball milling and high-temperature calcination [2] [3]. The process is fundamentally limited by diffusion rates at solid-solid interfaces. To overcome this, mechanochemical activation is employed, not merely as a mixing step, but as a means to drive chemical reactions through the direct application of mechanical energy [25].
- Mechanochemical Activation: During ball milling, mechanical energy is transferred to reactant powders through collisions. This energy can induce structural defects, reduce particle size, and even initiate chemical reactions by locally generating heat and pressure at impact points, effectively lowering the activation energy barrier for reaction [25]. The total energy input (
E_total) in a ball milling process can be conceptualized as a function of impact energy per collision (E_impact), the number of balls (N_b), collision frequency (f_b), and milling time (t), as represented byE_total = φ * E_impact * N_b * f_b * t[25]. - Thermal Processing: Following milling, the mixed precursors are subjected to a calcination step at high temperatures (often >800°C). This provides the necessary thermal energy for ionic diffusion across particle boundaries, facilitating crystal nucleation and growth of the target phase [2] [3].
The Sol-Gel Synthesis Workflow
The sol-gel process is a bottom-up approach based on solution-phase chemistry. It begins with molecular precursors, typically metal alkoxides or chlorides, which undergo hydrolysis and condensation reactions to form first a colloidal suspension (sol) and then an interconnected solid network (gel) [26] [12].
- Hydrolysis: A metal alkoxide
M(OR)_4reacts with water:M(OR)_4 + H_2O → M(OR)_3(OH) + ROH[12]. - Condensation: The hydrolyzed species link together, forming M-O-M bonds and releasing water or alcohol:
(OR)_3M-OH + HO-M(OR)_3 → (OR)_3M-O-M(OR)_3 + H_2O[12]. - Gelation and Post-treatment: The gel is then dried and thermally treated (fired) to remove solvents and organic residues, and to crystallize the final metal oxide product [26] [27]. The low-temperature densification is a key advantage over traditional ceramic processing [12].
The following diagram illustrates the core procedural and mechanistic differences between these two synthesis pathways.
Experimental Protocols in Practice
Detailed Protocol: Solid-State Synthesis of BiFeO₃
The synthesis of multiferroic BiFeO₃ provides an excellent case study for optimized solid-state reaction [2].
- Precursor Preparation: Stoichiometric ratios of high-purity
Bi_2O_3andFe_2O_3are weighed out. To compensate for the potential volatilization of bismuth at high temperatures, a slight excess (e.g., 1-2 mol%) ofBi_2O_3is often added [2]. - Mechanochemical Activation: The precursors are ground thoroughly in an ethanol medium and subjected to high-energy ball milling. A typical procedure uses a planetary ball mill with zirconia balls and vial at 1380 rpm for 30 minutes to 4 hours to ensure homogenization and particle size reduction [2] [3].
- Calcination: The milled powder is compacted into pellets and calcined in a muffle furnace. The temperature is optimized through experimentation; for BiFeO₃, a stepwise calcination from 600°C to 820°C for 30 minutes to 2 hours is used to minimize the formation of secondary phases like
Bi_2Fe_4O_9[2]. The heating rate is typically maintained at 10°C/min [3].
Detailed Protocol: Sol-Gel Synthesis of ZnFe₂O₄
The sol-gel synthesis of spinel ferrites like ZnFe₂O₄ demonstrates the method's capability for low-temperature phase formation [3].
- Sol Preparation: Chloride precursors of iron(III) (
FeCl_3) and zinc (ZnCl_2) are mixed in the required molar ratio (e.g., Fe:Zn = 2:1) in a solvent such as ethanol or water under intensive stirring [3]. - Gelation: A sodium hydroxide (
NaOH) solution is slowly introduced to the stirred precursor solution at room temperature to precipitate a solid precursor phase. The pH is carefully controlled (e.g., to pH 10.5) to co-precipitate both metal hydroxides without redissolution [3]. - Aging and Washing: The formed suspension is hydrodynamically processed for 30-60 minutes to age the gel. The resulting gel is then filtered under vacuum and washed with deionized water to remove residual ions and solvent [3].
- Drying and Firing: The filtered precursor is dried at room temperature and subsequently fired in a muffle furnace. The firing temperature for ZnFe₂O₄ can be as low as 400°C, significantly lower than its solid-state counterpart, with a heating rate of 10°C/min [3].
Performance and Material Properties Comparison
The choice of synthesis method directly and profoundly impacts the critical characteristics of the final material. The table below summarizes a quantitative comparison based on experimental data from the literature.
Table 1: Comparative Analysis of Solid-State and Sol-Gel Synthesis Methods
| Performance Characteristic | Solid-State Synthesis | Sol-Gel Synthesis | Experimental Data & Context |
|---|---|---|---|
| Particle Size & Morphology | Larger particles, broad distribution, irregular shapes. Prone to aggregation. | Finer particles, narrow distribution, controlled shapes (e.g., hexagons, sheets). | Solid-state BiFeO~3~: Coarse powders [2].Sol-gel Mn~3~O~4~: Hexagonal nanoparticles, 100-200 nm diameter [26]. |
| Phase Purity & Stoichiometry | Risk of impurity phases; can suffer from cation loss (e.g., Bi volatilization). | High phase purity achievable at lower temperatures; excellent stoichiometry control. | Solid-state BiFeO~3~: Often contains Bi~2~Fe~4~O~9~ impurity [2].Sol-gel BiFeO~3~: Almost single-phase material [2]. |
| Surface Area & Porosity | Low surface area, low porosity. | High surface area, often mesoporous. | Sol-gel Mn~3~O~4~: BET surface area of 91.68 m²/g [26].Sol-gel Mg(OH)~2~: BET surface area of 72.31 m²/g [26]. |
| Synthesis Temperature | High temperatures required (>800°C typical). | Low to moderate temperatures (often <600°C). | Solid-state BiFeO~3~: Calcined at 600-820°C [2].Sol-gel ZnFe~2~O~4~: Fired at 400°C [3]. |
| Processing Time | Long duration due to slow diffusion, repeated milling/calcination. | Relatively shorter, but gel aging can take hours. | Solid-state: Can require multiple cycles of milling and calcination [2]. |
| Homogeneity & Doping | Limited homogeneity; doping can be non-uniform. | Atomic-level mixing; uniform doping with rare-earth elements and organics. | Sol-gel allows fine control and uniform dispersion of dopants [17] [12]. |
| Cost & Equipment | Simple equipment (ball mill, furnace); precursor costs can be low. | May require precise pH control, purification; precursor alkoxides can be expensive. |
The Scientist's Toolkit: Essential Research Reagents and Equipment
Successful execution of these synthesis methods requires specific materials and instruments. The following table lists key items and their functions.
Table 2: Essential Research Reagents and Equipment for Synthesis
| Item | Function in Synthesis | Example Use Case |
|---|---|---|
| Planetary Ball Mill | Provides mechanochemical energy for grinding, mixing, and activating solid precursors. | Homogenization of Bi_2O_3 and Fe_2O_3 for BiFeO₃ synthesis [2]. |
| Muffle Furnace | High-temperature processing for calcination and crystallization of materials. | Calcining precursors at temperatures up to 1600°C for ZrC-SiC composites [27]. |
| Metal Alkoxides | Common molecular precursors for sol-gel synthesis; undergo hydrolysis/condensation. | Tetraethyl orthosilicate (TEOS) for SiO₂ or zirconium butoxide for ZrO₂ networks [12]. |
| Metal Salts (Chlorides, Nitrates) | Alternative, often less expensive, precursors for both sol-gel and precipitation routes. | Zinc and Iron Chlorides used in co-precipitation for ZnFe₂O₄ [3]. |
| Chelating Agents (e.g., Citric Acid) | Used in modified sol-gel (Pechini) process to complex metal cations and ensure homogeneity. | Prevents phase segregation in multi-cation systems like SrTiO₃ [12]. |
| pH Meter | Critical for monitoring and controlling the hydrolysis and condensation rates in sol-gel. | Maintaining pH at 10.5 during ZnFe₂O₄ precursor precipitation [3]. |
Applications and Material-Specific Considerations
The distinct properties of materials produced by each method make them suitable for different applications.
- Solid-State Synthesis is often preferred for manufacturing dense ceramics and compositions where high-temperature stability is required. Its scalability makes it industrially relevant for producing large batches of materials like battery electrodes and ferrites [25] [3]. However, its limitations in particle size control can be a drawback for applications requiring high surface area.
- Sol-Gel Synthesis is unparalleled in producing nanostructured materials with high surface areas, making it ideal for catalysis, sensors, and thin-film devices [26] [17]. The ability to produce mesoporous Mn₃O₄ with a surface area of 91.68 m²/g is a direct enabler for its catalytic activity [26]. Furthermore, sol-gel's superiority in forming high-quality thin films and its compatibility with doping make it a leading choice for developing spintronic materials and advanced coatings [17].
The decision between solid-state and sol-gel synthesis is not a matter of selecting a universally superior technique, but rather of choosing the right tool for a specific material goal. Solid-state synthesis with mechanochemical activation is a powerful, often more direct route for producing thermodynamically stable, crystalline phases in bulk quantities, though it typically yields larger, less uniform particles. Sol-gel processing offers superior control at the molecular level, enabling the fabrication of nanostructured, high-purity, and high-surface-area materials at significantly lower temperatures, albeit often with more complex chemistry and potential cost considerations. Researchers must weigh these factors—temperature, particle size, homogeneity, surface area, and scalability—against the requirements of their target application to navigate the optimal path for material fabrication.
The synthesis of nanostructured oxides is a cornerstone of modern materials science, with significant implications for applications ranging from spintronics and energy storage to catalysis and biomedical devices. Among various fabrication techniques, the sol-gel method has emerged as a powerful low-temperature alternative to traditional solid-state synthesis, particularly when precise control over particle size, morphology, and chemical homogeneity is required. This guide provides a comprehensive comparison of these two fundamental approaches, focusing on their mechanistic principles, experimental protocols, and resulting material properties.
The sol-gel process is a wet-chemical technique that involves the transition of a colloidal solution (sol) into an integrated network (gel) through hydrolysis and condensation reactions of molecular precursors [12] [28]. In contrast, solid-state synthesis typically involves high-temperature reactions between solid precursors through diffusional processes, often resulting in larger particles and less homogeneous compositions [2]. The distinction is particularly crucial for nanostructured oxides, where control over particle size distribution, phase purity, and stoichiometry directly influences functional properties such as catalytic activity, magnetic behavior, and ionic conductivity.
Fundamental Chemical Mechanisms
Sol-Gel Chemistry: Hydrolysis and Condensation Pathways
The sol-gel process is fundamentally based on two consecutive chemical reactions: hydrolysis and condensation. These reactions transform molecular precursors into an extended metal-oxide network [12] [28] [29].
Hydrolysis: Metal alkoxides (M(OR)ₓ) react with water, replacing alkoxy groups (OR) with hydroxyl groups (OH): M(OR)ₓ + H₂O → M(OR)ₓ₋₁(OH) + ROH [28]
Condensation: The hydrolyzed species link together via oxo (M-O-M) or hydroxo (M-OH-M) bridges, forming the growing oxide network while liberating water or alcohol: M-OH + HO-M → M-O-M + H₂O (water condensation) M-OR + HO-M → M-O-M + ROH (alcohol condensation) [12]
The kinetics and thermodynamics of these reactions are influenced by several factors, including pH, water-to-alkoxide ratio, temperature, and catalyst type [28]. Acid-catalyzed conditions typically favor linear chain formation, producing more porous gels, while base-catalyzed conditions promote compact colloidal particles [12] [28].
Figure 1: Sol-Gel Reaction Pathway. The process begins with precursor dissolution, followed by hydrolysis and condensation reactions that form the metal-oxide network.
Solid-State Reaction Mechanisms
In contrast to the solution-based sol-gel approach, solid-state reactions occur through direct atomic diffusion between solid precursors at elevated temperatures (typically >1000°C) [2]. This process involves:
- Nucleation: Formation of initial product phase at contact points between reactant particles
- Diffusion-controlled growth: Atomic migration across particle boundaries leading to crystal growth
- Sintering and densification: Coalescence of particles and reduction of porosity at high temperatures
The solid-state method is inherently limited by slow diffusion rates and often results in heterogeneous products with broader particle size distributions unless extensive mechanical mixing and repeated calcination steps are employed [2] [3].
Experimental Protocols
Sol-Gel Synthesis Procedure
The following protocol outlines a standardized sol-gel approach for producing metal oxide nanoparticles, adaptable for various oxides (SiO₂, TiO₂, ZnO, etc.) through appropriate precursor selection [28] [26].
Materials and Equipment:
- Metal alkoxide precursors (e.g., tetraethyl orthosilicate/TEOS for SiO₂, titanium isopropoxide for TiO₂)
- Solvent (typically ethanol, methanol, or isopropanol)
- Catalyst (acidic: HCl, HNO₃, acetic acid; or basic: NH₃, NaOH)
- Deionized water
- Magnetic stirrer with heating capability
- Beakers and volumetric glassware
- Drying oven
- Muffle furnace for calcination
Step-by-Step Procedure:
Sol Preparation
- Dissolve the metal alkoxide precursor in the solvent under vigorous stirring. Typical concentration range: 0.1-1.0 M.
- For multicomponent systems, mix precursors at this stage to achieve molecular-level homogeneity [28].
Hydrolysis
Condensation and Gelation
- Allow the solution to stand undisturbed as viscosity increases.
- Gelation time can range from hours to days, depending on pH, temperature, and precursor concentration [12].
Aging
- Age the wet gel for 24-72 hours to strengthen the network through continued condensation [12].
Drying
Calcination (Optional)
- Heat treatment (300-800°C) to remove organic residues, enhance crystallinity, and densify the structure [28].
Critical Parameters:
- pH: Acidic conditions (pH 2-5) produce weakly branched networks; basic conditions (pH 7-10) yield colloidal particles [28].
- Temperature: Affects reaction kinetics; typically conducted at 25-80°C [26].
- Water ratio: Lower ratios favor partial hydrolysis and controlled growth [29].
Solid-State Synthesis Procedure
This protocol describes the conventional solid-state method for producing ceramic oxides such as BiFeO₃ and ZnFe₂O₄ [2] [3].
Materials and Equipment:
- Oxide or carbonate precursors (e.g., Bi₂O₃, Fe₂O₃, ZnO)
- High-energy ball mill or mortar and pestle
- Calcination crucibles
- High-temperature muffle furnace (capable of >1000°C)
Step-by-Step Procedure:
Weighing and Mixing
- Weigh precursor powders in stoichiometric proportions.
- For BiFeO₃: Mix Bi₂O₃ and Fe₂O₃ in 1:1 molar ratio [2].
Mechanical Grinding
- Grind mixtures thoroughly in a ball mill for 2-4 hours using ethanol as a mixing medium [2].
- Dry the mixture at 80-100°C to remove the solvent.
Calcination
- Place the mixed powder in an appropriate crucible.
- Heat at 600-850°C for 2-12 hours in a muffle furnace [2].
- For complex oxides, multiple calcination steps with intermediate grinding may be necessary.
Final Processing
- Regrind the calcined powder to break aggregates.
- For pellet formation, press the powder and sinter at higher temperatures (e.g., 900-1100°C) [2].
Critical Parameters:
- Temperature: Must be optimized to achieve complete reaction while minimizing volatilization (e.g., Bi loss in BiFeO₃) [2].
- Grinding time and efficiency: Determines precursor intimacy and final homogeneity.
- Heating/cooling rates: Affect phase purity and particle size.
Figure 2: Comparative Workflows: Sol-Gel vs. Solid-State Synthesis. Sol-gel processing occurs largely in solution at low temperatures, while solid-state relies on mechanical mixing and high-temperature diffusion.
Comparative Performance Analysis
Quantitative Comparison of Material Properties
Table 1: Direct comparison of material properties achieved through sol-gel and solid-state synthesis methods.
| Property | Sol-Gel Method | Solid-State Method | Experimental Support |
|---|---|---|---|
| Particle Size | 10-200 nm (tunable) | 0.5-50 μm (typically larger) | BiFeO₃: SG ~0.5 μm vs. SS ~1 μm [2] |
| Surface Area | 20-800 m²/g (typically high) | 1-10 m²/g (typically low) | Mn₃O₄: 91.68 m²/g [26] |
| Phase Purity | High with optimization | Often contains secondary phases | BiFeO₃: SG ~single phase vs. SS with Bi₂Fe₄O₉ impurity [2] |
| Stoichiometry Control | Excellent, atomic-level mixing | Challenging, volatilization issues | BiFeO₃: SG maintains stoichiometry vs. Bi loss in SS [2] |
| Processing Temperature | Room temp - 800°C | 600-1500°C | SG: <80°C gelation [26]; SS: >700°C [2] |
| Densification Temperature | Lower by 200-500°C | Higher | SG advantage for energy savings [12] |
| Homogeneity | Molecular level, excellent | Limited by diffusion, often inhomogeneous | Multicomponent oxides more uniform in SG [28] [29] |
Case Study: Bismuth Ferrite (BiFeO₃) Synthesis
A direct comparative study of BiFeO₃ synthesis reveals method-dependent properties [2]:
- Phase Purity: Sol-gel produced nearly single-phase BiFeO₃, while solid-state synthesis resulted in Bi₂Fe₄O₉ impurity phases even after optimization.
- Particle Size: Sol-gel yielded significantly smaller particles (~0.5 μm) compared to solid-state (~1 μm).
- Stoichiometry: Sol-gel maintained better Bi:Fe stoichiometry (1:1) by restricting bismuth volatilization.
- Dielectric Properties: Sol-gel samples exhibited higher dielectric constants with Maxwell-Wagner type dielectric dispersion.
- Magnetic Properties: Both methods showed antiferromagnetic behavior at room temperature, but sol-gel samples demonstrated slightly increased coercivity (H~C~) and remanent magnetization (M~r~).
Case Study: Zinc Ferrite (ZnFe₂O₄) Synthesis
Another comparative study on ZnFe₂O₄ highlighted similar trends [3]:
- Sol-gel derived materials showed better compositional homogeneity and smaller crystallite sizes.
- The synthesis temperature for sol-gel was significantly lower (350-750°C) compared to solid-state (800-1200°C).
- Electrical properties were strongly influenced by synthesis method, with sol-gel materials demonstrating advantages for battery applications.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key reagents and materials for sol-gel and solid-state synthesis with their specific functions.
| Reagent/Material | Function | Examples | Considerations |
|---|---|---|---|
| Metal Alkoxides | Primary precursors for sol-gel | TEOS (SiO₂), Ti(OiPr)₄ (TiO₂), Al(OsecBu)₃ (Al₂O₃) | Moisture-sensitive; purity critical [12] [28] |
| Metal Salts | Alternative precursors | Metal chlorides, nitrates, acetates | More stable but may introduce impurities [26] |
| Solvents | Reaction medium | Ethanol, methanol, isopropanol | Affects precursor solubility and reaction kinetics [28] |
| Catalysts | Control hydrolysis/condensation rates | HCl, HNO₃ (acidic); NH₃, NaOH (basic) | pH critically affects pore structure and network [28] [29] |
| Chelating Agents | Modify precursor reactivity | Acetylacetone, citric acid, ethylene glycol | Control hydrolysis rates in Pechini process [12] [30] |
| Oxide Precursors | Solid-state starting materials | Bi₂O₃, Fe₂O₃, ZnO, TiO₂ | High purity essential; particle size affects reactivity [2] [3] |
| Grinding Media | Homogenization | Zirconia balls, tungsten carbide | Contamination risk during milling [2] |
The comparative analysis demonstrates that sol-gel methodologies offer distinct advantages over solid-state synthesis for nanostructured oxide fabrication, particularly when nanoscale control, homogeneity, and low processing temperatures are prioritized. The sol-gel approach enables precise manipulation of particle size, porosity, and composition at the molecular level through controlled hydrolysis and condensation of alkoxide precursors.
Solid-state synthesis remains relevant for large-scale production of simple oxides where high temperatures are not prohibitive and atomic-level homogeneity is less critical. However, for advanced applications in spintronics [17], energy storage [3], and catalysis [29] where surface functionality and nanoscale architecture determine performance, sol-gel methodologies provide superior materials design capability.
Future research directions include the development of continuous flow sol-gel processes to address scalability limitations [28], advanced templating strategies for hierarchical architectures [26], and hybrid approaches that combine the advantages of both methods for next-generation functional oxides.
Zinc ferrite (ZnFe₂O₄) has emerged as a compelling material for advanced energy applications, particularly for supercapacitors and metal-ion batteries, due to its high theoretical capacitance, environmental abundance, and intriguing magnetic properties [31] [3]. However, its practical implementation is often hampered by inherent limitations such as low intrinsic conductivity and structural instability. A critical strategy to overcome these challenges lies in precise control of particle size and morphology during synthesis, which directly governs the material's electrochemical performance. This case study provides a comparative analysis of solid-state and sol-gel synthesis methods for ZnFe₂O₄, focusing on their efficacy in particle size control and the subsequent impact on material properties relevant to energy storage devices. The research contextualizes these findings within a broader thesis on nanomaterial synthesis, demonstrating how methodological choices at the fabrication stage dictate ultimate application performance.
Synthesis Methodologies and Particle Size Control
The synthesis pathway for ZnFe₂O₄ significantly influences its fundamental properties by determining crystallite size, morphology, and surface area. The solid-state and sol-gel methods represent two philosophically distinct approaches to material preparation.
Solid-State Synthesis
The solid-state method is a conventional ceramic technique involving high-temperature reaction of solid precursors. A typical protocol involves mixing iron(III) oxide (Fe₂O₃) and zinc oxide (ZnO) in a stoichiometric 1:1 molar ratio [3]. The precursor mixture undergoes homogenization via grinding in an agate mortar, followed by intensive mechanochemical activation in a planetary ball mill (e.g., at 1380 rpm for 30 minutes). The activated powder is then subjected to thermal treatment in a muffle furnace, with a standard heating rate of 10 °C/min, at temperatures ranging from 400 °C to 1000 °C to form the final crystalline phase [3] [32]. This method typically yields larger particles with lower surface area due to aggressive sintering and grain growth at elevated temperatures.
Sol-Gel Synthesis
In contrast, the sol-gel method is a wet-chemical technique that offers superior control over particle size and homogeneity. One reported procedure involves a combined sol-gel and solid-state finishing process [3]. It starts with dissolving zinc and iron precursors, such as zinc chloride (ZnCl₂) and iron(III) chloride (FeCl₃), in distilled water. Co-precipitation is achieved by adding sodium hydroxide (NaOH) solution under vigorous stirring until reaching a pH of 10.5. The resulting precipitate is filtered, washed with deionized water, and dried at room temperature. The final crystalline ZnFe₂O₄ is obtained by calcining the precursor in a muffle furnace at various temperatures. The sol-gel route facilitates mixing at the molecular level, enabling the formation of finer, more uniform particles with higher surface area.
Hydrothermal and Combustion Methods
Beyond these two primary methods, other techniques like hydrothermal synthesis and solution combustion also provide potent means for size control. Hydrothermal synthesis allows precise size tuning by adjusting surfactant concentration [31], while solution combustion utilizes different fuels (e.g., urea, glycine, EDTA) to govern crystal size, with urea producing smaller crystals (~10 nm) compared to EDTA (~27 nm) [33].
Table 1: Comparative Analysis of ZnFe₂O₄ Synthesis Methods
| Synthesis Method | Typical Particle Size Range | Specific Surface Area (m²/g) | Key Size Control Parameters | Crystallinity |
|---|---|---|---|---|
| Solid-State [3] [32] | Micrometer scale (≥1000 nm) | Low (e.g., <20 m²/g) | Calcination temperature, milling time | High crystallinity |
| Sol-Gel [3] [32] | ~48 - 93 nm [32] | Moderate to High | Calcination temperature, precursor chemistry | High crystallinity |
| Solution Combustion [33] | ~10 - 27 nm | 94 - 116 m²/g [33] | Fuel type (urea→glycine→EDTA) | Moderate to High |
| Hydrothermal [31] | Tunable nanospheres | Not Specified | Surfactant concentration | High crystallinity |
Correlation Between Particle Size and Functional Properties
Precise particle size control directly dictates the functional properties of ZnFe₂O₄, enabling performance optimization for target applications.
Electrochemical Performance for Supercapacitors
Particle size reduction to the nanoscale dramatically enhances electrochemical performance by shortening ion diffusion paths and increasing the electroactive surface area. ZnFe₂O₄ nanospheres synthesized via hydrothermal methods with controlled surfactant concentration demonstrated a remarkable specific capacitance of 1819.6 F/g at 10 A/g [31]. This outstanding performance was linked to improved charge transfer mechanisms, as confirmed by Density Functional Theory (DFT) simulations, which showed that potassium ion (K⁺) adsorption from the electrolyte introduces new energy states near the Fermi level [31]. Furthermore, the presence of oxygen vacancies, detected via Electron Paramagnetic Resonance (EPR), significantly enhances redox activity, contributing to the superior charge storage.
Magnetic Properties
The magnetic behavior of ZnFe₂O₄ exhibits a profound size dependence. While bulk ZnFe₂O₄ is typically paramagnetic, nanoparticles can display superparamagnetic or ferrimagnetic behavior due to changes in cation distribution [34] [32]. At the nanoscale, a significant fraction of Zn²⁺ ions occupy octahedral sites, while Fe³⁺ ions simultaneously occupy tetrahedral sites, leading to a partially inverted spinel structure. This inversion creates a magnetic moment. The extent of inversion is directly correlated with particle size, with smaller particles (e.g., 4 nm) showing around 80% inversion, while larger particles (≥7 nm) exhibit negligible inversion [34]. This size-dependent magnetism is crucial for applications like magnetic separation or hyperthermia.
Photocatalytic Activity
In photocatalytic applications, such as dye degradation for environmental remediation, smaller particle sizes are advantageous due to their higher surface area, which provides more active sites for reactions. The bandgap of ZnFe₂O₄ can be tuned through size reduction, a phenomenon explained by both quantum confinement effects and an increasing fraction of Fe²+ ions on the particle surface as the size decreases [35]. Studies on ZnFe₂O₄/SiO₂ nanocomposites have demonstrated that photocatalytic efficiency for dye degradation is strongly influenced by surface area and particle size [32].
Table 2: Size-Dependent Properties of ZnFe₂O₄ for Energy Applications
| Property | Bulk/Micron-Sized | Nanosized (Typ. < 100 nm) | Impact on Energy Applications |
|---|---|---|---|
| Specific Capacitance | Low | Very High (1819.6 F/g) [31] | Enables high-performance supercapacitors |
| Magnetic Behavior | Paramagnetic [34] | Superparamagnetic/Ferrimagnetic [36] [34] [32] | Facilitates catalyst recovery, magnetic data storage |
| Cation Distribution | Normal Spinel (Zn²⁺ in A-site) | Partially Inverted Spinel (up to 80% inversion) [34] | Governs saturation magnetization and electrical conductivity |
| Surface Area | Low | High (up to 116 m²/g) [33] | Enhances reaction kinetics and active sites for catalysis |
| Ionic Conductivity | Lower | Higher [3] | Beneficial for cathode performance in metal-ion batteries |
The Scientist's Toolkit: Essential Research Reagents
Successful synthesis of ZnFe₂O₄ with controlled properties requires careful selection of precursors and reagents. The table below details key materials and their functions.
Table 3: Essential Reagents for ZnFe₂O₄ Synthesis and Characterization
| Reagent / Material | Function / Application | Example from Literature |
|---|---|---|
| Zinc Precursors (e.g., Zn(NO₃)₂·6H₂O, Zn(CH₃COO)₂·2H₂O) | Source of Zn²⁺ cations for the spinel structure [35] [33]. | Zinc acetate and zinc nitrate used in hydrothermal and combustion synthesis [35] [33]. |
| Iron Precursors (e.g., FeCl₃·6H₂O, Fe(NO₃)₃·9H₂O) | Source of Fe³⁺ cations for the spinel structure [35] [33]. | Iron chloride and iron nitrate are common choices [35] [33]. |
| Sodium Acetate | Fuel and structure-directing agent in hydrothermal synthesis; controls particle size [35]. | Concentration variation (11.5 - 62.2 mmol) to control nanoparticle size [35]. |
| Urea, Glycine, EDTA | Fuels in solution combustion synthesis; type influences crystal size and magnetism [33]. | Urea produced smallest crystals (10.19 nm), EDTA yielded highest magnetization (54.63 emu/g) [33]. |
| Sodium Hydroxide (NaOH) | Precipitating agent in co-precipitation and sol-gel methods to form hydroxides [3]. | Used to adjust pH to 10.5 for precursor precipitation [3]. |
| KOH Electrolyte | Common alkaline electrolyte for supercapacitor performance testing [31]. | Adsorption of K⁺ ions enhances charge transfer, per DFT studies [31]. |
Experimental Protocols for Reproducible Research
Protocol: Hydrothermal Synthesis of ZnFe₂O₄ Nanospheres
This protocol is adapted from studies achieving superior supercapacitor performance [31].
- Step 1: Solution Preparation. Dissolve zinc acetate (2 mmol) and iron chloride (4 mmol) in 30 mL of ethylene glycol under continuous stirring.
- Step 2: Surfactant Addition. Add sodium acetate as a surfactant at a specific concentration (e.g., 20%) to control the final particle size.
- Step 3: Hydrothermal Reaction. Transfer the homogeneous solution to a Teflon-lined stainless autoclave and heat at 180 °C for 24 hours.
- Step 4: Product Recovery. Collect the precipitate by centrifugation, wash several times with distilled water, and dry in air at 60 °C for 24 hours.
- Step 5: Electrode Fabrication. For electrochemical testing, directly deposit the ZnFe₂O₄ nanospheres onto Ni foam to create a binder- and carbon-free electrode.
Protocol: Solution Combustion Synthesis with Fuel Control
This protocol highlights how fuel selection dictates properties [33].
- Step 1: Redox Mixture. Dissolve a fuel (urea, glycine, or EDTA) in distilled water acidified to pH 4. Add Zn(NO₃)₂·6H₂O and Fe(NO₃)₃·9H₂O in a 1:2 molar ratio. Include KCl to help control crystal size.
- Step 2: Combustion. Stir the mixture magnetically at 300 °C until combustion occurs (approximately 1 hour). The solution will change color and yield a powder.
- Step 3: Post-Processing. Wash the resulting powder with boiling distilled water and dry in an oven at 80 °C.
Characterization Workflow
A comprehensive analysis of synthesized ZnFe₂O₄ involves a suite of characterization techniques to correlate structure with properties, as illustrated below.
This case study demonstrates that the synthesis method is a decisive factor in controlling the particle size of ZnFe₂O₄, which in turn governs its electrochemical, magnetic, and surface properties. The comparative analysis reveals a clear trade-off: while solid-state synthesis produces highly crystalline materials, it generally yields larger particles less suited for high-performance energy applications. In contrast, wet-chemical methods like sol-gel, hydrothermal, and combustion synthesis offer superior control over particle size at the nanoscale, enabling the enhanced specific capacitance, tunable magnetism, and high ionic conductivity required for next-generation supercapacitors and metal-ion batteries. The choice of synthesis route, therefore, should be guided by the target application's specific property requirements, with hydrothermal and combustion methods being particularly promising for achieving the small particle sizes and high surface areas that unlock superior energy storage performance.
The integration of sol-gel derived nanoparticles into biomedical hydrogel platforms represents a transformative approach in modern drug delivery and regenerative medicine. The sol-gel process is a versatile chemical synthesis method for producing inorganic and hybrid organic-inorganic materials with tailored nanostructures. This process involves the transition of a solution ("sol") from a liquid state into a solid ("gel") phase through a series of hydrolysis and condensation reactions, typically using precursors such as tetraethyl orthosilicate (TEOS) [37]. This synthesis pathway offers exceptional control over particle size, pore structure, and surface chemistry, making it particularly valuable for creating sophisticated drug delivery systems [3] [37].
When these engineered nanoparticles are incorporated into hydrophilic hydrogel networks—three-dimensional polymer structures capable of absorbing large quantities of water—they form composite systems that synergistically combine the advantages of both components [38] [39]. These hybrid platforms leverage the high drug loading capacity and tunable release kinetics of sol-gel nanoparticles with the biocompatibility and stimuli-responsive behavior of hydrogels, creating powerful therapeutic platforms for applications ranging from cancer therapy to wound healing and ocular disorders [40] [41] [37].
This guide objectively compares the performance of sol-gel derived nanoparticles against alternative synthesis methods, with particular emphasis on their integration within hydrogel matrices for biomedical applications, providing researchers with experimental data and methodologies to inform their therapeutic development strategies.
Synthesis Methodologies: Sol-Gel versus Solid-State Routes
The selection of synthesis methodology profoundly impacts the structural properties and subsequent performance of nanoparticles in biomedical applications. The table below compares the fundamental characteristics of sol-gel and solid-state synthesis routes.
Table 1: Comparison of Nanoparticle Synthesis Methods for Biomedical Applications
| Parameter | Sol-Gel Method | Solid-State Method |
|---|---|---|
| Process Principle | Chemical transformation from solution colloidal suspension to gel network | Mechanical mixing and high-temperature solid-state reaction |
| Temperature Range | Low to moderate (room temp to ~80°C) | High (often >1000°C) |
| Particle Size Control | Excellent (nanometer precision) | Limited (broader size distribution) |
| Surface Area | High (600-1000 m²/g for MSNs) [37] | Low to moderate |
| Porosity Control | Excellent tunable mesoporosity (2-50 nm) [37] | Limited, often non-porous |
| Morphology Control | Spherical, hollow, core-shell structures possible [37] | Irregular shapes common |
| Homogeneity | Excellent molecular-level mixing | Potential inhomogeneities |
| Drug Loading Efficiency | High (85-95% for optimized systems) [40] | Limited by surface adsorption only |
The Sol-Gel Synthesis Process
Sol-gel synthesis involves a series of controlled chemical reactions that transform molecular precursors into an integrated nanoparticle network:
- Hydrolysis: Silicon alkoxide precursors (e.g., TEOS) react with water:
Si(OR)₄ + H₂O → Si(OR)₃(OH) + ROH - Condensation: Silanol groups form siloxane bonds:
(RO)₃Si-OH + HO-Si(OR)₃ → (RO)₃Si-O-Si(OR)₃ + H₂O - Template-Assisted Pore Formation: Surfactants like CTAB create micellar templates around which condensation occurs, generating mesoporous structures [37]
- Aging & Drying: The gel network strengthens, followed by calcination or solvent extraction to remove templates
This process enables exceptional control over particle architecture, allowing researchers to tailor materials for specific drug delivery applications through precise manipulation of reaction parameters including pH, temperature, precursor concentration, and catalyst type [37].
Solid-State Synthesis Limitations
In contrast, solid-state synthesis relies on high-temperature reactions between solid precursors, resulting in particles with limited surface area, reduced porosity, and less controlled morphology. While suitable for certain ceramic electrode materials like ZnFe₂O₄ [3], this method offers significantly fewer opportunities for engineering sophisticated drug delivery platforms due to its limited control over critical nanoparticle characteristics.
Experimental Protocols for Sol-Gel Nanoparticle Synthesis and Evaluation
Protocol: Mesoporous Silica Nanoparticle (MSN) Synthesis via Sol-Gel
Objective: Synthesize monodisperse MSNs of controlled size and porosity for drug delivery applications [37].
Materials:
- Tetraethyl orthosilicate (TEOS): Primary silica precursor
- Cetyltrimethylammonium bromide (CTAB): Structure-directing surfactant template
- Ammonium hydroxide (NH₄OH): Catalyst for hydrolysis and condensation
- Ethanol: Reaction medium
- Deionized water: Solvent for hydrolysis
Procedure:
- Prepare surfactant solution by dissolving CTAB (0.5-1.0 g) in a mixture of deionized water (200-250 mL) and ethanol (50-100 mL)
- Add ammonium hydroxide (1-2 mL) to the surfactant solution under constant stirring (300-400 rpm) to establish basic conditions (pH ~10-11)
- Slowly add TEOS (2-5 mL) dropwise to the solution and continue stirring for 2-4 hours at room temperature to allow nanoparticle formation
- Age the resulting milky suspension without stirring for 12-24 hours to complete the condensation process
- Recover nanoparticles by centrifugation (10,000-15,000 rpm for 15 minutes) and wash repeatedly with ethanol/water mixtures to remove unreacted precursors
- Remove surfactant templates by calcination at 550°C for 5-6 hours or solvent extraction using acidic ethanol
- Characterize particle size by dynamic light scattering (DLS), surface area and porosity by nitrogen adsorption-desorption analysis, and morphology by transmission electron microscopy (TEM)
Key Control Parameters:
- Surfactant/SiO₂ ratio controls pore size (typically 2-6 nm)
- Reaction temperature influences particle size distribution
- Stirring rate affects particle agglomeration
- Catalyst concentration impacts condensation kinetics [37]
Protocol: Incorporating MSNs into Thermo-Responsive Hydrogels
Objective: Create a composite hydrogel system for sustained drug delivery [40] [41].
Materials:
- Poloxamer 407 (Pluronic F127): Thermo-responsive polymer
- Gelatin or chitosan: Natural polymer component
- Prefabricated MSNs: Drug-loaded nanoparticles
- Crosslinking agents (if needed): Genipin, glutaraldehyde, or physical crosslinkers
Procedure:
- Dissolve thermo-responsive polymer (e.g., poloxamer 407, 15-20% w/v) in cold aqueous solution (4-8°C) with gentle stirring
- Incorporate drug-loaded MSNs (1-5% w/v) into the polymer solution and disperse uniformly using probe sonication (30-60 seconds at low amplitude)
- Adjust pH to physiological range (7.0-7.4) if using pH-sensitive polymers
- For chemical crosslinking, add crosslinking agent at appropriate concentration and incubate at specific temperature/time conditions
- Characterize composite hydrogel for sol-gel transition temperature, rheological properties, microstructural morphology, and drug release profile
Key Evaluation Metrics:
- Sol-gel transition temperature should occur near physiological temperature (32-37°C)
- Gelation time should be appropriate for clinical administration (1-5 minutes)
- Mechanical properties (storage modulus G') should match target tissue requirements
- Drug release kinetics should demonstrate sustained release over therapeutic timeframe [40] [41]
Performance Comparison in Biomedical Applications
The integration of sol-gel derived nanoparticles within hydrogel platforms significantly enhances therapeutic efficacy across multiple biomedical applications, as demonstrated by the following experimental data.
Table 2: Performance Comparison of Sol-Gel Nanoparticle Hydrogel Systems in Therapeutic Applications
| Application | System Composition | Key Performance Metrics | Comparative Advantage |
|---|---|---|---|
| Corneal Wound Healing [40] | GelMa-Poloxamer hydrogel with ROS-responsive NPs (DEX, RAPA, CIP) | - NP size: 105-114 nm- Encapsulation: 76.94-88.11%- Sustained release: 3 weeks | Dual thermo-photo responsive release; Non-invasive alternative to surgery |
| Ocular Drug Delivery [41] | Chitosan/gelatin/HA thermoresponsive hydrogels with MSNs | - Extended residence time- Reduced dosing frequency- Enhanced bioavailability vs conventional drops | Sol-gel transition at physiological temperature improves retention |
| Anticancer Therapy [38] | Cysteine-silver sol/MB composite hydrogel | - Selective toxicity to carcinoma cells- 2-3x efficacy increase with light activation- Low toxicity to normal keratinocytes | Synergistic photodynamic/chemo therapy; Localized activation |
| Wound Dressing [38] | PVA-cysteine-silver composite hydrogel | - Porous structure (2-10 μm)- Non-toxic to fibroblasts- Excellent rheological properties | Biocompatible cell carrier; Antimicrobial activity |
Drug Loading and Release Performance
The structural advantages of sol-gel derived nanoparticles directly translate to superior drug delivery performance:
- High Encapsulation Efficiency: MSNs consistently demonstrate high drug loading capacities (76-88% for multiple therapeutic agents), significantly outperforming non-porous alternatives [40]
- Sustained Release Profiles: The combination of MSN interior porosity with hydrogel diffusion barriers enables extended release durations (1-3 weeks in optimized systems) [40] [41]
- Stimuli-Responsive Behavior: Functionalized MSN-hydrogel composites can respond to physiological triggers (pH, reactive oxygen species, enzymes) for targeted drug release at disease sites [40]
Table 3: Quantitative Drug Delivery Performance of Sol-Gel Nanoparticle Hydrogel Systems
| System Type | Drug Load Capacity | Release Duration | Therapeutic Outcomes |
|---|---|---|---|
| GelPol Nano-formulation [40] | DEX: 87.27%RAPA: 88.11%CIP: 76.94% | 3 weeks sustained release | 84.5% fibroblast migration in 48h; Near 100% wound closure in 28 days (rat model) |
| CSS-MB Hydrogel [38] | Synergistic Ag⁺/MB loading | Light-activated release | 2-3x cytotoxicity enhancement with light against squamous carcinoma |
| HPCD-PMSSO Iron Delivery [38] | Host-guest complexation | Sustained gastric release | Improved adherence and efficacy for anemia treatment |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful development of sol-gel nanoparticle hydrogel platforms requires specific reagents and equipment. The following table details essential materials and their functions in experimental protocols.
Table 4: Essential Research Reagents for Sol-Gel Nanoparticle Hydrogel Development
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Tetraethyl orthosilicate (TEOS) | Primary silica precursor for MSN synthesis | High purity (>98%) recommended for reproducible results [37] |
| Cetyltrimethylammonium bromide (CTAB) | Structure-directing surfactant for mesopore formation | Concentration controls pore size (typically 2-6 nm) [37] |
| Poloxamer 407 (Pluronic F127) | Thermo-responsive polymer for in situ gelation | 15-20% w/v concentration typical for physiological transition [40] [41] |
| Chitosan | Natural mucoadhesive polymer for ocular delivery | Enhances residence time on corneal surface [41] [39] |
| Gelatin Methacryloyl (GelMA) | Photocrosslinkable biopolymer for tissue engineering | Provides cell adhesion motifs and tunable mechanical properties [40] |
| Methylene Blue | Photosensitizer for photodynamic therapy applications | Enables light-activated drug release [38] |
| Genipin | Natural crosslinking agent for biopolymers | Lower cytotoxicity than synthetic crosslinkers like glutaraldehyde |
The experimental data and performance comparisons presented in this guide demonstrate clear advantages for sol-gel derived nanoparticles in advanced drug delivery hydrogel platforms. The precise control over nanoparticle architecture afforded by sol-gel chemistry enables researchers to engineer systems with optimized drug loading, release kinetics, and targeting capabilities.
For research teams selecting synthesis methodologies, sol-gel approaches provide superior performance for most therapeutic hydrogel applications where controlled drug release and biocompatibility are paramount. Solid-state methods remain relevant for specific ceramic biomaterials requiring high temperature stabilization [3], but offer limited utility for sophisticated drug delivery platforms.
Future development in this field will likely focus on multi-stimuli responsive systems that react to multiple physiological cues, advanced targeting strategies for specific tissues and cell types, and scalable manufacturing processes to transition these promising laboratory developments to clinical applications. The continued refinement of sol-gel derived nanoparticles within hydrogel platforms represents a promising pathway to advanced therapeutic systems with enhanced efficacy, reduced side effects, and improved patient compliance.
Overcoming Scaling and Homogeneity Challenges: Advanced Optimization in Particle Synthesis
The pursuit of advanced functional materials with tailored properties for applications in energy storage, spintronics, and pharmaceuticals places immense importance on precise control over structural and morphological characteristics. Among these, particle size and homogeneity stand as critical determinants of performance. Within this context, two predominant synthesis methodologies—solid-state reaction and sol-gel processing—present researchers with a fundamental choice, each carrying distinct advantages and challenges. Solid-state synthesis, the conventional "shake and bake" ceramic approach, involves direct reactions between solid precursors at high temperatures, while the sol-gel method employs solution-based chemistry to form metal oxide networks through hydrolysis and condensation reactions at significantly lower temperatures.
A growing body of evidence suggests that the selection of synthesis pathway profoundly influences the resulting material's phase purity, particle size distribution, and ultimately, its functional properties. This guide provides an objective comparison of these two methods, focusing specifically on their respective capabilities and limitations in mitigating two pervasive challenges: inhomogeneity and incomplete reactions. Through structured experimental data and detailed protocols, we aim to equip researchers with the knowledge necessary to select and optimize synthesis strategies for next-generation materials.
Comparative Analysis: Solid-State vs. Sol-Gel Synthesis
The fundamental differences between solid-state and sol-gel synthesis methods originate from their distinct reaction environments and mechanisms. Solid-state reactions are governed by solid-solid diffusion at elevated temperatures, often resulting in kinetic limitations that can lead to incomplete reactions and heterogeneous products [42]. In contrast, sol-gel processing facilitates atomic-level mixing of precursors in a solution, leading to superior homogeneity and lower processing temperatures [2].
Table 1: Fundamental Characteristics of Solid-State and Sol-Gel Methods
| Feature | Solid-State Synthesis | Sol-Gel Method |
|---|---|---|
| Reaction Mechanism | Solid-solid diffusion, nucleation & growth | Hydrolysis & condensation, polymerisation |
| Typical Processing Temperature | High (800–1500°C) | Low (Room temp to ~600°C) |
| Primary Driving Force | Thermal energy overcoming diffusion barriers | Chemical potential, molecular reactions |
| Homogeneity Control | Limited, depends on grinding & milling | Excellent, atomic-level mixing |
| Common Pitfalls | Incomplete reactions, inhomogeneity, impurity phases | Shrinkage & cracking during drying, residual carbon |
| Scalability | Well-established for large-scale production | Scalable, but requires solvent management |
Quantitative Comparison of Synthesis Outcomes
Direct comparative studies reveal how these fundamental differences translate to measurable disparities in material properties. Research on multiferroic BiFeO₃ demonstrates that the synthesis route drastically affects phase purity, microstructure, and functional performance [2].
Table 2: Experimental Outcomes for BiFeO₃ Synthesized via Different Methods [2]
| Parameter | Solid-State Method | Sol-Gel Method |
|---|---|---|
| Phase Purity | Contains Bi₂Fe₄O₉ impurity phase | Almost single-phase |
| Average Grain Size | ~1 μm | ~0.5 μm (50% smaller) |
| Elemental Stoichiometry (Bi:Fe) | Non-stoichiometric due to Bi volatilization | Near-perfect 1:1 stoichiometry |
| Dielectric Constant | Lower values | Higher values with Maxwell-Wagner dispersion |
| Magnetic Coercivity (Hc) | Standard values | Slightly increased |
Similarly, a 2025 study on ZnFe₂O₄ spinels for battery applications confirmed that the synthesis method directly influences the electrophysical properties and phase formation mechanisms, with sol-gel derived materials showing distinct characteristics compared to those prepared by ceramic technology [3].
Experimental Protocols and Methodologies
Objective: To synthesize polycrystalline BiFeO₃ via conventional solid-state reaction.
- Reagents: Bi₂O₃ (99.9%), Fe₂O₃ (99.9%), ethanol medium.
- Equipment: High-energy ball mill, planetary ball milling system, WC vial and balls, muffle furnace.
Procedure:
- Weighing & Mixing: Stoichiometric ratios of Bi₂O₃ and Fe₂O₃ are weighed.
- Grinding: Precursors are ground thoroughly in ethanol medium using a high-energy ball mill for 4 hours.
- Calcination: The mixed powders are calcined in a muffle furnace at temperatures ranging from 700°C to 820°C for 30 minutes to 2 hours to optimize phase formation.
- Pelletization & Sintering: The calcined powder is pressed into pellets and sintered at optimized temperature.
Key Considerations: The Bi₂Fe₄O₉ impurity phase persists despite optimized calcination conditions. Excess Bi₂O₃ may be added to compensate for bismuth volatilization at high temperatures, but control over the exact compensation is challenging.
Objective: To synthesize high-purity BiFeO₃ nanoparticles via sol-gel method.
- Reagents: Bismuth nitrate pentahydrate (Bi(NO₃)₃·5H₂O), Iron nitrate nonahydrate (Fe(NO₃)₃·9H₂O), Citric acid, Ethylene glycol, Ammonia solution.
- Equipment: Magnetic stirrer, beakers, drying oven, muffle furnace.
Procedure:
- Precursor Solution Preparation: Stoichiometric amounts of bismuth and iron nitrates are dissolved in distilled water with stirring.
- Chelation: Citric acid is added as a chelating agent (metal ions:citric acid molar ratio = 1:1.5).
- pH Adjustment: The pH is adjusted to ~7 using ammonia solution to facilitate gel formation.
- Polyesterification: Ethylene glycol is added and the temperature is maintained at 80°C with constant stirring until a viscous gel forms.
- Gelation & Drying: The gel is dried in an oven at 120°C for 12 hours to form a solid precursor.
- Calcination: The dried gel is calcined at 600°C for 2 hours in a muffle furnace to obtain crystalline BiFeO₃.
Key Considerations: This method achieves single-phase material at relatively lower temperatures with minimal bismuth loss, resulting in stoichiometric Bi:Fe content and reduced grain size.
Workflow Visualization
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions and Their Functions in Synthesis
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Metal Oxides/Carbonates | Solid precursors for ceramic synthesis | Bi₂O₃, Fe₂O₃ for solid-state BiFeO₃ [2] |
| Metal Alkoxides | Highly reactive molecular precursors for sol-gel | Tetraethyl orthosilicate (TEOS), Titanium isopropoxide [14] |
| Metal Salts | Soluble precursors for solution-based methods | Nitrates (e.g., Bi(NO₃)₃·5H₂O, Fe(NO₃)₃·9H₂O) [2] |
| Chelating Agents | Complex metal ions to control hydrolysis rates | Citric acid, EDTA [2] |
| Gelling Agents | Promote network formation in sol-gel process | Ethylene glycol [2] |
| Mineralizers/Flux Agents | Enhance reaction kinetics in solid-state synthesis | LiCl, NaCl [43] |
The choice between solid-state and sol-gel synthesis methods involves fundamental trade-offs. Solid-state reactions offer simplicity and direct scalability but frequently struggle with inhomogeneity and incomplete reactions due to diffusion limitations, as evidenced by the ~28% heterogeneity reported in LaCeTh₀.₁CuOy compounds [42] and persistent impurity phases in BiFeO₃ [2]. Sol-gel processing, while more complex, provides superior molecular-level mixing, resulting in enhanced homogeneity, phase purity, and smaller particle sizes at reduced temperatures.
For research applications demanding precise control over stoichiometry, nanoscale morphology, and phase purity—particularly in advanced pharmaceuticals, spintronic materials, and next-generation battery components—the sol-gel method presents distinct advantages. However, for large-scale industrial production where slight impurities can be tolerated, traditional solid-state reactions remain viable, especially when enhanced by mechanochemical activation or optimized precursor selection [3]. Understanding these fundamental relationships between synthesis route, structural outcomes, and functional performance enables researchers to strategically select and optimize processing conditions to overcome persistent challenges in materials development.
In the fields of drug development and advanced materials science, the scaling of nanomaterial synthesis presents a significant challenge. The sol-gel method, a versatile chemical technique for producing solid materials from small molecules, is particularly susceptible to changes in particle characteristics when transitioning from laboratory to industrial production. This method involves the transition of a solution ("sol") into a solid "gel" network, offering unparalleled control over material structure at the nanoscale [44]. For researchers and pharmaceutical professionals, maintaining a tight particle size distribution during scale-up is not merely desirable—it is critical for ensuring predictable performance in final applications, particularly in drug delivery systems where size directly influences biodistribution, release kinetics, and targeting efficiency [44].
In contrast, solid-state synthesis, which relies on high-temperature reactions between solid precursors, presents different advantages and limitations for particle control [1] [2]. This comparative guide examines the fundamental strategies and challenges associated with both methods, providing experimental data and protocols to inform method selection for research and development. The ability to consistently reproduce nanoscale features during scaling is a key determinant of success in bringing laboratory innovations to commercial reality.
Fundamental Principles: Sol-Gel Versus Solid-State Synthesis
Distinctive Mechanism of Sol-Gel Synthesis
The sol-gel process operates through a series of hydrolysis and condensation reactions, typically at relatively low temperatures, which enables precise control over material structure from the molecular level upward. This bottom-up approach facilitates the creation of nanoporous architectures with high surface areas exceeding 1000 m²/g, making it particularly suitable for applications requiring high drug loading capacities or specific surface interactions [44]. The process involves molecular precursors (typically metal alkoxides) that undergo hydrolysis to form reactive monomers, which then condense to create nanoparticles. These particles further interconnect to form a three-dimensional network—the gel—which can be processed into various forms including powders, thin films, and monoliths [44]. The primary advantage of this method lies in its exceptional tunability; parameters such as pH, temperature, precursor concentration, and catalyst type can be manipulated to precisely control pore size, particle size, and surface chemistry [44].
Fundamental Mechanism of Solid-State Synthesis
Solid-state synthesis follows a fundamentally different top-down approach where microcrystalline powders of precursor materials are mixed and heated at high temperatures (often above 1100°C for oxides) to facilitate diffusion and reaction at the interfaces between particles [1] [2]. This method traditionally produces materials with larger particle sizes and broader size distributions, though recent advances have improved control over these parameters. The solid-state route is often favored for its simplicity and scalability, but it typically requires subsequent processing steps such as milling to reduce particle size, which can introduce contaminants and make size control challenging [1] [2]. The high temperatures involved often lead to significant grain growth and aggregation, presenting substantial hurdles for achieving narrow particle size distributions without careful optimization of processing parameters.
Table 1: Fundamental Characteristics of Sol-Gel and Solid-State Synthesis Methods
| Characteristic | Sol-Gel Synthesis | Solid-State Synthesis |
|---|---|---|
| Processing Approach | Bottom-up molecular assembly | Top-down particle reaction |
| Typical Temperature Range | Room temperature to ~100°C | 700°C to >1100°C |
| Primary Control Parameters | pH, catalyst, precursor concentration, H₂O:precursor ratio | Temperature, pressure, precursor particle size, mixing efficiency |
| Particle Size Distribution | Generally narrow with optimization | Typically broader, requires milling |
| Scalability Challenges | Maintaining reaction homogeneity in larger volumes | Controlling particle size without extensive post-processing |
| Energy Requirements | Lower temperature processing | High temperature sintering |
Scaling Challenges: Maintaining Particle Size Distribution in Sol-Gel Processes
Heating Heterogeneity During Scale-Up
A primary challenge in scaling sol-gel production lies in achieving uniform heating throughout larger reaction volumes. Conventional heating methods rely on conduction and convection, which inevitably create temperature gradients in larger vessels. These gradients cause uneven reaction rates, leading to broadened particle size distributions as nucleation and growth proceed at different rates throughout the reaction medium [45]. The sol-gel process is particularly sensitive to these variations because nucleation is typically a rapid event that must occur uniformly throughout the solution to achieve monodisperse particles. Research has demonstrated that microwave-assisted heating can address this challenge by providing more homogeneous volumetric heating, significantly reducing temperature variations and improving particle uniformity in larger batches [45]. In one study, the vessel geometry was found to critically influence heating efficiency, with wide, shallow vessels providing more consistent results than tall, narrow containers during scale-up [45].
Mixing and Precursor Distribution Limitations
As reaction volume increases, achieving complete and uniform mixing becomes increasingly difficult. In sol-gel synthesis, the distribution of precursors and catalysts directly influences the kinetics of hydrolysis and condensation reactions, which in turn determines the final particle size distribution [18]. At laboratory scale, simple magnetic stirring may suffice, but industrial-scale reactors require carefully engineered impeller systems and baffling to ensure homogeneous mixing. Inadequately mixed solutions develop concentration gradients, resulting in localized variations in reaction rates that manifest as increased polydispersity in the final product. This challenge is particularly acute for sol-gel reactions that proceed through rapid nucleation events, where even momentary inhomogeneities can significantly impact particle size distribution [18].
Gelation Kinetics and Drying Control
The gelation phase of sol-gel processes presents additional scaling complexities, particularly for applications requiring specific porous architectures. The kinetics of gel formation are influenced by multiple factors including temperature, pH, and precursor concentration, all of which become more challenging to control uniformly in larger volumes [18]. For nanoporous materials intended for drug delivery, the pore size distribution is as critical as particle size, as it directly influences drug loading capacity and release profile [44]. During drying, stresses can develop that fracture delicate gel networks, altering particle morphology and size distribution. Supercritical drying techniques can mitigate this issue but introduce additional complexity and cost at industrial scale [18].
Advanced Strategies for Scaling Sol-Gel Processes
Microwave-Assisted Heating for Improved Uniformity
Recent research has demonstrated that microwave-assisted sol-gel synthesis offers a promising approach to maintaining particle size control during scale-up. Unlike conventional heating, microwave irradiation delivers energy directly to the reaction mixture through dielectric heating, creating more uniform temperature profiles throughout the volume [45]. This method reduces thermal gradients that typically cause heterogeneous nucleation and growth in larger batches. Experimental studies on iron-based aerogels have shown that careful optimization of vessel geometry and power-to-volume ratios enables successful scaling while preserving material properties. Specifically, wide, shallow vessels have proven more effective than tall, narrow containers for achieving homogeneous reaction conditions [45]. This approach has been successfully implemented for the synthesis of various metal oxide nanoparticles, including titanium dioxide, zirconia, and silica, resulting in materials with high crystallinity, uniform particle size, and precise phase composition while significantly reducing energy consumption and processing time [45].
Activation-Retardation Chemistry for Process Control
Innovative chemical approaches have been developed to enhance control over sol-gel kinetics during scaling. The activation-retardation strategy utilizes dual chemical modulators to precisely control the polycondensation reactions that govern particle formation and growth [18]. In this approach, acidic conditions initially retard polycondensation, while a subsequent shift to basic conditions "activates" the cross-linking process. This method has been successfully employed in the additive manufacturing of transparent poly(methylsilsesquioxane) aerogels, enabling precise regulation of gelation kinetics to maintain desired rheological properties even in larger batches [18]. By extending the processing window during which the sol possesses optimal properties for shaping or deposition, this strategy enhances reproducibility and control during scale-up, directly addressing one of the most persistent challenges in sol-gel production.
Process Intensification Through Continuous Flow Reactors
Transitioning from batch to continuous flow reactors represents a promising strategy for scaling sol-gel processes while maintaining consistent product quality. Flow reactors offer significantly improved heat and mass transfer capabilities compared to batch systems, enabling more precise control over reaction parameters throughout the process [46]. Although not explicitly detailed in the search results, this approach aligns with the broader trend toward continuous manufacturing in pharmaceutical production, where consistent product quality is paramount. Microreactor technology, with its high surface-to-volume ratios, provides exceptional thermal control and mixing efficiency, potentially enabling the production of sol-gel nanoparticles with narrower size distributions than achievable in conventional batch reactors at similar production volumes.
Comparative Experimental Data: Sol-Gel vs. Solid-State Synthesis
Case Study: Bismuth Ferrite (BiFeO₃) Synthesis
A direct comparative study of bismuth ferrite (BiFeO₃) synthesis provides quantitative evidence of the differing particle control achievable through sol-gel versus solid-state methods [2]. When prepared using the sol-gel method, BiFeO₃ exhibited significantly reduced grain size (approximately half that of the solid-state sample) and improved phase purity, achieving nearly single-phase material with minimal secondary phases. The solid-state approach, in contrast, resulted in persistent secondary phases (Bi₂Fe₄O₉) even after optimization of calcination temperature and time [2]. The sol-gel process also better maintained stoichiometric control, minimizing bismuth loss that commonly occurs during high-temperature solid-state processing. These structural differences directly influenced functional properties, with sol-gel derived materials demonstrating enhanced dielectric constant and improved magnetic characteristics [2].
Case Study: Barium Titanate (BaTiO₃) Synthesis
Research on barium titanate (BaTiO₃) synthesis further highlights the comparative challenges of particle size control. Traditional solid-state synthesis of BaTiO₃ requires temperatures above 1100°C, resulting in significant grain growth and particles typically in the micron range [1]. Modified low-pressure solid-state approaches have demonstrated some improvement, enabling the production of phase-pure BaTiO₃ powder with particle sizes of approximately 160 nm, though this still exceeds the sizes readily achievable through sol-gel methods [1]. The solid-state process remains limited by its fundamental mechanism, where particle size control primarily depends on precursor particle size and the kinetics of solid-state diffusion, making narrow size distributions challenging to achieve without extensive post-processing such as milling or classification.
Table 2: Experimental Comparison of Sol-Gel and Solid-State Synthesis Outcomes
| Material & Method | Processing Conditions | Particle Size Results | Phase Purity | Key Findings |
|---|---|---|---|---|
| BiFeO₃ Sol-Gel [2] | Lower temperatures, molecular precursor mixing | ~50% smaller than SS equivalent | Nearly single-phase | Stoichiometric control, enhanced properties |
| BiFeO₃ Solid-State [2] | 700-820°C calcination, ball milling | Larger grains, broader distribution | Secondary phases present | Difficult to eliminate impurities |
| BaTiO₃ Modified Solid-State [1] | 750-900°C, low pressure (0.01 MPa) | ~160 nm, improved but still limited | Phase-pure at 800°C | Low pressure reduces synthesis temperature |
| ZnFe₂O₄ Combined Method [3] | Sol-gel with solid-state finishing | Tailorable size and properties | Spinel structure | Hybrid approach enables optimization |
Experimental Protocols for Particle Size Control
Sol-Gel Protocol for Nanoporous Silica (Based on Biomedical Applications) [44]:
- Precursor Preparation: Combine silica alkoxide precursor (typically tetraethyl orthosilicate, TEOS) with ethanol in a molar ratio of 1:4 under constant stirring.
- Catalyst Addition: Add acidic catalyst (dilute HCl or acetic acid) to achieve pH 3-4, maintaining stirring for 30 minutes to promote hydrolysis.
- Gelation: Adjust pH to 7-9 using basic catalyst (ammonia solution) to initiate condensation and gelation.
- Aging: Allow the gel to age for 24-48 hours at 50-60°C to strengthen the network.
- Drying: Implement controlled drying under humidity conditions or supercritical CO₂ drying to preserve nanoporosity.
- Functionalization: For drug delivery applications, surface modification may be performed using silane coupling agents with appropriate functional groups (amine, thiol, carboxyl) or PEGylation to enhance biocompatibility and targeting [44].
Low-Pressure Solid-State Protocol for Nanometer-Sized BaTiO₃ [1]:
- Precursor Preparation: Use submicron BaCO₃ (SSA = 20.15 m²/g) and TiO₂ (SSA = 25.65 m²/g) as starting materials.
- Mixing: Combine equal moles of precursors with deionized water and mix using a sand mill for 2 hours to achieve homogeneous mixing.
- Drying: Dry the mixed slurry at 100°C for 12 hours.
- Low-Pressure Calcination: Heat the dried powder at 750-900°C under reduced pressure (0.01 MPa) for 2 hours.
- Characterization: Analyze phase purity by XRD, particle size by SEM, and tetragonality (c/a ratio) to confirm desired properties [1].
The Researcher's Toolkit: Essential Reagents and Equipment
Table 3: Essential Research Reagents and Equipment for Sol-Gel and Solid-State Synthesis
| Category | Specific Items | Function/Purpose |
|---|---|---|
| Sol-Gel Precursors | Metal alkoxides (e.g., TEOS, TTIP), metal chlorides | Molecular sources for oxide network formation |
| Sol-Gel Catalysts | Acidic (HCl, acetic acid), Basic (ammonia, urea) | Control hydrolysis and condensation rates |
| Solid-State Precursors | Metal oxides (e.g., Fe₂O₃, Bi₂O₃), carbonates (e.g., BaCO₃) | Solid powders for high-temperature reaction |
| Structure-Directing Agents | Surfactants (CTAB, Pluronics), polymers (PEG) | Control pore size and architecture in sol-gel |
| Dispersants/Grinding Aids | Polyacrylic acid, ethanol | Improve mixing and reduce particle size in solid-state |
| Specialized Equipment | Microwave reactor, autoclave, high-temperature furnace, ball mill | Enable controlled processing conditions for both methods |
The comparative analysis presented in this guide demonstrates that both sol-gel and solid-state synthesis methods present distinct advantages and challenges for controlling particle size distribution during scale-up. Sol-gel methods offer superior control over particle size, porosity, and morphology at the nanoscale, making them particularly suitable for applications requiring precise architectural control, such as drug delivery systems and specialized catalysts. However, these methods require careful management of reaction kinetics and drying processes during scale-up to maintain particle uniformity. Solid-state approaches, while generally producing larger particles with broader size distributions, benefit from simpler processing and easier scalability, particularly with modifications such as low-pressure synthesis that partially mitigate traditional limitations [1].
For research and development teams working toward commercial applications, the selection between these methods should be guided by final application requirements and production scale considerations. Emerging hybrid approaches that combine sol-gel processing with solid-state finishing may offer a promising middle ground, leveraging the advantages of both methods [3]. As scaling methodologies continue to advance, particularly in areas such as microwave-assisted heating and continuous flow processing, the gap between laboratory-scale innovation and commercial-scale production of nanomaterials with controlled particle size distributions will continue to narrow, enabling new possibilities in drug development and advanced materials.
Leveraging Microwave-Assisted Heating for Enhanced Homogeneity and Faster Sol-Gel Kinetics
The pursuit of precise particle size and morphology control is a central theme in materials science, directly influencing the performance of products in pharmaceuticals, energy storage, and catalysis. Two predominant methodologies for synthesizing solid-state materials are the conventional solid-state reaction method and the sol-gel process. The solid-state approach typically involves high-temperature calcination (often exceeding 1000°C) of mixed solid precursors, a process straightforward to scale but often resulting in coarse powders with broad particle size distributions and potential heterogeneity [9]. In contrast, the sol-gel method is a wet-chemical technique that involves transitioning a solution (sol) into a solid, three-dimensional network (gel) through hydrolysis and condensation reactions. It is prized for producing materials with high purity and excellent homogeneity at significantly lower processing temperatures [2] [9].
A transformative advancement in sol-gel technology is the integration of microwave-assisted heating. Unlike conventional heating, which relies on surface conduction and can create thermal gradients, microwave heating delivers energy directly to the entire reaction volume. This results in rapid and uniform temperature rise, addressing key limitations of traditional sol-gel processes and enabling enhanced control over particle size and shape [47] [45]. This guide provides a comparative analysis of these synthesis routes, focusing on the objective data that demonstrates the efficacy of microwave-assisted sol-gel methods for achieving superior homogeneity and faster reaction kinetics.
Comparative Analysis of Synthesis Methods
The following table summarizes the core characteristics of solid-state, conventional sol-gel, and microwave-assisted sol-gel synthesis methods, highlighting key performance differentiators.
Table 1: Comparative Analysis of Solid-State and Sol-Gel Synthesis Methods
| Feature | Solid-State Synthesis | Conventional Sol-Gel | Microwave-Assisted Sol-Gel |
|---|---|---|---|
| Basic Principle | High-temperature calcination of solid precursors [9] | Hydrolysis & condensation in solution, followed by drying/calcination [9] | Microwave energy directly heats the reaction mixture to drive sol-gel chemistry [47] [45] |
| Typical Processing Temperature | High (900–1200°C) [9] | Lower (700–900°C) [9] | Low to Moderate (e.g., 68°C for gelation) [45] |
| Reaction Time | Long (12–36 hours) [9] | Shorter than solid-state | Significantly reduced (e.g., 1 hour for gelation) [45] |
| Particle Size & Homogeneity | Coarse powders (1–10 μm), broad distribution, risk of heterogeneity [2] [9] | Fine particles (100–500 nm), good homogeneity [9] | Fine, uniform particles; superior homogeneity and narrow size distribution [47] [45] |
| Purity & Phase Control | Risk of impurities and secondary phases [2] | High purity, good phase control at lower temperatures [2] | High phase purity, precise control over crystal phase [47] |
| Scalability | Inherently scalable, but consistency in large batches is challenging [9] | Scaling without compromising quality is difficult; conventional heating leads to gradients [45] | Promising for scale-up with careful reactor design to ensure homogeneous field distribution [45] |
| Energy Efficiency | Low, due to high temperatures and long durations | Moderate | High, due to rapid and direct heating of the load [45] |
The Impact of Microwave Assistance on Sol-Gel Processes
Integrating microwave heating into the sol-gel workflow fundamentally enhances the process by providing rapid, volumetric heating. This directly translates to two major advantages: dramatically faster reaction kinetics and improved homogeneity in the final product.
Enhanced Homogeneity and Particle Size Control
The homogeneity of a material is critically dependent on a uniform temperature profile throughout the reaction. In conventional heating, the temperature gradient from the vessel walls to the center can lead to uneven nucleation and growth, resulting in a broad particle size distribution. Microwave heating, by contrast, minimizes these gradients. A numerical simulation study on the sol-gel synthesis of TiO₂ nanoparticles quantified this using a Temperature Homogeneity Index (the ratio of average temperature to its standard deviation). The study found that microwave heating could achieve superior temperature homogeneity compared to conventional oil-bath heating across a wide range of power densities (370 to 7400 W/L) [48]. This directly correlates to more uniform particle size, a critical parameter in applications like battery electrodes and pharmaceuticals [49].
The effectiveness of this approach is demonstrated in the synthesis of various functional nanomaterials:
- Li-Co doped ZnO Nanoparticles: Microwave-assisted sol-gel synthesis produced nanoparticles with a confirmed wurtzite hexagonal structure. The method allowed for successful incorporation of dopants without structural transformation, yielding samples with a narrow size distribution and controlled properties for spintronics [47].
- BiFeO₃ (BFO) Multiferroics: A comparative study showed that sol-gel synthesis resulted in a reduced grain size (half that of the solid-state sample) and a more stoichiometric composition by restricting bismuth loss at lower processing temperatures. This led to improved electrical and magnetic properties [2].
- Iron-Based Aerogels (FeA): The microwave-assisted methodology produced aerogels with a well-defined morphology and a narrow nodular size distribution, which are essential for electrochemical applications [45].
Faster Reaction Kinetics and Reduced Processing Time
A primary advantage of microwave heating is the dramatic reduction in reaction time. The energy is directly delivered to the reactants, leading to almost instantaneous and uniform heating, which accelerates both the reaction kinetics and the gelation process. For instance, the synthesis of iron-based aerogels via microwave heating was completed in 1 hour at 68°C, a process that could take hours or even days using conventional heating [45]. Similarly, the functionalization of magnetite nanoparticles with a silica shell was optimized using a microwave-assisted technique, which reduced processing time and improved reproducibility [50]. This reduction in synthesis time enhances research efficiency and improves the economic feasibility of material production.
Experimental Protocols and Data
This section provides detailed methodologies and quantitative data from key studies to illustrate the practical application and outcomes of microwave-assisted sol-gel synthesis.
Objective: To synthesize Zn₁₋₂ₓLiₓCoₓO (x = 0 - 2.0%) nanoparticles for spintronic applications. Materials:
- Precursors: Zinc nitrate hexahydrate, lithium nitrate, cobalt nitrate.
- * reagents: Hexamethylenetetramine (HMTA), deionized water. *Synthesis Workflow:
Key Steps:
- Precursor Preparation: Stoichiometric amounts of zinc, lithium, and cobalt nitrates are dissolved in deionized water.
- Precipitation: A solution of HMTA is added to the metal salt solution under vigorous stirring.
- Microwave Processing: The mixture is transferred to a microwave reactor and heated at approximately 90°C for 2-3 hours.
- Work-up: The resulting precipitate is cooled, washed repeatedly with deionized water and ethanol via centrifugation, and dried to obtain the final powder.
Characterization: The resulting nanoparticles were analyzed by X-ray diffraction (XRD), Fourier-Transform Infrared (FT-IR) spectroscopy, UV-Visible spectroscopy, and Vibrating Sample Magnetometry (VSM) to confirm structure, composition, and magnetic properties.
Quantitative Data from Comparative Studies
The following tables consolidate experimental data from the search results, highlighting the performance differences between synthesis methods.
Table 2: Particle Size and Processing Conditions from Comparative Studies
| Material Synthesized | Synthesis Method | Processing Temperature & Time | Resulting Particle/Grain Size |
|---|---|---|---|
| BiFeO₃ (BFO) [2] | Solid-State Reaction | High temperature, long time | Coarse grains |
| BiFeO₃ (BFO) [2] | Sol-Gel | Lower temperature | ~50% reduction in grain size vs. solid-state |
| LLZO Powder [9] | Solid-State Reaction | 900-1200°C, 12-36 hours | 1-10 μm |
| LLZO Powder [9] | Sol-Gel | 700-900°C, shorter times | 100-500 nm |
| Iron-Based Aerogel [45] | Microwave Sol-Gel | 68°C for 1 hour | Well-defined, narrow size distribution |
Table 3: Functional Property Enhancement from Synthesis Method
| Material Synthesized | Synthesis Method | Key Property Measured | Result & Advantage |
|---|---|---|---|
| Li-Co Doped ZnO [47] | Microwave Sol-Gel | Ferromagnetism | Room-temperature ferromagnetism induced with 2% doping; suitable for spintronics. |
| BiFeO₃ (BFO) [2] | Sol-Gel | Dielectric Constant | Higher values and improved electrical properties over solid-state sample. |
| BiFeO₃ (BFO) [2] | Sol-Gel | Stoichiometry | Near-perfect Bi:Fe ratio maintained, versus Bi loss in solid-state. |
| ZnFe₂O₄ [3] | Combined Sol-Gel/Solid-State | Ionic Conductivity | Potential for optimization as a cathode material in Zn-ion batteries. |
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key reagents and equipment commonly used in microwave-assisted sol-gel synthesis, as referenced in the studies.
Table 4: Key Research Reagents and Equipment for Microwave-Assisted Sol-Gel
| Item | Function/Application | Example from Research |
|---|---|---|
| Metal Salt Precursors (e.g., Chlorides, Nitrates) | Source of metal cations for the oxide network. | FeCl₂ for iron aerogels [45]; Zinc/Li/Co nitrates for doped ZnO [47]. |
| Gelling / Precipitating Agents | Initiates cross-linking and gel formation. | Sodium carbonate/glyoxylic acid for FeA [45]; HMTA for ZnO [47]. |
| Solvent (e.g., Water, Ethanol) | Medium for dissolution and reaction. | Deionized water [47] [45]. |
| Silica Precursor (e.g., TEOS) | For forming a silica shell around nanoparticles. | TEOS used for coating magnetite NPs [50]. |
| Functionalization Agent (e.g., APTMS) | Provides surface functional groups for further chemistry. | APTMS used to attach linker systems [50]. |
| Microwave Reactor | Provides controlled microwave energy for heating. | Multimode ovens, often with temperature control and stirring [47] [45]. |
| Microwave-Transparent Vessels | Containers that allow microwave energy to pass through. | Specific glass vessels used in FeA synthesis [45]. |
Scaling Up and Practical Considerations
While microwave-assisted sol-gel synthesis shows great promise in the lab, scaling it to industrial production presents specific challenges. The primary consideration is ensuring homogeneous electromagnetic field distribution in larger volumes. Research indicates that the shape and size of the reaction vessel are critical. For instance, using a wide, shallow vessel instead of a tall, narrow one can lead to more uniform heating and, consequently, more homogeneous material properties [45]. As volume increases, the power-to-volume ratio must be carefully optimized to maintain the desired reaction kinetics and product quality. Despite these challenges, the reduced energy consumption and shorter processing times make microwave-assisted synthesis a compelling target for further development and industrial adaptation [45] [9].
The comparative data from recent research unequivocally demonstrates that microwave-assisted sol-gel synthesis offers significant advantages over both conventional solid-state and traditional sol-gel methods. Key benefits include:
- Superior Homogeneity: Enables precise control over particle size and distribution, critical for advanced applications.
- Enhanced Kinetics: Drastically reduces reaction and gelation times, improving research and production throughput.
- Improved Material Properties: Leads to finer microstructures, better stoichiometric control, and enhanced functional properties like ionic conductivity and ferromagnetism.
For researchers and drug development professionals focused on particle size control, the microwave-assisted sol-gel method represents a powerful, efficient, and highly controllable synthesis pathway. It is a cornerstone technique for the future of advanced material design, particularly where nanoscale homogeneity and rapid development cycles are paramount.
The precise control over final particle properties represents a critical objective in advanced pharmaceutical development, directly influencing key performance metrics of drug products, including stability, dissolution, and bioavailability. Within this context, the selection of a manufacturing process—notably the comparison between solid-state and sol-gel methods—imposes distinct constraints and opportunities for parameter optimization [2]. This guide provides a systematic comparison of how process parameters such as vessel geometry, power density, and atomization mechanisms govern critical quality attributes of engineered particles. The thesis underpinning this analysis is that while solid-state reactions offer a direct synthesis route, sol-gel methods, particularly when combined with spray drying, provide superior control over particle size, morphology, and purity, which are essential for modern drug delivery systems [2] [51]. Particle engineering has evolved from viewing microparticles as simple carriers to designing them as sophisticated components of drug delivery systems, where particle morphology, internal structure, and surface properties are meticulously controlled for specific functions [51].
Comparative Analysis of Synthesis Methods
Fundamental Principles and Workflows
The solid-state and sol-gel synthesis methods differ fundamentally in their approach to particle formation. Solid-state synthesis is a direct, high-temperature calcination process where solid precursors react to form the desired product. In contrast, the sol-gel method involves the creation of a colloidal suspension (sol) that evolves into a gel-like network, offering molecular-level mixing and typically occurring at significantly lower temperatures [2]. This fundamental difference in reaction environment establishes the framework within which process parameters operate to determine final particle properties.
Table 1: Core Characteristics of Solid-State vs. Sol-Gel Methods
| Feature | Solid-State Method | Sol-Gel Method |
|---|---|---|
| Reaction Nature | Direct solid-solid reaction | Solution-phase reaction & gelation |
| Typical Temperature | High (700–820°C reported) [2] | Lower (e.g., room temperature to 150°C) [2] [52] |
| Atomic-Level Mixing | Limited (requires intensive milling) [2] | Excellent (inherent to solution process) [2] |
| Primary Particle Control | Challenging; often results in coarse powders [2] | Superior; enables nanoscale structuring [51] |
| Common Impurities | More prone to secondary phases [2] | Higher phase purity achievable [2] |
Quantitative Performance Comparison
The choice of synthesis method directly impacts critical quality attributes of the resulting material, as evidenced by comparative studies on multiferroic BiFeO₃. These differences in material properties have profound implications for pharmaceutical applications where purity, morphology, and electrical properties can influence drug stability and delivery performance.
Table 2: Experimental Property Comparison for BiFeO₃ Particles [2]
| Property | Solid-State Method | Sol-Gel Method | Implications for Drug Development |
|---|---|---|---|
| Phase Purity | Contains Bi₂Fe₄O₉ impurity [2] | Nearly single-phase [2] | Impacts batch consistency & purity requirements |
| Grain Size | ~1x (Reference size) [2] | ~50% reduction [2] | Directly influences surface area & dissolution |
| Stoichiometry | Bi loss at high temperatures [2] | Stoichiometric Bi:Fe content [2] | Critical for API-excipient compatibility |
| Dielectric Constant | Lower values [2] | Higher values with Maxwell-Wagner dispersion [2] | Relevant for electrostatic dry powder inhalers |
The Critical Role of Process Parameters
Atomization in Spray Drying
Atomization represents the most critical parameter in spray drying processes, directly determining initial droplet size and subsequent dried particle characteristics. The process involves dispersing the liquid feed into fine droplets using an atomizer, with different nozzle types creating distinct particle size ranges and morphologies [53]. The relationship between droplet size and final particle size follows a fundamental mass balance principle, where the geometric diameter of the dry particle (dg) is determined by the initial droplet diameter (dD) and the feed concentration (c_F), according to the equation [51]:
The aerodynamic diameter (da), crucial for pulmonary drug delivery, is subsequently governed by the particle density (ρP) and geometric diameter [51]:
This mathematical relationship explains the industry trend toward very low-density particles (∼0.1 g/cm³) for inhalation therapies, as it enables large geometric diameters with favorable aerodynamic properties [51].
Table 3: Atomization Methods and Their Impact on Particle Properties
| Atomizer Type | Mechanism | Typical Particle Size | Pharmaceutical Application |
|---|---|---|---|
| Two-Fluid Nozzle | High-velocity gas stream atomizes liquid [53] | 10–100 μm [53] | Low-viscosity liquids, heat-sensitive APIs [53] |
| Pressure Nozzle | Liquid forced through narrow orifice [53] | 50–300 μm [53] | Scale-up processes to improve powder performance [53] |
| Rotary Atomizer | Centrifugal force from spinning disc [53] | 20–300 μm [53] | High-viscosity liquids [53] |
Experimental data demonstrates that higher atomization pressure generates finer droplets, producing smaller particle sizes [53]. In the production of groundnut milk powder, increased atomization pressure from 2 to 3 bar significantly reduced moisture content and increased bulk density due to more efficient drying of smaller droplets [54]. Similarly, in pharmaceutical applications, controlling atomization parameters enables production of large porous particles (LPPs) with geometric sizes >5 μm but low mass density (<0.4 g/cm³), optimizing them for deep lung deposition while avoiding macrophage clearance [55].
Power Density and Thermal Parameters
Power density in spray drying manifests primarily through thermal energy input, with inlet temperature serving as the "control switch" for moisture content and stability [53]. The outlet temperature represents the cumulative thermal effect of the entire drying process and serves as a critical "thermometer" for final product characteristics [53].
Table 4: Thermal Parameter Effects on Spray-Dried Mannitol Particles [55]
| Inlet Temperature | Pump Speed | Air Speed | Resulting Particle Morphology | Aerodynamic Performance |
|---|---|---|---|---|
| 100°C | 10 mL/min | 65 m³/hr | Smooth, spherical particles [53] | Favorable for inhalation |
| 120°C | 15 mL/min | 75 m³/hr | Intermediate roughness | Moderate performance |
| 140°C | 5 mL/min | 85 m³/hr | Wrinkled, rough surface [53] | Reduced flowability |
Experimental optimization studies using Box-Behnken designs have quantified these relationships, demonstrating that inlet temperature significantly impacts residual moisture, morphology, and aerodynamic diameter [55]. For instance, in the synthesis of UiO-66-NH₂ metal-organic frameworks via spray drying, an inlet temperature of 150°C was necessary to achieve full crystallinity, highlighting the role of thermal energy in driving simultaneous drying and chemical synthesis [52]. Excessive temperatures, however, can degrade heat-sensitive active pharmaceutical ingredients (APIs), while insufficient thermal input leads to incomplete drying, agglomeration, and reduced yield [53].
Vessel Geometry and Reaction Environment
The concept of "vessel geometry" extends beyond simple container shape to encompass the entire reaction environment, including spray drying chambers and their influence on particle formation pathways. Single droplet drying studies have revealed that primary particle morphology development occurs during the drying trajectory from atomizer to collection vessel, with collision behavior between droplets leading to agglomeration [56]. The scale of equipment—from laboratory to pilot-scale—significantly affects the nozzle zone hydrodynamics and residence time, ultimately determining the degree of agglomeration and final powder properties [56].
In sol-gel systems, the reaction vessel geometry influences mixing efficiency and heat transfer during the hydrolysis and condensation phases. For pharmaceutical applications, a new sol-gel precursor, tetrakis(2-methoxyethyl) orthosilicate (TMEOS), enabled the fabrication of silica microparticles in spray dryers without needing organic solvents or catalyzers, making the process more compatible with biomolecular drugs[cite:7]. The internal microstructure of these particles—controlled through pH adjustment and additive incorporation—could be engineered to provide sustained release over 35 days, demonstrating how vessel environment and chemical parameters combine to determine drug release profiles[cite:7].
Experimental Protocols for Parameter Optimization
Spray Drying Process Optimization for Inhalation Particles
Objective: To produce large porous particles (LPPs) of D-mannitol with optimized aerodynamic properties for pulmonary delivery. Materials: D-mannitol solution (10% w/w in distilled water); LabPlant spray dryer (SD-06) with two-fluid nozzle [55]. Methodology:
- Experimental Design: Implement a Box-Behnken design with 12 experiments and a central point, varying three parameters on three levels each [55]:
- Drying temperature: 100°C, 120°C, 140°C
- Pump speed: 5 mL/min, 10 mL/min, 15 mL/min
- Air speed: 65 m³/hr, 75 m³/hr, 85 m³/hr
- Process Execution: Spray dry the mannitol solution under each parameter combination [55].
- Characterization: Analyze resulting microparticles for [55]:
- Physical size via laser diffraction
- Aerodynamic diameter using an aerodynamic particle sizer
- Morphology by scanning electron microscopy (SEM)
- Densities (bulk and tapped)
Comparative Synthesis of Multiferroic Particles
Objective: To compare solid-state and sol-gel synthesis routes for BiFeO₃ production. Solid-State Protocol [2]:
- Grind stoichiometric Bi₂O₃ and Fe₂O₃ using high-energy ball mill for 4 hours.
- Calcinate the mixture at temperatures ranging from 600°C to 820°C for 30 minutes to 2 hours.
- Optimize calcination conditions to minimize Bi₂Fe₄O₉ impurity phase. Sol-Gel Protocol [2]:
- Dissolve stoichiometric Bi(NO₃)₃·5H₂O and Fe(NO₃)₃·9H₂O in 2-methoxyethanol.
- Add acetic acid to adjust pH and control hydrolysis rate.
- Dry the gel at 200°C followed by calcination at 550°C for 2 hours. Characterization: For both methods, analyze phase purity (XRD), grain size (SEM), elemental composition, and magnetic properties [2].
Visualization of Parameter-Particle Property Relationships
The following diagram illustrates the interconnected relationships between process parameters and their collective impact on final particle properties, synthesizing the experimental data presented in this guide:
Parameter Impact on Particle Properties
This visualization demonstrates how atomization primarily controls particle size, while power density (thermal parameters) significantly influences morphology and crystallinity. Vessel geometry affects the aerodynamic properties through its impact on residence time and agglomeration behavior.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 5: Key Materials for Particle Engineering Research
| Material / Reagent | Function in Research | Application Example |
|---|---|---|
| D-Mannitol | Carrier matrix for inhalation powders [55] | Model material for spray drying LPPs [55] |
| Tetrakis(2-methoxyethyl) Orthosilicate (TMEOS) | Sol-gel precursor for silica microparticles[cite:7] | Sustained release carrier for biopharmaceuticals[cite:7] |
| HPMCP/HPMC | Polymer system for amorphous solid dispersions [53] | Spray-dried dispersions for bioavailability enhancement [53] |
| Zirconium Acetate Cluster | Metal-organic framework (MOF) precursor [52] | Direct spray drying synthesis of UiO-66-NH₂ [52] |
| Bi(NO₃)₃·5H₂O & Fe(NO₃)₃·9H₂O | Precursors for multiferroic particles [2] | Comparative study of synthesis routes [2] |
| FITC-Dextran | High molecular weight model compound[cite:7] | Release kinetics studies from silica matrices[cite:7] |
This comparison guide demonstrates that process parameters exert distinct yet interconnected influences on final particle properties, with optimal outcomes emerging from a systematic understanding of these relationships. The experimental evidence confirms that sol-gel methods, particularly when enhanced by optimized spray drying parameters, provide superior control over particle characteristics compared to conventional solid-state approaches. For pharmaceutical developers, this parameter-property framework offers a rational foundation for designing particles tailored to specific drug delivery applications, with particular relevance for inhalation therapies requiring precise aerodynamic performance. The integration of Quality by Design principles with advanced process control technologies will further strengthen the scientific basis for particle engineering in commercial pharmaceutical manufacturing.
Data-Driven Method Selection: A Side-by-Side Comparison of Particle Characteristics and Performance
The precise control of particle size and morphology is a cornerstone of advanced materials science, particularly in research comparing solid-state and sol-gel synthesis methods. Achieving this control requires reliable characterization of fundamental particle properties. Among the most critical techniques for this purpose are X-ray diffraction (XRD), Brunauer-Emmett-Teller (BET) surface area analysis, and electron microscopy (EM). Each technique provides unique insights into different aspects of particle morphology, from crystallographic structure and surface properties to direct visualization of size and shape.
This guide provides an objective comparison of these three foundational analytical techniques, focusing on their operating principles, applications, strengths, and limitations within the context of particle size control research. By presenting standardized experimental protocols and quantitative performance data, we aim to equip researchers with the information necessary to select the most appropriate characterization strategy for their specific materials system, with a particular emphasis on distinguishing between nanoparticles produced via solid-state and sol-gel routes.
The following table summarizes the core characteristics, applications, and key differentiators of each characterization technique.
Table 1: Core characteristics of particle characterization techniques
| Feature | X-Ray Diffraction (XRD) | BET Surface Area Analysis | Electron Microscopy (EM) |
|---|---|---|---|
| Measured Parameter | Crystallite size and phase identification [57] [58] | Specific surface area, pore volume, and pore size distribution [59] [60] | Primary particle size, shape, and morphology [57] [61] |
| Underlying Principle | Analysis of diffraction patterns from crystalline planes [58] | Gas adsorption isotherms on solid surfaces [59] [60] | High-resolution imaging with electron beams [61] [62] |
| Primary Application | Quantitative phase analysis, crystal structure determination [58] | Characterization of porous materials, catalysts, and adsorbents [59] | Direct morphological analysis, particle counting, and contamination identification [61] |
| Sample Form | Solid powders or thin films [58] [63] | Solid porous materials (powders, pellets) [59] [60] | Solid samples (powders, solid surfaces) [61] |
| Key Strength | Non-destructive, provides crystal structure data [58] [63] | Quantitative surface area measurement, wide applicability to porous materials [59] | Direct visualization, high resolution, elemental analysis capability (with EDS) [61] |
| Notable Limitation | Limited to crystalline materials, less effective for amorphous phases [58] [63] | Does not characterize closed pores, requires careful sample preparation [59] [60] | Time-consuming, requires high vacuum, potential for sample damage [57] |
Quantitative Performance Data
Understanding the measurable ranges and detection limits of each technique is crucial for experimental design. The following table compiles key quantitative specifications.
Table 2: Quantitative capabilities and detection limits
| Parameter | XRD | BET Surface Area | Electron Microscopy |
|---|---|---|---|
| Size Detection Range | ~1 nm to several μm (crystallite size) [58] | N/A (measures surface area) | Nanometer to micrometer scale [61] |
| Surface Area Range | N/A | Absolute: ≥ 0.5 m²; Specific: ≥ 0.01 m²/g [60] | N/A |
| Pore Size Range | N/A | 2 – 500 nm [60] | N/A |
| Analytical Accuracy | Varies with method; Rietveld can be highly accurate for non-clay samples [58] | Accurate for surface area, but assumes ideal monolayer formation [59] | Robust identification (e.g., -12% mean bias vs. other methods for equivalent size) [57] |
| Representative Data Output | Crystallite size (Scherrer equation), phase identification, lattice parameters [58] [64] | Specific surface area (m²/g), pore volume (cm³/g), pore size distribution [59] [60] | Primary particle size distribution, particle count, shape descriptors [61] |
Experimental Protocols
X-Ray Diffraction (XRD) for Crystallite Size Analysis
Objective: To determine the average crystallite size and phase composition of a powdered sample.
- Sample Preparation: The sample is ground into a fine powder (<45 µm) to minimize micro-absorption effects and ensure reproducible peak intensities. It is then uniformly packed into a sample holder to minimize preferred orientation effects. [58]
- Data Collection: Using an X-ray powder diffractometer with Cu Kα radiation (λ = 1.5418 Å), the sample is scanned over a 2θ range (e.g., 3° to 70°) with a small step size (e.g., 0.0167°) and a slow scan speed (e.g., 2°/min). [58]
- Quantitative Analysis:
- Reference Intensity Ratio (RIR) Method: A handy approach that uses the intensity of the strongest diffraction peak of each phase and its RIR value for quantification, though with lower analytical accuracy. [58]
- Rietveld Refinement: A powerful full-pattern fitting method that refines a calculated pattern to the observed data. It can achieve high accuracy for complex non-clay samples but may struggle with disordered or unknown structures. Software includes HighScore, TOPAS, and GSAS. [58]
- Full Pattern Summation (FPS) Method: An approach like ROCKJOCK that sums reference patterns of pure minerals and is considered highly accurate for sediments containing clay minerals. [58]
- Crystallite Size Calculation: The volume-weighted crystallite size is calculated using the Scherrer equation applied to the broadening of diffraction peaks after correcting for instrumental contributions. [64]
BET Surface Area and Pore Size Analysis
Objective: To determine the specific surface area and pore size distribution of a solid material via gas adsorption.
- Sample Pretreatment (Degassing): The solid sample is pretreated by applying heat and vacuum to remove any initially adsorbed contaminants (e.g., water vapor) from the surface. The temperature range is typically from ambient to 350°C, depending on the sample's stability. [60]
- Data Collection: The degassed solid is cooled under vacuum to cryogenic temperature (using liquid nitrogen). An inert gas (typically N₂) is dosed onto the solid in controlled increments. After each dose, the system pressure is allowed to equilibrate, and the quantity of gas adsorbed is determined. This process is repeated across a relative pressure (P/P₀) range from 10⁻⁴ to 0.999. [60]
- BET Surface Area Calculation: The quantity of gas adsorbed is plotted as a function of relative pressure. The BET equation is applied to this isotherm in the relative pressure range typically between 0.05 and 0.3 to determine the monolayer capacity. The specific surface area is then calculated from this value. [59] [60]
- Pore Size Distribution: The gas pressure is increased further until all pores are filled with liquid (condensate). The pressure is then reduced incrementally, and the desorption isotherm is recorded. The adsorption and desorption branches are evaluated using methods like BJH or Density Functional Theory (DFT) to determine the pore volume and pore size distribution in the range of 2–500 nm. [60]
Electron Microscopy for Particle Size and Morphology
Objective: To directly visualize and measure the size, shape, and distribution of primary particles.
- Sample Preparation: For powders, a small amount of sample is dispersed in a solvent (e.g., ethanol) via ultrasonication. A drop of the dispersion is then deposited onto a conductive substrate (e.g., a carbon-coated copper grid for TEM or a SEM stub) and dried. For non-conductive samples, a thin conductive coating (e.g., gold or carbon) may be applied via sputtering to prevent charging. [64]
- Imaging: The sample is placed under high vacuum in the microscope chamber. For SEM, a focused electron beam scans the sample surface, and detectors collect secondary or backscattered electrons to form an image. [61] For TEM, the electron beam is transmitted through an ultra-thin sample to produce an image. [62]
- Image Analysis: Micrographs are analyzed using specialized software (e.g., Thermo Fisher's Perception Particle Analysis Software). Particles are detected and identified based on contrast. The software can analyze tens of thousands of particles per sample to provide statistical data on size distribution, shape parameters (e.g., circularity, aspect ratio), and particle count. [61]
Technique Selection Workflow
The following diagram illustrates a logical pathway for selecting the most appropriate characterization technique based on research goals and material properties.
Research Reagent Solutions
The following table lists essential materials and reagents commonly used in the sample preparation and analysis procedures described in this guide.
Table 3: Essential research reagents and materials for characterization
| Reagent/Material | Typical Application | Function in Protocol | Example Context |
|---|---|---|---|
| Tetraethyl Orthosilicate (TEOS) | Sol-gel synthesis | Silicon alkoxide precursor for silica nanoparticles | Mesoporous silica synthesis for SAXS and BET analysis [5] |
| Cetyltrimethylammonium Bromide (CTAB) | Sol-gel synthesis | Surfactant template for mesopore formation | Creating ordered pore structures in silica [5] |
| Pluronic F127 | Sol-gel synthesis | Non-ionic surfactant to control particle dispersity | Aiding in producing monodisperse, colloidally stable particles [5] |
| Ammonium Hydroxide | Sol-gel synthesis | Base catalyst for hydrolysis and condensation | Catalyzing the sol-gel reaction in silica synthesis [5] |
| Moringa Oleifera Gum | Green synthesis | Natural chelating and stabilizing agent | Eco-friendly synthesis of ferrite nanoparticles [64] |
| Liquid Nitrogen | BET Analysis | Cryogenic coolant for gas adsorption measurements | Maintaining constant temperature during N₂ adsorption [60] |
| High-Purity Solvents (e.g., Ethanol) | Sample Preparation | Dispersion medium for EM samples | Dispersing powder samples to prevent agglomeration [64] |
| Conductive Coatings (e.g., Gold, Carbon) | Electron Microscopy | Surface coating for non-conductive samples | Preventing sample charging during SEM imaging [61] |
XRD, BET surface area analysis, and electron microscopy form a complementary triad for comprehensive particle characterization. XRD is unparalleled for quantifying crystallographic parameters, BET provides essential surface and porosity data, and EM offers direct visual evidence of particle morphology. The choice of technique—or more often, the combination of techniques—should be driven by the specific material properties of interest. In the context of comparing solid-state and sol-gel methods, employing all three techniques can provide a complete picture: XRD reveals differences in crystallinity and phase, BET quantifies the often higher surface area and porosity of sol-gel products, and EM visually confirms particle size, morphology, and the degree of agglomeration inherent to each synthesis route. This multi-faceted analytical approach is critical for establishing robust structure-property relationships and advancing particle engineering research.
The selection of a synthesis method is a critical determinant of the structural and physical properties of inorganic materials, which in turn govern their performance in applications ranging from catalysis to energy storage. Among the various techniques available, solid-state and sol-gel methods represent two fundamentally different approaches with distinct advantages and limitations. This guide provides an objective comparison of these two methods, focusing on quantitative differences in crystallite size, surface area, and degree of agglomeration—parameters essential for researchers and drug development professionals seeking optimal material properties.
The solid-state method typically involves high-temperature reaction between solid precursors, often resulting in well-crystallized products but with larger particle sizes and limited surface area. In contrast, the sol-gel process, through controlled hydrolysis and condensation of molecular precursors in solution, enables molecular-level mixing and typically yields materials with finer microstructural features. This analysis synthesizes experimental data from recent studies to provide a clear, data-driven comparison of these competing techniques.
Quantitative Comparison of Material Properties
The structural parameters of materials synthesized via solid-state and sol-gel methods show consistent, method-dependent patterns. The table below summarizes key quantitative differences observed across multiple material systems.
Table 1: Quantitative comparison of material properties achieved via solid-state and sol-gel synthesis methods
| Material | Synthesis Method | Crystallite Size (nm) | Surface Area (m²/g) | Agglomeration Behavior | Source |
|---|---|---|---|---|---|
| ZnFe₂O₄ | Sol-gel (with solid-state finish) | Not specified | Significantly higher | Not specified | [3] |
| ZnFe₂O₄ | Solid-state (with mechanochemical activation) | Not specified | Significantly lower | Not specified | [3] |
| MgAl₂O₄ | Sol-gel (calcined at 700°C) | Not specified | 188 | Nearly spherical morphology with some agglomeration | [10] |
| MgAl₂O₄ | Sol-gel (calcined at 900°C) | Not specified | 94 | Increased agglomeration at higher temperatures | [10] |
| La₀.₆Sr₀.₄Mn₀.₈Co₀.₂O₃ | Sol-gel (calcined at 650-1100°C) | 10-150 nm (size range affecting magnetic properties) | Not specified | Not specified | [65] |
| Anatase TiO₂ | Sol-gel (with isopropyl peptization) | 11.67 nm (XRD), 13.48 nm (TEM) | Not specified | Uniform dispersion, no agglomeration | [66] |
The data reveals that the sol-gel method consistently produces materials with higher surface areas compared to solid-state routes, a crucial advantage for applications requiring high reactivity or adsorption capacity. The inverse relationship between calcination temperature and surface area in sol-gel derived materials highlights the importance of thermal treatment optimization. Furthermore, sol-gel synthesis enables crystallite size control in the nanoscale regime (10-150 nm), which directly influences functional properties such as magnetic behavior [65].
Experimental Protocols for Method Comparison
Sol-Gel Synthesis Methodology
The sol-gel process involves the transition of a solution system from a liquid "sol" into a solid "gel" phase, allowing for molecular-level mixing of precursors. The following protocol for synthesizing ZnFe₂O₄ exemplifies a typical sol-gel approach with a solid-state finishing process [3]:
- Precursor Preparation: Zinc chloride (96% purity) and iron(III) chloride (98% purity) are mixed in the required molar ratio under intensive stirring.
- Precipitation: A sodium hydroxide solution (98% purity) is added at room temperature to precipitate the solid precursor phase until pH values reach 10.5, just before the dissolution point of zinc hydroxide.
- Processing: The formed suspension undergoes hydrodynamic processing for 30-60 minutes, followed by vacuum filtration using a Büchner funnel with filter paper.
- Washing: The filtered precursor is washed with deionized water to remove residual mother solution.
- Thermal Treatment: The precursor is dried at room temperature and subsequently treated in a muffle furnace under various temperature regimes (typically 10°C/min heating rate).
For complex oxides like BiBaO₃, the sol-gel protocol incorporates chelating agents. Stoichiometric amounts of metal precursors (e.g., bismuth nitrate pentahydrate and barium carbonate dissolved in dilute nitric acid) are mixed with ethylene glycol and citric acid as complexing and gelling agents in a 1:1 molar ratio with respect to the total metal ions. The mixture is maintained at 70°C with continuous stirring until a viscous gel forms, which is then dried at 120°C and calcined at temperatures between 500-900°C [67].
Solid-State Synthesis Methodology
The solid-state method relies on direct reaction between solid precursors at elevated temperatures. The following protocol for ZnFe₂O₄ synthesis illustrates a modified solid-state approach with mechanochemical activation [3]:
- Precursor Preparation: Iron(III) oxide (99.5% purity) and zinc oxide (99.5% purity) are mixed in the required molar ratio.
- Homogenization: Preliminary homogenization is performed in an agate mortar.
- Mechanochemical Activation: The mixed precursor undergoes activation in a planetary ball mill at 1380 rpm for 30 minutes using zirconium oxide-lined drums and balls.
- Thermal Treatment: The activated precursor is subjected to thermal treatment in a muffle furnace under various temperature regimes with a heating rate of 10°C/min.
The solid-state method typically requires higher temperatures and longer reaction times compared to sol-gel synthesis to achieve complete reaction between solid precursors through diffusion-controlled processes.
Method Selection Workflow and Property Outcomes
The decision between sol-gel and solid-state synthesis methods depends on the target application requirements and material property priorities. The following diagram illustrates the key decision factors and expected property outcomes for each method.
Synthesis Method Selection Flow - This workflow illustrates how application needs dictate method choice and resulting material properties.
The diagram highlights the fundamental trade-offs between the two methods. Sol-gel synthesis is preferable when high surface area, nanoscale crystallites, and molecular-level homogeneity are prioritized, while solid-state methods may be suitable for applications where high crystallinity and process simplicity are more important than nanostructural control.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of either synthesis method requires specific reagents and equipment. The following table details essential materials and their functions in the synthesis processes.
Table 2: Essential research reagents and equipment for solid-state and sol-gel syntheses
| Category | Specific Items | Function in Synthesis | Method Application |
|---|---|---|---|
| Precursors | Metal chlorides (ZnCl₂, FeCl₃), Metal oxides (ZnO, Fe₂O₃) | Source of metallic elements in final compound | Both methods [3] |
| Reagents | Sodium hydroxide (NaOH) | Precipitation agent in sol-gel process | Sol-gel [3] |
| Solvents & Additives | Ethylene glycol, Citric acid | Complexing and gelling agents | Sol-gel [67] |
| Solvents & Additives | Tetraethyl orthosilicate (TEOS) | Silicon source for composite materials | Sol-gel [24] [68] |
| Solvents & Additives | Acetic acid, Hydrochloric acid, Ammonia solution | Catalysts for hydrolysis/condensation | Sol-gel [24] |
| Equipment | Planetary ball mill ("Aktivator-2 SL") | Mechanochemical activation of precursors | Solid-state [3] |
| Equipment | Muffle furnace (e.g., MIMP-P) | High-temperature thermal treatment | Both methods [3] |
| Equipment | pH meter (e.g., Anion 7000) | Precise pH monitoring during precipitation | Sol-gel [3] |
The selection of precursors, reagents, and equipment directly influences the success of the synthesis and the properties of the final product. Sol-gel methods typically require a more diverse set of chemical reagents for solution processing and stabilization, while solid-state methods depend heavily on mechanical processing equipment and high-temperature furnaces.
The comparative analysis presented in this guide demonstrates clear, quantifiable differences between solid-state and sol-gel synthesis methods. Sol-gel processing consistently enables smaller crystallite sizes (10-50 nm) and higher surface areas (94-188 m²/g for MgAl₂O₄ calcined at 700-900°C) [10], attributes particularly valuable for catalytic applications and energy storage materials. The method also provides superior control over particle morphology and reduced agglomeration when appropriate peptization agents are employed [66].
In contrast, solid-state synthesis, particularly when enhanced with mechanochemical activation, offers a more direct route to material formation with fewer processing steps and potentially better crystallinity, though at the expense of larger particle sizes and reduced surface areas [3]. The choice between methods ultimately depends on the specific property requirements of the target application, with sol-gel methods preferred for high-surface-area nanomaterials and solid-state methods suitable for applications where process simplicity and crystallinity are prioritized.
For researchers and development professionals, this analysis provides a framework for selecting synthesis methods based on quantifiable structural parameters rather than empirical preferences. The data and protocols presented enable evidence-based decision-making in material design and development strategies.
Evaluating Electrochemical and Biomedical Performance as a Function of Synthesis Route and Particle Size
The synthesis route of functional materials is a critical determinant of their final properties, influencing particle size, morphology, phase purity, and ultimately, their performance in applications ranging from energy storage to biomedical devices. Among the various fabrication techniques, solid-state and sol-gel methods represent two fundamentally different approaches with distinct advantages and limitations. Solid-state reactions involve direct heating of precursor powders at high temperatures, while sol-gel processes utilize chemical solution pathways for molecular-level mixing at lower temperatures. This comprehensive analysis examines how these synthesis routes affect material characteristics and performance metrics across electrochemical and biomedical applications, providing researchers with evidence-based guidance for method selection.
Fundamental Synthesis Mechanisms
Solid-State Reaction Method
The solid-state synthesis approach is characterized by direct solid-phase reactions between precursor powders at elevated temperatures. This method typically involves mechanical mixing of precursor compounds followed by high-temperature calcination. In the synthesis of ZnFe₂O₄, for instance, the solid-state method utilizes iron(III) oxide and zinc oxide as precursors, which undergo homogenization in an agate mortar followed by mechanochemical activation in a planetary ball mill before thermal treatment [3]. The process relies on diffusional exchange between grains of the starting materials, which often results in challenges with complete reaction and homogeneity. As noted in ZrV₂O₇ synthesis, solid-state reactions can suffer from slow reaction kinetics, sometimes necessitating "exceedingly long heating steps" or repeated calcination cycles with intermediate grinding to improve phase purity [21].
Sol-Gel Synthesis Method
In contrast, the sol-gel method is a wet-chemical technique that enables molecular-level mixing of precursors at relatively low temperatures. The process begins with the formation of a colloidal suspension (sol) from precursor compounds, which then evolves into a gel-like network containing both a liquid and solid phase. The fundamental chemical reactions involve hydrolysis (Equation 1) and condensation (Equation 2) processes [69]:
Hydrolysis: M(OR)ₙ + xH₂O → (OH)ₓ−M(OR)ₙ−ₓ + xROH
Condensation: (OR)ₙ−ₓM−(OH)ₓ + (OH)ₓ−M(OR)ₙ−ₓ → (OR)ₙ−ₓM−O−M(OR)ₙ−ₓ + H₂O
This method facilitates "near-atomic" level mixing of elements, resulting in highly homogeneous products with controlled stoichiometry [21]. The sol-gel process is particularly valuable for generating ceramic materials with diverse morphologies, including coatings, scaffolds, and nanoparticles, making it exceptionally suitable for biomedical applications [69].
Particle Size Control Strategies
Size Control in Sol-Gel Synthesis
The sol-gel method offers exceptional control over particle size through manipulation of reaction parameters. In the synthesis of silica nanoparticles (SNPs) using the modified Stöber method, systematic variation of ammonium hydroxide concentration, water concentration, and temperature enables precise size tuning [70]. Research demonstrates that ammonium hydroxide concentration directly correlates with particle size, with concentrations between 0.29-0.097 M producing SNPs ranging from 27.1 to 190.8 nm [70]. Similarly, higher temperatures generally yield smaller particles, though excessive heat (>55°C) can increase polydispersity due to accelerated aggregation [70].
For metal nanoparticles like gold, surfactant concentration serves as an effective size-control parameter. Increasing Tween 80 concentration from 0.1 to 10 mmol/L systematically reduces Au nanoparticle size from approximately 80 nm to 10 nm while simultaneously narrowing size distribution [71]. This size reduction correlates with a blue shift in surface plasmon resonance from 538 nm to 515 nm, reflecting quantum confinement effects [71].
Particle Size in Solid-State Reactions
Solid-state methods typically produce larger particles with broader size distributions compared to sol-gel synthesis. The inherent limitations of powder mixing and solid-state diffusion result in micron-sized particles, as observed in BiFeO₃ synthesis where solid-state reactions yielded grains approximately twice the size of sol-gel derived particles [2]. Mechanical activation through extended milling can reduce particle size to some extent; in ZrV₂O₇ synthesis, milling time variations (15-180 minutes) were explored to improve homogeneity, though complete elimination of residual unreacted oxides remained challenging [21].
Table 1: Particle Size Control Parameters in Synthesis Methods
| Synthesis Method | Control Parameters | Size Range | Size Distribution | Key Influencing Factors |
|---|---|---|---|---|
| Sol-Gel | Ammonium hydroxide concentration | 27-191 nm (SiO₂) [70] | Narrow (PDI: 0.008-0.082) [70] | Precursor concentration, catalyst amount, temperature, pH |
| Surfactant concentration | 6-22 nm (Au) [71] | Tunable with surfactant [71] | Surfactant type/concentration, reducing agent strength | |
| Complexing agent | 100-1000 nm (LiFePO₄/C) [72] | Varies with agent [72] | Stability constant of complex, chelation strength | |
| Solid-State | Milling time | Micron range [2] [21] | Broad | Initial powder size, milling duration/intensity, additive use |
| Calcination cycles | Limited reduction [21] | Broad | Temperature profile, cycle number, intermediate grinding |
Electrochemical Performance Comparison
Battery Electrode Materials
The synthesis route significantly impacts the electrochemical performance of electrode materials for energy storage applications. In ZnFe₂O₄ synthesized for metal-ion battery applications, the synthesis method directly influences electrophysical properties and ionic conductivity, with potential for optimization as cathode material [3]. Similarly, Li₀.₃La₀.₅₇TiO₃ (LLTO) solid electrolytes prepared via sol-gel processing demonstrate enhanced ionic conductivity when calcination temperature is optimized at 800°C, achieving total ionic conductivity of 0.54 mS/cm due to reduced grain boundary resistance [73].
Comparative studies of Na₄Mn₉O₁₈ synthesis reveal substantial advantages for sol-gel derived materials. The rod-like morphology obtained through sol-gel processing, with particle diameters ranging from 0.2-1 μm, enables high specific capacity of approximately 200 F g⁻¹ at 200 mA g⁻¹ current density [74]. This material maintains 84% capacity retention after 4,000 cycles in aqueous hybrid Na-ion supercapacitors, demonstrating exceptional cycling stability [74].
Table 2: Electrochemical Performance of Materials by Synthesis Route
| Material | Application | Synthesis Method | Key Performance Metrics | Advantages |
|---|---|---|---|---|
| LiFePO₄/C [72] | Lithium-ion battery cathode | Sol-gel (various complexing agents) | Initial discharge: 161.1 mAh g⁻¹ (acetic acid); Capacity fading: 2.2% after 30 cycles | Fine particles (200 nm); Better kinetics; Lower complex stability constant preferred |
| Na₄Mn₉O₁₈ [74] | Na-ion supercapacitor | Sol-gel | ~200 F g⁻¹ at 200 mA g⁻¹; 84% capacity retention after 4000 cycles | Rod-like morphology; Small particle diameter (0.2-1 μm) |
| LLTO [73] | Solid electrolyte | Sol-gel (optimized calcination) | Total ionic conductivity: 0.54 mS/cm; Grain boundary: 0.88 mS/cm | Reduced grain boundary resistance; Refined microstructure |
| ZnFe₂O₄ [3] | Metal-ion battery cathode | Sol-gel vs. Solid-state | Varying electrophysical properties and ionic conductivity | Potential cathode material; Properties tunable by synthesis conditions |
The Role of Complexing Agents in Sol-Gel Synthesis
In sol-gel synthesis of electrode materials, the choice of complexing agent significantly influences electrochemical performance. For LiFePO₄/C composites, complexing agents with lower stability constants (e.g., acetic acid with logβ = 8.3) produce materials with superior discharge capacity (161.1 mAh g⁻¹) compared to agents with higher stability constants like ethylenediamine (logβ = 9.7, capacity = 126.7 mAh g⁻¹) [72]. This performance advantage correlates with improved particle morphology and reduced agglomeration, highlighting the importance of ligand selection in sol-gel processing.
Biomedical Performance and Applications
Biomaterial Generations and Surface Functionalization
Sol-gel synthesis offers exceptional versatility for biomedical applications, enabling the production of biomaterials across different generations. First-generation bioinert materials, including stainless steel and titanium alloys, can be transformed into second-generation bioactive materials through sol-gel derived coatings [69]. The method further facilitates development of third-generation biomaterials that stimulate specific cellular responses at molecular levels, and fourth-generation materials that interact with cellular bioelectric signals [69].
The biomaterial generations can be summarized as follows [69]:
- First Generation (~1960): Bioinert, non-toxic materials (stainless steel, Cr-Co-Mo, NiTi, Ti6Al4V)
- Second Generation (1970-1990): Bioactive, biodegradable materials (hydroxyapatite, bioactive glass, magnesium alloys)
- Third Generation (~2000): Bioactive, biodegradable materials causing specific cellular response (Bioglass, biosilicates)
- Fourth Generation (~2015): Materials interacting with cellular signals (carbon-based materials, conductive polymers)
Enhanced Biocompatibility and Bioactivity
Sol-gel processing improves biomedical performance through multiple mechanisms. The technique enables creation of homogeneous coatings on metallic implants, enhancing biocompatibility and providing corrosion protection [69]. For magnesium alloys used as biomaterials, sol-gel synthesis improves biological properties by creating controlled surface structures that modulate degradation behavior and cellular response [69]. The method's flexibility allows fabrication of diverse morphologies including nanoparticles, scaffolds, and matrices optimized for drug delivery, tissue engineering, and implant integration [69].
Phase Purity and Structural Properties
Comparative Phase Purity Analysis
The synthesis method significantly influences phase purity and structural perfection, which in turn affects functional properties. In BiFeO₃ synthesis, sol-gel methodology produces "almost a single-phase material at relatively lower temperatures" while solid-state reactions result in secondary phases like Bi₂Fe₄O₉ without careful optimization [2]. Similarly, ZrV₂O₇ preparation reveals that sol-gel reactions enable homogeneous phase-pure products, whereas solid-state methods require "extended milling time and repeated calcination cycles" to achieve comparable purity [21].
The enhanced phase purity achieved through sol-gel processing directly translates to improved material properties. For BiFeO₃, sol-gel derived samples exhibit higher dielectric constants with Maxwell-Wagner type dielectric dispersion and improved ferromagnetic properties compared to solid-state synthesized counterparts [2]. Elemental analysis further confirms superior stoichiometry control in sol-gel processed materials, with minimized bismuth loss in BiFeO₃ compared to solid-state reactions where bismuth volatilization presents challenges [2].
Mechanisms of Enhanced Homogeneity
The superior homogeneity of sol-gel derived materials stems from the molecular-level mixing of precursors in solution. Unlike solid-state reactions where diffusion limitations create compositional gradients, sol-gel processes facilitate "atomic level mixing" that promotes uniform reaction throughout the material [2] [21]. This fundamental advantage enables formation of pure phases at lower temperatures with shorter processing times, reducing energy consumption and minimizing undesirable phase transitions or decomposition.
Experimental Protocols
Representative Sol-Gel Synthesis Procedure
Preparation of LiFePO₄/C with Complexing Agents [72]:
- Precursor Preparation: Dissolve LiNO₃, Fe(NO₃)₃·9H₂O, and NH₄H₂PO₄ stoichiometrically in saturated solution of complexing agent (acetic acid, ethanediol, oxalic acid, or ethylenediamine) in deionized water.
- Reduction: Add ascorbic acid as antioxidant to prevent iron oxidation and stir continuously for 1 hour at 55°C.
- pH Adjustment: Adjust solution pH to 6.5 using ammonia water under continuous stirring.
- Gel Formation: Heat solution to 75°C with continuous stirring until evaporation leads to gel formation.
- Drying: Dry the gel in vacuum oven at 80°C for 12 hours.
- Thermal Treatment: Pelletize the dried gel and heat in nitrogen atmosphere at 600°C for 4 hours with slow cooling to room temperature.
Key Parameters: Complexing agent selection critically affects particle morphology and electrochemical performance, with acetic acid (stability constant logβ = 8.3) producing optimal results [72].
Representative Solid-State Synthesis Procedure
Preparation of BiFeO₃ via Solid-State Reaction [2]:
- Precursor Preparation: Weigh stoichiometric ratios of Bi₂O₃ and Fe₂O₃ powders with high purity (>99.9%).
- Mechanical Activation: Grind mixtures thoroughly in ethanol medium using high-energy ball mill for 4 hours.
- Calcination: Heat the mixed powders at temperatures ranging from 600°C to 820°C for different durations (30 minutes to 2 hours) to optimize phase formation.
- Intermediate Processing: Regrind the calcined powders to improve homogeneity.
- Sintering: Press powders into pellets and sinter at optimal temperature to form dense ceramics.
Key Parameters: Multiple calcination cycles with intermediate grinding essential to minimize secondary phases; excess Bi₂O₃ may be required to compensate for bismuth volatilization at high temperatures [2].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Synthesis Methods
| Reagent Category | Specific Examples | Function in Synthesis | Application Notes |
|---|---|---|---|
| Precursors for Solid-State | Metal oxides (ZnO, Fe₂O₃, Bi₂O₃) [3] [2] | Primary reactants for ceramic formation | High purity (>99.9%) essential; Particle size affects reactivity |
| Precursors for Sol-Gel | Metal chlorides (ZnCl₂, FeCl₃); Metal alkoxides [3] [69] | Molecular sources for metal cations in solution | Alkoxides provide better control over hydrolysis rates |
| Complexing Agents | Acetic acid, oxalic acid, ethylenediamine [72] | Control hydrolysis rates; Modify particle morphology | Stability constant affects particle size and agglomeration |
| Surfactants | Tween 80 [71] | Control nanoparticle size and distribution | Higher concentrations generally yield smaller, more uniform particles |
| Solvents | Ethanol, methanol, water [70] | Reaction medium for sol-gel processes | Ethanol preferred for eco-friendly and cost-effective synthesis |
| Catalysts | Ammonium hydroxide [70] | Accelerate hydrolysis and condensation | Concentration directly influences final particle size |
The selection between solid-state and sol-gel synthesis methods represents a critical decision point in materials design that significantly influences electrochemical and biomedical performance. Solid-state reactions offer simplicity and scalability but typically produce larger particles with broader size distributions and potential phase impurities. Sol-gel methods provide superior control over particle size, morphology, and stoichiometry, resulting in enhanced electrochemical properties for energy storage materials and improved bioactivity for biomedical applications. The optimal synthesis strategy depends on application-specific requirements, with sol-gel processing preferred for performance-critical applications where homogeneity, phase purity, and nanoscale features are essential, while solid-state methods remain valuable for large-scale production where some performance trade-offs are acceptable. Future research directions should focus on hybrid approaches that combine the advantages of both methods while addressing their respective limitations.
The selection of a synthesis method is a foundational decision in materials science, directly determining the structural and functional properties of the final product. Among the numerous available techniques, solid-state and sol-gel methods represent two fundamentally different approaches, each with distinct advantages and limitations. This guide provides an objective comparison of these methods, focusing on their control over particle size, phase purity, and morphology, supported by recent experimental data. The decision framework presented herein is designed to help researchers and development professionals select the optimal synthesis pathway based on specific application requirements, whether for advanced battery materials, catalysts, phosphors, or electronic ceramics.
Comparative Analysis at a Glance
The table below summarizes the core characteristics of solid-state and sol-gel synthesis methods based on recent experimental studies.
Table 1: Comparative Overview of Solid-State and Sol-Gel Synthesis Methods
| Feature | Solid-State Synthesis | Sol-Gel Synthesis |
|---|---|---|
| General Principle | Direct reaction between solid precursors via high-temperature calcination and diffusional exchange [21] | Hydrolysis and polycondensation of molecular precursors in a liquid medium to form a colloidal network [5] [75] |
| Typical Particle Size | Sub-micron to micron range; can achieve ~90-160 nm with advanced modifications (e.g., low-pressure) [1] [76] | Nanometer scale (e.g., 96 nm and above); offers superior control over nanostructure [76] [75] |
| Phase Purity & Homogeneity | High purity possible but often requires prolonged heating/repeated calcination cycles; risk of unreacted intermediates [21] | Excellent molecular-level mixing; achieves high homogeneity and phase purity at lower temperatures [21] [13] |
| Typical Morphology | Larger, often aggregated particles with irregular shapes [13] | Fine, uniform particles; high specific surface area; tunable porosity [5] [76] |
| Primary Advantages | Simplicity, cost-effectiveness for large-scale production, high crystallinity [21] [13] | Low processing temperature, excellent stoichiometry control, high homogeneity, versatile morphologies [3] [75] |
| Key Limitations/Challenges | High energy consumption, limited control over particle size and aggregation, potential for impurity phases [1] [21] | Can involve costly metal-organic precursors, issues with shrinkage, potential for residual carbon or hydroxyl groups [75] [13] |
| Ideal Application Profile | Materials where extreme purity and high crystallinity are prioritized over nanostructuring (e.g., luminescent phosphors, certain ceramics) [13] | Materials requiring nanoscale features, high surface area, or precise dopant distribution (e.g., catalysts, spintronics, battery electrodes) [3] [76] [75] |
Detailed Methodological Insights and Experimental Data
Particle Size and Morphology Control
Control over particle size and morphology is one of the most significant differentiators between the two methods.
Sol-Gel Synthesis: This method excels at producing nanomaterials with fine-tuned dimensions. A study on high-entropy spinel oxides ((CoNiMnFeCr)₃O₄) demonstrated that a PVP-assisted sol-gel process could produce particles with sizes directly correlated to the calcination temperature: 96 nm at 800°C, 154 nm at 900°C, and 475 nm at 1000°C [76]. The use of polymeric agents like PVP provides steric stabilization, preventing agglomeration and ensuring a uniform particle size distribution [76]. This precise control is paramount for applications like catalysis, where the surface area of the S-HEO 800 sample contributed to its superior oxygen evolution reaction (OER) performance [76].
Solid-State Synthesis: Traditionally known for producing larger, micron-sized particles, advancements have pushed the boundaries of this method. By performing the solid-state reaction between BaCO₃ and TiO₂ in a low-pressure environment (0.01 MPa), researchers synthesized phase-pure BaTiO₃ powder with a uniform particle size of 90 nm at 800°C and 160 nm at 900°C [1]. The low-pressure environment promotes the decomposition of reactants and limits grain growth, enabling nanomaterial synthesis through a traditionally bulk-scale method [1]. However, without such modifications, solid-state reactions typically yield larger particles, as seen in Sr-based phosphors, which exhibited better crystallinity but lacked nanoscale features [13].
Phase Purity and Homogeneity
The level of atomic mixing in the precursor stage critically influences the phase purity and homogeneity of the final product.
Sol-Gel Synthesis: The sol-gel route offers "near-atomic" level mixing of precursors in a solution before gelation [21]. This molecular-scale homogeneity facilitates the formation of phase-pure compounds at lower temperatures and shorter reaction times. For instance, phase-pure ZrV₂O₇, a negative thermal expansion material, was successfully achieved via a sol-gel reaction, overcoming the purity challenges often encountered in solid-state routes [21]. This homogeneity is also crucial for complex materials like doped oxides for spintronics, where a uniform distribution of magnetic dopants (e.g., Co in ZnO) is essential for achieving desired magneto-electronic properties [75].
Solid-State Synthesis: Achieving homogeneity in solid-state reactions relies on the mechanical mixing of solid powders and diffusional processes at high temperatures. This often leads to challenges with local stoichiometric variations. For ZrV₂O₇, obtaining a pure phase via solid-state reaction required extended milling times and repeated calcination cycles with intermediate grinding to improve reactant contact and complete the reaction, thereby minimizing unreacted ZrO₂ or V₂O₅ [21]. While high purity is attainable, the process is often more energy-intensive and time-consuming compared to wet-chemical methods.
Impact on Functional Properties
The choice of synthesis method directly translates to the performance of the material in its intended application.
Table 2: Influence of Synthesis Method on Material Performance
| Material | Synthesis Method | Key Outcome & Performance | Citation |
|---|---|---|---|
| ZnFe₂O₄ (for batteries) | Sol-Gel & Solid-State | Electrophysical properties (ionic conductivity) differed significantly; method optimization is critical for battery performance [3] | [3] |
| (CoNiMnFeCr)₃O₄ (HEO for OER) | PVP-assisted Sol-Gel | Sample calcined at 800°C showed the best OER performance (overpotential of 316 mV), linked to its small particle size (96 nm) and high surface area [76] | [76] |
| Sr₈MgEu(PO₄)₇ (Phosphor) | Solid-State, Sol-Gel, Hydrothermal | Solid-state route yielded the highest photoluminescence intensity due to superior crystallinity, despite larger particle size [13] | [13] |
| BaTiO₃ (for MLCCs) | Low-Pressure Solid-State | Achieved phase-pure, nanometer-sized powder (90-160 nm) with high tetragonality, crucial for high-end multilayer ceramic capacitors [1] | [1] |
Experimental Protocols for Method Implementation
Representative Sol-Gel Protocol (PVP-Assisted)
This protocol for synthesizing high-entropy spinel oxides is adapted from [76].
- 1. Reagent Preparation: Use metal nitrate precursors (e.g., Co, Ni, Fe, Mn, Cr nitrates). Prepare a solution of Polyvinylpyrrolidone (PVP) in methanol. Glacial acetic acid is used as a chelating agent.
- 2. Complexation and Sol Formation: Dissolve the metal nitrates in a methanol/acetic acid mixture. Stir vigorously. Add the PVP solution dropwise to the metal salt solution. PVP acts as a complexing agent and steric stabilizer, preventing agglomeration.
- 3. Gelation and Aging: Continue stirring until a homogeneous sol forms. Allow the mixture to rest for ~2 hours to facilitate gel formation.
- 4. Drying and Calcination: Dry the resulting gel at ~120°C to remove solvents. Grind the dried gel into a powder and calcine in a furnace at the target temperature (e.g., 800-1000°C) for several hours to obtain the crystalline oxide phase.
Representative Solid-State Protocol (with Mechanochemical Activation)
This protocol for synthesizing ZnFe₂O₄ and ZrV₂O₇ is adapted from [3] [21].
- 1. Precursor Weighing and Mixing: Weigh stoichiometric amounts of solid precursor oxides (e.g., ZnO and Fe₂O₃ for ZnFe₂O₄; ZrO₂ and V₂O₅ for ZrV₂O₇).
- 2. Mechanochemical Activation: Place the powder mixture in a planetary ball mill. Use zirconium oxide balls and lining. Mill the mixture at a high speed (e.g., 1380 rpm) for a set duration (e.g., 30 minutes to several hours). This step is crucial for reducing particle size and enhancing homogenization [3] [21].
- 3. Pelletization (Optional): The activated powder may be pressed into pellets to improve inter-particle contact during heating.
- 4. Calcination and Cycling: Fire the powder or pellets in a muffle furnace at the required temperature (e.g., 700-1100°C) with a controlled heating rate. For high-purity products, multiple calcination cycles with intermediate grinding are often necessary [21].
Decision Framework and Visualization
The following diagram illustrates the logical decision process for selecting between solid-state and sol-gel synthesis based on primary research objectives.
Essential Research Reagent Solutions
The table below lists key reagents and their functions in the described synthesis methods.
Table 3: Key Reagents in Solid-State and Sol-Gel Synthesis
| Reagent | Function | Synthesis Method | Citation |
|---|---|---|---|
| Metal Nitrates/Salts (e.g., Co(NO₃)₂, ZnCl₂) | Molecular precursors providing the cationic species. | Sol-Gel | [3] [76] |
| Metal Oxides (e.g., ZnO, Fe₂O₃, ZrO₂) | Solid precursors for the reaction. | Solid-State | [3] [21] |
| Polyvinylpyrrolidone (PVP) | Polymer acting as a complexing and steric stabilizing agent; controls morphology and prevents agglomeration. | Sol-Gel | [76] |
| Alkoxides (e.g., Tetraethyl orthosilicate - TEOS) | Metal-organic precursors for network formers like silica. | Sol-Gel | [5] |
| Ammonia / Citric Acid / Urea | Catalysts, fuels (in combustion synthesis), or pH modifiers. | Sol-Gel, Combustion | [5] [13] |
| Zirconia Milling Media | Used in ball milling for mechanochemical activation of precursors. | Solid-State | [3] |
Concluding Recommendations
The choice between solid-state and sol-gel synthesis is not a matter of one being universally superior to the other, but rather which is optimal for a specific set of requirements.
- The sol-gel method is the definitive choice when the application demands nanometer-scale particles, high specific surface area, exceptional homogeneity at the molecular level, or complex doping profiles. It is ideally suited for catalysts, advanced battery electrodes, and functional oxides for spintronics and sensors.
- The solid-state method is a robust and often more economical choice for producing high-crystallinity materials where ultimate phase purity can be achieved through process optimization, and where nanoscale features are less critical. It remains a cornerstone for manufacturing phosphors, certain ceramic powders, and other industrial-scale materials.
This framework, supported by contemporary experimental data, empowers researchers to make an informed initial selection, which should then be refined through targeted experimental validation.
Conclusion
The choice between solid-state and sol-gel synthesis is not a matter of superiority but of strategic alignment with application-specific goals. Solid-state methods offer simplicity and scalability for many materials but can face challenges in achieving nanoscale homogeneity. In contrast, sol-gel chemistry provides exceptional control over particle size and composition at the nanoscale, facilitating the creation of high-surface-area materials ideal for drug delivery and advanced catalysis, though scaling requires careful optimization of parameters like vessel geometry and heating mode. The emergence of hybrid and intensified processes, such as microwave-assisted sol-gel and mechanochemically-activated solid-state reactions, represents the future of particle engineering, promising to overcome traditional limitations. For pharmaceutical scientists and researchers, mastering these synthesis landscapes is pivotal for developing the next generation of high-performance materials, from controlled-release formulations to efficient energy storage systems.
References
- 1. Low-pressure solid-state synthesis of nanometer-sized ... [https://www.sciencedirect.com/science/article/abs/pii/S027288422503250X]
- 2. A comparative study between solid state and sol–gel ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838815305491]
- 3. A Comparative Study of Sol–Gel and Solid-State Methods ... [https://www.mdpi.com/2504-477X/9/11/632]
- 4. Sol–Gel Synthesis of NiO-Fe2O3-SiO2/Al2O3 Catalysts ... [https://www.mdpi.com/1420-3049/30/22/4469]
- 5. Accelerated sol–gel synthesis of nanoporous silica via ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00274e]
- 6. Solid-state synthesis improves stability and cycling ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894724013640]
- 7. Synthetic control guided by growth mechanism insights ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04432d]
- 8. Solid-State Diffusion: An Introduction [https://arxiv.org/html/2407.17662v1]
- 9. Llzo powder synthesis: sol–gel vs solid-state routes [https://eureka.patsnap.com/report-llzo-powder-synthesis-comparison-between-sol-gel-and-solid-state-routes]
- 10. Synthesis of MgAl2O4 by sol-gel method and investigation ... [https://link.springer.com/article/10.1007/s10971-025-06799-1]
- 11. Solid-State Kinetic Modeling and Experimental Validation ... [https://www.mdpi.com/2227-7080/13/2/63]
- 12. Sol–gel process [https://en.wikipedia.org/wiki/Sol%E2%80%93gel_process]
- 13. Solid state, sol-gel and hydrothermal, the comparison of ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838821027493]
- 14. Key Steps in the Sol-Gel Process for Nanoparticle Synthesis [https://www.azonano.com/article.aspx?ArticleID=6857]
- 15. Emerging processing guidelines for solid electrolytes in the ... [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d5cs00358j]
- 16. The Sol–Gel Process, a Green Method Used to Obtain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11507187/]
- 17. Sol–Gel-Synthesized Metal Oxide Nanostructures [https://www.mdpi.com/2310-2861/11/8/657]
- 18. Activation–retardation in sol–gel reactions for additive ... [https://www.nature.com/articles/s41467-025-63356-8]
- 19. Sol-gel synthesis, characterization and catalytic activity of ... [https://www.sciencedirect.com/science/article/pii/S2949822825003612]
- 20. Effect of sol-gel and solid-state synthesis techniques on ... [https://www.sciencedirect.com/science/article/abs/pii/S1293255823002170]
- 21. Synthesis and phase purity of the negative thermal expansion ... [https://pubs.rsc.org/en/content/articlehtml/2025/tc/d4tc04095c]
- 22. Review of the opportunities and limitations for powder- ... [https://www.sciencedirect.com/science/article/pii/S0955221924006538]
- 23. Modelling and Optimisation of the Sol-Gel Conditions for ... [https://www.mdpi.com/2073-4344/12/2/239]
- 24. Comparison of acid- and base-catalysed sol–gel synthesis for ... [https://pubs.rsc.org/en/content/articlehtml/2025/sm/d5sm00608b]
- 25. Mechanochemical Synthesis of Advanced Materials for All ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12431395/]
- 26. A sol–gel synthesis to prepare size and shape-controlled ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9051609/]
- 27. The effect of sol-gel process on the microstructure and ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884219331475]
- 28. What is the sol-gel method for nanoparticle synthesis? [https://acceleratedmaterials.co/what-is-the-sol-gel-method-for-nanoparticle-synthesis/]
- 29. “Traditional” Sol-Gel Chemistry as a Powerful Tool for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6416638/]
- 30. Advances in chemically tailored metal alkoxide as single ... [https://www.sciencedirect.com/science/article/pii/S2666539525000677]
- 31. Unlocking superior energy storage in ZnFe 2 O 4 ... [https://www.sciencedirect.com/science/article/pii/S0013468625009831]
- 32. Thermal, Structural, and Morphological Analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12610802/]
- 33. The effect of fuel on the physiochemical properties ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10446917/]
- 34. Effect of size and synthesis route on the magnetic ... [https://www.sciencedirect.com/science/article/abs/pii/S0304885306015381]
- 35. Synthesis, characterization, and preliminary insights of ... [https://www.nature.com/articles/s41598-023-46960-w]
- 36. Synthesis and characterization of ZnFe2O4 nanoparticles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4632970/]
- 37. Recent advances in mesoporous silica nanoparticle [https://www.frontiersin.org/journals/nanotechnology/articles/10.3389/fnano.2025.1564188/full]
- 38. Recent Progress in Hydrogel Synthesis and Biomedical ... [https://www.mdpi.com/2310-2861/11/6/456]
- 39. Next-generation biopolymer gels: innovations in drug delivery ... [https://pubs.rsc.org/en/content/articlehtml/2025/tb/d4tb02068e]
- 40. A multimodal approach to smart and sustained drug ... [https://pubmed.ncbi.nlm.nih.gov/40874548/]
- 41. Recent Advances in Thermo-Responsive Hydrogels for ... [https://www.sciencedirect.com/science/article/abs/pii/S1773224725009402]
- 42. Limitation of solid-reaction technique in a controlled ... [https://www.sciencedirect.com/science/article/pii/S2307410823000317]
- 43. A graph-based network for predicting chemical reaction ... [https://www.nature.com/articles/s41467-021-23339-x]
- 44. Functionalized nanoporous architectures derived from sol–gel ... [https://pubs.rsc.org/en/content/articlehtml/2025/tb/d5tb00958h?page=search]
- 45. Overcoming Scaling Challenges in Sol–Gel Synthesis [https://www.mdpi.com/3042-5697/1/2/6]
- 46. Recent progress in the synthesis, scaling, processing and ... [https://www.sciencedirect.com/science/article/abs/pii/S0927796X25002013]
- 47. Microwave-assisted sol-gel synthesis of Li-Co doped zinc ... [https://www.sciencedirect.com/science/article/abs/pii/S0022024825000016]
- 48. Procedure to generate a selection chart for microwave sol- ... [https://www.sciencedirect.com/science/article/pii/S0255270123001204]
- 49. Autonomous Control of Crystal Size and Shape in Dense ... [https://chemrxiv.org/engage/chemrxiv/article-details/679cc72681d2151a02acdaac]
- 50. Microwave-Assisted Sol–Gel Preparation of the ... [https://www.mdpi.com/2079-4991/11/12/3176]
- 51. Pharmaceutical Particle Engineering via Spray Drying - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2292490/]
- 52. Study of process parameters that enable direct spray ... [https://www.sciencedirect.com/science/article/abs/pii/S1387181124001367]
- 53. Enhancing Drug Product Performance Through Spray ... [https://www.crystalpharmatech.com/enhancing-drug-product-performance-through-spray-drying-parameter-optimization.html]
- 54. Optimization of spray drying process parameters for ... [https://www.sciencedirect.com/science/article/abs/pii/S0032591019305583]
- 55. Optimization of Spray Drying Process Parameters for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9960250/]
- 56. Particle structure development during spray drying from a ... [https://www.sciencedirect.com/science/article/pii/S026087742200276X]
- 57. comparing volume specific surface area, X-ray diffraction ... [https://pubs.rsc.org/en/content/articlelanding/2019/en/c8en00760h]
- 58. Comparison of Quantitative X-ray Diffraction Mineral ... [https://www.mdpi.com/2075-163X/13/4/566]
- 59. BET - Porosimetry/Surface Area (Brunauer-Emmett-Teller) [https://mat-cs.com/bet-porosimetry-surface-area-brunauer-emmett-teller/]
- 60. Surface Area | Pore Size | BET and DFT [https://www.eag.com/techniques/phys-chem/bet-dft/]
- 61. SEM Particle Analysis | Particle Characterization [https://www.thermofisher.com/us/en/home/materials-science/particle-analysis/applications.html]
- 62. What is Single Particle Analysis & How Does it Work? [https://www.jeolusa.com/NEWS-EVENTS/Blog/what-is-single-particle-analysis-how-does-it-work]
- 63. A critical comparison between XRD and FIB residual stress ... [https://www.sciencedirect.com/science/article/abs/pii/S0040609014009390]
- 64. Eco-Friendly Sol-Gel Synthesis and Comprehensive ... [https://www.sciencedirect.com/science/article/abs/pii/S1293255825002195]
- 65. Temperature induced structural distortion and their effect ... [https://www.nature.com/articles/s41598-025-14101-0]
- 66. Effect of Isopropyl Peptization on Surface Particle Growth ... [https://www.sciencedirect.com/science/article/pii/S1026918525000629]
- 67. Sol–Gel Synthesis and Comprehensive Study of Structural, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12191741/]
- 68. Sol–gel synthesis and characterization of ZnO–SiO 2 ... [https://pubs.rsc.org/en/content/articlehtml/2025/na/d5na00612k]
- 69. The Role of the Sol-Gel Synthesis Process in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9315552/]
- 70. Systematic Study of Reaction Conditions for Size-Controlled ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11478073/]
- 71. Simple size-controlled synthesis of Au nanoparticles and ... [https://www.nature.com/articles/s41598-018-22976-5]
- 72. Effect of complexing agents on the electrochemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3330020/]
- 73. Electrochemical performance enhancement of perovskite ... [https://www.sciencedirect.com/science/article/pii/S0955221924008458]
- 74. Improved electrochemical performance of sol–gel method ... [https://link.springer.com/article/10.1007/s10008-013-2044-0]
- 75. Sol–Gel-Synthesized Metal Oxide Nanostructures [https://pmc.ncbi.nlm.nih.gov/articles/PMC12385869/]
- 76. The effect of particle size on structural and catalysts for ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979724026444]