Latent Space Bayesian Optimization: Accelerating Advanced Materials and Drug Discovery
This article explores the transformative potential of Bayesian Optimization (BO) within latent spaces for accelerating the discovery and design of novel materials and molecules.
Latent Space Bayesian Optimization: Accelerating Advanced Materials and Drug Discovery
Abstract
This article explores the transformative potential of Bayesian Optimization (BO) within latent spaces for accelerating the discovery and design of novel materials and molecules. We first establish the foundational principles of BO and the necessity of latent representations for navigating complex, discrete scientific spaces. The core of the article details cutting-edge methodologies, including graph neural network encodings of chemical space and multi-level optimization frameworks that balance exploration and exploitation. We further address critical troubleshooting and optimization challenges, such as the curse of dimensionality from expert knowledge and the selection of surrogate models. Finally, the article provides a rigorous validation of these techniques through benchmarking studies and comparative analyses of multi-task and deep Gaussian processes against conventional approaches, offering a comprehensive guide for researchers and drug development professionals seeking to implement these efficient optimization strategies.
The Foundations of Bayesian Optimization and Latent Space Representations
Bayesian Optimization (BO) is a powerful, sample-efficient strategy for optimizing black-box functions that are expensive to evaluate, a common scenario in materials science research and development [1]. By building a probabilistic surrogate model of the objective function and using an acquisition function to guide the search, BO can find optimal solutions with a minimal number of experiments [2]. This approach has become a cornerstone technique for accelerating materials discovery and design, enabling researchers to navigate complex, high-dimensional design spaces that include compositions, processing parameters, and microstructural variables [1] [3].
Recent advances have extended BO capabilities to handle the mixed quantitative and qualitative variables inherent in materials design problems [2]. Furthermore, the integration of BO with latent space representations has emerged as a particularly promising direction, allowing for the optimization of structured and discrete materials such as molecules and crystal structures by working in a continuous, meaningful latent space [4] [5]. This primer introduces the core concepts of Bayesian optimization with a specific focus on its application in materials science, detailing practical protocols and highlighting how latent space approaches are transforming the field.
Fundamental Principles and Algorithmic Workflow
Core Components of Bayesian Optimization
The Bayesian Optimization framework consists of two fundamental components:
Surrogate Model: Typically a Gaussian Process (GP), which provides a probabilistic distribution over the possible functions that fit the observed data. For a set of observations, the GP can make predictions for new points with associated uncertainty estimates [6]. The model uses a covariance function (kernel) to capture the similarity between data points, which is crucial for modeling complex material property relationships [2].
Acquisition Function: A criterion that uses the surrogate model's predictions to select the next point to evaluate by balancing exploration (sampling in uncertain regions) and exploitation (sampling where the model predicts high performance) [6]. Common acquisition functions include Expected Improvement (EI), Knowledge Gradient, and Upper Confidence Bound (UCB) [7].
The BO Cycle in Materials Design
The following workflow diagram illustrates the iterative BO process adapted for materials design:
Advanced BO Methodologies for Materials Science
Latent Space Bayesian Optimization
Latent Space BO addresses the challenge of optimizing structured, discrete, or hard-to-enumerate materials search spaces by leveraging deep generative models like variational autoencoders (VAEs) [4] [5]. These models map complex structured inputs (e.g., molecules, crystal structures) into a continuous latent space where standard BO techniques can be applied more effectively.
Key Advancement: The Correlated latent space Bayesian Optimization (CoBO) method introduces Lipschitz regularization, loss weighting, and trust region recoordination to minimize the inherent discrepancy between the latent space and objective function space, particularly around promising areas [4]. This approach has demonstrated strong performance in discrete data optimization tasks such as molecule design and arithmetic expression fitting.
Implementation Consideration: A significant challenge is that the latent space often remains high-dimensional. The LOL-BO algorithm adapts trust region concepts to the structured input setting by reformulating the encoder to serve both as a global encoder for the deep autoencoder and as a deep kernel for the surrogate model within a trust region, better aligning local optimization in the latent space with local optimization in the input space [5].
Multi-Objective and Target-Oriented BO
Materials design frequently involves balancing multiple, often competing objectives. Multi-objective BO identifies Pareto-optimal solutions representing the best trade-offs between objectives [1] [8].
Hierarchical Multi-Objective Optimization: The BoTier framework implements a tiered objective structure that reflects practical experimental hierarchies, where primary objectives (e.g., reaction yield) are prioritized over secondary objectives (e.g., minimizing expensive reagent use) [8]. This approach uses a composite scalarization function that ensures subordinate objectives contribute only after superordinate objectives meet satisfaction thresholds.
Target-Oriented Optimization: Many materials applications require achieving specific property values rather than simply maximizing or minimizing properties. Target-oriented BO (t-EGO) employs a target-specific Expected Improvement (t-EI) acquisition function that samples candidates based on their potential to reduce the difference from the target value, significantly improving efficiency for finding materials with predefined properties [7].
Handling Mixed Variable Types
Materials design naturally involves both quantitative variables (e.g., temperatures, concentrations) and qualitative variables (e.g., material types, processing methods). The Latent Variable GP (LVGP) approach maps qualitative factors into underlying numerical latent variables with strong physical justification, providing an inherent ordering and structure that captures complex correlations between qualitative levels [2]. This method enables more accurate modeling and efficient optimization compared to traditional dummy variable encoding approaches.
Experimental Protocols and Applications
Protocol: Multi-Objective Optimization of Biodegradable Magnesium Alloys
This protocol outlines the methodology for applying BO to design magnesium alloys with optimized mechanical properties and corrosion resistance [1].
- Objective: Synergistically optimize Ultimate Tensile Strength (UTS), Elongation (EL), and Corrosion Potential (Ecorr) of biodegradable Mg alloys.
- Design Variables: Alloy compositions (wt.%) and process parameters (extrusion temperature, extrusion ratio).
- Data Collection: Compile dataset from published literature and experimental results.
- Model Training: Train machine learning models (e.g., XGBoost) on the collected data to predict properties based on compositions and process parameters.
- BO Implementation:
- Construct multi-objective BO framework using Gaussian Process surrogates.
- Employ acquisition function to navigate the high-dimensional design space.
- Iteratively suggest candidate alloys for experimental validation.
- Experimental Validation: Prepare suggested alloy compositions, conduct tensile tests, and perform electrochemical measurements to verify predictions.
- Model Interpretation: Apply SHAP (SHapley Additive exPlanations) to interpret the machine learning model and identify critical factors influencing properties.
Protocol: Microstructure-Aware Bayesian Materials Design
This protocol incorporates microstructural descriptors as latent variables to enhance the mapping from design variables to material properties [3].
- Objective: Optimize material properties by explicitly incorporating microstructural information in the BO framework.
- Microstructural Characterization: Quantify microstructural features using descriptors such as grain size, phase fractions, and spatial correlations.
- Dimensionality Reduction: Apply Active Subspace Method to identify key subspaces within the latent microstructural space that most influence property variability.
- Latent-Space-Aware BO: Integrate reduced microstructural descriptors as latent variables in the Gaussian Process model to improve probabilistic modeling and design performance.
- Validation: Compare optimization performance between traditional (microstructure-agnostic) and microstructure-aware BO approaches.
Application Examples and Performance Metrics
Table 1: Bayesian Optimization Applications in Materials Science
| Material System | Design Variables | Target Properties | BO Method | Key Results | Citation |
|---|---|---|---|---|---|
| Biodegradable Mg Alloys | Composition, Extrusion parameters | UTS, EL, Ecorr | Multi-objective BO | Identified high-performance alloys; Experimental validation | [1] |
| Shape Memory Alloys | Composition | Transformation temperature | Target-oriented BO (t-EGO) | Ti0.20Ni0.36Cu0.12Hf0.24Zr0.08 with ΔT = 2.66°C from target in 3 iterations | [7] |
| Quasi-random Solar Cells | Pattern parameters, Material selection | Light absorption | LVGP-BO | Concurrent materials selection and microstructure optimization | [2] |
| Hybrid Organic-Inorganic Perovskites | Material constituents | Device performance | LVGP-BO | Combinatorial search for optimal compositions | [2] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for BO-Driven Materials Research
| Reagent/Material | Function in Experimental Protocol | Example Application |
|---|---|---|
| Elemental Metal Powders | Starting materials for alloy synthesis via arc melting or powder metallurgy | Mg alloy development [1] |
| High-Temperature Furnaces | For homogenization and thermal processing of alloy samples | Control of microstructure evolution [3] |
| Extrusion Equipment | Thermo-mechanical processing to refine microstructure and improve properties | Mg alloy processing [1] |
| Electrochemical Workstation | Corrosion potential (Ecorr) measurements to assess corrosion resistance | Evaluation of biodegradable alloys [1] |
| Universal Testing Machine | Mechanical property characterization (UTS, Elongation) | Validation of predicted mechanical properties [1] |
| Microscopy Equipment | Microstructural characterization (grain size, phase distribution) | Quantification of microstructural descriptors [3] |
Implementation Considerations and Computational Framework
Software and Computational Tools
The following diagram illustrates the computational workflow integrating various software tools for implementing BO in materials research:
Best Practices and Practical Recommendations
Initial Experimental Design: Begin with space-filling designs (e.g., Latin Hypercube Sampling) to build an initial surrogate model when no prior data exists [2].
Kernel Selection: Choose appropriate covariance functions for the Gaussian Process based on the nature of the design space. Matérn kernels often work well for materials properties with moderate smoothness.
Dimensionality Management: For high-dimensional problems (e.g., multi-component alloys), incorporate dimensionality reduction techniques or active subspaces to improve BO efficiency [3].
Constraint Handling: Incorporate known physical constraints or domain knowledge directly into the BO framework to avoid exploring infeasible regions of the design space.
Batch Optimization: When parallel experimental capabilities exist, implement batch BO approaches to suggest multiple candidates for simultaneous evaluation.
Uncertainty Quantification: Leverage the probabilistic nature of BO to quantify and communicate uncertainty in predictions, which is crucial for experimental planning and decision-making.
Bayesian Optimization represents a paradigm shift in materials discovery, enabling efficient navigation of complex design spaces with minimal experimental iterations. The integration of latent space approaches further extends these capabilities to structured materials design problems, offering powerful new strategies for accelerating the development of advanced materials with tailored properties.
Why Latent Space? Navigating High-Dimensional and Discrete Scientific Landscapes
Latent spaces—low-dimensional representations learned from high-dimensional data—are revolutionizing how researchers navigate complex scientific problems. In fields ranging from materials science to drug development, these compressed embeddings capture the essential, underlying features of data, transforming intractable problems into manageable ones. The core premise is that while scientific data may be high-dimensional and discrete in its raw form (e.g., molecular structures, microstructural images, or clinical patient profiles), its true structure often resides on a much lower-dimensional manifold. By projecting this data into a latent space, scientists can perform efficient optimization, identify meaningful patterns, and make predictions that would be impossible in the original high-dimensional space. This approach is particularly powerful when integrated with Bayesian optimization (BO), creating a framework for data-driven experimental design and discovery that explicitly accounts for uncertainty in sparse-data regimes common in scientific research.
Theoretical Foundations
The Mathematical Framework of Latent Space Embeddings
Latent space approaches fundamentally rely on learning a mapping from a high-dimensional observation space (\mathcal{X} \subseteq \mathbb{R}^d) to a lower-dimensional latent space (\mathcal{Z} \subseteq \mathbb{R}^{d'}) where (d' \ll d). This mapping (f: \mathcal{X} \to \mathcal{Z}) preserves essential structural relationships while discarding redundant information. In scientific applications, this process typically involves either:
- Dimensionality reduction techniques (e.g., PCA, autoencoders) that compress while preserving variance
- Generative model embeddings (e.g., VAEs, diffusion models) that learn the data distribution
- Task-specific embeddings optimized for particular scientific objectives
The mathematical power of this approach stems from the manifold hypothesis, which posits that most high-dimensional scientific data actually lies on or near a low-dimensional manifold. Latent space identification effectively parameterizes this manifold, enabling efficient navigation and optimization.
Bayesian Optimization in Latent Space
Bayesian optimization provides a principled framework for global optimization of expensive black-box functions. When combined with latent space representations, it becomes particularly powerful for scientific applications. The standard BO process in latent space involves:
- Learning a latent representation (z = f(x)) of the scientific data
- Constructing a probabilistic surrogate model (typically Gaussian Process) in latent space
- Using an acquisition function to select promising candidates for evaluation
- Updating the model with new data and repeating
This approach addresses the "curse of dimensionality" that plagues high-dimensional optimization, as the surrogate model operates in a lower-dimensional space where data is less sparse and relationships are more easily learned.
Applications Across Scientific Domains
Materials Science and Microstructure Design
In materials science, latent space approaches have enabled microstructure-aware design, moving beyond traditional composition-property relationships to explicitly incorporate structural information.
Table 1: Latent Space Applications in Materials Design
| Application Area | Key Latent Variables | Performance Improvement | Reference |
|---|---|---|---|
| Thermoelectric Materials | Grain size, phase distribution, defect concentration | Enhanced conversion efficiency | [3] |
| Advanced Alloys | Grain boundaries, phase distributions | Improved strength-toughness balance | [3] |
| Polymer-Bonded Explosives | Statistical microstructure descriptors | Accurate shock prediction with reduced simulation | [3] |
The microstructure-aware Bayesian materials design framework demonstrates how latent microstructural descriptors create a more direct pathway through the Process-Structure-Property-Performance (PSPP) chain, traditionally a fundamental challenge in materials science [3]. By treating microstructural features as tunable design parameters rather than emergent by-products, researchers can more efficiently navigate toward materials with desired properties.
Drug Development and Treatment Personalization
In pharmaceutical research, latent space approaches have shown particular promise for treatment personalization, especially for complex disorders where traditional subgrouping approaches fail.
Table 2: Treatment Selection Performance in Major Depressive Disorder
| Method | Personalization Paradigm | Improvement over Random Allocation | Patient Cohort |
|---|---|---|---|
| Fully Personalized | Individual-level ITE estimation | Not specified | [9] |
| Sub-grouping | Cluster-level optimization | Not specified | [9] |
| DPNN (Latent-Space Prototyping) | Balanced personalization & prototyping | 8% absolute, 23% relative | 4,754 MDD patients [9] |
The Differential Prototypes Neural Network (DPNN) exemplifies how latent space prototyping strikes a balance between fully personalized and sub-grouping paradigms [9]. By identifying "actionable prototypes" in latent space—groups that differ in their expected treatment responses—this approach achieved clinically significant improvements for Major Depressive Disorder patients, addressing the heterogeneity that has long challenged psychiatric treatment optimization.
Molecular Design and Protein Engineering
Modern generative AI models have created new opportunities for latent space optimization in molecular design. Sample-based approaches like diffusion and flow matching models can generate diverse molecular structures, while latent space optimization enables efficient navigation toward molecules with desired properties.
The surrogate latent space approach allows researchers to define custom latent spaces using example molecules, creating low-dimensional Euclidean embeddings that maintain biological relevance while being convenient for optimization tasks [10]. This method has shown particular promise in protein generation, where it enabled successful generation of proteins with greater length than previously feasible, demonstrating the scalability of latent space approaches for complex biomolecular design problems.
Experimental Protocols and Methodologies
Protocol 1: Microstructure-Aware Bayesian Optimization for Materials Design
Objective: Identify material processing parameters that yield optimal properties by incorporating microstructural descriptors as latent variables.
Materials and Reagents:
- Material precursor compounds (composition-dependent)
- Processing equipment (e.g., furnaces, mills)
- Characterization tools (e.g., SEM, EBSD, XRD)
- Computational resources for simulation and modeling
Procedure:
- Data Collection and Feature Extraction
- Collect historical data on processing parameters, microstructural features, and properties
- Extract relevant microstructural descriptors using image analysis (grain size, phase distribution, etc.)
- Normalize all features to zero mean and unit variance
Latent Space Construction
- Apply Active Subspace Method to identify dominant directions in microstructure space
- Construct latent variables (z = W^T \cdot \text{microstructure features}) where (W) contains eigenvectors of the gradient covariance matrix
- Validate latent space preservation of property-relevant information
Bayesian Optimization Loop
- Initialize Gaussian Process surrogate model linking processing parameters to properties through latent space
- For each iteration: a. Select next processing parameters using Expected Improvement acquisition function b. Execute experimental processing (e.g., heat treatment, deformation) c. Characterize resulting microstructure d. Measure target properties e. Update surrogate model with new data
- Continue until convergence or resource exhaustion
Validation and Interpretation
- Validate optimal materials through independent replication
- Interpret active subspaces to identify microstructural features most critical to performance
- Document process-structure-property relationships revealed by the optimization
Troubleshooting:
- If optimization stagnates, consider expanding the latent dimensionality
- For noisy property measurements, increase the regularization in the GP model
- If experimental variance is high, implement batch sampling to account for uncertainty
Protocol 2: Latent-Space Treatment Personalization for Clinical Applications
Objective: Identify optimal treatment assignments for individual patients based on latent patient prototypes.
Materials and Data Requirements:
- Patient baseline characteristics (demographic, clinical, biomarker)
- Treatment response data
- Computational infrastructure for deep learning model training
- Validation cohort data
Procedure:
- Data Preprocessing and Integration
- Clean and normalize patient baseline characteristics
- Handle missing data using appropriate imputation methods
- Encode categorical variables and ensure data quality
DPNN Model Architecture and Training
- Implement neural network with parallel structure for prototype identification and outcome prediction
- Configure multifaceted loss function balancing prediction accuracy and prototype cohesiveness
- Train model using historical patient-treatment-outcome data
- Validate prototype quality and predictive performance on holdout set
Treatment Assignment Optimization
- For new patient, encode baseline characteristics into latent space
- Identify relevant prototypes and their membership weights
- Compute personalized outcome predictions for each available treatment
- Select treatment with highest predicted probability of success
Validation and Model Updating
- Implement prospective validation in clinical setting
- Continuously monitor treatment outcomes and model performance
- Update model with new data following appropriate validation protocols
Ethical Considerations:
- Ensure diverse representation in training data to avoid biased recommendations
- Maintain physician oversight of all treatment decisions
- Implement transparent documentation of model limitations and uncertainties
Computational Tools and Implementation
Workflow Visualization: Microstructure-Aware Materials Design
Diagram 1: Microstructure-aware Bayesian optimization workflow for materials design
Workflow Visualization: Latent-Space Treatment Personalization
Diagram 2: Latent-space treatment personalization using differential prototyping
Research Reagent Solutions
Table 3: Essential Computational Tools for Latent Space Research
| Tool Category | Specific Solutions | Function | Application Examples |
|---|---|---|---|
| Generative Models | Diffusion Models, Flow Matching, VAEs | Learn latent representations from high-dimensional data | Molecular design, microstructure generation [10] |
| Optimization Frameworks | Bayesian Optimization, CMA-ES | Efficient navigation in latent space | Materials design, treatment optimization [3] [9] |
| Dimensionality Reduction | Active Subspaces, PCA, Autoencoders | Construct lower-dimensional latent spaces | Microstructure descriptor compression [3] |
| Surrogate Modeling | Gaussian Processes, Bayesian Neural Networks | Probabilistic modeling in latent space | Predicting material properties, treatment outcomes [11] |
| Inversion Tools | DDIM, Probability Flow ODE | Map data to latent representations | Protein design, molecular optimization [10] |
Challenges and Future Directions
Despite promising results, latent space approaches face several significant challenges. The distribution mismatch between original and reconstructed spaces can lead to suboptimal performance, particularly in high-dimensional Bayesian optimization [11]. Methods like HiPPO-based space consistency aim to address this by preserving kernel relationships during latent space construction, but general solutions remain elusive. Interpretability presents another challenge—while latent spaces enable efficient optimization, understanding what specific features they capture requires additional analysis techniques like active subspaces.
Future work will likely focus on improving the robustness of latent space representations, developing better methods for handling multi-modal data, and creating more interpretable latent representations. The integration of physical constraints and domain knowledge into latent space learning represents another promising direction, particularly for scientific applications where fundamental principles are known. As generative AI models continue to advance, their integration with latent space optimization will likely open new frontiers in materials design, drug development, and scientific discovery.
Bayesian optimization (BO) is a powerful strategy for the global optimization of expensive, black-box functions, making it particularly suited for advanced materials research and drug development where physical experiments or complex simulations are costly and time-consuming [12] [13]. The core challenge it addresses is finding the global optimum of a function whose analytical form is unknown and whose derivatives are unavailable, with as few evaluations as possible [13] [14]. This is achieved through a synergistic interplay of three key components: a surrogate model that statistically approximates the black-box function, an acquisition function that guides the selection of future experiments by balancing exploration and exploitation, and an active learning loop that iteratively updates the model with new data [12] [13]. Within materials science, this framework has been successfully applied to tasks such as discovering shape memory alloys with specific transformation temperatures [7] and identifying novel phase-change memory materials with superior properties [15].
Surrogate Models: Gaussian Processes and Beyond
The surrogate model forms the probabilistic foundation of BO, providing a computationally cheap approximation of the expensive objective function and quantifying the uncertainty of its own predictions [12] [13].
Gaussian Process Models
Gaussian Processes (GPs) are the most widely used surrogate models in Bayesian optimization. A GP defines a prior over functions and can be updated with data to form a posterior distribution. For a set of data points (\mathcal{D}{1:t} = {(\mathbf{x}1, y),...,(\mathbf{x}t, y_t)}), the GP posterior predictive distribution at a new point (\mathbf{x}) is characterized by a mean (\mu(\mathbf{x})) and variance (\sigma^2(\mathbf{x})) [13]. The mean function provides an estimate of the objective, while the variance represents the model's uncertainty. This explicit uncertainty quantification is crucial for the function of acquisition functions. GPs are distinguished by their mathematical explicitness, flexibility, and straightforward uncertainty quantification [12].
Advanced and Adaptive Surrogate Models
While GPs are powerful, their performance can be challenged by high-dimensional design spaces or non-smooth objective functions. Consequently, more adaptive and flexible Bayesian models have been explored as surrogates to enhance the BO framework's robustness and efficiency [12].
- Bayesian Additive Regression Trees (BART): BART is an ensemble-learning-based method that fits unknown functional patterns through a sum of small trees. It is equipped with automatic feature selection techniques, making it particularly useful when the objective function is very complex [12].
- Bayesian Multivariate Adaptive Regression Splines (BMARS): BMARS is a flexible nonparametric approach based on product spline basis functions. It can effectively model non-smooth functions with sudden transitions, a common occurrence in many materials science challenges where phase changes occur [12].
- Correlated Latent Space Models (CoBO): For structured or discrete data, such as molecular structures, optimization can be performed in a latent space learned by deep generative models like variational autoencoders. The CoBO framework introduces techniques to strengthen the correlation between distances in the latent space and distances in the objective function, minimizing the inherent gap that can lead to suboptimal solutions [4].
Table 1: Comparison of Surrogate Models for Bayesian Optimization.
| Model Type | Key Features | Best-Suited Problems | Performance Notes |
|---|---|---|---|
| Gaussian Process (GP) | Explicit uncertainty, smooth interpolation [12]. | Low-to-moderate dimensional, smooth objective functions. | Benchmark method; performance can degrade with high dimensions or non-smoothness [12]. |
| BART | Ensemble of trees, automatic feature selection [12]. | Complex, high-dimensional functions with interactions. | Enhanced search efficiency and robustness compared to GP on complex test functions (e.g., Rastrigin) [12]. |
| BMARS | Spline-based, nonparametric, handles non-smoothness [12]. | Functions with sudden transitions or non-smooth patterns. | Superior to GP-based methods on non-smooth objectives; efficient in high dimensions [12]. |
| Correlated Latent BO | Operates in generative model's latent space [4]. | Structured/discrete data (e.g., molecules, chemical formulas). | Effectively optimizes discrete structures by learning a correlated latent representation [4]. |
Acquisition Functions: Guiding the Experiment
The acquisition function (u(\mathbf{x})) is the decision-making engine of BO. It uses the surrogate model's posterior to compute the utility of evaluating a candidate point (\mathbf{x}), balancing the need to explore regions of high uncertainty (to reduce model error) and exploit regions with promising predicted values (to refine the optimum) [13]. The next point to evaluate is chosen by maximizing the acquisition function: (\mathbf{x}{t+1} = \operatorname{argmax}{\mathbf{x}} u(\mathbf{x})) [13].
Common Acquisition Functions
Several acquisition functions have been developed, each with a slightly different mechanism for balancing exploration and exploitation.
Expected Improvement (EI): EI measures the expected amount by which the observation at (\mathbf{x}) will improve upon the current best observation (f(\mathbf{x}^+)). It is one of the most widely used acquisition functions and can be evaluated analytically under the GP surrogate [13] [16]: [ \operatorname{EI}(\mathbf{x}) = \begin{cases} (\mu(\mathbf{x}) - f(\mathbf{x}^+) - \xi)\Phi(Z) + \sigma(\mathbf{x})\phi(Z) &\text{if}\ \sigma(\mathbf{x}) > 0 \ 0 & \text{if}\ \sigma(\mathbf{x}) = 0 \end{cases} ] where (Z = \frac{\mu(\mathbf{x}) - f(\mathbf{x}^+) - \xi}{\sigma(\mathbf{x})}), and (\Phi) and (\phi) are the CDF and PDF of the standard normal distribution, respectively. The parameter (\xi) controls the exploration-exploitation trade-off, with higher values leading to more exploration [13].
Upper Confidence Bound (UCB): Also known as the lower confidence bound for minimization, UCB is a straightforward function that combines the mean and uncertainty of the prediction [16]: [ \operatorname{UCB}(\mathbf{x}) = \mu(\mathbf{x}) + \beta \sigma(\mathbf{x}) ] Here, (\beta) is a parameter that controls the weight given to exploration.
Probability of Improvement (PI): PI measures the probability that a new sample will improve upon the current best value [16]: [ \operatorname{PI}(x) = \Phi\left( \frac{\mu(x) - f(x^+) - \xi}{\sigma(x)} \right) ]
Specialized Acquisition Functions for Materials Science
The standard acquisition functions are designed for finding global maxima or minima. However, many materials applications require finding a material with a specific target property value, not merely an optimum.
Target-Oriented Expected Improvement (t-EI): Developed for target-specific property values, t-EI aims to sample candidates whose property value is closer to a predefined target (t) than the current best candidate [7]. It is defined as: [ t-EI = E\left[\max (0, |y{t.min} - t| - |Y - t| )\right] ] where (y{t.min}) is the value in the training set closest to the target, and (Y) is the predicted random variable. This method has been shown to require significantly fewer experimental iterations to reach a target value compared to reformulating the problem as a minimization of (|y-t|) using standard EI [7].
Multi-Objective and Constrained Acquisition Functions: In real-world materials design, it is often necessary to optimize multiple properties simultaneously or subject to constraints. Multi-objective acquisition functions (MOAF) seek a Pareto-front solution, while constrained EGO (CEGO) uses a constrained expected improvement (CEI) to incorporate feasibility [7].
Table 2: Key Acquisition Functions and Their Applications in Research.
| Acquisition Function | Mathematical Formulation | Primary Use-Case | ||||
|---|---|---|---|---|---|---|
| Expected Improvement (EI) | (\operatorname{EI}(\mathbf{x}) = (\mu - f^+ - \xi)\Phi(Z) + \sigma\phi(Z)) [13] [16] | General-purpose global optimization (default choice). | ||||
| Upper Confidence Bound (UCB) | (\operatorname{UCB}(\mathbf{x}) = \mu(\mathbf{x}) + \beta \sigma(\mathbf{x})) [16] | Optimization with an explicit exploration parameter. | ||||
| Probability of Improvement (PI) | (\operatorname{PI}(x) = \Phi\left( \frac{\mu(x) - f(x^+) - \xi}{\sigma(x)} \right)) [16] | Finding the region of the optimum quickly; less used than EI. | ||||
| Target-oriented EI (t-EI) | (E\left[\max (0, | y_{t.min} - t | - | Y - t | )\right]) [7] | Finding a material with a specific target property value. |
| Multi-Objective AF (MOAF) | Seeks Pareto-front solution for multiple acquisition values [7]. | Simultaneously optimizing multiple material properties. |
The Active Learning Loop in Practice
The active learning loop is the iterative procedure that integrates the surrogate model and acquisition function into a closed-loop experimental design system [15]. A typical implementation of this loop for materials research is as follows [13]:
- Initialization: A small initial dataset is collected, often using a space-filling design like a Latin Hypercube Sample (LHS) to get a rough initial model of the design space [14].
- Model Fitting: A surrogate model (e.g., a GP) is trained on all data collected so far.
- Acquisition Optimization: The acquisition function (e.g., EI) is computed based on the surrogate's predictions and is then maximized to propose the next experiment (\mathbf{x}_{t+1}).
- Experiment Execution: The proposed experiment is performed (e.g., a new material composition is synthesized and characterized), and the objective function value (y_{t+1}) is obtained.
- Data Update: The new data pair ((\mathbf{x}{t+1}, y{t+1})) is added to the training dataset.
- Iteration: Steps 2-5 are repeated until a stopping criterion is met, such as the exhaustion of an experimental budget, convergence, or the discovery of a material satisfying the target criteria.
This closed-loop system has been embodied in platforms like the Autonomous System for Materials Exploration and Optimization (CAMEO), which operates at synchrotron beamlines to autonomously discover new materials, demonstrating a ten-fold reduction in the number of experiments required [15].
Diagram 1: The Bayesian Optimization Active Learning Loop.
Application Notes & Protocols
Protocol 1: Discovering a Shape Memory Alloy with a Target Transformation Temperature
Objective: To discover a Ni-Ti-based shape memory alloy (SMA) with a phase transformation temperature as close as possible to a target of 440°C for use as a thermostatic valve material [7].
Research Reagent Solutions:
- Elemental Precursors: High-purity Nickel (Ni), Titanium (Ti), Copper (Cu), Hafnium (Hf), and Zirconium (Zr) for alloy synthesis.
- Synthesis Platform: An arc melter or a high-throughput synthesis robot for rapid sample preparation.
- Characterization Tool: A differential scanning calorimeter (DSC) for accurate measurement of the phase transformation temperature.
Methodology:
- Initial Design: Prepare a small set (e.g., 5-10) of initial Ni-Ti-X alloy samples with compositions selected via a space-filling design over the feasible composition space.
- Characterization: Measure the transformation temperature for each initial sample using DSC.
- Model Configuration: Implement a BO loop using a GP surrogate model and the target-oriented Expected Improvement (t-EI) acquisition function, with the target value (t) set to 440°C.
- Active Learning Loop: For each iteration: a. Model Training: Train the GP model on all available (composition, temperature) data. b. Suggestion: Propose the next alloy composition to synthesize by maximizing the t-EI function. c. Synthesis & Measurement: Synthesify the proposed alloy and measure its transformation temperature. d. Update: Add the new data point to the training set.
- Termination: Stop the loop after a fixed budget of experiments (e.g., 10-15 iterations) or when a material with a transformation temperature within an acceptable tolerance (e.g., ±5°C) is found.
Outcome: This protocol led to the discovery of SMA (\text{Ti}{0.20}\text{Ni}{0.36}\text{Cu}{0.12}\text{Hf}{0.24}\text{Zr}_{0.08}) with a transformation temperature of 437.34°C, only 2.66°C from the target, within just 3 experimental iterations [7].
Protocol 2: Autonomous Discovery of a Novel Phase-Change Material
Objective: To find a Ge-Sb-Te (GST) ternary composition with the largest possible change in optical bandgap ((\Delta E_g)) between its amorphous and crystalline states for superior photonic switching devices [15].
Research Reagent Solutions:
- Materials Library: A composition spread wafer library covering the Ge-Sb-Te ternary system, fabricated via combinatorial sputtering.
- Characterization Setup: A synchrotron beamline for high-throughput X-ray diffraction (XRD) to determine the crystal structure and phase map, and an ellipsometer for measuring optical properties.
- Automation Framework: The CAMEO algorithm installed on the beamline's control computer for real-time, closed-loop operation.
Methodology:
- Prior Integration: Incorporate any prior knowledge (e.g., from ellipsometry spectra) into the initial phase mapping model.
- Dual-Objective Acquisition: Implement the CAMEO algorithm, which uses a specialized acquisition function, (g), that balances two objectives: maximizing knowledge of the phase map (P(\mathbf{x})) and optimizing the functional property (F(\mathbf{x})) (here, (\Delta Eg)). The next point is selected as (\mathbf{x}* = \mathrm{argmax}_{\mathbf{x}} \, g(F(\mathbf{x}), P(\mathbf{x}))) [15].
- Closed-Loop Execution: The algorithm autonomously: a. Analyzes the latest XRD data to update the phase map. b. Selects the most informative composition to measure next based on the dual objective. c. Directs the beamline to perform the measurement.
- Human-in-the-Loop: Optionally, a human expert can monitor the process and provide guidance, which the algorithm can incorporate.
- Validation: Promising compositions identified by the autonomous loop are then validated by fabricating and testing actual device prototypes.
Outcome: This protocol resulted in the discovery of a novel, stable epitaxial nanocomposite phase-change material at a phase boundary, which exhibited an optical contrast up to three times larger than the well-known Ge(2)Sb(2)Te(_5) (GST225) compound [15].
Table 3: Quantitative Performance of Bayesian Optimization Methods.
| Optimization Method / Strategy | Test Problem / Application | Key Performance Metric | Result |
|---|---|---|---|
| Target-Oriented BO (t-EGO) [7] | Synthetic functions & 2D materials database (HER catalysts). | Average iterations to reach target (vs. EGO/MOAF). | Required 1 to 2 times fewer experimental iterations [7]. |
| BO with BART/BMARS [12] | Rosenbrock & Rastrigin function optimization. | Minimum observed value vs. number of function evaluations. | Showed faster decline and better performance than GP-based methods, especially with small initial datasets [12]. |
| CAMEO [15] | Discovery of phase-change memory material (Ge-Sb-Te system). | Number of experiments required for discovery. | Achieved a ten-fold reduction in the number of experiments required compared to traditional methods [15]. |
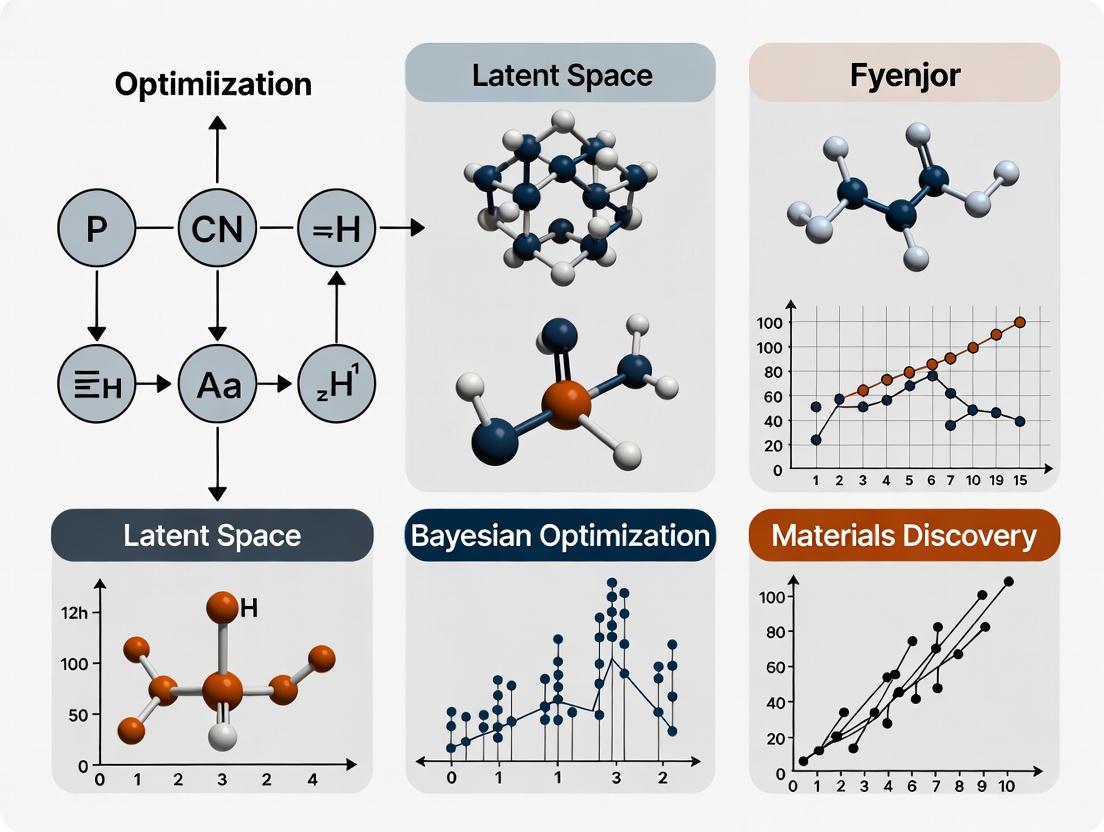
The exploration of chemical space for molecular and materials discovery is fundamentally a challenge of navigating discrete, combinatorial structures. However, key computational methodologies, particularly Bayesian optimization (BO), operate most effectively within continuous domains. This application note examines the critical need for and advantages of creating continuous representations of discrete chemical structures to accelerate discovery campaigns. We detail how latent space Bayesian optimization frameworks address this representational challenge, enabling efficient navigation of vast molecular search spaces. Within the context of an overarching thesis on Bayesian optimization in latent space for materials research, we provide specific protocols for implementing these approaches, including quantitative performance comparisons and detailed workflows for representing discrete molecular graphs as continuous vectors suitable for probabilistic modeling and optimization.
The set of possible molecules and materials is fundamentally discrete and combinatorially vast. Individual molecular structures are distinct, separate entities, much like the distinct values that define discrete data [17]. However, the properties and functions of these materials often depend on continuous physical phenomena. This creates a fundamental tension: how can we efficiently search a discrete, high-dimensional chemical space using optimization frameworks that typically require continuous input representations?
Bayesian optimization (BO) has emerged as a powerful, sample-efficient framework for guiding materials discovery within an active learning loop, particularly when experiments or simulations are expensive [7] [18]. BO relies on a probabilistic surrogate model, such as a Gaussian Process, to model an unknown objective function and an acquisition function to decide which experiments to perform next. However, the performance of BO is heavily influenced by the representation of the input material [18]. A fixed, high-dimensional discrete representation can lead to poor performance due to the curse of dimensionality, while an overly simplified representation may lack the chemical detail necessary to predict performance accurately.
Consequently, there is a pressing need for continuous representations of these discrete structures. A continuous representation embeds discrete objects (like molecules) into a continuous space where similarities and distances are meaningfully preserved. This allows for the application of powerful continuous optimization techniques, such as BO, to problems of a discrete nature. The following sections detail the methodologies, applications, and protocols for successfully implementing this paradigm.
Theoretical Foundations: Bridging the Discrete-Continuous Divide
Discrete vs. Continuous Mathematical Frameworks
- Discrete Structures: Molecules and crystalline materials are naturally represented as discrete graphs (atoms as nodes, bonds as edges) or combinatorial sets of building blocks. These structures are countable and possess distinct, separable states [19] [17].
- Continuous Representations: These are function representations that map positional coordinates or latent variables to a response value. They offer inherent advantages for optimization, including resolution flexibility, inherent smoothness, and parameter efficiency [20]. In machine learning, this often involves creating a smooth, continuous latent space where similar molecules are clustered together.
Latent Space Bayesian Optimization
The core solution involves compressing discrete chemical structures into a smooth, continuous latent space where Bayesian optimization can be performed efficiently.
- Multi-level Bayesian Optimization: One advanced approach uses transferable coarse-grained models to compress chemical space into varying levels of resolution. Discrete molecular spaces are first transformed into smooth latent representations. Bayesian optimization is then performed within these latent spaces, using simulations to calculate target properties. This multi-level approach effectively balances exploration (at lower resolutions) and exploitation (at higher resolutions) [21].
- Joint Composite Latent Space BO (JoCo): For complex, high-dimensional outputs, the JoCo framework jointly trains neural network encoders and probabilistic models to adaptively compress both high-dimensional inputs and outputs into manageable latent representations. This enables effective BO on these compressed representations [22].
- Feature Adaptive Bayesian Optimization (FABO): Instead of relying on a fixed representation, FABO dynamically identifies the most informative features influencing material performance at each BO cycle. This enables autonomous exploration of the search space with minimal prior information about the best representation [18].
Application Notes and Performance Metrics
The following table summarizes the performance of various latent space BO methods across different molecular and materials optimization tasks, as reported in the literature.
Table 1: Performance Comparison of Continuous Representation Methods in Bayesian Optimization
| Method | Core Approach | Application Domain | Reported Performance | Key Advantage |
|---|---|---|---|---|
| Multi-level BO with Hierarchical Coarse-Graining [21] | Transforms discrete molecules into multi-resolution latent spaces for BO. | Enhancing phase separation in phospholipid bilayers. | Effective navigation of chemical space for free energy-based optimization. | Balances combinatorial complexity & chemical detail. |
| Target-Oriented BO (t-EGO) [7] | Uses acquisition function t-EI to sample candidates based on distance to a target property value. | Discovering shape memory alloys with a target transformation temperature. | Achieved a temperature difference of only 2.66°C from target in 3 experimental iterations. | Superior for finding materials with specific target properties, not just optima. |
| Feature Adaptive BO (FABO) [18] | Dynamically adapts material representations throughout BO cycles. | MOF discovery for CO2 adsorption and band gap optimization. | Outperforms BO with fixed representations, especially in novel tasks. | Automatically identifies relevant features without prior knowledge. |
| Joint Composite Latent BO (JoCo) [22] | Jointly compresses high-dimensional input and output spaces into latent representations. | High-dimensional BO in generative AI, molecular design, and robotics. | Outperforms state-of-the-art methods on a variety of simulated and real-world problems. | Effectively handles high-dimensional input and output spaces. |
Detailed Experimental Protocols
Protocol 1: Multi-Level Bayesian Optimization with Coarse-Graining
This protocol is adapted from methods used to optimize molecules for enhancing phase separation in phospholipid bilayers [21].
1. Problem Definition:
- Objective: Identify molecules that maximize or minimize a target property (e.g., free energy of binding, phase separation propensity).
- Search Space: Define the discrete set of molecular structures or modifications to be considered.
2. Multi-Resolution Representation:
- Step 1: Hierarchical Coarse-Graining. Develop or apply transferable coarse-grained models that represent molecules at multiple levels of resolution (e.g., atomistic, united-atom, ultra-coarse-grained).
- Step 2: Latent Space Transformation. For each resolution level, use an encoder (e.g., a variational autoencoder or other neural network) to transform the discrete molecular representation into a continuous latent vector. The latent space should be smooth, meaning small changes in the latent vector correspond to small changes in the molecular structure and its properties.
3. Bayesian Optimization Loop:
- Step 3: Initialization. Populate a dataset with an initial set of molecules and their measured properties. This can be a small, random set or based on prior knowledge.
- Step 4: Lower-Resolution Exploration. Use a lower-fidelity, coarse-grained model to perform Bayesian optimization in the corresponding latent space. This step is computationally cheaper and aims to identify promising neighborhoods in the chemical space.
- Step 5: Higher-Resolution Exploitation. Use the promising regions identified at the coarse level to guide a more focused BO campaign in a higher-resolution latent space. The surrogate model at this level uses more accurate (and expensive) simulations or experiments.
- Step 6: Iteration and Selection. Iterate between exploration and exploitation across resolution levels until a convergence criterion is met (e.g., no significant improvement after a set number of iterations). The best-performing molecule from the high-fidelity evaluations is selected.
Diagram 1: Multi-level BO workflow
Protocol 2: Target-Oriented Bayesian Optimization for Specific Properties
This protocol is designed for discovering materials with a specific target property value, rather than simply a maximum or minimum, as demonstrated in the discovery of shape memory alloys [7].
1. Problem Definition:
- Objective: Find a material
xsuch that its propertyy(x)is as close as possible to a predefined target valuet(e.g., a transformation temperature of 440°C). - Search Space: Define the compositional or structural space of materials (e.g., the proportions of Ti, Ni, Cu, Hf, Zr in an alloy).
2. Gaussian Process Modeling:
- Step 1: Model Training. Train a Gaussian Process (GP) surrogate model using the available data. The input is the material representation (e.g., composition), and the output is the measured property
y. - Model Specification: A GP is defined as
f(x) ~ GP(0, k(x, x')), wherekis a kernel function. The predictive meanμ(x)and varianceσ²(x)for a new candidatexare given by:μ(x) = k_x(K + σ²_εI)⁻¹yσ²(x) = k(x,x) - k_x(K + σ²_εI)⁻¹k_xᵀ
3. Target-Oriented Acquisition:
- Step 2: Calculate Target-specific Expected Improvement (t-EI). Instead of the standard Expected Improvement (EI), use the t-EI acquisition function.
- Definition: Let
y_t.minbe the current property value closest to the targett. The improvement is defined asI = max( |y_t.min - t| - |Y - t|, 0 ), whereYis the random variable of the GP prediction atx. The expected improvement is then:t-EI(x) = E[I] - Step 3: Candidate Selection. Select the next material to test by maximizing the t-EI function:
x_next = argmax t-EI(x).
4. Iteration:
- Step 4: Experiment and Update. Synthesize and test the candidate
x_next, measure its propertyy_next, and add the new data point(x_next, y_next)to the training dataset. Update the GP model and repeat from Step 2 until a material satisfying the target criterion is found.
Protocol 3: Feature Adaptive Bayesian Optimization (FABO)
This protocol is used when the optimal representation of a material is not known in advance, as in the discovery of metal-organic frameworks (MOFs) [18].
1. Initialization:
- Step 1: Define a Complete Feature Pool. Start with a high-dimensional, comprehensive set of features that describe the materials. For MOFs, this could include chemical descriptors (e.g., Revised Autocorrelation Calculations - RACs) and pore geometric characteristics.
2. Bayesian Optimization Cycle:
- Step 2: Data Labeling. Perform an experiment or simulation to measure the property of interest for an initial set of materials.
- Step 3: Feature Selection. Using only the data acquired during the BO campaign so far, apply a feature selection method to identify the most relevant features.
- Method A: Maximum Relevancy Minimum Redundancy (mRMR). Selects features that maximize relevance to the target
y(e.g., using F-statistic) while minimizing redundancy with already-selected features. - Method B: Spearman Ranking. A univariate method that ranks features based on the absolute value of their Spearman rank correlation coefficient with the target variable.
- Method A: Maximum Relevancy Minimum Redundancy (mRMR). Selects features that maximize relevance to the target
- Step 4: Surrogate Model Update. Train the GP surrogate model using the currently selected, reduced feature set.
- Step 5: Candidate Selection. Use a standard acquisition function (e.g., Expected Improvement or Upper Confidence Bound) to select the next material to test based on the updated model.
- Step 6: Iterate. Repeat steps 2-5 until the optimization budget is exhausted or performance converges.
Diagram 2: Feature adaptive BO cycle
The Scientist's Toolkit: Research Reagent Solutions
This table outlines key computational "reagents" and their functions in constructing continuous representations for Bayesian optimization.
Table 2: Essential Components for Continuous Representation workflows
| Tool / Method | Type | Primary Function | Example Use Case |
|---|---|---|---|
| Gaussian Process (GP) [7] [18] | Probabilistic Model | Serves as a surrogate model to predict material properties and quantify uncertainty. | Core to all BO protocols for regression and uncertainty estimation. |
| Variational Autoencoder (VAE) | Neural Network | Encodes discrete structures (e.g., molecular graphs) into a continuous latent vector; can decode vectors back to structures. | Creating the latent space for multi-level BO [21] and JoCo [22]. |
| Coarse-Grained Molecular Model [21] | Simplified Physical Model | Provides a low-fidelity, computationally cheap representation of a molecule for initial screening. | The lower-resolution level in multi-level BO. |
| Molecular Descriptors (e.g., RACs) [18] | Feature Vector | Numerically encodes chemical and topological aspects of a molecule into a fixed-length vector. | Forms the initial feature pool in FABO for representing MOFs and molecules. |
| Maximum Relevancy Minimum Redundancy (mRMR) [18] | Feature Selection Algorithm | Dynamically selects an informative and non-redundant subset of features from a large pool. | Adapting the representation in the FABO protocol. |
| Target-specific Expected Improvement (t-EI) [7] | Acquisition Function | Guides the search towards candidates whose predicted property is close to a specific target value. | Core component of the target-oriented BO protocol. |
Methodologies and Real-World Applications in Chemistry and Biomedicine
The exploration of chemical space, estimated to contain over 10^60 drug-like molecules, represents one of the most significant challenges in modern materials research and drug discovery [23]. This vastness necessitates sophisticated computational approaches that can efficiently navigate molecular structures and properties. Graph neural networks (GNNs) and autoencoders have emerged as transformative technologies for molecular representation, directly addressing the inherent graph-based structure of molecules where atoms constitute nodes and bonds form edges. When integrated with Bayesian optimization (BO) frameworks, these encoding techniques enable accelerated materials discovery by constructing informative latent spaces that dramatically reduce the dimensionality and complexity of molecular design challenges.
Key Architectures for Molecular Representation
Graph Autoencoders for Molecular Generation
Autoencoders have proven effective as deep learning models that can function as both generative models and representation learning tools for downstream tasks. Specifically, graph autoencoders with encoder and decoder implemented as message-passing networks generate permutation-invariant graph representations—a critical property for handling molecular structures [24]. However, this approach faces significant challenges in decoding graph structures from single vectors and requires effective permutation-invariant similarity measures for comparing input and output graphs.
Recent innovations address these limitations through transformer-based message passing graph decoders. These architectures leverage global attention mechanisms to create more robust and expressive decoders compared to traditional graph neural network decoders [24]. The precision of graph matching during training has been shown to significantly impact model behavior and is essential for effective de novo molecular graph generation [24].
The Transformer Graph Variational Autoencoder (TGVAE) represents another architectural advancement that employs molecular graphs as direct input, capturing complex structural relationships more effectively than string-based models like Simplified Molecular Input Line Entry System (SMILES) [25]. This approach combines transformers, GNNs, and variational autoencoders (VAEs) to generate chemically valid and diverse molecular structures while addressing common training issues like over-smoothing in GNNs and posterior collapse in VAEs [26] [25].
Kolmogorov-Arnold Graph Neural Networks (KA-GNNs)
A novel framework called Kolmogorov-Arnold GNNs (KA-GNNs) integrates Kolmogorov-Arnold networks (KANs) into the three fundamental components of GNNs: node embedding, message passing, and readout [27]. This integration replaces conventional multi-layer perceptrons with learnable univariate functions on edges, enabling more accurate and interpretable modeling of complex molecular functions.
The KA-GNN architecture employs Fourier-series-based univariate functions within KANs to enhance function approximation capabilities. This approach effectively captures both low-frequency and high-frequency structural patterns in graphs, enhancing the expressiveness of feature embedding and message aggregation [27]. Theoretical analysis demonstrates that this Fourier-based KAN architecture possesses strong approximation capability for any square-integrable multivariate function, providing solid mathematical foundations for molecular property prediction [27].
Two architectural variants have been developed: KA-Graph Convolutional Networks (KA-GCN) and KA-Graph Attention Networks (KA-GAT). Experimental results across seven molecular benchmarks show that KA-GNNs consistently outperform conventional GNNs in prediction accuracy and computational efficiency while offering improved interpretability by highlighting chemically meaningful substructures [27].
Table 1: Key Architectural Innovations in Molecular Graph Representation
| Architecture | Core Innovation | Advantages | Applications |
|---|---|---|---|
| Transformer-based Graph Decoder [24] | Global attention mechanisms in decoding | Robustness, expressivity, improved graph matching | De novo molecular graph generation |
| TGVAE [26] [25] | Combines transformer, GNN, and VAE | Chemical validity, diversity, handles graph inputs | Drug discovery, molecular design |
| KA-GNN [27] | Integrates KAN modules into GNN components | Accuracy, parameter efficiency, interpretability | Molecular property prediction |
| DeeperGAT-VAE [26] | Lightweight, deep graph-attention blocks | Prevents over-smoothing, works on small datasets | Small-data molecular generation |
Experimental Protocols and Workflows
Protocol 1: Active Learning for Electrolyte Discovery
This protocol demonstrates how machine learning can explore massive chemical spaces with minimal initial data, achieving efficient molecular discovery through iterative experimentation [28].
Workflow Overview:
- Initialization: Begin with a small set of 58 experimentally validated data points covering diverse molecular structures.
- Model Training: Train an active learning model on available data points to predict electrolyte performance.
- Candidate Selection: Use the model to select promising electrolyte candidates from a virtual search space of 1 million potential molecules.
- Experimental Validation: Synthesize predicted high-performing electrolytes and test in actual battery systems, measuring cycle life and discharge capacity.
- Data Integration: Feed experimental results back into the model for refinement.
- Iteration: Repeat steps 2-5 through seven active learning campaigns, testing approximately 10 electrolytes per campaign.
- Final Selection: Identify top-performing electrolytes that rival state-of-the-art performance.
Key Considerations:
- Address model uncertainty through experimental verification at each cycle
- Focus on real-world performance metrics rather than computational proxies
- Overcome human bias toward previously studied chemical spaces
Protocol 2: KA-GNN Implementation for Property Prediction
This protocol details the implementation of Kolmogorov-Arnold Graph Neural Networks for molecular property prediction, combining the strengths of GNNs and KANs [27].
Workflow Overview:
- Data Preparation:
- Curate molecular datasets with annotated properties
- Represent molecules as graphs with node features (atomic number, radius) and edge features (bond type, length)
Model Configuration:
- Select KA-GCN or KA-GAT variant based on task requirements
- Initialize Fourier-based KAN layers with specified harmonic components
- Configure node embedding, message passing, and readout components with KAN modules
Training Procedure:
- Implement residual connections in KAN layers for stable training
- Use task-specific loss functions (e.g., mean squared error for regression, cross-entropy for classification)
- Employ adaptive optimization algorithms with gradient clipping
Interpretation and Analysis:
- Visualize learned KAN functions to identify important molecular features
- Analyze attention weights in KA-GAT for substructure importance
- Validate chemically meaningful patterns in latent representations
Implementation Details:
- Replace fixed activation functions with learnable Fourier series
- Initialize node embeddings by concatenating atomic features with neighborhood bond features
- Use residual KAN connections in message passing steps
- Implement graph-level readout through adaptive pooling and KAN transformations
Bayesian Optimization in Latent Chemical Space
Joint Composite Latent Space Bayesian Optimization (JoCo)
The JoCo framework addresses the challenge of optimizing high-dimensional composite functions common in molecular design, where ( f = g \circ h ), with ( h ) mapping to high-dimensional intermediate outputs [29]. Traditional Bayesian optimization struggles with high-dimensional spaces, but JoCo jointly trains neural network encoders and probabilistic models to adaptively compress both input and output spaces into manageable latent representations.
This approach enables effective BO on compressed representations, significantly outperforming state-of-the-art methods for high-dimensional problems with composite structure [29]. Applications include optimizing generative AI models with text prompts as inputs and complex outputs like images, molecular design problems, and aerodynamic design with high-dimensional output spaces of pressure and velocity fields.
Microstructure-Aware Bayesian Materials Design
A novel microstructure-sensitive BO framework enhances materials discovery efficiency by explicitly incorporating microstructural information as latent variables [3]. This approach moves beyond traditional chemistry-process-property relationships to establish comprehensive process-structure-property mappings.
The methodology employs active subspace methods for dimensionality reduction to identify influential microstructural features, reducing computational complexity while maintaining accuracy [3]. Case studies on Mg(2)Sn(x)Si(_{1-x}) thermoelectric materials demonstrate the framework's ability to accelerate convergence to optimal material configurations with fewer iterations.
Table 2: Bayesian Optimization Frameworks for Latent Space Exploration
| Framework | Core Approach | Dimensionality Handling | Application Scope |
|---|---|---|---|
| JoCo [29] | Joint encoding of inputs/outputs | Adaptive compression | High-dimensional composite functions |
| Microstructure-Aware BO [3] | Active subspace methods | Dimensionality reduction | Materials design with structural descriptors |
| Conformal Prediction [23] | Mondrian conformal predictors | Uncertainty-calibrated selection | Virtual screening of billion-molecule libraries |
Performance Benchmarks and Quantitative Analysis
Molecular Generation Performance
Recent advancements in graph-based autoencoders demonstrate significant improvements in generation metrics. The TGVAE and DeeperGAT-VAE models achieve high validity, uniqueness, diversity, and novelty while reproducing key drug-like property distributions [26]. The incorporation of SMILES pair-encoding rather than character-level tokens captures larger chemically relevant substructures, supporting generation of more diverse and novel molecules [26].
Evaluation against PubChem confirms that SMILES pair-encoding greatly expands the space of scaffolds and fragments unseen in public databases, significantly broadening accessible chemical space [26].
Virtual Screening Acceleration
Machine learning-guided docking screens enable rapid virtual screening of billion-compound libraries. The combination of conformal prediction with molecular docking achieves more than 1,000-fold reduction in computational cost compared to traditional structure-based virtual screening [23].
In application to G protein-coupled receptors (important drug targets), this approach successfully identified ligands with multi-target activity tailored for therapeutic effect [23]. The CatBoost classifier with Morgan2 fingerprints provided optimal balance between speed and accuracy, screening 3.5 billion compounds with high efficiency.
Table 3: Performance Benchmarks of Molecular Machine Learning Approaches
| Method | Dataset/Task | Key Metrics | Performance |
|---|---|---|---|
| KA-GNN [27] | 7 molecular benchmarks | Prediction accuracy | Consistent outperformance vs. conventional GNNs |
| Active Learning [28] | Electrolyte discovery (1M library) | Experimental validation | 4 new electrolytes rivaling state-of-the-art |
| ML-Guided Docking [23] | 3.5B compound library | Computational efficiency | 1000-fold cost reduction |
| Conformal Prediction [23] | 8 protein targets | Sensitivity/Precision | 0.87-0.88 sensitivity at ~10% library screening |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Computational Tools for Molecular Representation Learning
| Tool/Component | Function | Implementation Example |
|---|---|---|
| Graph Neural Networks | Learn molecular representations directly from graph structure | Message passing networks [24] |
| Kolmogorov-Arnold Networks | Learnable activation functions for improved expressivity | Fourier-based KAN layers [27] |
| Variational Autoencoders | Generative modeling and latent space learning | TGVAE, DeeperGAT-VAE [26] [25] |
| Active Learning Frameworks | Efficient exploration of chemical space with minimal data | Iterative experiment-model loops [28] |
| Conformal Prediction | Uncertainty-calibrated molecular screening | Mondrian conformal predictors [23] |
| Bayesian Optimization | Sample-efficient black-box optimization | JoCo framework [29] |
| Molecular Descriptors | Feature representation for machine learning | Morgan fingerprints, CDDD, RoBERTa embeddings [23] |
| Docking Software | Structure-based virtual screening | Molecular docking calculations [23] |
The integration of graph neural networks and autoencoders for molecular representation has created powerful frameworks for encoding chemical space into meaningful latent representations. Architectures such as KA-GNNs, transformer-based graph autoencoders, and deep graph variational autoencoders demonstrate significant advantages over traditional methods in terms of accuracy, efficiency, and interpretability. When combined with Bayesian optimization in latent spaces, these approaches enable accelerated discovery of novel materials and drug candidates by efficiently navigating vast chemical landscapes. The continued development of these methodologies, particularly with emphasis on uncertainty quantification, interpretability, and integration with experimental validation, promises to further transform materials research and drug discovery pipelines.
Molecular discovery within the vast chemical space remains a significant challenge due to the immense number of possible molecules and limited scalability of conventional screening methods [21] [30] [31]. The combinatorial complexity of atomic arrangements creates a search space too large for exhaustive exploration through traditional experimental or computational approaches. To address this challenge, researchers have developed multi-level Bayesian optimization (BO) with hierarchical coarse-graining, an active learning-based method that uses transferable coarse-grained models to compress chemical space into varying levels of resolution [31]. This approach effectively balances the competing demands of combinatorial complexity and chemical detail by employing a funnel-like strategy that progresses from low-resolution exploration to high-resolution exploitation.
Framed within the broader context of Bayesian optimization in latent space for materials research, this methodology represents a significant advancement for computational molecular discovery. By transforming discrete molecular spaces into smooth latent representations and performing Bayesian optimization within these spaces, the technique enables efficient navigation of chemical spaces for free energy-based molecular optimization [21]. The multi-level approach has demonstrated particular effectiveness in optimizing molecules to enhance phase separation in phospholipid bilayers, showcasing its potential for drug development and materials science applications [30] [31].
Theoretical Framework
Hierarchical Coarse-Graining of Chemical Space
Coarse-graining addresses the complexity of chemical space by grouping atoms into pseudo-particles or beads, effectively compressing the vast combinatorial possibilities into manageable representations [31]. This process consists of two fundamental steps: mapping groups of atoms to beads, and defining interactions between these beads based on underlying atomistic fragments. The resolution of coarse-graining can be varied through both steps, with lower resolutions assigning larger groups of atoms to single beads and employing fewer transferable bead types for interactions.
The hierarchical approach employs multiple coarse-grained (CG) models with varying resolutions, all using the same atom-to-bead mapping but differing in the assignment of transferable bead types [31]. Higher-resolution models feature more bead types, capturing finer chemical details while still reducing combinatorial complexity compared to the atomistic level. This reduction enables enumeration of all possible CG molecules corresponding to specific regions of chemical space at each resolution. Critically, the hierarchical model design allows systematic mapping of higher-resolution molecules to lower resolutions, creating an interconnected framework for navigating chemical space.
Table: Coarse-Graining Resolution Levels and Characteristics
| Resolution Level | Number of Bead Types | Chemical Detail | Combinatorial Complexity | Primary Function |
|---|---|---|---|---|
| Low Resolution | Fewer bead types | Limited structural information | Reduced complexity | Broad exploration |
| Medium Resolution | Moderate bead types | Balanced detail | Moderate complexity | Guided optimization |
| High Resolution | More bead types (e.g., 96 in Martini3) | Fine chemical details | Higher complexity | Detailed exploitation |
Latent Space Representation and Bayesian Optimization
The transformation of discrete molecular structures into continuous latent representations represents a crucial step in enabling efficient chemical space exploration [31]. This encoding is typically achieved through graph neural network (GNN)-based autoencoders, with each coarse-graining resolution encoded separately. The resulting smooth latent space ensures meaningful molecular similarity measures essential for subsequent Bayesian optimization, where molecules with similar properties are positioned close to each other in the latent representation.
Bayesian optimization operates within these latent spaces, using molecular dynamics simulations to calculate target free energies of coarse-grained compounds [31]. The multi-level approach effectively balances exploration and exploitation across resolutions, with lower resolutions facilitating broad exploration of chemical neighborhoods and higher resolutions enabling detailed optimization. This Bayesian framework provides an intuitive mechanism for combining information from different resolutions into the optimization process, relating to multi-fidelity BO approaches but utilizing varying coarse-graining complexities rather than different evaluation costs [31].
Experimental Protocols
Protocol 1: Establishing Hierarchical Coarse-Grained Models
Purpose: To define multiple coarse-grained models with varying resolutions for representing chemical space.
Materials and Reagents:
- Atomistic molecular structures
- Coarse-graining software (e.g., Martini3 [31])
- Computational resources for molecular dynamics simulations
Procedure:
- Define High-Resolution Model: Begin with establishing the high-resolution CG model, specifying available bead types based on relevant elements and chemical fragments from atomistic chemical space. The Martini3 CG model with 32 bead types per bead size (96 total, excluding water and divalent ions) serves as an appropriate starting point [31].
- Establish Medium and Low Resolutions: Create medium and low-resolution models using the same atom-to-bead mapping as the high-resolution model, but with progressively fewer bead types. This ensures systematic mapping between resolutions.
- Validate Transferability: Confirm that interaction parameters remain transferable across the chemical space of interest, enabling consistent force field application.
- Enumerate CG Molecules: For each resolution level, enumerate all possible CG molecules corresponding to the target region of chemical space, leveraging the reduced complexity at lower resolutions.
Troubleshooting Tips:
- If transferability validation fails, review bead type definitions and ensure consistent mapping conventions.
- For enumeration challenges at higher resolutions, consider constraining the chemical space or employing sampling rather than exhaustive enumeration.
Protocol 2: Latent Space Embedding of Coarse-Grained Structures
Purpose: To embed coarse-grained structures into a continuous latent space for Bayesian optimization.
Materials and Reagents:
- Enumerated CG structures from Protocol 1
- Graph neural network-based autoencoder framework
- Computational resources for deep learning
Procedure:
- Structure Representation: Represent each CG molecule as a graph where nodes correspond to beads and edges represent connections.
- Encoder Training: Train separate GNN-based encoders for each resolution level to map CG structures to latent vectors. Use reconstruction loss to ensure meaningful representations.
- Latent Space Validation: Verify that the latent space preserves chemical similarity by measuring distances between related molecular structures.
- Smoothness Assessment: Confirm the latent space provides smooth transitions between molecular structures, enabling effective Bayesian optimization.
Troubleshooting Tips:
- If reconstruction accuracy is poor, increase encoder capacity or adjust hyperparameters.
- For inadequate chemical similarity preservation, consider incorporating metric learning approaches into the training objective.
Protocol 3: Multi-Level Bayesian Optimization for Molecular Discovery
Purpose: To identify optimal molecular compounds through multi-level Bayesian optimization.
Materials and Reagents:
- Latent space representations from Protocol 2
- Molecular dynamics simulation software
- Free energy calculation methods (e.g., thermodynamic integration)
- Bayesian optimization framework
Procedure:
- Initial Sampling: Perform initial sampling across all resolution levels to build preliminary Gaussian process models.
- Lower-Resolution Exploration: Focus on lower resolutions for broad exploration of chemical space, using acquisition functions (e.g., Expected Improvement) to select promising candidates for evaluation.
- Information Transfer: Transfer neighborhood information from lower resolutions to guide optimization at higher resolutions.
- Higher-Resolution Exploitation: Concentrate computational resources on promising regions identified at lower resolutions, using higher-resolution models for detailed optimization.
- Target Calculation: For each candidate molecule selected by the Bayesian optimization, calculate target properties (e.g., free energy differences for phase separation enhancement) using molecular dynamics simulations.
- Iterative Refinement: Update Gaussian process models with new data and iterate the optimization process until convergence or resource exhaustion.
Troubleshooting Tips:
- If optimization stagnates, adjust the balance between exploration and exploitation parameters.
- For excessive computational costs at higher resolutions, implement early stopping criteria for unpromising candidates.
Research Reagent Solutions
Table: Essential Computational Tools and Resources
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Martini3 Coarse-Grained Model [31] | Provides high-resolution CG force field | Use as starting point; 32 bead types per bead size |
| Graph Neural Network Autoencoder | Creates latent space representations | Train separately for each resolution level |
| Molecular Dynamics Software | Calculates target free energies | Use for phase separation assessment |
| Gaussian Process Model | Surrogate for objective function | Models structure-property relationships |
| Bayesian Optimization Framework | Guides molecular selection | Implements acquisition functions across resolutions |
| Thermodynamic Integration | Computes free energy differences | Quantifies phase separation behavior |
Visualization of Workflows
Multi-Level Bayesian Optimization Workflow
Workflow Overview: This diagram illustrates the complete multi-level Bayesian optimization process with hierarchical coarse-graining. The workflow begins with establishing multiple coarse-grained models at different resolutions, which are then encoded into latent representations. Bayesian optimization progresses from low-resolution exploration to high-resolution exploitation, with molecular dynamics simulations providing target property calculations at each stage. The iterative process continues until convergence, identified optimal molecules with enhanced target properties.
Chemical Space Embedding and Navigation
Chemical Space Compression: This visualization depicts the transformation of complex atomistic chemical space into manageable coarse-grained representations and subsequent continuous latent spaces. The hierarchical coarse-graining compresses chemical information at multiple resolution levels, while the autoencoder creates smooth latent representations that enable effective Bayesian optimization. The process demonstrates how discrete molecular structures become navigable through continuous optimization techniques.
Application Notes
Case Study: Enhancing Phase Separation in Phospholipid Bilayers
The multi-level Bayesian optimization approach with hierarchical coarse-graining has been successfully demonstrated by optimizing small molecules to enhance phase separation in ternary lipid bilayers [31]. This application exemplifies the methodology's effectiveness for molecular design problems where target properties can be expressed as free-energy differences.
Implementation Details:
- Objective Function: Free-energy difference quantifying phase separation behavior of molecules inserted into ternary lipid bilayers
- Coarse-Graining Resolution: Three levels sharing the same atom-to-bead mapping but differing in available bead types
- Evaluation Method: Molecular dynamics simulation-based free-energy calculations
- Optimization Metric: Phase separation enhancement capability of designed molecules
Performance Outcomes:
- The multi-level approach effectively identified relevant chemical neighborhoods
- The method outperformed standard Bayesian optimization applied at a single resolution level
- The funnel-like strategy provided both optimal compounds and insight into relevant chemical space neighborhoods
Guidelines for Method Selection
When to Use Multi-Level Bayesian Optimization with Hierarchical Coarse-Graining:
- Molecular design problems with vast chemical spaces requiring efficient navigation
- Optimization targets that can be expressed as free-energy differences or computed via molecular dynamics
- Scenarios with sufficient computational resources for multi-resolution evaluation
- Problems where understanding chemical neighborhood relationships provides additional value
Alternative Approaches:
- Single-Level Bayesian Optimization: Suitable for less complex chemical spaces or when computational resources are limited
- Generative Methods: Appropriate when substantial training data exists for the specific molecular design domain
- High-Throughput Screening: Viable when evaluation costs are low and chemical space is constrained
Multi-level Bayesian optimization with hierarchical coarse-graining represents a significant advancement in computational molecular discovery, effectively addressing the fundamental challenge of navigating vast chemical spaces. By integrating transferable coarse-grained models at multiple resolutions with latent space embeddings and Bayesian optimization, this approach balances combinatorial complexity with chemical detail in a manner superior to single-fidelity methods.
The methodology's demonstration in enhancing phase separation in phospholipid bilayers underscores its potential for drug development and materials science applications, particularly for optimization targets expressible as free-energy differences. The provided protocols, reagent specifications, and visualization frameworks offer researchers practical resources for implementing this approach in their molecular discovery pipelines.
As Bayesian optimization in latent space continues to evolve within materials research, hierarchical coarse-graining stands as a powerful strategy for accelerating the discovery of novel compounds with tailored properties, potentially transforming development timelines across pharmaceutical and materials science domains.
Bayesian optimization (BO) is a powerful strategy for efficiently optimizing expensive black-box functions, making it particularly valuable in materials science and drug discovery where experiments or simulations are costly and time-consuming. Traditional BO approaches typically focus on finding the global maximum or minimum of an objective function. However, many scientific applications require identifying materials or compounds with a specific target property value rather than an extreme value. For instance, a catalyst might exhibit peak activity when an adsorption free energy is zero, or a shape memory alloy may need a precise phase transformation temperature for a specific application [7].
Target-Oriented Bayesian Optimization (t-EGO) addresses this critical need. It is a specialized BO variant designed to minimize the number of experimental iterations required to find a material or molecule whose properties hit a predefined target value. By strategically sampling candidates that allow properties to approach the target from either above or below, t-EGO offers a more efficient pathway for designing materials with predefined specific properties, significantly accelerating research and development cycles [7].
Core Methodology of t-EGO
The fundamental innovation of t-EGO lies in its novel acquisition function, the target-specific Expected Improvement (t-EI). This function directly guides the search toward the target value, in contrast to standard Expected Improvement (EI), which seeks improvement over a current best value (like a minimum or maximum) [7].
The t-EI Acquisition Function
The t-EI acquisition function is mathematically defined to maximize the expected reduction in the deviation from the target property. For a target value ( t ), and the closest observed value in the training dataset ( y{t.min} ), the improvement at a point ( \mathbf{x} ) is defined as the reduction in the absolute difference from the target, provided the new value is closer. Formally, t-EI is expressed as: [ t\text{-}EI = \mathbb{E}[\max(0, |y{t.min} - t| - |Y - t|)] ] where ( Y ) is the random variable representing the predicted property value at point ( \mathbf{x} ), typically modeled as normally distributed ( Y \sim \mathcal{N}(\mu, s^2) ) with mean ( \mu ) and variance ( s^2 ) from a Gaussian Process (GP) surrogate model [7].
This formulation ensures that the algorithm prioritizes candidate points where the predicted property value is expected to fall closest to the target ( t ), actively balancing the exploration of uncertain regions with the exploitation of areas likely to hit the target.
Comparative Analysis of Acquisition Functions
The table below summarizes how t-EI compares to other common acquisition functions used in Bayesian optimization.
Table 1: Comparison of Acquisition Functions in Bayesian Optimization
| Acquisition Function | Core Objective | Key Mathematical Formulation | Best Suited For | ||||
|---|---|---|---|---|---|---|---|
| t-EI (Target-oriented) | Minimize deviation from a specific target value ( t ) | ( \mathbb{E}[\max(0, | y_{t.min} - t | - | Y - t | )] ) | Finding materials with a precise property value |
| EI (Expected Improvement) | Find the global minimum | ( \mathbb{E}[\max(0, y_{min} - Y)] ) | Performance maximization or cost minimization | ||||
| PI (Probability of Improvement) | Maximize probability of exceeding current best | ( \mathbb{P}(Y > f^*) ) | Simple optimization, can get stuck in local optima | ||||
| UCB (Upper Confidence Bound) | Balance mean performance and uncertainty | ( \mu(\mathbf{x}) + \kappa \sigma(\mathbf{x}) ) | Exploration-focused tasks |
Quantitative Performance and Applications
Efficiency Benchmarks
The performance of t-EGO has been rigorously tested against other BO strategies, including standard EGO, Multi-Objective Acquisition Functions (MOAF), and Pure Exploitation (PureExp). Statistical results from hundreds of repeated trials on synthetic functions and materials databases demonstrate t-EGO's superior efficiency [7].
Table 2: Performance Comparison of t-EGO Against Other BO Methods
| Optimization Method | Relative Experimental Iterations to Reach Target | Key Characteristic | Performance in Small Data Regime | ||
|---|---|---|---|---|---|
| t-EGO | 1.0x (Baseline) | Directly minimizes distance to target | Excellent, highly data-efficient | ||
| EGO / MOAF | 1.5x - 2.0x more | Reformulates problem as minimization of | y-t | Less efficient, suboptimal sampling | |
| Constrained EGO (CEGO) | Variable, typically higher | Uses constrained EI | Performance depends on constraint handling | ||
| Pure Exploitation (PureExp) | Highest, prone to failure | Uses model mean prediction only, ignores uncertainty | Poor, high risk of stalling in wrong region |
These benchmarks show that t-EGO can require approximately 1 to 2 times fewer experimental iterations than EGO or MOAF strategies to reach the same target, an advantage that is especially pronounced when the initial training dataset is small [7].
Application in Materials Design: A Case Study
A notable application of t-EGO was the discovery of a thermally-responsive shape memory alloy for use as a thermostatic valve material. The target was a specific phase transformation temperature of 440 °C [7].
- Objective: Identify a SMA composition with a transformation temperature as close as possible to 440°C.
- Implementation: The t-EGO framework was employed, using a Gaussian Process to model the relationship between alloy composition and transformation temperature. The t-EI acquisition function guided the selection of which alloy composition to synthesize and test next.
- Outcome: In only 3 experimental iterations, the alloy
Ti₀.₂₀Ni₀.₃₆Cu₀.₁₂Hf₀.₂₄Zr₀.₀₈was identified. Its measured transformation temperature was 437.34 °C, achieving a remarkable difference of only 2.66 °C (0.58% of the search range) from the target [7]. This success underscores t-EGO's practical utility in achieving precise property targets with minimal experimental overhead.
Experimental Protocol: Implementing t-EGO for Materials Design
This protocol details the steps for applying t-EGO to discover a material with a target property, using the shape memory alloy (SMA) case study as a reference.
Pre-Experiment Planning
- Define Design Space and Target: Establish the bounds of your search space (e.g., compositional ranges for alloying elements) and the precise target property value ( t ) (e.g., 440 °C transformation temperature).
- Select Initial Training Data: Acquire a small initial dataset. This can be from historical data, literature, or a small set of purpose-designed experiments (e.g., 5-10 samples selected via Latin Hypercube Sampling).
- Configure Bayesian Optimization Model: Choose a Gaussian Process surrogate model with a suitable kernel (e.g., Matérn 5/2). Set the t-EI acquisition function as the core of the optimization loop.
Step-by-Step Procedure
- Characterize Initial Samples: Synthesize and characterize the initial set of alloy compositions from the training data. Measure the property of interest (transformation temperature) for each using Differential Scanning Calorimetry (DSC).
- Train the Surrogate Model: Use the accumulated dataset (composition, property) to train the GP model. The model will learn to predict the transformation temperature ( \mu(\mathbf{x}) ) and its uncertainty ( s(\mathbf{x}) ) for any composition within the design space.
- Optimize t-EI and Propose Next Experiment: Calculate the t-EI acquisition function over the entire design space. The composition ( \mathbf{x} ) that maximizes t-EI is the one predicted to most likely bring the property closer to the target, weighted by uncertainty.
- Execute Experiment and Validate: Synthesize and characterize the top candidate composition proposed by t-EI using the same protocols (e.g., arc-melting, homogenization, DSC).
- Update Dataset and Model: Add the new (composition, property) data point to the training dataset.
- Check Stopping Criterion: Repeat steps 2-5 until a candidate meets the target property within a specified tolerance (e.g., ±5 °C) or until the experimental budget is exhausted.
- Report Final Candidate: Once the loop is terminated, report the best-performing material composition and its properties.
Figure 1: The t-EGO experimental workflow for iterative materials design.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key computational and experimental resources essential for implementing a t-EGO campaign in a materials science context.
Table 3: Essential Tools and Resources for t-EGO-driven Materials Research
| Tool / Resource | Type | Primary Function in t-EGO Workflow |
|---|---|---|
| Gaussian Process Regression Model | Computational Model | Serves as the surrogate model to predict material properties and uncertainties from design variables. |
| t-EI Acquisition Function | Algorithm | The core component that calculates the expected utility of evaluating a candidate to reach the target value. |
| Bayesian Optimization Library | Software | Provides the computational backbone (e.g., BoTorch, GPyOpt) for implementing the BO loop. |
| High-Throughput Synthesis | Experimental Platform | Enables rapid preparation of material samples (e.g., alloy compositions) proposed by the algorithm. |
| Differential Scanning Calorimetry | Characterization Tool | Measures key thermal properties like phase transformation temperatures for validation. |
| Density Functional Theory | Computational Simulation | Can be used as a high-fidelity, albeit expensive, "experiment" within the loop to compute properties. |
Integration with Latent Space Bayesian Optimization
The t-EGO methodology is highly compatible with and can be enhanced by Bayesian optimization in latent spaces, a key approach for handling structured and complex materials like molecules and microstructures.
Optimization in Generative Model Latent Spaces
For designing complex molecules, the direct optimization in the high-dimensional and discrete molecular space is challenging. Latent Space Bayesian Optimization (LSBO) uses a generative model, such as a Variational Autoencoder (VAE), to map discrete structures into a continuous, low-dimensional latent space [32]. Standard BO, including t-EGO, can then be performed in this latent space. However, a known challenge in LSBO is the value discrepancy problem, where the reconstruction gap between the latent and input spaces leads to suboptimal performance [33]. Recent advances, such as using Normalizing Flows which provide one-to-one mapping between input and latent space, aim to resolve this issue and can be integrated with t-EGO for more robust molecular optimization [33].
Incorporating Physical and Microstructural Knowledge
t-EGO can be advanced beyond a pure black-box approach by incorporating domain knowledge. Physics-informed BO integrates known physical laws into the GP, for example, by using physics-infused kernels or an augmented mean function, transforming the problem into a more efficient "gray-box" optimization [34]. Furthermore, microstructure-aware Bayesian optimization explicitly includes microstructural descriptors (e.g., grain size, phase distribution) as latent variables in the model [3]. This creates a more informed mapping from composition/processing to final properties, potentially improving the efficiency of finding a material with a target property by leveraging the fundamental Process-Structure-Property (PSP) linkages.
Figure 2: t-EGO integrated with latent space and physical knowledge for enhanced materials design.
This application note details a case study on the use of a multi-level Bayesian optimization (BO) framework to efficiently discover small molecules that enhance phase separation in phospholipid bilayers. The protocol demonstrates how performing BO within hierarchically coarse-grained (CG) latent spaces accelerates navigation of vast chemical spaces (CS) to identify candidates that promote a target free-energy difference. By integrating reduced complexity at lower resolutions with chemical detail at higher resolutions, the outlined methodology achieved superior performance compared to standard single-level BO, successfully identifying an optimal molecule within a constrained experimental budget [35].
The discovery of novel molecules that modulate lipid bilayer properties holds significant promise for therapeutic and materials science applications. A key challenge in this domain is the immense size of chemical space, which makes exhaustive screening computationally intractable [35]. Bayesian optimization has emerged as a powerful, data-efficient strategy for guiding molecular design, particularly when integrated within active learning loops [3] [7]. This case study focuses on a specific application: optimizing a small molecule to promote phase separation in a ternary lipid bilayer, a process linked to the formation of lipid rafts and other membrane domains [36] [35].
The traditional trial-and-error approach to material development is increasingly inadequate for meeting the accelerated pace of innovation [3]. This protocol leverages a multi-level Bayesian optimization framework that utilizes transferable coarse-grained models to compress chemical space into varying levels of resolution. This funnel-like strategy balances combinatorial complexity and chemical detail, enabling efficient exploration and identification of target compounds [35]. The property of interest is quantified as a free-energy difference, calculated from molecular dynamics (MD) simulations, which characterizes the molecule's efficacy in promoting phase separation [35].
Methodologies & Workflow
Multi-Level Bayesian Optimization Framework
The core of this protocol is a multi-level BO framework that integrates information from multiple coarse-grained representations of chemical space.
The following diagram illustrates the integrated, multi-stage workflow for molecule optimization.
Key Computational Components
The optimization relies on several key computational components working in concert.
Table 1: Key Components of the Multi-Level BO Framework
| Component | Description | Function in Workflow |
|---|---|---|
| Coarse-Grained (CG) Models | Molecular representations where atom groups are mapped to pseudo-particles ("beads") [35]. | Reduces combinatorial complexity of chemical space; multiple resolutions (Low: 15, Med: 45, High: 96 bead types) are defined hierarchically [35]. |
| Chemical Space (CS) Encoding | Graph Neural Network (GNN)-based autoencoder transforms discrete molecular graphs into continuous latent representations [35]. | Enables meaningful similarity measures between molecules, a prerequisite for effective Bayesian optimization [35]. |
| Surrogate Model | Gaussian Process (GP) models the relationship between a molecule's latent representation and its target property [3]. | Provides a probabilistic prediction of molecule performance and quantifies associated uncertainty to guide the search [3] [35]. |
| Acquisition Function | Target-specific Expected Improvement (t-EI) recommends the most informative candidate for the next evaluation [7]. | Balances exploration (testing uncertain regions) and exploitation (testing promising regions) to find molecules with a specific target property [7] [35]. |
Experimental Protocol: MD Simulation for Free Energy Calculation
The following protocol details the steps for evaluating a candidate molecule's ability to promote phase separation, which serves as the objective function for the BO.
Subject Area: Cell biology, Molecular biology, Biophysics [37]. Experimental Goal: To quantify the phase separation propensity of a candidate molecule inserted into a model lipid bilayer via molecular dynamics simulation.
Materials and Reagents
Table 2: Research Reagent Solutions for MD Simulations
| Reagent / Material | Function / Description | Notes |
|---|---|---|
| Coarse-Grained Candidate Molecule | The molecule being optimized; structure is defined by its CG bead types and bonded parameters [35]. | Topology and parameters are defined by the Martini3 force field and the hierarchical mapping scheme [35]. |
| Ternary Lipid Bilayer | A model membrane; e.g., comprising DOPC, DPPC, and Cholesterol, which can exhibit liquid-ordered (L₀) and liquid-disordered (L𝑑) phase coexistence [36]. | Serves as the model system to test the molecule's effect on phase behavior. Lipid composition can be adjusted. |
| Martini3 Force Field | A versatile, transferable coarse-grained force field used to simulate molecular interactions [35]. | Provides parameters for bonded and non-bonded interactions between CG beads. Key for achieving chemical accuracy. |
| Molecular Dynamics Software | Software such as GROMACS [38] or LAMMPS for performing the simulations. | Handles the numerical integration of equations of motion for the molecular system. |
| Free Energy Calculation Method | A computational method, such as thermodynamic integration or free-energy perturbation, to compute the free-energy difference (ΔG) [35]. | ΔG of insertion quantifies the molecule's preference for a specific membrane phase, thus measuring its propensity to enhance phase separation [35]. |
Step-by-Step Procedure
System Setup a. Insert Molecule: Place the CG candidate molecule into a pre-equilibrated ternary lipid bilayer, ensuring it is embedded within the lipid tail region. Solvate the entire system with CG water. b. Neutralize System: Add an appropriate number of ions to neutralize the system's net charge.
Energy Minimization a. Run an energy minimization step (e.g., using the steepest descent algorithm) to remove any steric clashes and high-energy contacts in the initial configuration.
System Equilibration a. Perform equilibration simulations in the NPT ensemble (constant Number of particles, Pressure, and Temperature). b. Apply position restraints on the lipid and molecule beads initially, gradually releasing them over the course of the equilibration phase to allow the system to relax.
Production Simulation a. Run a production MD simulation for a sufficient duration (e.g., ≥1 µs in CG time) to observe phase behavior and collect adequate statistics for free-energy analysis. Ensure the simulation is conducted in the NPT ensemble.
Free Energy Analysis a. Using the trajectory from the production run, compute the free-energy difference (ΔG) associated with moving the candidate molecule between different membrane environments (e.g., from the L𝑑 to the L₀ phase) [35]. b. This calculated ΔG value is the key performance metric fed back into the Bayesian optimization loop.
Results & Data Analysis
The multi-level BO framework was validated by optimizing a small molecule (up to 4 CG beads) to enhance phase separation in a ternary lipid bilayer.
Performance Comparison of Optimization Strategies
The multi-level approach was benchmarked against a standard, single-level BO.
Table 3: Performance Comparison of Multi-Level vs. Single-Level BO
| Optimization Strategy | Key Principle | Performance Outcome |
|---|---|---|
| Multi-Level BO (Proposed) | Integrates surrogate models from multiple CG resolutions. Optimization shifts from lower (for exploration) to higher (for exploitation) resolutions [35]. | Outperformed standard BO, efficiently identifying relevant chemical neighborhoods and the optimal molecule. Demonstrated superior data efficiency [35]. |
| Standard Single-Level BO | Conducts optimization using a single, fixed chemical space representation (e.g., only the high-resolution Martini3 model) [35]. | Less efficient at navigating the vast chemical space compared to the multi-level approach. Required more iterations to find the optimal candidate [35]. |
Logical Workflow of the Multi-Level Strategy
The efficiency of the multi-level strategy stems from its hierarchical, funnel-like exploration of chemical space.
Discussion
This case study demonstrates that a multi-level Bayesian optimization strategy, which leverages hierarchically coarse-grained latent spaces, is a powerful and efficient method for optimizing molecular structures towards a specific biophysical property. The success of this approach hinges on several key factors.
First, the use of transferable coarse-grained models, such as Martini3, is critical for compressing the immense chemical space into a tractable size while retaining essential chemical information [35]. This compression allows for the enumeration of molecules and the creation of a smooth, encoded latent space necessary for the Gaussian process model. Second, the multi-fidelity nature of the information flow—where lower-resolution explorations guide higher-resolution optimizations—mimics a scientific discovery process, first identifying promising regions broadly before focusing on the most precise details [35]. This is a more efficient use of computational resources than single-level optimization. Finally, the choice of a target-oriented acquisition function (t-EI) is particularly suited for problems where the goal is not simply to maximize or minimize a property, but to find a molecule with a property as close as possible to a specific target value [7].
This application note has provided a detailed protocol for using multi-level Bayesian optimization to design molecules that modulate lipid bilayer phase separation. The outlined methodology successfully integrates hierarchical coarse-graining, latent space encoding, and target-oriented Bayesian optimization to navigate chemical space with high data efficiency. The provided workflow, from defining CG models to running and analyzing MD simulations, offers a practical guide for researchers aiming to apply similar strategies to other free-energy-based molecular optimization challenges in materials science and drug development.
The design of Shape Memory Alloys (SMAs) with specific transformation temperatures remains a formidable challenge in materials science, particularly for high-temperature applications in aerospace, biomedical devices, and solid-state actuation. Traditional Edisonian approaches are often inefficient in navigating the vast, high-dimensional compositional spaces of multi-component alloy systems. This case study examines the successful application of Bayesian optimization (BO) in latent space to discover SMAs with transformation temperatures precisely tuned to a predefined target. We detail the methodology, experimental protocols, and results of a campaign that identified a novel NiTi-based SMA with a transformation temperature within 2.66 °C of the target, demonstrating the power of this data-driven framework for accelerated materials discovery [7].
Methodological Framework: Bayesian Optimization in Latent Space
The core of this case study involves two advanced BO strategies that operate within learned latent representations of the materials' compositional space, moving beyond simple parameter tuning to a more fundamental design approach.
Target-Oriented Bayesian Optimization (t-EGO)
A key innovation for achieving target-specific properties is the t-EGO (target-oriented Efficient Global Optimization) algorithm [7]. Unlike standard BO that seeks minima or maxima, t-EGO is explicitly designed to converge on a specific target value.
Acquisition Function: The algorithm uses a target-specific Expected Improvement (t-EI) acquisition function. For a target property value ( t ), and the current closest value ( y{t.min} ), t-EI is defined as: ( t-EI = E[ max(0, |y{t.min} - t| - |Y - t|) ] ) where ( Y ) is the predicted property value for a candidate material. This function directly rewards candidates whose predicted properties are closer to the target than the current best candidate [7].
Advantage: This formulation allows the algorithm to sample from both above and below the target value, more efficiently narrowing in on the desired property. In the featured case, t-EGO required fewer experimental iterations than conventional extremum-seeking BO methods to reach the same target [7].
Generative Inversion for Inverse Design
Another powerful latent space strategy is the Generative Adversarial Network (GAN) inversion framework [39]. This method performs BO within the latent space of a pre-trained generative model.
Framework Components: The framework integrates a generator (( G )) that maps a latent vector ( \mathbf{z} ) to a material design (composition and processing parameters), and a surrogate predictor (( f )) that maps the design to predicted properties [39].
Latent Space Optimization: Given a target property ( \mathbf{y}t ), gradient-based optimization is performed in the latent space to find a vector ( \mathbf{z}^* ) that minimizes the loss ( \mathcal{L} = \| f(G(\mathbf{z})) - \mathbf{y}t \|^2 ). The output is a novel, realistic material design ( \mathbf{x}^* = G(\mathbf{z}^*) ) predicted to exhibit the target property [39].
This approach effectively inverts the design process, moving directly from a property target to an optimal material composition.
The following workflow diagram illustrates the iterative Bayesian optimization process in latent space for target-oriented materials discovery:
Experimental Protocols and Workflow
This section outlines the standard protocols for the computational and experimental steps involved in the BO-driven discovery of SMAs.
Computational Design Protocol
Objective: To computationally suggest the most promising alloy candidate for the next experimental iteration.
Step 1: Dataset Curation Collect and clean historical data on SMA compositions and their corresponding transformation temperatures. The dataset used in the featured t-EGO study was constructed from literature and contained 82 data points for the martensitic transformation temperature (TM) across various SMA families [40] [7].
Step 2: Feature Engineering Calculate features for each alloy composition. These can be elemental properties (e.g., atomic radius, electronegativity, valence electron concentration) or physical properties of the elements (e.g., melting point, density). The t-EGO study used a set of 15 physical features obtained through weighted averaging of the constituent elements' properties [41].
Step 3: Surrogate Model Training Train a surrogate model (e.g., Gaussian Process (GP), Random Forest, Deep Gaussian Process (DGP)) on the curated dataset to map features or latent representations to the transformation temperature. The t-EGO study employed a GP, while other advanced frameworks utilized DGP or Multi-Task GP (MTGP) to model correlations between multiple target properties [7] [42] [43].
Step 4: Bayesian Optimization Loop
- Prediction & Uncertainty Quantification: Use the trained surrogate model to predict the mean (( \mu )) and standard deviation (( s )) of the transformation temperature for all candidates in the design space.
- Candidate Selection: Apply the t-EI acquisition function to identify the single or batch of candidates that maximizes the expected improvement towards the target temperature.
- Recommendation: Output the top candidate(s) for experimental synthesis and validation [7].
Experimental Synthesis and Characterization Protocol
Objective: To synthesize and characterize the suggested alloy, providing accurate data for the BO feedback loop.
Step 1: Alloy Synthesis
- Weighing: Weigh high-purity (typically >99.9%) raw elements according to the calculated atomic percentages (at.%) [39].
- Arc Melting: Synthesize the alloy buttons using a vacuum arc melter. Melt the constituents under an inert argon atmosphere. Flip and re-melt the buttons several times (e.g., 5 times) to ensure chemical homogeneity [39].
- Homogenization: Seal the alloy buttons in evacuated quartz tubes and perform a solution heat treatment (homogenization). A representative protocol is annealing at 1050 °C for 72 hours, followed by quenching in ice water [39].
Step 2: Microstructural and Phase Analysis
Step 3: Transformation Temperature Measurement
- Differential Scanning Calorimetry (DSC): This is the standard technique for measuring transformation temperatures.
- Prepare a small sample (e.g., 20-50 mg) from the homogenized alloy.
- Load the sample into the DSC apparatus.
- Run controlled heating and cooling cycles (e.g., between 0 °C and 500 °C at a rate of 10 °C/min) under a nitrogen purge.
- Determine the characteristic transformation temperatures (Martensite Start ( Ms ), Martensite Finish ( Mf ), Austenite Start ( As ), Austenite Finish ( Af )) from the DSC thermogram using the tangent method. The transformation temperature (TM) is often taken as the peak temperature of the endothermic or exothermic reaction [7] [44].
- Electrical Resistivity (Optional): Measure electrical resistance as a function of temperature. The martensitic transformation is marked by a distinct change in resistance, providing complementary data on transformation behavior and hysteresis [44].
- Differential Scanning Calorimetry (DSC): This is the standard technique for measuring transformation temperatures.
Key Findings and Data Analysis
The application of the aforementioned protocols in a target-oriented BO campaign yielded the following quantitative results.
Table 1: Composition and Target Property Results of the Discovered SMA [7]
| Alloy System | Composition (at.%) | Target Transformation Temperature | Achieved Transformation Temperature | Deviation from Target |
|---|---|---|---|---|
| Ni-Ti-Cu-Hf-Zr | Ti0.20Ni0.36Cu0.12Hf0.24Zr0.08 | 440 °C | 437.34 °C | 2.66 °C |
Table 2: Performance Comparison of Bayesian Optimization Variants for Multi-Objective Materials Discovery [42] [43]
| BO Method | Surrogate Model | Key Features | Reported Advantages |
|---|---|---|---|
| cGP-BO | Conventional Gaussian Process | Models each property independently | Baseline method |
| MTGP-BO | Multi-Task Gaussian Process | Models correlations between multiple output properties | Improved prediction quality by sharing information across correlated tasks [42] |
| hDGP-BO | Hierarchical Deep Gaussian Process | Stacks multiple GP layers; captures complex, non-linear relationships | Most robust and efficient performance; accelerates discovery by exploiting property correlations [42] [43] |
| t-EGO | Gaussian Process | Uses target-specific acquisition function (t-EI) | High efficiency in finding materials with specific target properties, not just optima [7] |
The Scientist's Toolkit: Research Reagent Solutions
This table lists the essential materials, equipment, and software central to conducting this type of research.
Table 3: Essential Reagents, Equipment, and Computational Tools for SMA Discovery
| Category | Item | Specification / Function |
|---|---|---|
| Raw Materials | High-Purity Elements (Ni, Ti, Hf, Zr, Cu, etc.) | Purity >99.9% to ensure accurate composition and avoid impurity effects [39]. |
| Synthesis Equipment | Vacuum Arc Melter | Creates an inert atmosphere for melting and alloying constituent elements without oxidation [39]. |
| Tube Furnace & Quartz Tubes | Used for homogenization heat treatments to achieve a uniform chemical microstructure [39]. | |
| Characterization Equipment | Differential Scanning Calorimeter (DSC) | Measures the heat flow associated with phase transformations to determine critical temperatures [7] [44]. |
| X-ray Diffractometer (XRD) | Identifies the crystal structure and phases present in the synthesized alloy [39] [44]. | |
| Computational Tools | Gaussian Process (GP) / Deep GP Software | Serves as the surrogate model for predicting properties and quantifying uncertainty (e.g., in Python with GPyTorch or scikit-learn) [7] [42] [43]. |
| Bayesian Optimization Framework | Implements the optimization loop and acquisition functions (e.g., BoTorch, GPyOpt) [7]. |
This case study demonstrates that Bayesian optimization in latent space is a powerful and efficient paradigm for the inverse design of functional materials. The success of the t-EGO algorithm in discovering a Ni-Ti-Cu-Hf-Zr shape memory alloy with a transformation temperature precisely tuned to 440 °C underscores the potential of target-oriented BO to drastically reduce the number of expensive experimental iterations required. Furthermore, advanced surrogate models like Deep Gaussian Processes show promise in handling the complexity and multi-objective nature of real-world materials design problems. These data-driven strategies represent a significant leap forward from traditional methods, enabling a more rational and accelerated path to designing materials with pre-defined properties.
Overcoming Practical Challenges and Optimizing Performance
Bayesian Optimization (BO) is a powerful, sample-efficient strategy for global optimization of expensive black-box functions. However, its application in high-dimensional spaces, such as those encountered in materials science and drug development, presents significant challenges. The "curse of dimensionality" causes performance to degrade sharply as dimensions increase, leading to sparse data, poor surrogate model scalability, and acquisition functions that struggle to balance exploration and exploitation [11]. Furthermore, the integration of valuable expert knowledge, while potentially beneficial, can inadvertently introduce pitfalls that hinder the optimization process if not properly calibrated. This application note examines these common pitfalls and outlines validated protocols to mitigate them, specifically within the context of Latent-Space Bayesian Optimization (LSBO) for materials research.
The High-Dimensionality Challenge & The Latent Space Solution
A principal challenge in high-dimensional BO is the exponential growth of the search space volume with dimensionality (d). This sparsity of data drastically reduces surrogate model accuracy, while the computational complexity of Gaussian Processes (GPs) scales poorly as (O(t^3)) with the number of observations (t) [11]. The kernel function also becomes less discriminative in high dimensions, and the acquisition function's landscape turns highly multimodal, complicating the search for the global optimum [11].
Latent-Space Bayesian Optimization (LSBO) has emerged as a promising solution. This approach uses dimensionality reduction (DR) techniques to map the high-dimensional input space (\mathbb{X} \subseteq \mathbb{R}^d) to a low-dimensional latent space (\mathbb{Z} \subseteq \mathbb{R}^{d'}) where (d' \ll d), and performs the core BO routine within this compressed representation [45] [11]. The following table summarizes and compares the two primary DR mechanisms used in LSBO.
Table 1: Comparison of Dimensionality Reduction Mechanisms for Bayesian Optimization
| Mechanism | Description | Key Assumption | Advantages | Limitations |
|---|---|---|---|---|
| Random Embeddings (e.g., REMBO [45]) | Uses a random linear projection matrix (A \in \mathbb{R}^{D \times d}) to map points from a low-dimensional space (\mathcal{Y}) to the high-dimensional space (\mathcal{X}) via (x = p_{\mathcal{X}}(Ay)). | The objective function has a low effective dimension (d \ll D) [45] [46]. | Simple to implement; theoretical guarantees when effective dimension is known [45]. | Projection can map points outside feasible bounds; performance drops if effective dimension is mis-specified or subspace is not axis-aligned [45] [46]. |
| Variational Autoencoders (VAEs) [45] [47] [11] | Uses a neural network-based encoder-decoder pair to learn structured, non-linear data manifolds. The encoder ( \mu\phi ) maps data to latent space; the decoder ( \mu\theta ) reconstructs it. | The high-dimensional data resides on or near a lower-dimensional non-linear manifold [45]. | Captures complex, non-linear data structures; superior performance versus random embeddings [45]. | Risk of over-exploration in latent space and functional distribution mismatch between latent and original space [47] [11]. |
Common Pitfalls and Mitigation Protocols
Pitfall 1: Over-exploration in Latent Space
Problem Description: When performing continuous optimization in the latent space of a VAE, a common failure mode is over-exploration. The optimizer can propose latent points (z) that, when decoded, yield unrealistic or invalid solutions in the original space (e.g., non-synthesizable molecules) [47]. This occurs because the optimizer exploits the decoder to generate structures that score well on the objective function but lie outside the distribution of valid, training data-like candidates.
Mitigation Protocol: Latent Exploration Score (LES) The LES method constrains the BO process to regions of the latent space that decode to high-probability, realistic data points [47].
- Principle: LES leverages the VAE decoder's built-in approximation of the data distribution. It does not require additional model training, architectural changes, or access to the original training data, making it suitable for use with pre-trained models [47].
- Implementation: The LES is calculated and used as a penalty within the acquisition function to discourage sampling from low-probability regions.
- Experimental Workflow: The following diagram illustrates the integration of LES into a standard VAE-BO loop.
Pitfall 2: Functional Distribution Mismatch
Problem Description: The standard VAE loss function combines a reconstruction error with a Kullback-Leibler (KL) divergence term that regularizes the latent space. However, this formulation primarily ensures accurate data reconstruction and a smooth latent space; it does not guarantee that the relationships between data points (as captured by the GP kernel) are preserved after the encoding [11]. This leads to a "functional distribution mismatch," where the GP surrogate in the latent space is a poor model for the objective function in the original space, misleading the optimization.
Mitigation Protocol: HiPPO-based Space Consistency (HiBBO) The HiBBO framework introduces an additional constraint to the VAE training objective to enforce consistency between the functional distributions in the original and latent spaces [11].
- Principle: It uses HiPPO (High-order Polynomial Projection Operators) to create rich, long-term representations of the sequence of observations from both the original and latent spaces. A consistency loss is then applied between these HiPPO representations, which indirectly ensures the preservation of kernel distances and relationships between data points [11].
- Implementation: This method adds a HiPPO-based consistency term to the standard VAE loss function during training.
- Experimental Workflow: The protocol for training a VAE with HiPPO-based consistency is outlined below.
Pitfall 3: Miscalibrated Expert Knowledge and Representations
Problem Description: Incorporating expert knowledge through hand-crafted feature descriptors (e.g., chemical fingerprints) is common but requires extensive, time-consuming domain expertise to engineer and does not transfer well between scientific domains [48]. Conversely, using pre-trained Large Language Models (LLMs) as general-purpose feature extractors provides vast prior knowledge but often results in uncalibrated uncertainty estimates. LLMs can be overconfident and "hallucinate" suggestions, making them unreliable for high-stakes experimental optimization [48].
Mitigation Protocol: GOLLuM (Gaussian Process Optimized LLMs) The GOLLuM framework unifies LLMs and BO by jointly optimizing the LLM's embeddings with the GP surrogate [48].
- Principle: Instead of using static LLM embeddings, GOLLuM transforms textual descriptions of experiments into embeddings that are dynamically updated. Learning signals from the GP surrogate (trained on experimental outcomes) flow back to update the LLM, causing it to organize its latent space based on experimental performance rather than textual similarity [48].
- Implementation: This creates a bidirectional loop where the LLM generates representations for the GP, and the GP's performance feedback fine-tunes the LLM's embedding space.
- Key Insight: The ratio of the GP's learned lengthscale ((\ell)) to the average pairwise distance between points in the search space is a geometric metric that strongly correlates with optimization success, providing a guide for representation quality [48].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Latent-Space Bayesian Optimization
| Tool / Reagent | Type | Function in LSBO | Example Use-Case |
|---|---|---|---|
| Variational Autoencoder (VAE) [45] [47] | Deep Generative Model | Learns a non-linear, low-dimensional latent representation of the high-dimensional input data (e.g., molecular structures). | Encoding molecular graphs or material compositions into a continuous latent space for optimization [45] [47]. |
| Gaussian Process (GP) [45] [11] | Probabilistic Surrogate Model | Models the unknown objective function in the latent space, providing predictions and uncertainty estimates to guide the acquisition function. | Serving as the surrogate for the objective function (e.g., drug efficacy) within the latent space of a VAE [11]. |
| Latent Exploration Score (LES) [47] | Optimization Constraint | Penalizes the acquisition function to keep the search within regions of the latent space that decode to valid, high-probability data points. | Preventing the suggestion of invalid molecules in drug discovery campaigns [47]. |
| HiPPO (High-order Polynomial Projection Operators) [11] | Representation Learning Module | Enforces consistency between the original and latent functional distributions during VAE training, improving GP surrogate fidelity. | Aligning the latent space of a material structure VAE with the target property function for more reliable BO [11]. |
| GOLLuM Framework [48] | LLM-BO Integration | Dynamically adapts LLM embeddings based on experimental feedback from the GP, creating performance-oriented representations from text. | Optimizing reaction conditions for organic synthesis based on textual descriptions of chemical procedures [48]. |
Successfully applying Bayesian Optimization in high-dimensional materials and drug research landscapes requires a careful balance. While expert knowledge and latent space methods are powerful, practitioners must be wary of pitfalls like latent over-exploration, functional distribution mismatch, and miscalibrated representations. The protocols outlined herein—LES for constraining exploration, HiPPO for ensuring space consistency, and GOLLuM for calibrating knowledge-rich representations—provide a robust experimental framework to navigate these challenges. By integrating these mitigation strategies, researchers can enhance the reliability and efficiency of their optimization campaigns, accelerating the discovery of novel materials and therapeutic compounds.
In the context of Bayesian optimization (BO) for materials research, the surrogate model is a core component that approximates the expensive, black-box objective function (e.g., a material's property) based on available data. Its predictions and uncertainty estimates guide the selection of subsequent experiments, making its choice critical for the efficiency of the entire optimization campaign. This application note provides a comparative analysis of prominent surrogate models, including Gaussian Processes (GPs), Random Forests (RFs), and advanced variants like Deep Kernel Learning (DKL) and Bayesian Additive Regression Trees (BART). We detail their operational principles, performance characteristics, and provide structured protocols for their implementation in materials science and drug discovery.
Surrogate Model Fundamentals and Comparative Analysis
Core Model Architectures and Operating Principles
Gaussian Processes (GPs) define a non-parametric prior over functions, offering robust uncertainty quantification. A GP is fully specified by a mean function, (m(\mathbf{x})), and a covariance kernel, (k(\mathbf{x}, \mathbf{x}')). Given a dataset ( \mathcal{D} = \{\mathbf{X}n, \mathbf{y}n\} ), the posterior predictive distribution at a new point ( \mathbf{x} ) is Gaussian with closed-form expressions for the mean and variance, facilitating intuitive uncertainty estimation [49] [12]. The choice of kernel (e.g., Matérn, Radial-Basis Function) imbues the GP with properties such as smoothness and periodicity.
Random Forests (RFs) for regression are ensemble methods that aggregate predictions from multiple decision trees. While not inherently probabilistic, they can provide uncertainty estimates through methods like the jackknife-based bootstrap or by computing the empirical variance of predictions across the individual trees. Their key strengths lie in handling high-dimensional and mixed data types without stringent assumptions on the underlying functional form [50] [12].
Advanced Variants have emerged to address limitations of standard models:
- Deep Kernel Learning (DKL) combines a deep neural network for feature learning with a GP for probabilistic inference. The neural network transforms raw, high-dimensional inputs (e.g., molecular graphs) into a lower-dimensional latent space, upon which a GP kernel operates. This allows for automatic descriptor generation while retaining principled uncertainty quantification [51].
- Bayesian Additive Regression Trees (BART) is a Bayesian ensemble method that fits the unknown function as a sum of small regression trees. A regularization prior keeps the effect of each tree small, allowing BART to capture complex, non-smooth response surfaces and interactions without overfitting [12].
- Rank-Based Bayesian Optimization (RBO) replaces the standard regression surrogate with a model trained to learn the relative ordering of candidates. This approach can be more robust to rough landscapes and activity cliffs common in chemical datasets [52].
Quantitative Performance Comparison
The table below summarizes the performance characteristics of different surrogate models as observed in materials and chemistry optimization studies.
Table 1: Comparative Performance of Surrogate Models in Materials and Chemistry Domains
| Model | Key Strengths | Key Limitations | Reported Performance |
|---|---|---|---|
| Gaussian Process (GP) | Excellent uncertainty quantification; well-calibrated posteriors; mathematical explicitness [49] [12]. | Performance degrades with high-dimensional features (>20); assumes smooth function; kernel choice is critical [51] [12]. | ~12x more efficient than random search in finding oxide with largest bandgap [51]. Outperformed by more flexible models on complex functions [12]. |
| Random Forest (RF) | Handles high-dimensional and mixed data; requires less tuning; robust to irrelevant features [50] [12]. | Standard versions lack native, well-calibrated uncertainty estimates for BO [12]. | Used for property prediction from NMR data; performance degraded on noisy, unseen test data without denoising [50]. |
| Deep Kernel Learning (DKL) | Automatic feature learning from complex inputs (e.g., graphs); strong performance with complex descriptor spaces [51]. | Higher computational cost than standard GP; may underperform when hand-crafted descriptors are strongly correlated with target [51]. | Up to 2x more efficient than standard GP in searching for oxides with largest bandgap [51]. |
| BART / BMARS | Highly flexible; models non-smooth functions and complex interactions; built-in variable selection [12]. | Less common in standard BO libraries; computational cost can be higher than GP. | Demonstrated enhanced search efficiency and robustness on benchmark functions (Rosenbrock, Rastrigin) and real materials science cases compared to GP-based methods [12]. |
| Rank-Based Models (RBO) | Robust to activity cliffs and rough landscapes; relative ranking can be easier to learn than exact values [52]. | Novel approach; broader performance landscape not yet fully established. | Showed similar or improved optimization performance compared to regression surrogates on rough chemical datasets [52]. |
Model Selection Protocol
The following workflow outlines a systematic protocol for selecting a surrogate model based on the problem characteristics.
Experimental Protocols
Protocol 1: Standard Implementation of GP-Based BO
This protocol details the steps for setting up and running a standard GP-based Bayesian optimization loop, adaptable for materials and molecular design.
Table 2: Key Research Reagents and Computational Tools
| Item Name | Function/Description | Example/Notes |
|---|---|---|
| Design Space (X) | The discrete set of candidate materials or molecules to evaluate. | e.g., 922 oxides [51], a ZINC250k subset [52], or a combinatorial library [49]. |
| Objective Function (F(m)) | The expensive black-box function to optimize. | e.g., band gap, ionic conductivity, catalytic activity, or binding affinity. |
| Initial Dataset (D_ini) | A small set of randomly selected points from X with evaluated properties. | Typically 5-20 data points to build the initial surrogate model [51] [12]. |
| GP Surrogate Model | The probabilistic model that approximates F(m). | Use a Matérn 5/2 kernel. Implement with GPyTorch [52] or scikit-learn. |
| Acquisition Function | Guides the selection of the next experiment by balancing exploration and exploitation. | Common choices: Expected Improvement (EI) [12], Upper Confidence Bound (UCB) [51]. |
| Optimizer for Acquisition | Solves the inner optimization problem to find the point maximizing the acquisition function. | Random search or L-BFGS-B for discrete spaces; multi-start optimization for continuous spaces. |
Procedure:
- Problem Formulation: Define the molecular or materials search space ( \mathcal{M} ) and the objective function ( F(m) ) to maximize or minimize [49].
- Initialization: Select ( n_{ini} ) (e.g., 10) points uniformly at random from ( \mathcal{M} ) and evaluate ( F(m) ) for these points to form the initial dataset ( \mathcal{D}_{ini} = \{ (mi, yi) \}_{i=1}^{n_{ini}} ) [51] [12].
- BO Loop: For ( k = 1 ) to the maximum number of iterations (or until convergence), repeat: a. Model Training: Train the GP surrogate model on the current dataset ( \mathcal{D} ). This involves optimizing the kernel hyperparameters by maximizing the marginal log-likelihood [49] [12]. b. Acquisition Optimization: Using the trained GP's posterior, compute the acquisition function ( \alpha(m) ) over the search space. Identify the next candidate ( m_{next} ) that maximizes ( \alpha(m) ): ( m_{next} = \arg \max_m \alpha(m) ) [12]. c. Evaluation: Evaluate the expensive objective function ( y_{next} = F(m_{next}) ) (via experiment or simulation). d. Data Augmentation: Augment the dataset: ( \mathcal{D} = \mathcal{D} \cup \{ (m_{next}, y_{next}) \} ).
- Output: Return the best-performing candidate found during the optimization process.
Protocol 2: Implementing BO with a Flexible Surrogate (BART)
This protocol is adapted for using BART as a surrogate, which is particularly suited for non-smooth response surfaces.
Procedure:
- Steps 1 & 2: Follow the same Problem Formulation and Initialization as in Protocol 1.
- Model Training: Instead of a GP, train a BART model on the current dataset ( \mathcal{D} ). BART uses a Bayesian backfitting Markov Chain Monte Carlo (MCMC) algorithm to sample from the posterior distribution of the sum-of-trees model [12].
- Posterior Calculation: To compute the acquisition function, draw samples from the BART posterior predictive distribution. The mean prediction can be taken as the average of these samples, and the uncertainty can be quantified by the variance or quantiles of the samples [12].
- Acquisition Optimization & Evaluation: Proceed as in Steps 3b-3d of Protocol 1, using the BART-derived mean and uncertainty to calculate the acquisition function.
- Output: Return the best-performing candidate.
Protocol 3: High-Dimensional Optimization with Adaptive Subspaces (MolDAIS)
For high-dimensional descriptor spaces, the MolDAIS framework uses sparsity to identify relevant subspaces [49].
Procedure:
- Featurization: Represent each molecule ( m ) in the search space using a comprehensive library of molecular descriptors, creating a high-dimensional feature vector ( \mathbf{x} \in \mathbb{R}^d ) [49].
- Initialization: Select an initial dataset and evaluate the objective function, as in previous protocols.
- BO Loop with Sparse Surrogate: a. Model Training: Train a Gaussian Process surrogate that incorporates a sparsity-inducing prior (e.g., the Sparse Axis-Aligned Subspace (SAAS) prior) on the descriptor weights. This prior automatically forces the model to ignore irrelevant dimensions [49]. b. Subspace Identification: The trained model inherently identifies a low-dimensional, property-relevant subspace by assigning significant length-scales only to a sparse set of descriptors. c. Acquisition & Evaluation: Optimize the acquisition function in the full descriptor space, leveraging the sparse model for efficient computation. Evaluate the candidate and update the dataset [49].
The selection of a surrogate model is a pivotal decision that directly influences the efficiency of Bayesian optimization in materials science and drug discovery. While Gaussian Processes remain a gold standard for their uncertainty quantification in low-dimensional, smooth problems, advanced models like BART, BMARS, and DKL offer superior performance for non-smooth, high-dimensional, or complex structured data. The emerging paradigm of rank-based optimization presents a compelling alternative for navigating rough landscapes like those found in molecular activity data. The provided protocols and decision framework offer researchers a practical guide for selecting and implementing the most appropriate surrogate model for their specific optimization challenge.
Bayesian optimization (BO) has emerged as a powerful strategy for the global optimization of expensive, black-box functions, finding particular utility in fields with high experimental costs such as materials research and drug discovery. The efficiency of BO hinges on its use of a surrogate model, typically a Gaussian process (GP), to approximate the unknown objective function, and an acquisition function to guide the sequential selection of experimental samples. The acquisition function is the core mechanism that balances exploration (sampling in regions of high uncertainty) and exploitation (sampling where the surrogate model predicts high performance). This balance is crucial for achieving rapid convergence to optimal solutions, especially when working within strict experimental budgets. Within the specific context of materials research, where scientists often navigate complex, high-dimensional latent spaces representing material compositions, processing parameters, and microstructural descriptors, the choice of an appropriate acquisition function becomes paramount. This guide details the predominant acquisition functions, provides structured comparisons, and outlines specific protocols for their application in materials research.
The Core Mechanism of Acquisition Functions
The acquisition function, denoted as ( a(\mathbf{x}) ), uses the predictive distribution of the GP surrogate model to compute a utility score for any candidate point ( \mathbf{x} ) in the design space. The next point to evaluate is chosen by maximizing this function: ( \mathbf{x}{\text{next}} = \arg \max{\mathbf{x}} a(\mathbf{x}) ). The GP provides a predictive mean ( \mu(\mathbf{x}) ) (which estimates the function value) and a predictive variance ( \sigma^2(\mathbf{x}) ) (which quantifies the uncertainty at that point). Different acquisition functions combine these two quantities in distinct ways to manage the exploration-exploitation trade-off [53].
- Exploitation is driven by the mean ( \mu(\mathbf{x}) ). Given two points with similar uncertainty, the one with a higher predicted mean will have a higher utility for an exploitative acquisition function.
- Exploration is driven by the variance ( \sigma(\mathbf{x}) ). Given two points with similar predicted means, the one with higher predictive uncertainty will be preferred by an explorative acquisition function.
The following diagram illustrates the general Bayesian optimization workflow, highlighting the central role of the acquisition function.
Quantitative Comparison of Acquisition Functions
The table below summarizes the mathematical formulations, primary characteristics, and ideal use cases for several fundamental and advanced acquisition functions.
Table 1: Summary of Key Acquisition Functions
| Acquisition Function | Mathematical Formulation | Exploration-Exploitation Balance | Best Suited For |
|---|---|---|---|
| Upper Confidence Bound (UCB) | ( a(\mathbf{x}) = \mu(\mathbf{x}) + \kappa \sigma(\mathbf{x}) ) | Explicit, tunable via ( \kappa ) parameter [53]. | Problems where a pre-defined balance is acceptable; simple to implement. |
| Probability of Improvement (PI) | ( a(\mathbf{x}) = \Phi\left(\frac{\mu(\mathbf{x}) - f(\mathbf{x}^+)}{\sigma(\mathbf{x})}\right) ) | Greedy exploitation; can get stuck in local optima. | Finding a quick, local improvement from the best-known point ( f(\mathbf{x}^+) ). |
| Expected Improvement (EI) | ( a(\mathbf{x}) = \mathbb{E}[\max(f(\mathbf{x}) - f(\mathbf{x}^+), 0)] ) | Balanced, with a theoretical basis [54]. | General-purpose global optimization; the most widely used function. |
| EI-Hull-Area/Volume | Based on expected increase in convex hull area/volume [54]. | Exploratory, targets diverse high-performance candidates. | Materials discovery for mapping phase diagrams and ground-state lines. |
| Threshold-Driven UCB-EI (TDUE) | Dynamically switches from UCB to EI based on model uncertainty [55]. | Adaptive; starts explorative (UCB), becomes exploitative (EI). | High-dimensional material design spaces requiring efficient navigation. |
Advanced Strategies and Novel Developments
Targeted Discovery with Bayesian Algorithm Execution (BAX)
Moving beyond simple optimization, the BAX framework allows researchers to target specific subsets of the design space that meet complex, user-defined criteria. For instance, a goal might be to find all material compositions that simultaneously satisfy a minimum tensile strength and a maximum thermal conductivity. BAX translates such goals, expressed as algorithms, into efficient acquisition functions like InfoBAX, MeanBAX, and a hybrid SwitchBAX without requiring custom function design [56]. This is particularly valuable for identifying multiple promising candidate materials, thereby mitigating the risk associated with the long-term failure of a single "optimal" material.
Multi-Level and Microstructure-Aware Bayesian Optimization
For navigating vast chemical spaces, a multi-level Bayesian optimization strategy using hierarchical coarse-graining has been developed. This approach compresses the chemical space into different levels of resolution, performing exploration at lower resolutions (coarse-grained models) and exploitation at higher resolutions (fine-grained models). This funnel-like strategy efficiently identifies optimal compounds and relevant neighborhoods in the chemical space [21].
Furthermore, a paradigm shift is occurring toward microstructure-aware Bayesian optimization. Traditional methods treat microstructures as emergent outcomes rather than design parameters. The latent-space-aware BO framework incorporates microstructural descriptors (e.g., grain size, phase distributions) as latent variables to enhance the mapping from processing parameters to final material properties, leading to a more informed and efficient search through the Process-Structure-Property-Performance (PSPP) chain [3].
Application Notes & Experimental Protocols
Protocol A: Mapping a Convex Hull for Phase Stability
Objective: To efficiently determine the convex hull of a multi-component alloy system (e.g., Co-Ni, Ni-Al-Cr) using cluster expansion and Bayesian optimization [54].
Materials & Computational Tools:
- Software: Density Functional Theory (DFT) code (e.g., VASP), cluster expansion software (e.g., CASM).
- Surrogate Model: Gaussian Process with a kernel appropriate for material composition.
- Acquisition Function: EI-Hull-Area/Volume or EI-Below-Hull.
Procedure:
- Initialization: Generate a small initial dataset (e.g., 32 configurations) across the composition space using DFT calculations.
- Surrogate Model Training: Train a cluster expansion model on the current dataset to predict formation energies.
- Candidate Selection: Using the trained model, predict the mean and variance of formation energies for a large pool of unseen configurations.
- Acquisition & Batch Selection: Calculate the EI-Hull-Area score for batches of candidate configurations. The score measures the expected contribution of a batch to increasing the area (for binaries) or volume (for ternaries) of the predicted convex hull. Select the batch with the highest score.
- Expensive Evaluation: Perform DFT calculations on the selected batch of configurations to obtain their true formation energies.
- Iteration: Augment the dataset with the new results and repeat steps 2-5 until convergence. Convergence is typically assessed by the change in the ground-state line error (GSLE), a metric quantifying the difference between the current and target convex hulls [54].
- Validation: The final convex hull is considered accurate when the GSLE falls below a pre-defined threshold (e.g., close to zero).
Table 2: Key Performance Metrics from Convex Hull Mapping (Adapted from [54])
| Acquisition Function | Number of DFT Calculations to Reach Low GSLE | Key Advantage |
|---|---|---|
| EI-Hull-Area | ~78 (Most Efficient) | Maximizes information across the entire composition range. |
| EI-Below-Hull | ~87 | Prioritizes configurations close to the current hull. |
| EI-Global-Min | >87 (Least Efficient) | Focuses on the single lowest energy, missing hull diversity. |
| Genetic Algorithm (GA-CE-Hull) | ~82 | Well-established, but requires more user interaction. |
Protocol B: Threshold-Driven Adaptive Bayesian Optimization
Objective: To dynamically balance exploration and exploitation for optimizing a material property (e.g., thermoelectric efficiency) in a high-dimensional design space [55].
Materials & Computational Tools:
- Software: Bayesian optimization package (e.g., GPyOpt, BoTorch).
- Surrogate Model: Gaussian Process.
- Acquisition Functions: UCB and EI, with a switching policy.
Procedure:
- Initialization: Start with a space-filling design of experiments (DoE) to build an initial GP model.
- Uncertainty Monitoring: At each iteration ( t ), after updating the GP model, calculate the average predictive uncertainty (e.g., mean variance) over the entire design space or a representative sample.
- Dynamic Switching Policy:
- Phase 1 (Exploration): Use the UCB acquisition function ( a(\mathbf{x}) = \mu(\mathbf{x}) + \kappa \sigma(\mathbf{x}) ). This ensures broad exploration of the design space.
- Phase 2 (Exploitation): Monitor the average uncertainty. Once it drops below a pre-defined threshold ( \tau ), indicating the model has gained sufficient global knowledge, switch to the EI acquisition function. EI then focuses on refining and confirming promising areas.
- Evaluation & Iteration: Evaluate the selected sample, update the GP model, and repeat the process until the optimum is found or the experimental budget is exhausted.
The workflow for this adaptive strategy is depicted below.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Item / Software | Function / Role in Bayesian Optimization |
|---|---|
| Gaussian Process (GP) Regression | The core surrogate model that provides probabilistic predictions (mean and variance) for the black-box function. |
| Density Functional Theory (DFT) Codes | The "expensive experiment" used to calculate material properties like formation energy, serving as ground-truth data. |
| Cluster Expansion Software | A surrogate model that maps atomic configurations to properties, used with BO for phase stability analysis. |
| Expected Improvement (EI) | A versatile, widely-used acquisition function for general-purpose optimization tasks. |
| UCB & EI Hybrids (e.g., TDUE-BO) | Adaptive acquisition strategies for complex, high-dimensional design spaces [55]. |
| Specialized Acquisitions (EI-Hull-Area) | Acquisition functions tailored for specific scientific goals like mapping phase diagrams [54]. |
| Bayesian Algorithm Execution (BAX) | A framework for defining and solving complex target subset problems beyond pure optimization [56]. |
| High-Throughput Experimentation | Automated platforms that integrate with BO to physically execute the suggested experiments. |
Handling Multi-Objective and Constrained Optimization Problems
In materials research and drug development, optimizing processes often requires balancing multiple, competing objectives—such as maximizing product yield and purity while minimizing cost, energy consumption, or usage of expensive reagents—under strict safety and operational constraints. Constrained Multi-Objective Optimization Problems (cMOOPs) present a significant challenge, as the goal is not to find a single optimal solution, but a set of optimal trade-offs known as the Pareto Front (PF) [57]. Bayesian Optimization (BO) has emerged as a powerful, sample-efficient framework for navigating such complex design spaces, especially when experiments or simulations are expensive [58]. This document details advanced algorithms and practical protocols for applying Bayesian optimization to cMOOPs within materials science, with a focus on latent space representations.
Key Algorithms and Theoretical Advances
Recent algorithmic innovations more effectively manage the exploration-exploitation trade-off in cMOOPs by integrating Bayesian optimization with other optimization paradigms and novel theoretical insights.
Evolution-Guided Bayesian Optimization (EGBO)
The Evolution-Guided Bayesian Optimization (EGBO) algorithm synergistically combines an evolutionary algorithm's selection pressure with the probabilistic modeling of Bayesian optimization. Specifically, it integrates the q-Noisy Expected Hypervolume Improvement (qNEHVI) acquisition function with selection mechanisms from evolutionary algorithms [57].
This hybrid approach addresses a key limitation of standard qNEHVI-BO, which can lead to over-exploration and sampling wastage in constrained settings [57]. The evolutionary component guides the population of candidate solutions toward the Pareto Front, while the Bayesian component ensures efficient sampling and uncertainty quantification. EGBO has demonstrated significant performance improvements, including better coverage of the Pareto Front and a superior ability to propose feasible solutions compared to state-of-the-art qNEHVI alone [57].
COMBOO: Optimistic Constraints Estimation
The COMBOO algorithm is a novel approach that theoretically and empirically improves sample efficiency for cMOOPs. Its core innovation lies in optimistic constraint estimation to balance feasible region discovery with objective space optimization [59].
COMBOO actively learns the level-set of multiple unknown constraint functions while simultaneously performing multi-objective optimization within the estimated feasible region. This method moves beyond the heuristics used by earlier constrained multi-objective BO algorithms, enabling a more principled and sample-efficient search, as demonstrated on synthetic benchmarks and real-world applications [59].
Latent Space Optimization with GOLLuM
The GOLLuM framework reframes Large Language Model (LLM) fine-tuning as a Gaussian Process (GP) marginal likelihood optimization problem via deep kernel methods [58]. This is particularly relevant for optimizing textual or structured descriptions of materials or chemical reactions.
GOLLuM introduces LLM-based deep kernels, which are jointly optimized with the GP. The LLM provides a rich, flexible latent space from textual descriptions of experiments, while the GP models this space with principled uncertainty quantification [58]. This joint optimization induces a contrastive structure in the embedding space, effectively separating regions by performance ("the good, the bad, and the ugly") without any explicit contrastive loss. This leads to well-structured latent spaces, improved uncertainty calibration, and more efficient sampling [58].
Table 1: Summary of Advanced Bayesian Optimization Algorithms for cMOOPs.
| Algorithm | Core Mechanism | Key Advantages | Demonstrated Applications |
|---|---|---|---|
| Evolution-Guided BO (EGBO) | Hybrid of qNEHVI and evolutionary selection pressure [57] | Improved PF coverage; better handling of infeasible regions; mitigates over-exploitation [57] | Seed-mediated silver nanoparticle synthesis [57] |
| COMBOO | Optimistic estimation of constraint level-sets [59] | Theoretical sample efficiency guarantees; balanced learning of constraints and objectives [59] | Synthetic benchmarks; real-world applications [59] |
| GOLLuM | Joint training of LLM-based deep kernels and GP [58] | Leverages flexible LLM latent space; requires no specialized features; induces structured embeddings [58] | Buchwald-Hartwig reaction optimization; molecular property optimization [58] |
Quantitative Performance Benchmarks
Empirical evaluations across diverse problems highlight the performance gains offered by these advanced methods.
In one study, the EGBO algorithm demonstrated a significant hypervolume improvement over state-of-the-art qNEHVI across various synthetic multi-objective problems [57]. Hypervolume is a key metric that combines the convergence and diversity of the discovered Pareto Front.
For latent space optimization, the GOLLuM framework nearly doubled the discovery rate of high-performing reactions in the Buchwald-Hartwig reaction optimization, achieving 43% coverage of the top 5% reactions in just 50 iterations, compared to 24% coverage using static LLM embeddings [58]. It also showed a 14% improvement over domain-specific representations without requiring specialized feature engineering [58].
Table 2: Selected Quantitative Performance Results from Literature.
| Algorithm / Study | Metric | Performance | Baseline for Comparison |
|---|---|---|---|
| EGBO [57] | Hypervolume Improvement | Significant improvement | State-of-the-art qNEHVI |
| GOLLuM (Buchwald-Hartwig) [58] | Coverage of top 5% reactions | 43% | 24% (static LLM embeddings) |
| GOLLuM (vs. domain-specific) [58] | Performance improvement | +14% | Domain-specific representations |
| Materials Benchmark [60] [61] | Computational Scale | 494,498 simulations (206 CPU days) | Provides realistic benchmark surrogate |
Experimental Protocols
Protocol 1: Multi-Objective Optimization of Silver Nanoparticle Synthesis
This protocol outlines the application of EGBO for a three-objective optimization of seed-mediated silver nanoparticle synthesis in a self-driving lab [57].
Research Reagent Solutions
Table 3: Key Reagents for Silver Nanoparticle Synthesis Protocol.
| Reagent | Function / Role in Synthesis |
|---|---|
| Silver Seeds (10 nm, 0.02 mg mL⁻¹) | Nucleation centers for nanoparticle growth; costliest reactant [57]. |
| Silver Nitrate (AgNO₃, 15 mM) | Source of silver ions for particle growth [57]. |
| Ascorbic Acid (AA, 10 mM) | Reducing agent, converts silver ions to metallic silver [57]. |
| Trisodium Citrate (TSC, 15 mM) | Stabilizing and capping agent, influences particle shape and stability [57]. |
| Polyvinyl Alcohol (PVA, 5 wt%) | Stabilizing polymer, prevents aggregation [57]. |
Step-by-Step Procedure
Define Decision Variables and Objectives:
- Variables: Set the five flow rates (Q) for reagents: Silver Seeds, AgNO₃, AA, TSC, PVA. The domain for each is [0.6, 24] µL min⁻¹. A water flow rate is adjusted to maintain a total aqueous flow of 120 µL min⁻¹ [57].
- Objective 1 (y₁): Maximize spectral similarity to a target UV/Vis signature, quantified by the cosine similarity between the final absorbance spectrum and the target spectrum, capped at a maximum value [57].
- Objective 2 (y₂): Maximize reaction rate, defined as the maximum ratio of the absorbance peak to the residence time over the reaction period [57].
- Objective 3 (y₃): Minimize seed usage, calculated as 1 - (Q_seed / 120) [57].
Define Constraints:
Initialize the Self-Driving Lab:
- Use a microfluidic droplet generator to create reacting droplets and a line-scan hyperspectral imaging system for in-situ UV/Vis spectral measurement [57].
Configure and Run EGBO:
- Initialize the optimizer with the defined variables, objectives, and constraints.
- For each iteration, EGBO suggests a batch of candidate flow rates.
- The automated platform executes the experiments, and the hyperspectral sensor collects data.
- Objectives and constraints are computed from the absorbance maps.
- The algorithm updates its models and selects the next batch of candidates based on the EGBO acquisition function. This loop continues for a set number of iterations or until performance targets are met.
Protocol 2: Latent Space Optimization with GOLLuM for Chemical Reactions
This protocol describes how to use the GOLLuM framework to optimize chemical reactions using text-based representations [58].
Step-by-Step Procedure
Problem Templating:
- Define a standardized text template for the optimization task. For a reaction, this includes placeholders for parameters like reagents, catalysts, and conditions.
- Example:
"Reaction: {reagent_A} with {reagent_B}, Catalyst: {catalyst}, Temperature: {temp} C, Solvent: {solvent}."
Generate Latent Embeddings:
- Process the templated text descriptions through a pre-trained LLM to generate a fixed-dimensional embedding vector for each unique parameter set: ( \mathbf{x} = \text{LLM}(t) \in \mathbb{R}^d ) [58].
Configure the Deep Kernel Gaussian Process:
- Construct a GP using a deep kernel, where the kernel function operates on the LLM-generated embeddings: ( k{\theta,\phi}(\mathbf{x}, \mathbf{x}') = k{\theta}(g{\phi}(\mathbf{x}), g{\phi}(\mathbf{x}')) ). Here, ( g_{\phi} ) is the LLM embedding model with trainable parameters ( \phi ) [58].
Joint Optimization via Marginal Likelihood:
- Instead of keeping LLM embeddings static, jointly optimize the LLM parameters ( \phi ) and the GP hyperparameters ( \theta ) by maximizing the GP marginal likelihood. This step adapts the latent space specifically for the optimization task [58].
Bayesian Optimization Loop:
- Use an acquisition function (e.g., Expected Improvement) on the trained GP to select the most promising candidate to evaluate next.
- Execute the experiment or simulation to get the objective function value(s).
- Update the dataset and retrain the joint LLM-GP model. Iterate until the optimization budget is exhausted.
The Scientist's Toolkit: Benchmarking and Evaluation
Robust evaluation requires benchmarks that mirror real-world complexity. The Materials Science Optimization Benchmark Dataset for Multi-Objective, Multi-Fidelity Optimization of Hard-Sphere Packing Simulations provides a key resource [60] [61].
- Scale: The dataset comprises 494,498 simulations, representing 206 CPU days of computational effort [60] [61].
- Features: It encapsulates characteristics common to industrial chemistry and materials science tasks, including high noise levels, multiple fidelities, multiple objectives, linear constraints, non-linear correlations, and failure regions [60] [61].
- Usage: This benchmark enables the testing of optimization algorithms on a surrogate model that closely mimics the behavior of a real, expensive-to-evaluate simulation, providing a "Turing test" for optimization methods in a materials science context [60] [61].
Strategies for Data-Efficient Optimization with Limited Experimental Budgets
Data-efficient optimization strategies are paramount in materials science and drug development, where physical experiments and high-fidelity simulations are resource-intensive. Bayesian optimization (BO) stands as a powerful framework for guiding experimentation within an active learning loop to minimize the number of required measurements, especially when training data is limited [7]. This document details protocols and application notes for implementing these strategies, framed within the context of Bayesian optimization in latent space for materials research.
Core Optimization Strategies and Performance
Quantitative Comparison of Classification Strategies
A comprehensive performance analysis of 100 classification strategies was conducted across 31 distinct tasks in chemical and materials science [62]. The table below summarizes the key findings regarding the most data-efficient algorithms.
Table 1: Performance of Data-Efficient Classification Strategies
| Strategy Category | Specific Algorithms | Key Findings | Typical Domain Applications |
|---|---|---|---|
| Active Learning (AL) | Neural Network-based AL, Random Forest-based AL | Most efficient across diverse tasks; effective in low-data regimes [62]. | Phase behavior classification, solubility prediction, stability assessment [62]. |
| Bayesian Optimization (BO) | Target-Oriented EGO (t-EGO), Constrained EGO (CEGO) | Superior for finding target-specific properties; t-EGO requires 1-2x fewer experiments than standard EGO [7]. | Designing shape memory alloys, catalysts with specific adsorption energy [7]. |
| Constrained BO | PHOENICS, GRYFFIN | Handles interdependent, non-linear, and non-compact constraints intuitively [63]. | Optimizing chemical syntheses under constrained flow conditions [63]. |
Quantifying Classification Task Complexity
The complexity of a classification task, and thus the difficulty of data-efficient optimization, can be rationalized through task metafeatures. The most significant predictor of complexity is the noise-to-signal ratio [62]. This metric helps researchers anticipate the required experimental budget and select an appropriate strategy.
Experimental Protocols
Protocol 1: Target-Oriented Bayesian Optimization (t-EGO)
This protocol is designed for discovering materials with a specific target property value, rather than simply a maximum or minimum [7].
1. Problem Formulation:
- Define Target (t): Specify the desired property value (e.g., a transformation temperature of 440°C, a hydrogen adsorption free energy of 0 eV) [7].
- Define Design Space: Identify the parameter space to be searched (e.g., compositional space for an alloy).
2. Initial Data Collection:
- Select and characterize a small, space-filling initial set of candidates (e.g., via Latin Hypercube Sampling) to build an initial model.
3. Iterative Optimization Loop:
- Modeling: Train a Gaussian Process (GP) model using all acquired data (property
y) [7]. - Acquisition with t-EI: Calculate the Target-specific Expected Improvement (t-EI) for all candidates in the design space. t-EI is defined as:
t-EI = E[max(0, |y_t.min - t| - |Y - t|)]wherey_t.minis the current measurement closest to the targett, andYis the predicted property value from the GP model [7]. - Next Experiment Selection: Choose the candidate material or condition with the highest t-EI value.
- Experiment & Update: Perform the experiment, measure the property, and add the new data point to the training set.
4. Termination:
- The loop continues until a candidate is found within an acceptable tolerance of the target
tor the experimental budget is exhausted.
Protocol 2: Active Learning for Constraint Classification
This protocol is for building a data-efficient classifier to identify materials that satisfy a critical constraint (e.g., synthesizability, stability, solubility), thereby avoiding wasted resources on non-viable candidates [62].
1. Task Definition:
- Define Constraint: Establish the binary classification criterion (e.g., "soluble" vs. "insoluble", "stable" vs. "unstable").
- Select Molecular Representation: Choose an appropriate featurization (e.g., physico-chemical descriptors, fingerprints).
2. Initial Batch Selection:
- Use a sampler (e.g., random sampler) to select a small initial batch of points from the task domain for testing.
3. Active Learning Loop:
- Model Training: Train a classifier (e.g., Neural Network, Random Forest) on the currently labeled data.
- Uncertainty Quantification: Use the trained model to predict labels and, crucially, uncertainties for all unlabeled points in the domain.
- Batch Selection: Select the next batch of points with the highest prediction uncertainty.
- Experiment & Update: Perform experiments to label the new batch and add them to the training data.
4. Termination:
- The loop repeats until a pre-defined accuracy is achieved or the experimental budget for classification is used.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Components for a Bayesian Optimization Workflow
| Item / Solution | Function / Role | Example Use-Case |
|---|---|---|
| Gaussian Process (GP) Model | Serves as the probabilistic surrogate model that predicts material properties and quantifies uncertainty across the design space. | Core to all BO protocols; provides the mean and variance for acquisition function calculation [7] [64]. |
| Acquisition Function (e.g., t-EI, EI) | Balances exploration and exploitation to recommend the most informative next experiment. | t-EI guides the search towards a specific target value, while EI seeks the global optimum [7]. |
| Constrained Optimization Algorithm | Handles known experimental and design constraints to ensure recommended candidates are feasible. | Algorithms like PHOENICS and GRYFFIN prevent suggestions that violate synthesizability or safety limits [63]. |
| Molecular Descriptor Set | Translates chemical structures into a numerical feature vector for machine learning models. | Used in classification tasks to represent small molecules or polymers for predicting solubility or toxicity [62]. |
| Multi-Fidelity Modeling | Integrates data from low-cost (but noisy) simulations and high-cost (accurate) experiments to further reduce resource expenditure. | Combines results from fast force-field calculations with precise DFT data to accelerate discovery [65]. |
Benchmarking, Validation, and Comparative Analysis of BO Frameworks
Benchmarking BO Performance Across Diverse Experimental Materials Domains
Bayesian optimization (BO) has emerged as a powerful, data-efficient strategy for guiding autonomous and high-throughput experiments in materials science. It is particularly valuable for optimizing objectives that are costly, slow, or difficult to evaluate experimentally. However, the performance of BO is not universal; it depends critically on the choice of its components—the surrogate model and the acquisition function—and the nature of the materials system being studied. Therefore, comprehensive benchmarking across diverse experimental domains is essential to provide practical guidelines for researchers. This application note synthesizes recent benchmarking studies to quantify BO performance and provides detailed protocols for its effective implementation, with a specific focus on the emerging paradigm of latent-space-aware BO for materials research.
Quantitative Benchmarking of BO Performance
Benchmarking studies have quantified the performance of BO using metrics that compare its performance against a baseline strategy, typically random sampling. The two most prominent metrics are the Acceleration Factor (AF) and the Enhancement Factor (EF) [66] [67].
- Acceleration Factor (AF): Defined as the ratio of the number of experiments required for a reference strategy (e.g., random sampling) to achieve a given performance target compared to the active learning (e.g., BO) campaign: ( AF = n{ref} / n{AL} ). A higher AF indicates a more efficient AL process.
- Enhancement Factor (EF): Defined as the improvement in performance after a fixed number of experiments: ( EF = (y{AL} - y{ref}) / (y^* - y{ref}) ), where ( y{AL} ) is the performance of the AL campaign, ( y_{ref} ) is the reference performance, and ( y^* ) is the maximum possible performance.
A literature survey has revealed that reported AF values have a median of 6 and tend to increase with the dimensionality of the search space. In contrast, EF values consistently peak after approximately 10–20 experiments per dimension of the parameter space [67].
Performance of Surrogate Models
A key finding from cross-domain benchmarking is that the choice of surrogate model significantly impacts optimization efficiency. The following table summarizes the performance and characteristics of common surrogate models based on an evaluation across five diverse experimental materials datasets [66].
Table 1: Performance and Characteristics of Surrogate Models for Bayesian Optimization
| Surrogate Model | Theoretical Basis | Relative Performance | Key Advantages | Key Limitations / Considerations |
|---|---|---|---|---|
| Gaussian Process (GP) with Isotropic Kernel | Probabilistic model with stationary kernel. | Commonly used but outperformed by anisotropic models. | Simple, provides uncertainty estimates. | Assumes uniform smoothness across all dimensions; less robust. |
| GP with Anisotropic Kernel (ARD) | Probabilistic model with Automatic Relevance Determination. | Most robust performance; superior to isotropic GP. | Learns feature sensitivity; excellent uncertainty quantification. | Higher computational cost; requires careful hyperparameter tuning. |
| Random Forest (RF) | Ensemble of decision trees. | Comparable to GP-ARD; a strong alternative. | No distribution assumptions; lower time complexity; less sensitive to initial hyperparameters. | Uncertainty estimates are less native than GP. |
Advanced BO Formulations for Targeted Goals
Beyond standard optimization, specialized BO formulations have been developed for specific materials design objectives.
Table 2: Advanced Bayesian Optimization Formulations
| BO Formulation | Core Objective | Key Feature | Demonstrated Application |
|---|---|---|---|
| Target-Oriented BO (t-EGO) | Find materials with a specific target property value, not just a maximum/minimum. | Uses target-specific Expected Improvement (t-EI) that minimizes deviation from a target. | Discovery of a shape memory alloy with a transformation temperature within 2.66°C of the target in only 3 experiments [7]. |
| Multi-Fidelity BO (MFBO) | Reduce total optimization cost by leveraging information sources of different accuracy and cost. | Integrates low-fidelity (cheap, less accurate) and high-fidelity (expensive, accurate) data into a single model. | Can accelerate optimization, but performance depends on the cost ratio and informativeness of the low-fidelity source [68] [69]. |
| Latent-Space-Aware BO | Incorporate microstructure or other latent descriptors into the optimization loop. | Uses microstructural features as latent variables to enhance the mapping from process parameters to properties. | Shown to improve design performance by making the traditionally "latent space agnostic" BO aware of critical intermediate variables [3]. |
Detailed Experimental Protocols
Protocol 1: Standard Pool-Based Active Learning Benchmark
This protocol outlines the standard framework for benchmarking BO algorithms using historical experimental datasets [66].
1. Problem Formulation:
- Define the materials optimization objective as a minimization or maximization problem.
- Identify the independent input features (e.g., composition, processing parameters).
2. Data Preparation:
- Obtain a historical dataset where the objective has been evaluated for a large set of input conditions. This dataset serves as the discrete "ground truth" pool.
- Normalize all objective values and input features for consistent modeling.
3. Benchmarking Simulation:
- Initialization: Randomly select a small number of initial data points (e.g., 5-10) from the pool to form the initial training set.
- Iterative Loop: For a fixed number of iterations (or until a performance target is met):
- Model Training: Train the surrogate model (e.g., GP with anisotropic kernel, Random Forest) on the current training set.
- Candidate Selection: Using the acquisition function (e.g., EI, PI, LCB), evaluate all remaining points in the pool. Select the point that maximizes the acquisition function.
- Update: Add the selected point and its objective value from the pool to the training set. Record the best objective value found so far.
4. Performance Evaluation:
- Repeat the entire simulation multiple times with different random seeds for the initial sample to obtain statistical results.
- For each run, plot the best observed objective value against the number of experiments.
- Calculate the Acceleration Factor (AF) and Enhancement Factor (EF) by comparing the BO trajectory to that of a random sampling strategy.
Fig 1. Standard pool-based BO benchmarking workflow.
Protocol 2: Latent-Space-Aware Bayesian Optimization
This protocol extends standard BO by incorporating microstructural or other latent descriptors, moving from a purely black-box model to a microstructure-aware design framework [3].
1. Enhanced Data Collection:
- For each experimental observation, record not only the input parameters (chemistry, processing) and the final property but also quantitative microstructural descriptors (e.g., grain size, phase fractions, morphology metrics).
2. Latent Variable Processing:
- Apply dimensionality reduction techniques, such as the Active Subspace Method, to the latent microstructural descriptors. This identifies the key subspaces within the latent space that have the most significant influence on the variability of the objective function.
3. Model Integration:
- Construct a surrogate model that maps the original input parameters and the reduced latent variables to the material properties. This model more accurately captures the Process-Structure-Property (PSP) relationship.
- Alternatively, the latent variables can be used to define a more informative kernel for a Gaussian Process.
4. Optimization Loop:
- The BO algorithm operates on the combined space of input parameters and active latent dimensions.
- The acquisition function proposes the next experiment by considering both the potential to improve the property and to refine the understanding of the structure-property linkage.
Fig 2. Latent-space-aware BO workflow incorporating microstructure.
The Scientist's Toolkit: Research Reagent Solutions
This section details key computational and experimental "reagents" essential for implementing the protocols described above.
Table 3: Essential Tools for Benchmarking and Deploying Bayesian Optimization
| Tool / Resource | Function / Description | Relevance to Protocol |
|---|---|---|
| Benchmarking Datasets [66] | Publicly available experimental datasets (e.g., carbon nanotube-polymer blends, perovskites, silver nanoparticles) for validating BO performance. | Protocol 1: Serves as the "ground truth" pool for benchmarking simulations. |
| Gaussian Process (GP) Library (e.g., GPyTorch, scikit-learn) | Software library for building GP surrogate models, including support for anisotropic (ARD) kernels. | Protocol 1 & 2: Core component for building the probabilistic surrogate model. |
| Random Forest Library (e.g., scikit-learn) | Software library for building ensemble tree-based models, which can serve as an efficient surrogate. | Protocol 1: An alternative surrogate model that is computationally efficient and robust. |
| Acquisition Function (e.g., EI, t-EI) | The decision-making policy that selects the next experiment based on the surrogate's predictions. | Protocol 1 & 2: Critical for guiding the iterative optimization process. |
| Dimensionality Reduction Tool (e.g., Active Subspace Method) | Algorithm to identify the most influential directions in a high-dimensional latent space (e.g., microstructure descriptors). | Protocol 2: Key for processing latent variables and making the optimization tractable. |
| Automated Experimentation Platform | A self-driving lab (SDL) that integrates the BO algorithm with robotic hardware to execute experiments autonomously. | Protocol 1 & 2: Enables full closed-loop, experimental validation of the optimized results. |
Bayesian optimization (BO) has established itself as a powerful, sample-efficient framework for navigating complex materials design spaces, particularly when experimental data is scarce and evaluations are costly. The paradigm of conducting research within a latent space—often defined by microstructural descriptors or low-dimensional embeddings—further enhances its efficiency by reducing the effective dimensionality of the problem. This application note provides a consolidated overview of quantitative performance metrics and detailed experimental protocols for evaluating acceleration and enhancement factors in latent-space-aware Bayesian optimization for materials research. Designed for researchers and scientists, this document synthesizes the most recent methodological advances, supported by structured data and actionable workflows.
Quantitative Performance Metrics
The efficacy of optimization algorithms is measured by their sample efficiency, convergence speed, and success rate in achieving a target. The following tables summarize key quantitative metrics reported in recent literature for various BO strategies and their hybrids.
Table 1: Performance Metrics of Bayesian Optimization and Hybrid Methods
| Method | Key Feature | Test Environment | Performance Metric | Result | Reference |
|---|---|---|---|---|---|
| Multi-level BO | Hierarchical coarse-graining | Molecular dynamics simulations | Search efficiency in chemical space | Enhanced phase separation in phospholipid bilayers | [21] |
| Reinforcement Learning (RL) | Model-based & on-the-fly strategies | Benchmark functions (Ackley, Rastrigin); High-entropy alloys | Performance in high-D spaces (D ≥ 6); p-value | Statistically significant improvement over BO (p < 0.01) | [70] |
| Target-Oriented BO (t-EGO) | Targets specific property value | Synthetic functions; Shape memory alloys | Experimental iterations to reach target | Fewer iterations (1-2x) vs. EGO/MOAF; Target temp within 2.66°C in 3 runs | [7] |
| BO with Floor Padding | Handles experimental failure | Virtual data; SrRuO3 film growth | Success rate; Quality of optimized material | Achieved record residual resistivity ratio (80.1) in 35 runs | [71] |
| Threshold-Driven UCB-EI (TDUE-BO) | Dynamic UCB-to-EI switching | Three material datasets | Convergence efficiency; RMSE | Superior convergence and lower RMSE vs. EI/UCB | [55] |
Table 2: Latent-Space-Aware Bayesian Optimization Metrics
| Method | Latent Space Definition | Dimensionality Reduction | Reported Advantage | |
|---|---|---|---|---|
| Microstructure-aware BO | Microstructural descriptors (grain size, phase fractions) | Active Subspace Method | Improved mapping from design variables to properties; Enhanced GP modeling | [3] |
| Latent-Space-Aware BO | Microstructural features | Active Subspace Method | Quantifiable improvement over latent-space-agnostic methods | [3] |
Detailed Experimental Protocols
Protocol 1: Target-Oriented Bayesian Optimization (t-EGO)
Application: Discovering materials with a property value close to a specific target (e.g., transformation temperature, adsorption free energy=0) [7].
Workflow:
- Problem Formulation: Define the design variables
x(e.g., composition, processing parameters) and the target property valuet. - Initial Data Collection: Acquire a small initial dataset
{x_i, y_i}through a sparse design of experiments. - Model Training: Train a Gaussian Process (GP) regression model on the collected data, using the raw property values
yas the target. - Candidate Selection with t-EI: Calculate the target-specific Expected Improvement (t-EI) for all candidates in the design space. The t-EI acquisition function is defined as:
t-EI = E[max(0, |y_t.min - t| - |Y - t|)]wherey_t.minis the property value in the current dataset closest to the targett, andYis the GP's probabilistic prediction at an unknown pointx[7]. - Experiment and Update: Select the candidate
xwith the maximum t-EI value for the next experiment. Measure its true propertyy, add the new(x, y)pair to the dataset, and update the GP model. - Stopping Criterion: Iterate until a material is found where
|y - t|is within a pre-defined tolerance.
Protocol 2: Microstructure-Aware Latent-Space BO
Application: Accelerating materials discovery by explicitly incorporating microstructural information into the Bayesian optimization loop [3].
Workflow:
- Data Generation & Feature Extraction: For each set of process parameters, generate a material (via simulation or experiment) and characterize its microstructure. Extract a high-dimensional vector of microstructural descriptors (e.g., via 2-point spatial correlations).
- Dimensionality Reduction: Apply the Active Subspace Method (ASM) to the high-dimensional latent space of microstructural descriptors. This identifies the dominant directions in the latent space that most influence the target property, creating a reduced, informative representation [3].
- Model Training: Train a Gaussian Process model. The input can be either:
- The original design variables (chemistry/processing) augmented with the dominant latent dimensions from Step 2.
- The dominant latent dimensions alone.
- Candidate Selection: Use a standard acquisition function (e.g., Expected Improvement) to propose the next experiment or simulation based on the GP model's predictions and uncertainties.
- Iterate: Run the experiment, extract the new microstructure, compute its latent representation, and update the GP model. The process iteratively refines the understanding of the process-structure-property relationship.
The Scientist's Toolkit: Research Reagent Solutions
This section lists key computational and experimental resources essential for implementing the described protocols.
Table 3: Essential Tools for Latent-Space Bayesian Optimization
| Category | Tool / Resource | Function in Workflow | |
|---|---|---|---|
| Core Modeling | Gaussian Process Regression (GPR) | Serves as the surrogate model to approximate the expensive objective function and quantify prediction uncertainty. | [70] [3] [71] |
| Acquisition Functions | Expected Improvement (EI), Upper Confidence Bound (UCB), t-EI | Guides the selection of the next experiment by balancing exploration and exploitation or targeting a specific value. | [70] [7] [55] |
| Dimensionality Reduction | Active Subspace Method (ASM) | Identifies dominant, property-relevant directions in a high-dimensional latent space (e.g., of microstructural features). | [3] |
| Handling Failures | Floor Padding Trick | Manages experimental failures by imputing the worst observed value, allowing the BO to learn from and avoid failed regions. | [71] |
| Accelerated Computing | NVIDIA ALCHEMI NIM Microservices | Provides GPU-accelerated microservices for conformer search and molecular dynamics, drastically increasing simulation throughput. | [72] |
| Real-Time Data | NVIDIA Holoscan Platform | Enables real-time, high-throughput processing of streaming data from advanced instruments (e.g., synchrotron beamlines). | [72] |
The discovery and optimization of new materials, such as high-entropy alloys (HEAs) or pharmaceutical compounds, often require navigating vast design spaces with multiple, correlated target properties. In this context, Bayesian optimization (BO) has emerged as a powerful, data-efficient strategy for steering experiments toward optimal candidates. The performance of BO is profoundly influenced by the choice of its surrogate model, which approximates the complex landscape of material properties. Conventional Gaussian Processes (cGPs) have been widely used but show limitations when modeling correlated, multi-output data. This analysis details the performance and implementation of two advanced surrogates—Multi-Task Gaussian Processes (MTGPs) and Deep Gaussian Processes (DGPs)—against cGPs, providing a structured framework for their application in materials research and drug development.
Theoretical Framework and Key Differentiators
Model Architectures and Mathematical Foundations
- Conventional Gaussian Process (cGP): A cGP places a prior directly over the function mapping inputs (e.g., alloy composition) to a single output (e.g., yield strength). It is characterized by a mean function and a covariance kernel, providing robust uncertainty quantification for individual tasks. However, in multi-objective settings, a common but suboptimal practice is to model each property using an independent cGP, thereby ignoring potential correlations between them [42].
- Multi-Task Gaussian Process (MTGP): MTGPs extend the cGP framework to model multiple correlated outputs (tasks) simultaneously. They use structured kernels to learn correlations between tasks, allowing for information transfer. For instance, data from a well-characterized property can improve predictions for a sparsely measured, correlated property. This makes MTGPs highly effective for tasks like jointly predicting a material's yield strength and hardness [42] [73].
- Deep Gaussian Process (DGP): DGPs introduce a hierarchical structure by composing multiple layers of GPs. This architecture enables the model to learn complex, non-linear data transformations internally. A DGP can capture hierarchical and non-stationary relationships, effectively modeling intricate composition-property maps in materials science. DGPs inherently account for input-dependent noise (heteroscedasticity) and can share information across tasks through deep, latent representations [74] [42] [75].
Comparative Strengths and Limitations
Table 1: Comparative analysis of Conventional GP, Multi-Task GP, and Deep GP.
| Feature | Conventional GP (cGP) | Multi-Task GP (MTGP) | Deep GP (DGP) |
|---|---|---|---|
| Handling of Correlated Properties | Models properties independently; ignores correlations [42]. | Explicitly models correlations between tasks for information sharing [42] [73]. | Learns hierarchical correlations and shared latent representations across tasks [74] [75]. |
| Uncertainty Quantification | Provides well-calibrated uncertainty for single tasks. | Quantifies uncertainty within and across correlated tasks. | Propagates and refines uncertainty through multiple layers, capturing complex uncertainty patterns [74]. |
| Data Efficiency | Less efficient for multi-task problems due to isolated learning. | More data-efficient for multi-output problems by leveraging task correlations [42]. | High data efficiency; can leverage information from auxiliary tasks or data sources [73]. |
| Handling Heteroscedastic/Noisy Data | Assumes uniform (homoscedastic) noise. | Can be adapted for task-specific noise. | Naturally handles heteroscedastic, non-stationary, and noisy data via its layered structure [73] [75]. |
| Model Complexity & Scalability | Lower complexity; well-established scaling techniques. | Moderate complexity; scaling depends on the number of tasks. | Higher computational complexity, but more flexible for modeling intricate relationships [74]. |
| Ideal Use Case | Single-objective optimization with simple landscapes. | Multi-objective optimization with moderately correlated properties. | Complex, hierarchical data; multi-fidelity optimization; highly non-linear and correlated property spaces [42] [75]. |
Performance Benchmarking in Materials Science
Recent studies have systematically benchmarked these models, particularly in the domain of high-entropy alloys (HEAs), where property correlations are pronounced.
Quantitative Performance Metrics
Table 2: Summary of quantitative performance findings from case studies on High-Entropy Alloys (HEAs).
| Study Focus / Model | Key Performance Findings |
|---|---|
| HEA Property Prediction (AlCoCrCuFeMnNiV system) [74] [73] | |
| Deep GP (with ML prior) | Outperformed other surrogates; effectively captured inter-property correlations and input-dependent uncertainty. |
| Conventional GP | Suboptimal for capturing correlations in multi-property datasets. |
| Encoder-Decoder NN / XGBoost | Lacked robust, native uncertainty quantification. |
| BO for Materials Discovery (FeCrNiCoCu system) [42] | |
| Hierarchical DGP (hDGP-BO) | Most robust and efficient in multi-objective optimization (e.g., minimizing CTE & maximizing BM). |
| Multi-Task GP-BO (MTGP-BO) | Showed advantages over cGP-BO by leveraging property correlations. |
| Conventional GP-BO (cGP-BO) | Suboptimal performance due to inability to utilize shared information across correlated properties. |
Protocol: Implementing GP Models for Multi-Property Prediction
This protocol outlines the steps for benchmarking GP models, as applied in recent HEA studies [74] [42] [73].
Step 1: Dataset Curation and Preprocessing
- Data Assembly: Compile a hybrid dataset from experimental and computational sources. For HEAs, this includes target properties (e.g., yield strength, hardness) and auxiliary computational properties (e.g., stacking fault energy, valence electron concentration) [73].
- Handling Heterotopic Data: The dataset will likely be heterotopic, meaning not all properties are measured for all samples. The model must be trained to handle missing output data gracefully [73] [75].
- Data Scaling: Standardize or normalize input features (e.g., elemental compositions) and target properties to a common scale for stable model training.
Step 2: Model Configuration and Training
- cGP Baseline: Implement a cGP for each property independently using a Matérn kernel (e.g., Matérn-5/2). Optimize kernel hyperparameters (length scale, variance) by maximizing the marginal likelihood [42] [75].
- MTGP Implementation: Employ a multi-task kernel, such as the intrinsic coregionalization model (ICM). The model will learn a covariance matrix between tasks, capturing their correlations during hyperparameter optimization [42].
- DGP Implementation: Construct a DGP with multiple hidden layers (e.g., 2-3 layers). Use variational inference for approximate posterior inference to make training computationally tractable. Consider infusing a machine-learned prior to guide learning [74] [75].
Step 3: Model Evaluation and Validation
- Performance Metrics: Evaluate models using predictive accuracy (e.g., Mean Absolute Error, R²) and the quality of uncertainty quantification (e.g., negative log predictive density, calibration plots) on a held-out test set [74] [73].
- Benchmarking: Systematically compare all models on their ability to fit the data, capture property correlations, and provide accurate, calibrated uncertainties.
Diagram 1: Workflow for materials discovery using GP surrogates and Bayesian optimization.
Application Notes for Bayesian Optimization in Latent Space
Integrating these surrogate models into a BO loop enables efficient materials discovery, particularly when operating in a latent space or dealing with multiple objectives.
Advanced BO Integration
- Multi-Objective and Cost-Aware BO: Real-world materials design involves balancing multiple, often conflicting, objectives. DGPs and MTGPs, integrated with acquisition functions like Expected Hypervolume Improvement (qEHVI), can efficiently identify Pareto-optimal solutions. Furthermore, a cost-aware DGP-BO framework can strategically balance expensive high-fidelity evaluations (e.g., experiments) with cheaper low-fidelity ones (e.g., computations), maximizing information gain per unit cost [75].
- BO in Latent Space: The hierarchical nature of DGPs makes them exceptionally suited for BO in latent spaces. The hidden layers of a DGP can be interpreted as learning a non-linear mapping from the original input space to a latent representation that is more informative for predicting the target properties. This effectively performs Bayesian optimization in a learned, problem-dependent latent space, which can dramatically accelerate the discovery process for complex materials [42] [75].
Protocol: DGP-BO for Multi-Objective Materials Discovery
This protocol details the application of DGP-BO for finding optimal compositions, as demonstrated in refractory HEA studies [42] [75].
Step 1: Problem Formulation
- Define Objectives: Identify the target properties to optimize (e.g., maximize bulk modulus and minimize coefficient of thermal expansion).
- Set Constraints: Define any compositional or property constraints.
- Specify Fidelities and Costs: If using a multi-fidelity approach, define the available data sources (e.g., high-fidelity experiment vs. low-fidelity simulation) and their associated evaluation costs.
Step 2: Initial Surrogate Model Training
- Build a Multi-Output DGP: Train a DGP surrogate on the available initial data (e.g., a small set of characterized alloys). The DGP should model all target properties and fidelities simultaneously.
- Architecture Choice: A 2-layer DGP is often a good starting point. The first layer learns a latent representation of the compositions, and the second layer maps this representation to the property predictions.
Step 3: The BO Iteration Loop
- Acquisition Function Optimization: Use the DGP's predictions (mean and uncertainty) to compute a multi-objective acquisition function like qEHVI.
- Cost-Aware Batching: For a batch of
qcandidates, the acquisition function is extended to favor queries that offer the best information gain per unit cost. - Heterotopic Querying: The batch may include candidates for which only cheaper, low-fidelity properties are evaluated, allowing for broader exploration.
- Evaluation and Model Update: Synthesize, simulate, or test the proposed candidate materials. Add the new data to the training set and update the DGP surrogate. Repeat until convergence to a Pareto front or exhaustion of the resource budget.
Diagram 2: Architectural differences between GP models for materials property prediction.
Table 3: Essential components for computational materials discovery campaigns.
| Category / Item | Specification / Example | Function in the Workflow |
|---|---|---|
| Computational Framework | ||
| Gaussian Process Library | GPyTorch, GPflow, scikit-learn | Provides the core algorithms for building and training cGP, MTGP, and DGP models. |
| Bayesian Optimization Platform | BoTorch, Ax Platform | Offers implementations of acquisition functions (e.g., qEHVI) and optimization loops. |
| Data Sources | ||
| High-Throughput Atomistics | LAMMPS, VASP | Generates high-fidelity computational data on material properties (e.g., bulk modulus, CTE) [42]. |
| Experimental Datasets | BIRDSHOT HEA Dataset [73] | Provides critical experimental measurements for model training and validation. |
| Material System | ||
| High-Entropy Alloys (HEAs) | Al-Co-Cr-Cu-Fe-Mn-Ni-V, Fe-Cr-Ni-Co-Cu [74] [42] | A complex model system with vast compositional space and correlated properties for benchmarking. |
| Key Properties | ||
| Mechanical & Thermal | Yield Strength, Hardness, Bulk Modulus (BM), Coefficient of Thermal Expansion (CTE) [42] [73] | The target objectives for optimization, often exhibiting trade-offs and correlations. |
The Impact of Anisotropic Kernels and Automatic Relevance Detection (ARD)
In the context of Bayesian optimization (BO) for materials research, the choice of surrogate model kernel critically determines the efficiency of navigating complex design spaces. Anisotropic kernels, particularly those employing Automatic Relevance Determination (ARD), extend standard isotropic kernels by assigning independent length-scale parameters to each input dimension [76]. This allows the model to automatically adapt to the varying sensitivity of the objective function to different input parameters, a crucial capability when optimizing high-dimensional materials formulations and processing conditions where the influence of each variable is rarely uniform [66] [77].
Within a Gaussian Process (GP), the standard isotropic Radial Basis Function (RBF) kernel uses a single length scale l for all dimensions, calculating covariance between two points x and x' as k(x, x') = σ² exp(-||x - x'||² / 2l²) [78]. In contrast, an anisotropic ARD kernel employs a separate length scale l_j for each dimension j, transforming the covariance function to k(x, x') = σ² exp(-½ Σj (xj - x'j)² / lj²) [66] [76]. The inverse length scale 1/l_j effectively measures the importance or "relevance" of the j-th input dimension; a small l_j (large 1/l_j) indicates the objective function is highly sensitive to changes in that dimension, whereas a large l_j suggests relative insensitivity [66] [79]. This automatic weighting enables the model to focus its modeling capacity on the most critical factors driving materials performance.
Performance Advantages and Quantitative Benchmarks
Comprehensive benchmarking across diverse experimental materials systems demonstrates that BO algorithms leveraging anisotropic kernels significantly outperform their isotropic counterparts.
Table 1: Benchmarking BO Surrogate Model Performance Across Materials Systems
| Materials System | Surrogate Model | Performance vs. Isotropic GP | Key Metric |
|---|---|---|---|
| Carbon nanotube-polymer blends [66] | GP with ARD | Comparable to RF, both outperform isotropic GP | Acceleration factor |
| Silver nanoparticles [66] | GP with ARD | Comparable to RF, both outperform isotropic GP | Enhancement factor |
| Lead-halide perovskites [66] | GP with ARD | Comparable to RF, both outperform isotropic GP | Acceleration factor |
| Additively manufactured polymers [66] | GP with ARD | Comparable to RF, both outperform isotropic GP | Enhancement factor |
| Polymer composites for 5G [77] | GP with ARD | Effective optimization in 8-dimensional space | Target property achievement |
The robustness of GP with anisotropic kernels makes it particularly suitable for high-dimensional problems common in materials science, such as optimizing polymer composites for 5G applications, where it successfully managed eight input parameters covering filler morphology, surface chemistry, and compounding process parameters [77]. Empirical results show that anisotropic kernels provide more accurate recreations of underlying response surfaces compared to isotropic kernels, with the degree of optimal anisotropy varying by specific dataset but full anisotropy generally providing substantial improvement [80].
Experimental Protocols and Implementation Guidelines
Protocol 1: Bayesian Optimization with ARD for Polymer Composite Fabrication
This protocol outlines the procedure for optimizing an eight-parameter polymer composite fabrication process, adapting the methodology that successfully developed low-thermal-expansion, low-dielectric-loss composites for 5G applications [77].
- Objective: Optimize filler and processing parameters to minimize coefficient of thermal expansion (CTE) and dielectric loss of perfluoroalkoxyalkane (PFA)/silica composites.
- Materials and Equipment:
- Matrix Material: Perfluoroalkoxyalkane (PFA) resin.
- Fillers: Silica particles of varied morphology (spherical, fibrous), size, and surface functionalization.
- Processing Equipment: Compounding mixer, hot press, thermal expansion analyzer, high-frequency dielectric spectrometer.
- Experimental Workflow:
- Define Optimization Space: Identify eight critical input parameters:
- Filler volume fraction (10-40%)
- Filler shape (aspect ratio: 1-10)
- Filler size (100 nm-10 μm)
- Surface functionalization type (e.g., methyltriethoxysilane, phenyltriethoxysilane)
- Surface coverage (10-100%)
- Compounding temperature (300-400°C)
- Compounding speed (50-200 rpm)
- Compounding time (5-30 min)
- Initialize with Design of Experiments: Select 10-15 initial data points using Latin Hypercube Sampling to cover the parameter space evenly.
- Establish BO Loop:
- Surrogate Model: Configure Gaussian Process with ARD Matérn 5/2 kernel.
- Acquisition Function: Employ Expected Improvement (EI).
- Iteration: Run 15-20 optimization cycles, synthesizing and characterizing one candidate per iteration.
- Characterization: Measure CTE (ASTM E831) and dielectric loss at GHz frequencies (IPC TM-650 2.5.5.13) for each composite.
- Validation: Synthesize and validate top-performing candidate identified by BO.
- Define Optimization Space: Identify eight critical input parameters:
Protocol 2: Target-Oriented Bayesian Optimization for Shape Memory Alloys
This protocol implements a target-specific BO approach (t-EGO) for discovering materials with predefined property values, based on the methodology that identified a shape memory alloy with a specific transformation temperature [7].
- Objective: Discover Ti-Ni-Cu-Hf-Zr shape memory alloy with austenite finish temperature Af = 440°C for thermostatic valve applications.
- Materials and Equipment:
- Raw Materials: High-purity Ti, Ni, Cu, Hf, Zr metals.
- Fabrication Equipment: Arc melter with water-cooled copper hearth, inert atmosphere system.
- Characterization Equipment: Differential scanning calorimetry (DSC), X-ray diffraction (XRD).
- Experimental Workflow:
- Define Composition Space: Constrain total to 100 atomic percent with varying Ti (15-25%), Ni (30-40%), Cu (10-15%), Hf (20-30%), Zr (5-10%).
- Initialize Dataset: Include 5-10 known compositions with measured Af temperatures from literature or preliminary experiments.
- Configure t-EGO:
- Surrogate Model: Gaussian Process with ARD kernel.
- Acquisition Function: Target-specific Expected Improvement (t-EI) using t-EI = E[max(0, |y_t.min - t| - |Y - t|)] where t is target temperature.
- Iterative Optimization:
- Train GP model on current dataset.
- Evaluate t-EI across composition space.
- Select highest t-EI composition for experimental testing.
- Arc melt new alloy, homogenize, and characterize by DSC.
- Add new (composition, Af) data point to dataset.
- Repeat until |Af - t| < 5°C achieved.
- Validation: Confirm transformation behavior and temperature consistency over 3 thermal cycles.
Table 2: Key Research Reagent Solutions for Materials Optimization
| Reagent/Material | Function in Optimization | Application Example |
|---|---|---|
| Silica Fillers (varying morphology/size) | Modifies thermal and dielectric properties of composite | PFA composite for 5G applications [77] |
| Surface Functionalization Agents (e.g., silanes) | Improves filler-matrix compatibility and dispersion | Methyltriethoxysilane for silica-PTFE interface [77] |
| High-Purity Metal Elements (Ti, Ni, Hf, Zr, Cu) | Base constituents for alloy composition space | Ti-Ni-Cu-Hf-Zr shape memory alloys [7] |
| Perfluoroalkoxyalkane (PFA) Resin | Low-dielectric-loss polymer matrix | 5G polymer composite matrix [77] |
Workflow Visualization
Figure 1. Bayesian optimization workflow with ARD for materials research.
Figure 2. Automatic relevance determination mechanism for high-dimensional optimization.
Technical Implementation and Researcher's Toolkit
Implementing anisotropic kernels requires attention to several technical considerations. For Gaussian Processes, the ARD framework can be applied to various kernel types including Matérn 5/2, Matérn 3/2, and RBF [66]. The additional hyperparameters (length scales per dimension) increase model flexibility but require careful estimation through evidence maximization (type-II maximum likelihood) to avoid overfitting [79]. For high-dimensional problems (>20 parameters), sparse ARD methods or integration with the Maximum Partial Dependence Effect (MPDE) may be necessary to enhance stability [81].
- Software Libraries: Popular BO implementations like GPyTorch, Scikit-learn, and BoTorch support ARD kernels through length-scale parameters for each dimension.
- Hyperparameter Initialization: Initialize length scales based on parameter ranges (e.g., set initial l_j to approximately 20% of parameter j's range).
- Convergence Monitoring: Track length scale evolution during optimization; converging to very large values indicates low relevance parameters that can potentially be fixed.
- Alternative Approaches: Random Forest models provide a non-parametric alternative free from distribution assumptions with comparable performance to GP+ARD in some materials domains [66].
The integration of anisotropic kernels with specialized acquisition functions like target-oriented EI further enhances BO's applicability to materials design problems where specific property values rather than simple maxima/minima are desired [7]. This combination provides a powerful framework for addressing the complex, multi-faceted optimization challenges prevalent in modern materials research and drug development.
Validation Through High-throughput Atomistic Simulations in HEA Design Spaces
Application Notes
The Role of Validation in a Bayesian Optimization Workflow
Within a thesis focused on Bayesian optimization (BO) in latent space for materials research, validation via high-throughput atomistic simulations is a critical step that bridges computational prediction and physical understanding. This process involves using simulations to rapidly and inexpensively generate data and verify the properties of candidate materials suggested by the BO loop. By doing so, it provides the essential "ground truth" that refines the probabilistic model, guiding the search towards regions of the design space that are not only high-performing but also physically plausible and synthetically accessible.
The integration addresses two central challenges in complex material systems like High-Entropy Alloys (HEAs):
- Vast Design Space: The enormous compositional and processing space of HEAs makes exhaustive experimental exploration impractical [82]. High-throughput simulations act as a powerful filter, evaluating thousands of potential compositions in silico to identify the most promising candidates for physical synthesis [83] [84].
- Data Generation for Correlated Properties: Advanced BO methods, such as Multi-Task Gaussian Process BO (MTGP-BO) and hierarchical Deep Gaussian Process BO (hDGP-BO), excel when they can leverage correlations between multiple material properties [42]. High-throughput atomistic simulations are ideal for generating the consistent, multi-property datasets needed to train these models, thereby accelerating the discovery of alloys that simultaneously satisfy several objectives, such as low thermal expansion and high bulk modulus [42].
Key Insights from Integrated Frameworks
Recent studies demonstrate the power of combining high-throughput simulation with machine learning and BO. For instance, a workflow for designing Cr-Co-Ni medium-entropy alloys used high-throughput molecular dynamics (MD) simulations to establish a "composition-performance" database, which was then used to train an artificial neural network (ANN) to predict optimal compositions with high strength and low cost [83]. This approach fundamentally shifts the paradigm from traditional "trial-and-error" to a data-driven methodology [83].
Furthermore, the concept of target-oriented Bayesian optimization has been developed for scenarios where the goal is not simply to maximize or minimize a property, but to achieve a specific target value [7]. For example, discovering a shape memory alloy with a transformation temperature of exactly 440°C. High-throughput simulations are crucial for validating the model's suggestions in such precise optimization tasks [7].
For problems involving high-dimensional input or output spaces, such as optimizing complex molecular structures, Joint Composite Latent Space Bayesian Optimization (JoCo) provides a framework that uses neural networks to compress these high-dimensional spaces into manageable latent representations, enabling efficient BO [29]. High-throughput simulations can validate the predictions made within this compressed latent space.
Experimental Protocols
Protocol 1: High-Throughput MD for Mechanical Properties
1. Objective: To rapidly screen the mechanical properties (e.g., ultimate tensile strength) across a vast compositional space of a model MEA/HEA system, such as CrₓCoᵧNi₁₀₀₋ₓ₋ᵧ [83].
2. Workflow Overview: The following diagram illustrates the integrated high-throughput simulation and Bayesian optimization workflow for HEA design.
3. Detailed Methodology:
- Model Construction: Build atomistic models for a wide range of compositions (e.g., 20 ≤ x ≤ 60 at.%, 20 ≤ y ≤ 60 at.%). A typical bulk sample may contain ~13,500 atoms with dimensions of approximately 53Å × 53Å × 53Å [83].
- Interatomic Potential: Employ suitable semi-empirical potentials or forcefields (e.g., the second-moment approximation of the tight-binding potential, SMTB) that accurately capture the interactions between the constituent elements [83].
- Simulation Conditions:
- Equilibration: First, equilibrate the system in an isothermal-isobaric (NPT) ensemble at the target temperature (e.g., 300 K) and zero pressure to achieve a stable configuration.
- Deformation: Apply uniaxial tensile deformation by imposing a constant strain rate (e.g., 0.0005 ps⁻¹) along a specific crystallographic direction (e.g., [100]) [83].
- Data Extraction: From the simulation, calculate the virial stress and monitor the stress-strain relationship. The ultimate tensile strength (UTS) is identified as the maximum stress value on the stress-strain curve before fracture [83].
Protocol 2: High-Throughput Calculation of Thermodynamic Properties
1. Objective: To compute fundamental thermodynamic and thermo-physical properties, such as the coefficient of thermal expansion (CTE) and bulk modulus (BM), for a large number of HEA compositions (e.g., within the Fe-Cr-Ni-Co-Cu system) [42].
2. Detailed Methodology:
- Simulation Approach: Use molecular dynamics simulations with a verified interatomic potential.
- Ensemble for CTE:
- Simulate the system in the NPT ensemble across a range of temperatures.
- Calculate the average lattice parameter at each temperature.
- The CTE is derived from the slope of the lattice parameter versus temperature plot.
- Ensemble for BM:
- The isothermal bulk modulus can be calculated from volume fluctuations in the NPT ensemble using the relation: ( BT = kB T \langle V \rangle / \langle \delta V^2 \rangle ), where ( k_B ) is Boltzmann's constant, T is temperature, ( \langle V \rangle ) is the average volume, and ( \langle \delta V^2 \rangle ) is the volume variance [42].
- Alternatively, BM can be determined by applying small deformations to the simulation cell and measuring the resulting stress.
- Data Analysis: These simulation-derived properties must be validated against experimental data where available to ensure the forcefield's accuracy. Studies have shown that simulation-derived properties like density and heat of vaporization can achieve a correlation coefficient (R²) of ≥ 0.97 with experimental measurements [85].
Quantitative Data from High-Throughput Studies
Table 1: Performance of Different Bayesian Optimization Methods in HEA Design
| BO Method | Key Feature | Application in HEA Design | Performance Advantage |
|---|---|---|---|
| cGP-BO (Conventional) | Models each property independently [42]. | Baseline for comparison. | Suboptimal as it ignores property correlations [42]. |
| MTGP-BO (Multi-Task) | Learns correlations between related properties (tasks) [42]. | Optimizing correlated targets (e.g., CTE & BM) [42]. | More efficient exploration by sharing information across tasks [42]. |
| hDGP-BO (Hierarchical Deep GP) | A hierarchical, more expressive model [42]. | Complex, multi-objective optimization in vast HEA spaces [42]. | Most robust and efficient, accelerates discovery by exploiting correlations [42]. |
| t-EGO (Target-Oriented) | Aims for a specific property value, not just min/max [7]. | Finding shape memory alloys with a target transformation temperature [7]. | Requires fewer iterations to hit a precise target compared to standard BO [7]. |
Table 2: Example High-Throughput Simulation Results for Cr-Co-Ni MEAs
| Composition (CrₓCoᵧNi₂) | Simulated UTS (GPa) | Predicted UTS by ANN (GPa) | Notes | Reference |
|---|---|---|---|---|
| Cr₂₀Co₂₀Ni₆₀ | Data from simulation | Predicted by model | Example of composition with high strength | [83] |
| Cr₅₀Co₂₀Ni₃₀ | Data from simulation | Predicted by model | Example of composition with low cost/density | [83] |
| Correlation (R²) | > 0.99 (between simulated and ML-predicted UTS) | Demonstrates high-fidelity validation | [83] |
The Scientist's Toolkit
Research Reagent Solutions
Table 3: Essential Computational Tools for HEA Discovery
| Item | Function in Workflow | Example/Note |
|---|---|---|
| Interatomic Potentials | Defines the energy and forces between atoms in a simulation. | E.g., SMTB potential for MEAs [83]; OPLS4 for molecular mixtures [85]. |
| MD Simulation Software | Engine for performing high-throughput atomistic simulations. | LAMMPS, GROMACS, AMBER. |
| Screening Dataset | A large, consistent set of in-silico formulations or compositions. | E.g., A dataset of ~30,000 solvent mixtures [85]. |
| Gaussian Process (GP) Library | Core probabilistic model for Bayesian Optimization. | GPy, GPflow, GPyTorch. |
| Variational Autoencoder (VAE) | For compressing high-dimensional spaces (latent space BO). | Used to encode complex process trajectories [86] or molecular structures [29]. |
| Active Learning Framework | Manages the iterative BO loop. | Custom Python scripts coordinating simulation, ML, and acquisition functions. |
Visualizing Multi-Task Bayesian Optimization
The following diagram illustrates the architecture of a Multi-Task Gaussian Process (MTGP), which is key to capturing correlations between different material properties during optimization.
Conclusion
Bayesian optimization in latent space represents a paradigm shift for efficient materials and molecular discovery, effectively transforming complex, discrete search problems into tractable continuous optimization. The integration of advanced encodings like GNNs, coupled with sophisticated strategies such as multi-level and target-oriented BO, demonstrably accelerates the identification of promising candidates while minimizing costly experiments. Key takeaways include the superiority of anisotropic surrogate models for robust performance, the critical need to avoid unnecessary dimensionality that complicates the optimization landscape, and the proven advantage of hierarchical models that exploit correlations between material properties. Looking forward, these computational strategies hold immense promise for biomedical research, from the rational design of drug-like molecules and novel biomaterials to the optimization of clinical formulations, ultimately paving the way for a more data-driven and accelerated path from laboratory concept to clinical application.
References
- 1. Bayesian optimization and explainable machine learning ... [https://www.sciencedirect.com/science/article/abs/pii/S1005030225003664]
- 2. Bayesian Optimization for Materials Design with Mixed Quantitative and Qualitative Variables | Scientific Reports [https://www.nature.com/articles/s41598-020-60652-9]
- 3. Microstructure-aware bayesian materials design [https://www.sciencedirect.com/science/article/abs/pii/S1359645425008730]
- 4. [2310.20258] Advancing Bayesian Optimization via Learning Correlated Latent Space [https://arxiv.org/abs/2310.20258]
- 5. [2201.11872] Local Latent Space Bayesian Optimization over Structured Inputs [https://arxiv.org/abs/2201.11872]
- 6. Bayesian Optimization of Functions over Node Subsets in ... [https://arxiv.org/html/2405.15119v2]
- 7. Materials design with target-oriented Bayesian optimization [https://www.nature.com/articles/s41524-025-01704-4]
- 8. BoTier : multi-objective Bayesian optimization with tiered ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00039d]
- 9. Treatment selection using prototyping in latent-space with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8589171/]
- 10. Define latent spaces by example: optimisation over the ... [https://arxiv.org/html/2509.23800v1]
- 11. HiPPO-based Space Consistency for High-dimensional ... [https://arxiv.org/html/2510.08965v1]
- 12. Bayesian optimization with adaptive surrogate models for ... [https://www.nature.com/articles/s41524-021-00662-x]
- 13. Bayesian optimization - Martin Krasser's Blog [http://krasserm.github.io/2018/03/21/bayesian-optimization/]
- 14. Chapter 7 Optimization | Surrogates [https://bookdown.org/rbg/surrogates/chap7.html]
- 15. On-the-fly closed-loop materials discovery via Bayesian ... [https://www.nature.com/articles/s41467-020-19597-w]
- 16. Acquisition functions — modAL documentation - Read the Docs [https://modal-python.readthedocs.io/en/latest/content/query_strategies/Acquisition-functions.html]
- 17. Discrete vs. Continuous Data: Differences and Examples [https://www.appinio.com/en/blog/market-research/discrete-vs-continuous-data]
- 18. Adaptive representation of molecules and materials in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11851078/]
- 19. Discrete mathematics [https://en.wikipedia.org/wiki/Discrete_mathematics]
- 20. Continuous Representation Methods, Theories, and ... [https://arxiv.org/html/2505.15222v2]
- 21. Navigating chemical space: multi-level Bayesian optimization ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d5sc03855c]
- 22. Joint Composite Latent Space Bayesian Optimization [https://proceedings.mlr.press/v235/maus24a.html]
- 23. Rapid traversal of vast chemical space using machine ... [https://www.nature.com/articles/s43588-025-00777-x]
- 24. Auto-encoding Molecules: Graph-Matching Capabilities ... [https://arxiv.org/abs/2503.00426]
- 25. Transformer graph variational autoencoder for generative ... [https://www.sciencedirect.com/science/article/pii/S0006349525000359]
- 26. Expanding Molecular Design with Graph Variational ... [https://chemrxiv.org/engage/chemrxiv/article-details/6829051ee561f77ed46d15e9]
- 27. Kolmogorov–Arnold graph neural networks for molecular ... [https://www.nature.com/articles/s42256-025-01087-7]
- 28. New AI model explores massive chemical space with minimal ... [https://pme.uchicago.edu/news/new-ai-model-explores-massive-chemical-space-minimal-data]
- 29. Joint Composite Latent Space Bayesian Optimization [https://arxiv.org/html/2311.02213v2]
- 30. multi-level Bayesian optimization with hierarchical coarse ... [https://www.sciencedirect.com/org/science/article/pii/S2041652025012477]
- 31. Navigating Chemical Space: Multi-Level Bayesian ... [https://arxiv.org/html/2505.04169v1]
- 32. Re-thinking the use of VAEs for Bayesian Optimisation ... [https://arxiv.org/html/2507.03910v1]
- 33. Latent Bayesian Optimization via Autoregressive ... [https://arxiv.org/abs/2504.14889]
- 34. A physics informed bayesian optimization approach for ... [https://www.nature.com/articles/s41524-023-01173-7]
- 35. Navigating chemical space: multi-level Bayesian optimization ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc03855c?page=search]
- 36. Phase Separation in Lipid Membranes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3062215/]
- 37. A protocol for mimicking lipid-mediated phase separation ... [https://www.sciencedirect.com/science/article/pii/S2666166722005895]
- 38. Refining coarse-grained molecular topologies: a Bayesian ... [https://www.nature.com/articles/s41524-025-01729-9]
- 39. Generative Inversion for Property-Targeted Materials Design [https://arxiv.org/html/2508.07798v1]
- 40. Machine Learning-Assisted Discovery of Empirical Rule for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12113529/]
- 41. Accelerated design of a novel wide thermal hysteresis NiTi ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838825008928]
- 42. Hierarchical Gaussian process-based Bayesian ... [https://www.sciencedirect.com/science/article/abs/pii/S1359645425002009]
- 43. Deep Gaussian Process-based Cost-Aware Batch ... [https://arxiv.org/html/2509.14408v1]
- 44. Design of High Temperature Ti-Pd-Cr Shape Memory ... [https://www.nature.com/articles/srep28244]
- 45. Nonlinear Dimensionality Reduction Techniques for ... [https://arxiv.org/html/2510.15435v1]
- 46. A simple approach to high-dimensional Bayesian ... [https://www.sciencedirect.com/science/article/abs/pii/S0020025524009708]
- 47. MITIGATING OVER-EXPLORATION IN LATENT SPACE ... [https://openreview.net/forum?id=0jm6gkAuYH]
- 48. Large language models as uncertainty-calibrated ... [https://arxiv.org/html/2504.06265v3]
- 49. Adaptive subspace Bayesian optimization over molecular ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00188a]
- 50. Bayesian optimization of biodegradable polymers via ... [https://www.nature.com/articles/s41529-025-00613-7]
- 51. Bayesian Optimization with Gaussian Processes Assisted by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12128098/]
- 52. Ranking over Regression for Bayesian Optimization and ... [https://arxiv.org/html/2410.09290v1]
- 53. How does Bayesian Optimization balance exploration with ... [https://stats.stackexchange.com/questions/509812/how-does-bayesian-optimization-balance-exploration-with-exploitation]
- 54. Bayesian optimization acquisition functions for accelerated ... [https://www.nature.com/articles/s41524-024-01391-7]
- 55. Accelerating material discovery with a threshold-driven ... [https://www.sciencedirect.com/science/article/pii/S2213846324002414]
- 56. Targeted materials discovery using Bayesian algorithm ... [https://www.nature.com/articles/s41524-024-01326-2]
- 57. Evolution-guided Bayesian optimization for constrained ... [https://www.nature.com/articles/s41524-024-01274-x]
- 58. GOLLuM: Gaussian Process Optimized LLMs [https://arxiv.org/html/2504.06265v1]
- 59. Constrained Multi-objective Bayesian Optimization through ... [https://arxiv.org/abs/2411.03641]
- 60. Materials science optimization benchmark dataset for multi- ... [https://www.sciencedirect.com/science/article/pii/S2352340923005875]
- 61. Materials Science Optimization Benchmark Dataset for ... [https://chemrxiv.org/engage/chemrxiv/article-details/640654476642bf8c8f1458ae]
- 62. Data efficiency of classification strategies for chemical and ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00298a]
- 63. Bayesian optimization with known experimental and ... [https://pubs.rsc.org/en/content/articlelanding/2022/dd/d2dd00028h]
- 64. [1506.01349] Bayesian optimization for materials design [https://arxiv.org/abs/1506.01349]
- 65. Advancing Bayesian Optimization: Efficient Strategies for ... [https://insane.iit.demokritos.gr/2025/02/13/advancing-bayesian-optimization-efficient-strategies-for-accelerated-materials-discovery/]
- 66. Benchmarking the performance of Bayesian optimization ... [https://www.nature.com/articles/s41524-021-00656-9]
- 67. Benchmarking self-driving labs - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00337g]
- 68. Best Practices for Multi-Fidelity Bayesian Optimization in ... [https://arxiv.org/html/2410.00544v3]
- 69. Best practices for multi-fidelity Bayesian optimization in ... [https://pubmed.ncbi.nlm.nih.gov/40702272/]
- 70. Unlocking the black box beyond Bayesian global ... [https://www.nature.com/articles/s41524-025-01639-w]
- 71. Bayesian optimization with experimental failure for high- ... [https://www.nature.com/articles/s41524-022-00859-8]
- 72. NVIDIA Accelerated Computing Enables Materials Discovery [https://blogs.nvidia.com/blog/ai-science-materials-discovery-sc25/]
- 73. Accurate and uncertainty-aware multi-task prediction of ... [https://www.nature.com/articles/s41524-025-01811-2]
- 74. Accurate and Uncertainty-Aware Multi-Task Prediction of ... [https://arxiv.org/abs/2506.14828]
- 75. Deep Gaussian Process-based Cost-Aware Batch ... [https://arxiv.org/html/2509.14408]
- 76. Anisotropic Gaussian Kernel [https://www.emergentmind.com/topics/anisotropic-gaussian-kernel]
- 77. Experiment-in-loop interactive optimization of polymer ... [https://pubs.rsc.org/en/content/articlehtml/2025/mh/d4mh01606h]
- 78. Kernels for Gaussian Processes | anders e [https://anhosu.com/post/kernels-r/]
- 79. Sparse methods for automatic relevance determination [https://www.sciencedirect.com/science/article/abs/pii/S0167278921000014]
- 80. Anisotropic Kernels for Coordinate-Based Meta-Analyses ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3919071/]
- 81. Sparse modeling based Bayesian optimization for ... [https://www.sciencedirect.com/org/science/article/pii/S2633540925002087]
- 82. A review on high-throughput development of high-entropy ... [https://www.oaepublish.com/articles/jmi.2022.41]
- 83. High-throughput simulation combined machine learning ... [https://www.sciencedirect.com/science/article/abs/pii/S1005030220307027]
- 84. High-Throughput Calculations for High-Entropy Alloys [https://www.frontiersin.org/journals/materials/articles/10.3389/fmats.2020.00290/full]
- 85. Leveraging high-throughput molecular simulations and ... [https://www.nature.com/articles/s41524-025-01552-2]
- 86. Bayesian optimization in continuous spaces via virtual ... [https://pubs.rsc.org/en/content/articlelanding/2022/dd/d2dd00065b]