LaMer Mechanism Revisited: Diffusion vs. Surface-Controlled Growth in Modern Nanomaterial Design
This article provides a comprehensive analysis of the LaMer mechanism, a foundational model describing burst nucleation and diffusion-controlled growth for monodisperse nanoparticles.
LaMer Mechanism Revisited: Diffusion vs. Surface-Controlled Growth in Modern Nanomaterial Design
Abstract
This article provides a comprehensive analysis of the LaMer mechanism, a foundational model describing burst nucleation and diffusion-controlled growth for monodisperse nanoparticles. We critically examine its principles, its evolution over 70 years, and the crucial distinction between diffusion-controlled and surface-integration-controlled (or interfacial) growth processes. Tailored for researchers, scientists, and drug development professionals, this review synthesizes historical context, modern validation studies, and competing models like Finke-Watzky. We explore methodological approaches for differentiating growth types, troubleshooting synthesis for size and morphology control, and discuss the implications of these crystallization pathways for the development of advanced nanomaterials and nanomedicines.
Deconstructing LaMer's Legacy: The Fundamentals of Burst Nucleation and Growth Control
The classical model for particle formation, first articulated by LaMer and Dinegar in their seminal 1950 paper, introduced the foundational concepts of 'burst nucleation' and 'diffusion-controlled growth' to explain the formation of monodispersed hydrosols [1] [2]. For decades, this model has served as the principal theoretical framework for understanding crystallization processes across diverse scientific and industrial fields, from materials science to pharmaceutical development [3] [4]. The model's enduring significance lies in its elegant explanation of how uniform particles can be synthesized through the temporal separation of nucleation and growth stages. This whitepaper examines the core principles of the original LaMer model, its quantitative foundations, and its critical evolution in modern research, particularly within the context of distinguishing between diffusion-controlled and surface-reaction-controlled growth mechanisms. As contemporary research continues to refine and challenge aspects of the classical model, understanding its original formulation remains essential for researchers investigating crystallization kinetics, nanoparticle synthesis, and polymorph control in drug development.
Core Principles of the 1950 LaMer Model
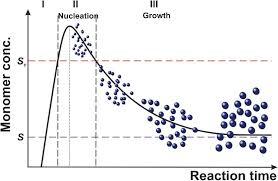
The LaMer model conceptualizes nanoparticle formation as a three-stage process driven by supersaturation, graphically represented as the now-famous LaMer curve [3]. The model's core premise is that temporal separation of the nucleation and growth phases is essential for achieving monodisperse particles.
The Three Stages of the LaMer Mechanism
Stage I: Precursor Production and Supersaturation: Monomers (atoms, ions, or molecules) are generated in solution, typically through chemical reduction or thermal decomposition, causing the monomer concentration to rise steadily. Once the concentration exceeds the equilibrium solubility (
C_s), the solution becomes supersaturated. However, nucleation does not occur immediately, allowing the monomer concentration to continue increasing well above the minimum supersaturation concentration (C_min^nu) required for nucleation [3].Stage II: Burst Nucleation: When the monomer concentration reaches a critical supersaturation threshold, nucleation occurs explosively [1]. LaMer described this nucleation rate as becoming "so exceedingly sensitive to an increase in concentration that the rate becomes effectively infinite" [1] [2]. This rapid, "burst" nucleation event depletes the monomer concentration rapidly until it falls below
C_min^nu, effectively quenching further nucleation. The cessation of nucleation after this brief burst is crucial for obtaining a uniform population of nuclei [3].Stage III: Diffusion-Controlled Growth: After nucleation is complete, the existing nuclei grow via diffusion-controlled monomer addition [1] [2]. Monomers from the bulk solution diffuse to and are incorporated into the particle surfaces. Growth continues until the monomer concentration decreases to the equilibrium solubility (
C_s), at which point the system reaches equilibrium and growth ceases [3].
Visualizing the LaMer Mechanism
The following diagram illustrates the relationship between monomer concentration and the stages of nucleation and growth, known as the LaMer curve, alongside the corresponding microscopic processes.
Quantitative Foundations: Assumptions and Rate Laws
The LaMer model is built upon specific quantitative foundations derived from Classical Nucleation Theory (CNT) and diffusion kinetics. A critical analysis of its mathematical assumptions reveals the model's strengths and limitations [1] [2].
Key Model Assumptions and Limitations
Table 1: Core Assumptions of the Original LaMer Model
| Assumption Category | Specific Premise | Modern Critical Analysis |
|---|---|---|
| Nucleation Kinetics | "Effectively infinite" nucleation rate at critical supersaturation [1] | Later studies show continuous nucleation often occurs; "burst" is not strictly necessary for monodispersity [5] [3] |
| Growth Mechanism | Purely diffusion-controlled growth; surface integration is instantaneous [2] | Growth is often co-controlled by diffusion and surface reaction kinetics [6] |
| Nucleation & Growth Separation | Clear temporal separation between nucleation (Stage II) and growth (Stage III) [3] | Overlap between nucleation and growth is commonly observed, challenging clear separation [5] |
| Growth Units | Atoms/ions/monomers are the sole building blocks [3] | Non-classical pathways involving particle-particle coalescence are significant [3] |
| Theoretical Foundation | Based on Classical Nucleation Theory (CNT) and Fickian diffusion [1] | CNT has limitations; alternative mechanisms like pre-nucleation clusters exist [4] |
Governing Rate Laws for Diffusion vs. Surface Control
The distinction between diffusion-controlled and surface-reaction-controlled growth is fundamental to crystallization research. Each mechanism follows a distinct rate law with characteristic temporal scaling.
Table 2: Key Rate Laws for Different Growth Control Mechanisms
| Process | Governing Equation | Rate-Limiting Step | Temporal Scaling (Size vs. Time) | Applicable Scenarios |
|---|---|---|---|---|
| Diffusion-Controlled Growth | dR/dt = D * ([C_b] - [C_s]) / R [6] [7] |
Mass transport of monomers to particle surface [7] | R² ∝ t (Parabolic Law) [6] |
Fast surface reaction kinetics, low monomer concentration, high viscosity |
| Surface-Reaction-Controlled Growth | dR/dt = k_s * ([C_b] - [C_s]) [6] |
Integration of monomers into crystal lattice at surface [6] | R ∝ t (Linear Law) [6] |
Slow surface integration, high monomer concentration, presence of growth inhibitors |
| Ostwald Ripening (Diffusion-Control) | LSW Theory: dR/dt ∝ 1/R² [6] |
Diffusion of monomers from small to large particles [6] | R̄³ ∝ t [6] |
Late-stage coarsening at low supersaturation |
| Ostwald Ripening (Interface-Control) | LSW Theory: dR/dt ∝ 1/R [6] |
Surface reaction/dissolution kinetics [6] | R̄² ∝ t [6] |
Coarsening where surface reaction is slower than diffusion |
The general solution for the growth rate dR/dt can be expressed as a combination of resistances [6]:
dR/dt = ([C_b] - [C_s]) / (R/D + 1/k_s)
where [C_b] is bulk concentration, [C_s] is surface solubility, D is diffusion coefficient, and k_s is surface reaction constant. This illustrates that the slower process exerts greater control over the overall growth rate.
Modern Experimental Validation & Protocol
Recent advances in characterization techniques and computational modeling have enabled direct experimental investigation of the LaMer mechanism's principles, particularly the distinction between diffusion and surface-controlled pathways.
Phase Field Simulation of Perovskite Crystallization
Advanced computational methods now allow for the direct simulation of crystallization processes, validating and extending LaMer's principles.
Objective: To simulate solution-based perovskite thin film formation and quantify the impact of evaporation rate on nucleation and growth dynamics [8].
Detailed Methodology:
- Model Setup: Implement a phase field (PF) model that couples fluid mechanics, evaporation, and crystallization dynamics. The model uses a free energy functional to describe the thermodynamics of the phase transition, with kinetic evolution governed by the Allen-Cahn and Cahn-Hilliard equations [8].
- Parameter Definition: Define initial simulation parameters including precursor concentration, solvent properties, evaporation rate constant (
k_evap), and crystallization rate constant (k_cryst). - Process Simulation:
- Simulate solvent evaporation, leading to supersaturation of the precursor.
- Model nucleation events when local supersaturation exceeds the critical threshold.
- Track crystal growth via monomer addition, with rates computed based on both diffusion and surface integration kinetics [8].
- Morphology Analysis: Quantify final film morphology characteristics including surface roughness, pinhole density, and grain size distribution as a function of the processing parameters [8].
Key Findings: The simulation recovers the experimentally observed transition from a porous, pinhole-rich film at low evaporation rates to a smooth, compact film at high evaporation rates. It identifies the ratio of evaporation rate to crystallization rate (k_evap/k_cryst) as the key parameter dictating final morphology, with higher ratios favoring high-quality films [8]. This finding provides a robust design rule for process optimization.
In Situ Monitoring of Gold Nanocrystal Synthesis
Objective: To experimentally observe the nucleation and growth stages during the synthesis of gold nanoparticles and determine the rate-controlling mechanism [3].
Detailed Methodology:
- Reaction Setup: Prepare an aqueous solution of chloroauric acid (HAuCl₄). Use a syringe pump to precisely inject the reducing agent (e.g., sodium citrate) under vigorous stirring [3].
- In Situ Monitoring:
- Use UV-Vis absorbance spectroscopy to track the formation of gold nuclei and plasmon peak evolution.
- Employ dynamic light scattering (DLS) to monitor the hydrodynamic radius growth in real-time.
- Utilize synchrotron-based SAXS/WAXS to obtain crystal structure and size information during early formation stages [3].
- Growth Kinetics Analysis:
- Plot nanoparticle radius (
R) versus time (t). - Fit the data to power-law equations:
R^n = K*t. - Determine the dominant growth mechanism by identifying the best-fit exponent:
n≈2suggests surface-reaction control, whilen≈3suggests diffusion control [6].
- Plot nanoparticle radius (
- Ex Situ Characterization: After reaction quenching, use TEM to analyze final particle size distribution and morphology [3].
Visualizing the Experimental Workflow
The following diagram outlines the general workflow for a modern nanocrystal synthesis and mechanistic analysis experiment, integrating both simulation and experimental approaches.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Studying Nucleation and Growth Mechanisms
| Reagent/Material | Function in Experiment | Specific Role in Mechanism Study |
|---|---|---|
| Chloroauric Acid (HAuCl₄) | Gold precursor for model nanocrystal synthesis [3] | Well-understood reduction kinetics allow isolation of nucleation/growth variables |
| Sodium Citrate | Reducing agent and weak stabilizer for gold nanoparticles [3] | Controls reduction rate, affecting supersaturation and nucleation burst characteristics |
| Alkanethiols | Strong-binding ligands for metal nanocrystals [5] | Modulate surface reaction kinetics; can switch growth from diffusion to surface-control |
| Polyvinylpyrrolidone (PVP) | Steric stabilizer and crystal habit modifier [3] | Selective facet binding alters surface integration energy, influencing growth mechanism |
| Microfluidic Reactors | Continuous flow platforms for nanocrystal synthesis [4] | Provide rapid, uniform mixing for precise separation of nucleation and growth stages |
| DMSO (Dimethyl Sulfoxide) | Coordinating solvent for perovskite precursors [8] | Forms intermediates that modulate supersaturation profile and crystallization pathway |
| Phase Field Modeling Software | Computational framework for simulating crystallization [8] | Decouples evaporation, diffusion, and surface integration kinetics for mechanistic insight |
Evolution Beyond the Classical Model
While the LaMer model remains foundational, modern research has significantly expanded our understanding of crystallization, identifying several non-classical pathways and more complex kinetic scenarios.
From "Burst Nucleation" to Continuous Mechanisms
The Finke-Watzky model and related subsequent work represent a major paradigm shift by modeling nucleation as a continuous process rather than a single burst event [5]. This mechanism, which involves slow continuous nucleation (A → B, rate constant k_1) alongside autocatalytic surface growth (A + B → 2B, rate constant k_2), can also produce narrow particle size distributions, challenging the long-held belief that "burst nucleation" was strictly necessary for monodispersity [5].
Particle-Mediated Non-Classical Growth
A significant advancement beyond the LaMer model is the documentation of particle-mediated growth pathways, where nanoparticles themselves act as building blocks [3]. This non-classical growth occurs via:
- Oriented Attachment (OA): Crystallographically aligned nanoparticles spontaneously attach and fuse, forming larger single crystals or mesocrystals [3].
- Non-Oriented Attachment: Random aggregation of nanoparticles followed by coalescence and recrystallization into larger structures [3].
- Mechanistic Complexity: These pathways introduce intermediate stages of aggregation and coalescence that are not accounted for in the classical atom-/monomer-addition model [3].
Coarsening Kinetics: Diffusion and Interface Control
The later stages of crystallization often involve Ostwald ripening, where larger particles grow at the expense of smaller ones to reduce overall surface energy. The Lifshitz-Slyozov-Wagner (LSW) theory describes this process, with the temporal power law exponent (n in R̄ⁿ ∝ t) indicating the rate-controlling mechanism: n=3 for diffusion-control and n=2 for interface-reaction-control [6]. In real systems, coarsening is often co-controlled by both diffusion and interface reactions, leading to kinetic behavior and particle size distributions that fall between the classical limits [6].
The original 1950 LaMer model, with its elegant formulation of 'burst nucleation' and 'diffusion-controlled growth', established a fundamental framework for understanding crystallization that remains relevant today. Its core insight—that temporal separation of nucleation and growth is key to achieving uniform particles—continues to guide synthetic strategies in fields ranging from nanomaterials to pharmaceutical development. However, modern research has revealed a more complex reality, where growth is often co-controlled by both diffusion and surface reactions, nucleation can be continuous rather than instantaneous, and non-classical pathways involving particle-particle interactions play significant roles. For today's researcher, the most powerful approach combines the conceptual clarity of the LaMer model with contemporary understanding of these multiple, coexisting mechanisms. This integrated perspective enables more precise control over particle size, morphology, and polymorphic outcome—objectives that lie at the heart of advanced materials design and drug development.
The "LaMer mechanism," introduced by LaMer and Dinegar in 1950, describes a pathway to synthesize monodisperse colloidal particles by separating the nucleation and growth stages [9]. This model posits an initial burst of homogeneous nucleation when monomer concentration surpasses a critical supersaturation level, followed by a diffusion-controlled growth phase that prevents further nucleation [3]. The theoretical underpinnings of this initial nucleation burst are rooted in Classical Nucleation Theory (CNT) and the broader concepts of Fluctuation Theory. These theories provide the fundamental framework for understanding how the first stable solid particles (nuclei) emerge from a supersaturated solution, a process central to controlling crystal size and morphology in applications ranging from pharmaceutical development to nanomaterials synthesis [10]. This guide examines the core principles of CNT and Fluctuation Theory, their quantitative formulations, and their critical role in the context of LaMer's mechanism.
The Fundamentals of Classical Nucleation Theory (CNT)
Thermodynamic and Kinetic Foundations
Classical Nucleation Theory is the primary theoretical model used to quantitatively study the kinetics of nucleation [11] [12]. Developed from the works of Gibbs, Volmer, Weber, Becker, and Döring, CNT provides a conceptual framework for understanding the first-order phase transition from a supersaturated solution to a solid crystal [11] [10].
The formation of a crystal nucleus is governed by a competition between bulk free energy gain and surface free energy cost [11]. In a supersaturated solution (Δμ = μsolute - μcrystal > 0), the formation of a solid cluster leads to a free energy loss of -nΔμ, where n is the number of molecules in the cluster. Conversely, creating a phase boundary with surface free energy γ leads to a free energy gain proportional to the cluster's surface area [10].
For a spherical cluster of radius r, the total free energy change is given by [12]:
ΔG = (4/3)πr³Δg_v + 4πr²σ
Where Δg_v is the free energy change per unit volume (negative in supersaturated solutions), and σ is the surface free energy per unit area.
This relationship produces the characteristic free energy profile shown in the diagram below, where ΔG reaches a maximum at the critical nucleus size [10] [12].
Diagram: Free energy landscape of nucleation showing the critical nucleus size as the barrier between unstable embryos and stable growing particles.
The Critical Nucleus and Energy Barrier
The critical nucleus size rc represents the threshold where the free energy barrier ΔG* is maximized [12]. Clusters smaller than rc (embryos) are unstable and tend to dissolve, while those larger than r_c are stable and likely to grow [10]. The critical radius and corresponding energy barrier are given by [12]:
r_c = 2σ/|Δg_v|
ΔG* = 16πσ³/(3|Δg_v|²)
The rate of nucleation R—the number of nuclei formed per unit volume per unit time—follows an Arrhenius-type dependence on this energy barrier [12]:
R = N_S Z j exp(-ΔG*/k_B T)
Where NS is the number of potential nucleation sites, Z is the Zeldovich factor, j is the monomer attachment rate, kB is Boltzmann's constant, and T is temperature [12].
Quantitative Framework: Key Parameters and Equations
Core Equations of Classical Nucleation Theory
Table 1: Fundamental Equations in Classical Nucleation Theory
| Parameter | Mathematical Expression | Physical Significance | Reference | ||
|---|---|---|---|---|---|
| Supersaturation | Δμ = kB T ln(S) where S = C/Csat | Driving force for nucleation; C is concentration, C_sat is saturation concentration | [10] | ||
| Critical Radius | r_c = 2σ/ | Δg_v | Size of nucleus with equal probability of growth and dissolution | [12] | |
| Nucleation Barrier | ΔG* = 16πσ³/(3 | Δg_v | ²) = (1/2)n*Δμ | Energy barrier that must be overcome for stable nucleus formation | [10] [12] |
| Nucleation Rate | R = NS Z j exp(-ΔG*/kB T) | Number of nuclei formed per unit volume per unit time | [12] | ||
| Free Energy Change | ΔG = -nΔμ + 6a²n²/³α (for cubic nucleus) | Total free energy change for cluster of n molecules | [10] |
Experimental Determinations of Nucleation Parameters
Table 2: Experimentally Accessible Nucleation Quantities
| Measurable Quantity | Experimental Approach | Information Obtained | Reference |
|---|---|---|---|
| Nucleation Rate (R) | Induction time measurements in small droplets | Effective nucleation rate under constant supersaturation | [13] |
| Nucleation Time (t_N) | Cumulative probability P(t) that nucleation has not occurred | Statistical distribution of nucleation times | [13] |
| Critical Supersaturation | Concentration at which rapid nucleation is first observed | Minimum driving force required for nucleation | [9] |
| Crystal Size Distribution | Analysis of final crystal populations | Insight into whether nucleation was burst-like or continuous | [10] |
Fluctuation Theory and the Molecular Basis of Nucleation
Fluctuation theory provides the statistical mechanical foundation for nucleation phenomena, explaining how random thermal motions of molecules can occasionally generate ordered clusters capable of surpassing the critical size [10]. In the pre-nucleation stage, microscopic crystallites continuously form and dissolve through stochastic fluctuations, as illustrated in computer simulations where the size of the largest crystallite fluctuates until one crosses the nucleation barrier [13].
The concept of pre-nucleation clusters (PNCs) represents an important development beyond classical CNT. These are thermodynamically stable, highly dynamic solute species that form independently of supersaturation level and lack a defined phase interface [11]. According to the non-classical two-step nucleation mechanism, crystalline nuclei appear inside pre-existing metastable clusters of dense liquid suspended in solution [10]. This mechanism helps explain several long-standing puzzles, including nucleation rates that are many orders of magnitude lower than theoretical predictions [10].
Connecting CNT to the LaMer Mechanism: Diffusion vs. Surface-Controlled Growth
The LaMer mechanism explicitly relies on CNT principles to achieve monodisperse particles. The model requires "effectively infinite nucleation" followed by "diffusion-controlled growth" [9]. The initial burst nucleation occurs when supersaturation reaches its maximum, creating a large number of nuclei simultaneously. Once nucleation depletes monomers below the critical concentration for nucleation (C_min^nu), the growth stage begins without further nucleation [3].
The transition from nucleation to growth is crucial for achieving monodispersity. As LaMer recognized, when the concentration of growth monomers falls below the minimum critical concentration required for nucleation, crystal development continues but nucleation ceases [4]. This creates the separation between nucleation and growth stages essential for obtaining uniform particles.
The growth phase itself can proceed through different pathways [4]:
- Diffusion-controlled growth: Occurs when monomer concentration falls below the critical nucleation concentration but remains available for growth; the growth rate is limited by diffusion of monomers to the crystal surface.
- Surface-process-controlled growth: Occurs when diffusion from bulk to growth surface is fast enough that surface integration processes control the growth rate.
The following diagram illustrates the complete LaMer process within the CNT framework:
Diagram: The three stages of the LaMer mechanism showing the relationship between monomer concentration and nucleation/growth processes.
Experimental Methodologies for Studying Nucleation
Quantitative Studies at Constant Supersaturation
The cleanest experimental data on crystal nucleation comes from studies of small droplets at constant supersaturation (isothermal crystallization) [13]. This approach eliminates complications from time-varying free energy barriers. Key methodological considerations include:
- Droplet-based experiments: Using many small, nominally identical droplets allows statistical analysis of nucleation times while ensuring that only one nucleation event occurs per droplet [13].
- Cumulative probability analysis: Plotting P(t)—the probability that nucleation has not occurred by time t—provides a robust way to analyze nucleation kinetics [13].
- Constant supersaturation maintenance: Temperature, pressure, and concentration must be carefully controlled to maintain constant driving force for nucleation [13].
When the effective nucleation rate is constant, P(t) follows a simple exponential decay: P(t) = exp(-kt), where k is the nucleation rate [13]. Deviations from this behavior indicate more complex nucleation mechanisms or time-dependent surfaces.
Advanced Characterization Techniques
Recent advances in experimental methods have enabled more direct observation of nucleation processes:
- In situ microscopy and spectroscopy: Techniques like high-speed atomic force and electron microscopy allow real-time monitoring of nucleation and crystal development processes [4].
- Computational modeling: Molecular dynamics simulations and density functional theory computations provide atomistic-level information on nucleation energetics and kinetics [4].
- Process intensification strategies: Microreactors and continuous flow systems provide better mixing, heat transfer, and process control for studying nucleation kinetics [4].
The Scientist's Toolkit: Essential Reagents and Methods
Key Research Reagent Solutions
Table 3: Essential Materials for Nucleation and Growth Studies
| Reagent/Material | Function in Nucleation Studies | Example Application | Reference |
|---|---|---|---|
| Small molecule organics | Model compounds for fundamental nucleation studies | Studying polymorph selection and nucleation kinetics | [10] |
| Proteins (e.g., Lysozyme) | Model systems for protein crystallization | Investigating two-step nucleation mechanism | [10] |
| Metal precursors | Source of monomers for nanocrystal synthesis | InAs quantum dot synthesis via continuous injection | [14] |
| Surfactants/Ligands | Control surface energy and colloidal stability | Tuning nucleation barriers and growth kinetics | [3] |
| Microfluidic devices | Enable precise supersaturation control | Studying nucleation kinetics at constant supersaturation | [4] [13] |
Classical Nucleation Theory and Fluctuation Theory provide the fundamental framework for understanding the initial stages of particle formation in the LaMer mechanism. While CNT successfully explains the basic dependence of nucleation on supersaturation and surface energy, recent research has revealed limitations in its quantitative predictive power and led to the development of non-classical pathways like the two-step mechanism [10].
Future research directions include:
- Developing more accurate computational models that bridge molecular-scale interactions with macroscopic nucleation rates [4]
- Exploring non-classical nucleation pathways involving pre-nucleation clusters and particle-mediated growth [3]
- Integrating advanced characterization techniques with theoretical modeling to obtain a more complete molecular-level understanding of nucleation [4]
- Applying nucleation control strategies to pharmaceutical development, where polymorphism and crystal size distribution critically impact drug efficacy and processing [10]
The continued refinement of nucleation theory remains essential for advancing materials synthesis, pharmaceutical development, and our understanding of phase transitions in both natural and engineered systems.
In the controlled synthesis of crystals, from pharmaceuticals to nanomaterials, determining the rate-limiting step of growth is a fundamental scientific and engineering challenge. The process is primarily governed by two distinct regimes: diffusion control and surface reaction control (often termed surface control). In the diffusion-controlled regime, the rate of crystal growth is limited by the physical transport of solute molecules or particles through the solution to the crystal surface. In contrast, the surface-reaction-controlled regime is limited by the kinetics of the incorporation of these solute entities into the crystal lattice at the interface [7] [15]. Understanding which regime dominates is essential for manipulating final crystal properties, including size, morphology, purity, and functionality. This knowledge is framed within a century of research on phase-change mechanisms, with LaMer's model of "diffusion-controlled growth" serving as a foundational, though often debated, concept [1] [3].
This whitepaper provides an in-depth technical guide to these two growth regimes. It details their theoretical foundations, describes experimental methodologies for their identification, and discusses their implications within the context of modern crystal growth research, particularly the evolution of thought beyond the classical LaMer mechanism.
Theoretical Foundations
The Diffusion-Controlled Regime
Diffusion-controlled reactions occur when the reaction rate is equal to the rate of transport of the reactants through the reaction medium [16]. In the context of crystal growth, this means that molecules (or ions, atoms) diffuse from the bulk solution to the crystal surface more slowly than they can be incorporated into the lattice.
The process can be modeled by considering the flux of solute B toward a growing crystal A. Under steady-state conditions, and assuming no intermolecular forces (U(r) ≈ 0), the resulting rate constant k for a bimolecular reaction is a combination of the intrinsic reaction rate constant k_r and the diffusion-controlled rate constant k_D [16]:
k = (k_D * k_r) / (k_r + k_D)
When the reaction at the surface is very fast (k_r >> k_D), the equation simplifies to k ≈ k_D, meaning the reaction is entirely diffusion-limited. The diffusion-limited rate constant for a spherical particle in a solution with low intermolecular forces is often approximated as k_D = 4πR_AB * D_AB, where R_AB is the encounter distance and D_AB is the mutual diffusion coefficient [16].
A key consequence of this regime is the formation of a depletion zone near the reactive surface, where the concentration of the solute is lower than in the bulk solution [7]. The overall growth rate is then governed by the diffusive flux onto the surface, J(t), which for a perfect sink (instantaneous surface reaction) is given by Fick's first law [7]:
J(t) = ∫_C dx -D (∂[A](x,t))/∂n
Table 1: Key Characteristics of Diffusion-Controlled Growth
| Parameter | Description | Mathematical Expression |
|---|---|---|
| Rate Law | Growth rate is proportional to the bulk concentration gradient. | J = -D ∇c |
| Depletion Zone | A region of lower solute concentration forms near the crystal surface. | c_surface < c_bulk |
| Viscosity Dependence | Growth rate is inversely proportional to solvent viscosity (η). | k_D ∝ 1/η [16] |
| Agitation Dependence | The observed growth rate is affected by stirring or agitation. | Rate increases with stirring [16] |
The Surface-Reaction-Controlled Regime
In this regime, the transport of solute to the surface is rapid, and the concentration at the interface is effectively equal to the bulk concentration. The rate-limiting step is the reaction at the surface itself, which involves processes such as the adsorption of solute, desolvation, surface diffusion, and integration into the crystal lattice [15].
This kinetic limitation is formally implemented via a boundary condition on the catalytic (growth) surface. The Collins and Kimball model replaces the perfect-sink Dirichlet condition ([A] = 0 at the surface) with a Robin or radiative boundary condition [7]:
-D (∂[A](x,t))/∂n = κ [A](x,t)
Here, κ is the surface reactivity, a measure of the intrinsic kinetic rate of the surface reaction. A low κ value signifies a slow surface incorporation step, leading to surface-reaction control. In this case, the surface concentration of the reactant, [A], is finite and greater than zero.
A prime example of this regime is the dissolution of calcite, where at higher pH values (lower acidity), the velocity of step-edge retreat is constant and controlled by the surface reaction kinetics rather than by the diffusion of protons to the surface [15].
Table 2: Key Characteristics of Surface-Reaction-Controlled Growth
| Parameter | Description | Implication |
|---|---|---|
| Rate Law | Growth rate is proportional to surface area and driving force (e.g., supersaturation). | Often follows a nonlinear function of supersaturation. |
| Surface Concentration | Solute concentration at the crystal surface is close to the bulk concentration. | c_surface ≈ c_bulk |
| Agitation Dependence | The observed growth rate is not affected by stirring or agitation. | Mixing has minimal effect on rate [16]. |
| Structural Sensitivity | Growth rate is highly dependent on crystal face, presence of defects, and additives. | Enables crystal morphology engineering. |
The LaMer Model and Its Modern Context
The LaMer model, introduced in 1950, is a cornerstone of particle formation theory. It describes the formation of monodisperse colloids through three stages: I) a gradual increase in monomer concentration, II) an "effectively infinite" burst of nucleation once a critical supersaturation (C_min^nu) is reached, and III) diffusion-controlled growth of the nuclei as the monomer concentration drops below the nucleation threshold [1] [3].
The model's postulation of "diffusion-controlled growth" has been widely cited and applied for decades. However, a critical analysis of 70 years of subsequent research reveals that the experimental evidence for purely diffusion-controlled growth is often lacking. Many systems demonstrate more complex behavior, involving a combination of diffusion, surface reaction kinetics, and non-classical pathways [1].
Modern research has moved "beyond the LaMer curve" to include non-classical growth mechanisms, where nanoparticles or clusters—not individual atoms or molecules—act as the primary building blocks for crystal growth via aggregation and coalescence [3]. This particle-mediated pathway represents a significant expansion of the classical theory and can lead to unique hierarchical structures.
Experimental Protocols for Regime Identification
Distinguishing between diffusion and surface-reaction control requires carefully designed experiments that probe the kinetics and spatial distribution of solute during growth.
Method 1: Agitation and Flow Rate Dependence
This is a classical test for diffusion control in a heterogeneous reaction [16].
- Principle: If the crystal growth rate increases with increased stirring speed or solution flow rate, the process is likely diffusion-controlled. Enhanced mixing reduces the thickness of the diffusion boundary layer, facilitating solute transport to the crystal surface. If the rate is independent of agitation, the process is surface-reaction-controlled.
- Protocol:
- Use a well-instrumented crystallizer (e.g., a jacketed reactor with controlled temperature).
- Prepare a supersaturated solution of the target compound (e.g., Aceclofenac in acetone or methyl acetate [17]).
- Introduce crystal seeds of known size and surface area.
- Measure the crystal growth rate (e.g., via in-situ image analysis or particle size monitoring) under different, precisely controlled stirring speeds (e.g., 100, 200, 300, 400 rpm).
- Maintain constant temperature and supersaturation throughout all experiments.
- Data Analysis: Plot the measured growth rate against stirring speed. A significant positive correlation indicates diffusion control. No correlation indicates surface reaction control.
Method 2: Direct Surface Observation with Liquid-Cell AFM
Atomic Force Microscopy (AFM) allows for direct, real-time observation of molecular-scale processes on a crystal surface, providing unambiguous evidence of the rate-determining step [15].
- Principle: By monitoring the velocity of step edges on a crystal surface under different conditions, one can determine the controlling regime. In surface-reaction control, step velocities are constant and anisotropic (dependent on crystallographic direction). A change in mechanism, such as the onset of step meandering and a sharp increase in velocity at very high driving forces, can signal a transition to diffusion control.
- Protocol (as applied to calcite dissolution [15]):
- Mount a freshly cleaved calcite (101̄4) crystal in a liquid-cell AFM.
- Use a flow-through cell to introduce solutions of varying pH (from neutral to strongly acidic) at a controlled flow rate.
- Under constant ambient conditions, image the crystal surface in real-time to observe the formation and growth of etch pits.
- Measure the retreat velocities of steps oriented along specific crystallographic directions (e.g., [4̄41]+ and [481̄]+) for each pH condition.
- Correlate the measured step velocities with the solution chemistry.
- Data Analysis: The study found that at pH 2.7 and above, step velocities were relatively constant and anisotropic, indicative of surface control. Below pH 2.7, a significant increase in velocity and the appearance of meandering steps indicated a shift toward diffusion control, as the surface reaction was no longer able to keep up with the high flux of protons [15].
Method 3: Crystal Regeneration and Micro-Mechanical Testing
This method investigates growth by studying the repair of crystal defects, providing insight into site-specific reactivity [17].
- Principle: A crystal is intentionally fractured, and its regrowth from the broken surface is observed. The propensity and morphology of regeneration under controlled supersaturation reveal the relative rates of transport and surface integration.
- Protocol (as applied to Aceclofenac [17]):
- Prepare single crystals of the model compound (e.g., Aceclofenac).
- Using a precision blade, cleave the crystal along a predetermined crystallographic plane (e.g., the (1 0 -1) facet for ACF).
- Immerse the broken crystal in a supersaturated solution of the same compound in different solvents (e.g., acetone and methyl acetate).
- Monitor the regeneration process over time using optical or electron microscopy to observe the regrowth morphology and direction.
- Optionally, add polymeric additives (e.g., Hydroxypropyl methyl cellulose, HPMC) to study their regulatory effect on growth kinetics and habit.
- Data Analysis: In the ACF study, crystals consistently regrew along the broken face, restoring their original morphology before further growth, demonstrating that the broken surface had higher reactivity. The different growth morphologies in different solvents also highlighted the role of solvent-surface interaction kinetics, a hallmark of surface-reaction influence [17].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagents and Materials for Crystal Growth Studies
| Reagent/Material | Function in Experiment | Example Use Case |
|---|---|---|
| Gold Nanoparticles | Acts as a nucleation catalyst and localized heat source when irradiated with lasers. | Used to seed and "draw" lead halide perovskite crystals on demand with laser pulses [18]. |
| Hydroxypropyl Methyl Cellulose (HPMC) | A polymeric additive that selectively binds to specific crystal faces, modifying surface kinetics and crystal habit. | Used to regulate the crystal morphology of Aceclofenac during regeneration experiments [17]. |
| Lead Halide Perovskite Precursors | Model system for studying crystallization kinetics with relevance to optoelectronics and photovoltaics. | Studied for non-classical particle-mediated growth pathways [3] and laser-induced crystallization [18]. |
| Calcite (CaCO₃) Single Crystals | A well-characterized model substrate for fundamental studies of dissolution and growth mechanisms. | Used in Liquid-Cell AFM to measure pH-dependent step-edge velocities and identify regime transitions [15]. |
| Controlled pH Solutions (e.g., HCl) | Modifies the chemical driving force (e.g., proton concentration) for dissolution or growth. | Essential for probing the transition from surface-reaction to diffusion control in calcite dissolution [15]. |
Implications for Research and Industry
The distinction between growth regimes is not merely academic; it has profound implications for controlling material properties in various applications.
- Pharmaceutical Development: Controlling the crystal habit of an Active Pharmaceutical Ingredient (API) is critical for its processing, stability, and bioavailability. Since crystal morphology is determined by the relative growth rates of different faces, a surface-reaction-controlled regime is typically targeted. Additives like HPMC can be designed to selectively bind to fast-growing faces, slowing their growth and producing a more desirable crystal shape, as demonstrated with Aceclofenac [17].
- Nanocrystal Synthesis: The pursuit of monodisperse nanoparticles for applications in catalysis, medicine, and electronics has its roots in the LaMer model. Achieving "burst nucleation" and controlled growth remains a key strategy. Furthermore, understanding and harnessing non-classical, particle-mediated pathways allows for the synthesis of complex hierarchical and mesocrystalline structures that are inaccessible through classical atomic addition alone [3].
- Advanced Materials Manufacturing: New techniques, such as using laser pulses to heat gold nanoparticles and locally induce crystallization, provide unprecedented spatiotemporal control. This method bypasses the randomness of traditional growth, allowing crystals to be "drawn" on demand [18]. The crystallization pathway itself can be optimized; for example, encouraging a dense liquid intermediate state has been shown to produce higher-quality nanocrystal superlattices faster than direct crystallization [19].
The dichotomy between diffusion-controlled and surface-reaction-controlled growth provides an essential framework for understanding and manipulating crystallization processes. While the classical LaMer model, with its emphasis on diffusion-controlled growth, laid the groundwork for modern colloid science, contemporary research has revealed a more complex and nuanced picture. The emergence of non-classical, particle-mediated pathways and the ability to observe growth in real-time at the molecular level have significantly expanded our toolkit.
For researchers and drug development professionals, the ability to definitively identify the governing growth regime through experiments like agitation tests and liquid-cell AFM is a critical skill. It directly informs the strategic levers—be they mixing, solvent choice, or additive design—that must be pulled to achieve precise control over crystal size, morphology, and structure. As crystal growth science continues to evolve, integrating these classical concepts with new mechanistic insights and data-centric approaches will undoubtedly lead to the next generation of advanced functional materials.
{# The User's Request}
You must use this exact title for the article. Do not modify or optimize it.
Competing Models and Mechanisms: From Turkevich to Finke-Watzky
An In-Depth Technical Guide on Nucleation and Growth Mechanisms for Material Scientists
The synthesis of monodisperse colloidal nanoparticles remains a cornerstone of advanced materials science, with profound implications for applications ranging from drug delivery to catalysis. For decades, the field has been guided by classical nucleation theory (CNT) and the influential model proposed by LaMer and Dinegar in 1950. This model postulates a distinctive mechanism of "burst nucleation" followed by diffusion-controlled growth to explain the formation of uniform particles [1]. The LaMer model schematically represents a scenario where the concentration of a monomer precursor increases to a critical supersaturation level, triggering an instantaneous, finite nucleation event. The subsequent decrease in monomer concentration below the nucleation threshold ensures that no new nuclei form, while existing nuclei grow exclusively through monomer diffusion from the solution bulk, ultimately yielding a monodisperse colloid [1] [2].
However, the past quarter-century has witnessed the emergence of compelling experimental data, particularly for metal nanoparticle systems, that challenge the universal applicability of the LaMer mechanism. These observations have spurred the development of alternative kinetic models that provide a more nuanced description of the formation process. This review provides a comprehensive technical analysis of the competing models and mechanisms that define the modern understanding of nanoparticle synthesis. We trace the historical development from the foundational work of Turkevich to the currently dominant Finke-Watzky (FW) two-step mechanism, critically examining the experimental evidence and methodological approaches that underpin our current mechanistic paradigms.
Historical Trajectory: From LaMer to Modern Mechanisms
The LaMer Model: Foundations and Postulates
The seminal 1950 LaMer model was conceived to explain the formation of monodispersed sulfur hydrosols. Its core premise is a physical separation of the nucleation and growth stages [1] [2]:
- Stage I (Precursor Formation): Generation of monomeric species from precursor compounds.
- Stage II (Burst Nucleation): Upon reaching a critical supersaturation, the system experiences an "exceedingly sensitive" or "effectively infinite" rate of nucleation, leading to a sudden, finite burst of stable nuclei [1].
- Stage III (Diffusion-Controlled Growth): The monomer concentration drops below the critical threshold for nucleation. The remaining monomers diffuse to and are incorporated onto the existing particle surfaces, leading to growth without further nucleation.
The model's elegance and intuitive explanation for monodispersity have contributed to its enduring popularity. However, a critical analysis of the 70 years of experimental data since its publication reveals that the concepts of "burst nucleation" and "diffusion-controlled growth" often lack sound, compelling experimental support, especially outside the original sulfur sol system [1].
The Turkevich Mechanism: Pioneering Gold Nanoparticle Synthesis
In the early 1950s, contemporaneous with LaMer, Turkevich and colleagues conducted groundbreaking work on the synthesis of colloidal gold, providing some of the first detailed electron microscope studies of nanoparticle formation [1] [20]. His investigations into the reduction of chloroauric acid (HAuCl₄) by citrate established a foundational synthesis protocol still used today. While Turkevich's work is often interpreted through the lens of the LaMer model, his careful observations—particularly of the gradual color changes and the presence of various intermediate sizes—hinted at a more complex reality. His research highlighted that nucleation and growth could be overlapping, kinetically controlled processes, setting the stage for future mechanistic debates [20] [21].
The Finke-Watzky Two-Step Mechanism: A Paradigm Shift
In 1997, Finke and Watzky introduced a minimal, two-step mechanism to describe the kinetics of transition metal nanoparticle formation. This model has since become one of the most highly cited and accepted mechanisms in the field [20]. The FW mechanism challenges the core LaMer assumption of temporally distinct nucleation and growth, proposing instead:
- Step A (Continuous Nucleation): Slow, continuous nucleation of metal atoms (A) to form stable nanoclusters (B).
A → B(rate constant = ( k_1 )) - Step B (Autocatalytic Surface Growth): Fast autocatalytic growth on the surface of existing nanoclusters (B).
A + B → 2B(rate constant = ( k_2 ))
The associated integrated rate equation (Eq. 2) describes the concentration of metal atoms in nanoclusters, [B], over time, where [A]₀ is the initial concentration of the metal precursor A [20]: [ [B]t = \frac{[A]0}{1 + \frac{k1}{k2[A]0}} \left( 1 - e^{-(k1 + k2[A]0)t} \right) ]
This model provides an excellent fit for a vast array of sigmoidal kinetic data for nanoparticle formation. A critical insight from this mechanism is that a "burst" nucleation event is not a prerequisite for achieving narrow, near-monodisperse particle size distributions. The balance between the continuous nucleation rate (( k1 )) and the autocatalytic growth rate (( k2 )) is sufficient to explain the observed size distributions [2] [20].
Table 1: Core Postulates of Competing Nucleation and Growth Models
| Feature | LaMer Model (1950) | Finke-Watzky (FW) Model (1997) |
|---|---|---|
| Nucleation Kinetics | "Burst" or "Instantaneous," then ceases | Slow, continuous throughout the reaction |
| Growth Mechanism | Diffusion-controlled monomer addition | Autocatalytic surface growth |
| Temporal Overlap | Nucleation and growth are discrete stages | Nucleation and growth occur concurrently |
| Key Evidence | Light scattering of sulfur sols [1] | Kinetic fits to sigmoidal data for metal NPs [20] |
| Mathematical Form | Complex, rarely-used differential equation [1] | Well-defined integrated rate equation (Eq. 2) |
Diagram 1: A workflow comparison of the fundamental postulates in the LaMer and Finke-Watzky models, highlighting their sequential versus concurrent views of nucleation and growth.
Critical Analysis of Competing Kinetic Models
The Redox-Crystallization (R-C) Model and a Critical Interplay
A notable episode in the evolution of formation mechanisms was the 2013 proposal of a "Redox-Crystallization (R-C)" model for gold nanoparticle formation. The authors of this model derived an integrated kinetic equation (Eq. 1) to describe their sigmoidal data. Interestingly, they noted their equation was mathematically identical to the FW integrated equation (Eq. 2), yet claimed the underlying chemical mechanism was "totally different" [20]. The R-C model defined its overall rate constants as ( k1 = \alpha k{01} ) and ( k2 = \alpha \epsilon [Mn]0 k{02} ), where ( \alpha ) accounts for a redox equilibrium and ( \epsilon ) represents the fraction of surface active sites.
A subsequent critical reanalysis demonstrated that the original kinetic data supporting the R-C model was fitted excellently by the well-precedented FW 2-step mechanism (R² = 0.9987) [20]. This finding, coupled with an unconventional and problematic kinetic analysis in the original R-C work—involving "cherry-picked" linear regions from zeroth-, first-, and second-order plots—led to the conclusion that the R-C model did not represent a novel mechanism but was, in fact, functionally identical to the FW mechanism [20]. This case underscores the critical importance of rigorous kinetic analysis and the principle of attempting to disprove alternative mechanisms before claiming novelty.
The JMAK Model: A Physical Perspective
The Johnson-Mehl-Avrami-Kolmogorov (JMAK) model, a classical model for physical phase transformations, has also been applied to nanoparticle formation. Its general form is ( x = 1 - e^{-kt^n} ), where ( x ) is the fraction transformed, ( k ) is the rate constant, and ( n ) is the Avrami exponent related to the transformation mechanism [21]. Research has shown that the JMAK model can efficiently characterize GNP formation kinetics, with the Avrami exponent ( n ) serving as an indicator of nucleation behavior (e.g., homogeneous vs. heterogeneous) and the geometric dimension of growth [21]. While the JMAK model is phenomenologically useful, its parameters are often less directly tied to specific chemical steps compared to the FW model. Furthermore, the FW model has been shown to provide the underlying chemical mechanism for the sigmoidal kinetics that the JMAK model describes mathematically [21].
Table 2: Quantitative Comparison of Model Parameters from Representative Gold Nanoparticle Formation Studies
| Model Applied | System Description | Reported Rate Constants | Avrami Exponent (n) | Key Analytical Method |
|---|---|---|---|---|
| Finke-Watzky (FW) | Chemical reduction of AuCl₄⁻ | ( k1 = 1.0 \times 10^{-3} \, \text{M}^{-1}\text{s}^{-1} ) ( k2 = 2.7 \, \text{M}^{-1}\text{s}^{-1} ) [20] | Not Applicable | UV-Vis Spectroscopy (SPR) |
| Redox-Crystallization (R-C) | Chemical reduction of AuCl₄⁻ | Overall constants derived from FW-equivalent math [20] | Not Applicable | UV-Vis Spectroscopy (SPR) |
| JMAK Model | Biosynthesis using C. Camphor | ( k = 0.15 \, \text{s}^{-1} ) (for n=2.1) [21] | ~2.1 | UV-Vis Spectroscopy (SPR) |
| JMAK Model | Chemical reduction by ascorbic acid | ( k = 0.11 \, \text{s}^{-1} ) (for n=2.9) [21] | ~2.9 | UV-Vis Spectroscopy (SPR) |
Experimental Protocols and the Scientist's Toolkit
Validating any kinetic model requires robust, time-resolved experimental data. Below are detailed methodologies for key experiments cited in this field.
Protocol for Time-Resolved Monitoring of Gold Nanoparticle Formation via UV-Vis Spectroscopy
This protocol is adapted from procedures used to generate kinetic data for the FW and R-C model analyses [20] [21].
- Objective: To obtain a sigmoidal absorbance-time (Abs-t) profile for the formation of gold nanoparticles (GNPs) by tracking the surface plasmon resonance (SPR) band.
- Materials:
- Precursor Solution: Chloroauric acid (HAuCl₄·4H₂O) in deionized water.
- Reducing Agent Solution: L-ascorbic acid or sodium citrate in deionized water.
- Stabilizing Agent (optional): Polyvinylpyrrolidone (PVP K-30).
- Apparatus: UV-Vis Spectrophotometer (e.g., Shimadzu UV-1800) equipped with a thermostatted cell holder.
- Procedure:
- Prepare a solution of the gold precursor (e.g., 0.25 mM HAuCl₄) in a quartz cuvette.
- Place the cuvette in the spectrophotometer, set to a constant temperature (e.g., 25°C).
- Initiate the reaction by rapidly adding a small, precise volume of the reducing agent solution (e.g., ascorbic acid) directly to the cuvette and mix quickly via pipetting or a built-in stirrer.
- Immediately start collecting absorbance data at a fixed wavelength corresponding to the SPR of GNPs (e.g., 526 nm or 540 nm). Data points should be collected at frequent intervals (e.g., every 1-5 seconds) until the absorbance reaches a plateau.
- For full spectral analysis, periodically scan the entire wavelength range (e.g., 400-800 nm).
- Data Analysis: The absorbance at the SPR wavelength is used as a proxy for the concentration of formed gold nanoparticles (B). This Abs-t data is then fitted to the integrated FW equation (Eq. 2) using non-linear regression software to extract the rate constants ( k1 ) and ( k2 ) [20] [21].
The Research Reagent Toolkit
Table 3: Essential Reagents and Materials for Nanoparticle Formation Kinetics Studies
| Reagent/Material | Typical Function in Experiment | Example from Cited Research |
|---|---|---|
| Chloroauric Acid (HAuCl₄) | Gold precursor; source of Au(III) ions. | Used as the primary metal precursor in chemical reduction studies [20] [21]. |
| L-Ascorbic Acid | Reducing agent; converts Au(III) to Au(0). | Employed as a chemical reductant in the R-C and JMAK model studies [21]. |
| Sodium Citrate | Reducing and stabilizing agent; confers negative charge to NPs. | Basis of the classic Turkevich synthesis method [20]. |
| Polyvinylpyrrolidone (PVP) | Capping or stabilizing agent; sterically stabilizes NPs and controls growth. | Used as a stabilizer in kinetics studies of gold nanoparticle formation [21]. |
| Foliar Aqueous Extract (e.g., C. Camphor) | Biogenic reducing/capping agent; contains phytochemicals that reduce metal ions. | Used in biosynthesis studies analyzed with the JMAK model [21]. |
| Quartz Cuvette | Container for reaction mixture during spectroscopic monitoring. | Standard for UV-Vis kinetics measurements [21]. |
Implications for Drug Development and Research
The mechanistic debate between LaMer's diffusion-controlled growth and the FW model's autocatalytic surface growth extends far beyond academic interest. For drug development professionals, the mechanism of particle formation has direct consequences on critical quality attributes of nanomedicines.
- Predictability and Control: A chemically realistic model like the FW mechanism enables better prediction and control over particle size distribution—a key factor in the biological performance, stability, and dose consistency of nanotherapeutic agents.
- Rational Optimization: Understanding that nucleation can be continuous (( k1 )) and growth autocatalytic (( k2 )) allows formulators to rationally adjust synthetic conditions (e.g., precursor addition rate, temperature, catalyst) to fine-tune the balance between these rates, thereby achieving the desired particle size.
- Solid-State Properties: The principles of nucleation and growth kinetics are equally critical in understanding and controlling the crystallization of active pharmaceutical ingredients (APIs). As demonstrated in studies of HCV drug analogues, minor molecular changes can drastically alter conformational preferences, crystal packing, and intermolecular interactions, leading to challenges like polymorphism and low aqueous solubility [22]. Physics-based modeling that explicitly considers 3D structure and crystal packing is essential for de-risking such challenges in drug development [22].
The journey from the LaMer model to the Finke-Watzky mechanism illustrates the dynamic and self-correcting nature of scientific inquiry. While the LaMer model provided an invaluable conceptual framework for understanding monodisperse particle formation, modern kinetic data, particularly for metal nanoparticles, increasingly supports a mechanism of continuous nucleation coupled with autocatalytic surface growth. The Finke-Watzky two-step model has emerged as a dominant, minimal mechanism capable of accounting for a wide range of sigmoidal formation kinetics. Its mathematical robustness and chemical plausibility make it a powerful tool for researchers aiming to achieve precise control over nanoparticle synthesis. As the field progresses, the integration of these kinetic models with advanced in situ characterization techniques and computational predictions will undoubtedly pave the way for the rational design of next-generation nanomaterials with tailor-made properties for drug development and beyond.
Seventy years ago, LaMer and Dinegar postulated a seminal model for particle formation that has fundamentally shaped the field of colloidal science and nanocrystal synthesis. Their 1950 paper, "Theory, Production and Mechanism of Formation of Monodispersed Hydrosols," introduced a conceptual framework describing how monodisperse particles might form through distinct stages of nucleation and growth [1]. This model proposed that an initial "effectively infinite" nucleation burst occurs when solute concentration reaches a critical supersaturation point, followed by a diffusion-controlled growth phase where existing particles grow without additional nucleation events [1] [3]. The LaMer model's significance lay in its potential explanation for monodisperse particle formation—a fundamental challenge in materials science. Its intuitive schematic representation, now famously known as the "LaMer curve," has become a cornerstone concept taught to generations of scientists and cited in nearly 2,000 papers as of 2019 [1]. This review critically examines the evolution of our understanding of this model over seven decades, focusing specifically on the central dichotomy between diffusion-controlled and surface-process-controlled crystal growth mechanisms within broader research on nanoparticle synthesis and drug development.
The Original LaMer Mechanism: Foundations and Principles
The classical LaMer mechanism conceptually describes thin film growth through two distinct pathways [23]:
- Diffusion-controlled growth: When the concentration of growing particles falls below the minimum critical concentration required for nucleation, crystal development continues but nucleation ceases
- Surface-process-controlled growth: When the diffusion of growth species from the bulk to the growth surface is sufficiently rapid, the surface process controls the growth rate
The original model visualized nanoparticle formation as a three-stage process [23], as illustrated in Figure 1. In Stage I, precursor decomposition or reaction increases the concentration of monomers (the basic building units) in solution until reaching supersaturation. In Stage II, once concentration exceeds the critical supersaturation point (Cmin), a burst of nucleation occurs, forming stable nuclei. In Stage III, the monomer concentration drops below Cmin, nucleation ceases, and existing particles grow primarily through monomer diffusion to the particle surface.
Table 1: Key Parameters in the Original LaMer Model
| Parameter | Symbol | Description | Role in Mechanism |
|---|---|---|---|
| Supersaturation | δ | Concentration exceeding solute solubility | Driving force for nucleation |
| Critical Supersaturation | Cmin | Minimum concentration for nucleation | Threshold for burst nucleation |
| Solubility Concentration | Cs | Equilibrium concentration | Determines growth termination |
| Monomer Concentration | Cm | Concentration of building units | Controls nucleation and growth rates |
The mathematical basis of the model relied on classical nucleation theory (CNT), where the free energy of particle formation (ΔG) is plotted against the radius of the assumed spherical particle [1]. Key variables in this framework included ΔGs (the free energy of the particle's surface), ΔGv (the free energy of the bulk crystal), and rc (the critical radius required for the particle to form without redissolution). The model assumed that "the rate of nucleation becomes effectively infinite" once the system reaches critical supersaturation, leading to the widely cited concepts of "instantaneous" or "burst" nucleation [1].
Critical Assessment of the LaMer Model: 70 Years of Evidence
Theoretical and Experimental Limitations
A comprehensive critical analysis of the 164 papers that provided substantive discussion of the LaMer model revealed significant theoretical and experimental limitations. The model's foundation in Classical Nucleation Theory (CNT) and fluctuation theory presented inherent constraints, as CNT assumes particles are spherical with a well-defined solid-liquid interface and uniform density—simplifications that often deviate from real systems [1]. The "effectively infinite nucleation" postulate was found to be problematic, as nucleation rates are always finite and measurable [1].
Experimental evidence gathered over decades has challenged the model's universal applicability. A critical review demonstrated that the concepts of "burst/instantaneous nucleation" and "diffusion-controlled growth" lack compelling experimental support across many material systems [1]. Even in silver halide nanoparticles, where the best evidence for the LaMer model was thought to exist, closer examination revealed inconsistencies [1]. Similarly, studies of semiconductor, metal, and metal-oxide nanoparticles frequently deviated from the predicted behavior [1].
The Diffusion vs. Surface-Control Dichotomy
The central dichotomy between diffusion-controlled and surface-process-controlled growth has been particularly scrutinized. Research indicates that most real systems operate under mixed control mechanisms rather than purely diffusion-limited growth [1]. The diffusion-controlled growth assumption requires that surface integration is significantly faster than monomer diffusion to the surface—a condition not universally satisfied across different material systems and synthesis conditions [23].
Table 2: Diffusion-Controlled vs. Surface-Controlled Growth Characteristics
| Characteristic | Diffusion-Controlled Growth | Surface-Process-Controlled Growth |
|---|---|---|
| Rate Determination | Monomer diffusion to surface | Surface integration kinetics |
| Size Dependence | Growth rate decreases with size | Less dependent on particle size |
| Temperature Dependence | Weak (diffusion coefficient) | Strong (activation energy dependent) |
| Agitation Effect | Significant impact | Minimal impact |
| Size Distribution | Narrowing (size-focusing) | Can lead to broadening |
Modern analyses of colloidal quantum dot synthesis reveal that maintaining diffusion-dependent growth requires precise control over monomer concentration (Cm). Too-rapid precursor injection can disturb diffusion-dependent growth, leading to secondary nucleation or interparticle ripening that compromises size uniformity [14]. This highlights a significant practical limitation in applying the classical model to complex synthetic systems.
Beyond LaMer: Modern Theoretical Frameworks
Non-Classical Nucleation and Growth Mechanisms
The evolution of understanding beyond the LaMer model has led to the development of non-classical nucleation and growth theories. The particle-mediated nucleation and growth model represents a fundamental paradigm shift, where nanoparticles or clusters serve as building units rather than individual atoms [3]. This "non-classical" model explains phenomena such as mesocrystal formation, oriented attachment, and the synthesis of hierarchical structures that cannot be adequately described by the classical LaMer curve [3].
In situ characterization techniques have revealed that nanoparticle formation often involves complex pathways including cluster aggregation, oriented attachment, and mesoscopic transformations [3]. These processes frequently operate alongside classical atom-mediated growth, creating hybrid growth pathways that combine multiple mechanisms. The critical observation of sudden size increases accompanied by decreases in cluster numbers provides direct evidence for particle-mediated growth mechanisms beyond LaMer's original conception [3].
Competing and Complementary Models
Several other theories have emerged to explain crystal growth phenomena that deviate from the LaMer mechanism:
Ostwald Ripening describes the dissolution of smaller particles and redeposition onto larger particles, driven by higher chemical potential and solubility of smaller crystals [23]. This thermodynamic process typically occurs in later growth stages and can lead to size defocusing rather than the focusing predicted by simple diffusion models.
Von Weimarn's Theory establishes empirical relationships between initial supersaturation and crystal size, proposing that average crystal size increases as initial relative supersaturation decreases [23]. This framework helps explain why moderate supersaturation often produces larger crystals than high supersaturation, contrary to simple expectations.
Pre-Nucleation Cluster (PNC) Theory challenges classical nucleation by proposing that stable clusters exist in solution before nucleation, serving as precursors to solid phases [24]. This model has been particularly influential in understanding biomineralization and polymorph selection.
Advanced Experimental Methodologies
Modern Characterization Techniques
Cutting-edge experimental methods have revolutionized our ability to probe nucleation and growth processes in real-time:
In situ Electron Microscopy: Advanced transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) with direct electron detectors enable direct observation of nucleation events and crystal growth at near-atomic resolution [25] [3]. These techniques have been instrumental in identifying non-classical growth pathways like oriented attachment.
In situ X-ray Absorption Spectroscopy (XAS): This technique provides element-specific information about local electronic structure and coordination geometry during nucleation, allowing researchers to track precursor conversion and cluster formation [25].
Atomic Force Microscopy (AFM): High-speed AFM can probe surface processes and growth mechanisms at the nanoscale, particularly useful for understanding surface-controlled growth [25].
Fast Scanning Calorimetry (FSC): This method enables precise investigation of crystallization kinetics over wide temperature ranges, revealing complex nucleation behavior and polymorph selection [4].
Process Intensification Strategies
Advanced synthesis approaches have emerged that enable better control over nucleation and growth:
Microreactor Technology: Microscale process intensification enables enhanced micromixing, reduced mixing times, and precise control over nucleation-growth processes, producing crystals with optimal form and structural stability [4].
Membrane Crystallization (MCr): This hybrid approach uses membranes as heterogeneous nucleation interfaces, providing superior control over crystal nucleation and enabling continuous crystallization intensification [4].
Continuous Injection Systems: Unlike traditional hot-injection methods, continuous precursor injection maintains constant monomer concentration (Cm), extending the size-focusing regime and enabling synthesis of larger nanocrystals with narrow size distributions [14].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Crystal Growth Studies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Poly(vinyl pyrrolidone) (PVP) | Surfactant & structure-directing agent | Shape-controlled metal nanocrystal synthesis |
| Tris(trimethylsilyl)arsine ((TMS)3As) | Precursor for covalent nanocrystals | InAs quantum dot synthesis |
| Aminoarsine Precursors | Less reactive arsenic source | Extended growth of InAs nanocrystals |
| Metal-Oxide Precursors | Oxidizers in solution combustion | Porous metal oxide nanomaterials |
| Architecture-Directing Agents (ADAs) | Colloidal nanocrystal framework formation | Ordered porous materials for energy devices |
| Ionic Liquids (PILs/SILs) | Green solvent media for crystallization | Potential-driven metal crystal growth |
Application Frontiers: From Perovskite Photovoltaics to Pharmaceutical Development
Perovskite Thin-Film Crystallization
The crystallization of perovskite thin films for photovoltaics represents an application where LaMer's principles have been extensively adapted and refined. Research shows that fast nucleation followed by slow crystallization improves perovskite thin film morphology [23]. Techniques like antisolvent extraction, hot-casting, vacuum quenching, and gas blowing facilitate rapid solvent removal to achieve high supersaturation, initiating rapid nucleation [23]. Additive engineering further modulates crystal morphology by slowing crystallization, leading to preferred grain growth and better film quality [23].
Pharmaceutical Polymorph Control
In pharmaceutical development, understanding nucleation and growth mechanisms is crucial for controlling polymorphism—where different crystal forms of the same drug substance exhibit different physical properties, solubility, and bioavailability [24]. The bimodal temperature dependency of crystallization rates in systems like polyamide 11 demonstrates how nucleation mechanisms influence polymorph selection, with high nucleation densities favoring metastable phases [4].
Visualizing the Evolutionary Pathway: From Classical to Modern Understanding
The following diagram illustrates the key evolutionary pathway in our understanding of crystal growth mechanisms, from the classical LaMer model to modern integrated perspectives:
Figure 1: Evolution of crystal growth understanding from classical to modern frameworks.
The 70-year evolution of our understanding of crystal growth mechanisms reveals a complex landscape far beyond LaMer's original conception. The simple dichotomy between diffusion-controlled and surface-process-controlled growth has given way to a sophisticated framework incorporating multiple simultaneous growth pathways, particle-mediated processes, and system-specific dominant mechanisms. Future research directions should focus on developing multi-scale models that integrate classical and non-classical theories, advancing in situ characterization techniques for real-time observation of growth processes, and designing intelligent synthesis platforms that dynamically adapt conditions to steer growth along desired pathways. The continued critical analysis of foundational models like LaMer's remains essential for driving innovation in nanomaterials synthesis, pharmaceutical development, and functional materials design.
Experimental Strategies for Differentiating and Applying Growth Mechanisms
Electrocrystallization—the process whereby metal cations are reduced at an electrode surface to form solid crystalline deposits—serves as a critical bridge between electrochemical synthesis and the fundamental science of phase formation [26]. The kinetics of nucleation and growth during this process directly determine the structural and functional properties of electrodeposited materials, ranging from catalytic nanoparticles to microelectronic components [27]. This technical guide examines the modeling of current-time transients to quantitatively analyze these kinetic processes, placing specific emphasis on their relationship to the broader context of crystal growth research, particularly the classical LaMer mechanism that describes diffusion-controlled growth for monodisperse particle formation [1].
The LaMer model, originally developed to explain the formation of monodisperse sulfur sols, posits a distinct separation between nucleation and growth phases [1]. According to this framework, a rapid, "burst" nucleation event occurs once supersaturation reaches a critical threshold, followed by a diffusion-controlled growth stage where solute diffuses to existing nuclei rather than forming new ones. In electrocrystallization, this concept manifests in the distinction between instantaneous nucleation (a fixed number of nuclei form rapidly) and progressive nucleation (nuclei continue to form over time) [28]. Current-time transient analysis provides the experimental methodology to distinguish these regimes and quantify their kinetic parameters in electrochemical systems.
Theoretical Foundations of Current-Time Transients
Fundamental Steps in Electrocrystallization
The electrocrystallization process comprises a complex sequence of steps that can be rate-limited by different physical phenomena. The microscopic process involves: (1) ion migration to the electrode surface, (2) ligand removal for complex ions, (3) electron transfer to generate adsorbed atoms, and (4) surface diffusion to crystallization sites or direct incorporation into growth centers [26]. The overall reaction rate is typically governed by the slowest step in this sequence, leading to two primary limiting cases with distinct current-time behaviors:
- Electrochemical polarization control occurs when the charge transfer reaction at the electrode interface is rate-limiting. In this regime, the current density follows the Butler-Volmer equation: ( J = J0 \left[ e^{\alphaa \eta zF/RT} - e^{-\alphac \eta zF/RT} \right] ), where ( J0 ) is the exchange current density, ( \eta ) is the overpotential, ( \alphaa ) and ( \alphac ) are charge transfer coefficients, ( z ) is the charge number, ( F ) is Faraday's constant, ( R ) is the gas constant, and ( T ) is temperature [26].
- Diffusion control becomes dominant when the mass transport of depositing ions through the solution to the electrode surface is rate-limiting. This condition frequently occurs at high overpotentials where electrochemical reactions would otherwise be exceedingly fast [26].
Table 1: Rate-Controlling Steps in Electrocrystallization
| Control Mechanism | Rate-Limiting Process | Current-Potential Relationship | Common Experimental Conditions |
|---|---|---|---|
| Electrochemical Polarization | Charge transfer at interface | Butler-Volmer equation | Low overpotential, high concentration |
| Diffusion Control | Mass transport to electrode | Cottrell equation (early time) | High overpotential, low concentration |
| Mixed Control | Combined charge and mass transfer | Complex integrated forms | Intermediate conditions |
The LaMer Connection: From Colloidal Synthesis to Electrocrystallization
The LaMer model provides a conceptual foundation for understanding how monodisperse particles can form through separated nucleation and growth stages [1]. In LaMer's original schematic, a rapid increase in monomer concentration leads to a brief "burst nucleation" event when supersaturation reaches a critical threshold, followed by a diffusion-controlled growth period where monomers are consumed by existing particles rather than forming new nuclei. This mechanism directly parallels the distinction between instantaneous and progressive nucleation in electrocrystallization, though the driving force in electrochemical systems is overpotential rather than chemical supersaturation.
Recent critical analyses of the LaMer model have revealed that the concepts of "burst nucleation" and "diffusion-controlled growth" lack comprehensive experimental validation in many nanoparticle systems [1]. This underscores the importance of rigorous electrochemical techniques like current-time transient analysis for quantitatively testing these fundamental growth hypotheses in controlled environments.
Experimental Methodologies and Protocols
Potentiostatic Current-Transient Technique
The potentiostatic method, employing a potential step with current monitoring, represents the most widely utilized approach for studying electrocrystallization kinetics [27]. The experimental protocol involves:
- Initial Condition Establishment: Maintain the working electrode at a potential where no deposition occurs (typically a potential positive of the reduction potential) for a sufficient time to establish a stable baseline.
- Potential Step Application: Apply a sudden cathodic potential step to a value where nucleation and growth proceed. The magnitude of this overpotential (η) directly controls the driving force for deposition.
- Current Monitoring: Record the current response with high temporal resolution throughout the nucleation and growth process. Modern potentiostats can achieve microsecond to millisecond resolution, capturing the initial stages of nucleus formation [29].
- Data Collection: Continue measurements until the current reaches a steady state or begins to decline due to complete surface coverage or diffusion layer overlap.
For systems with slow nucleation kinetics, an additional double-step potentiostatic method may be employed, where a high overpotential pulse initiates nucleation followed by a lower overpotential that allows growth without additional nucleation [28].
Advanced In Situ Characterization Techniques
Complementary techniques provide structural insights to correlate with electrochemical data:
- Time-Resolved Surface X-ray Diffraction (TRSXRD): This method captures structural changes at electrode surfaces with millisecond resolution. For copper deposition on Au(111), TRSXRD revealed a metastable state where hydrated Cu²⁺ ions accumulate at the outer Helmholtz plane before undergoing dehydration and incorporation into the crystal lattice [29].
- Time-Resolved Surface-Enhanced Infrared Spectroscopy (TRSEIRAS): This technique monitors molecular species and their bonding during electrocrystallization. During copper underpotential deposition, TRSEIRAS tracked the disappearance and reappearance of adsorbed (bi)sulfate anions, revealing co-adsorption dynamics with depositing copper atoms [29].
Modeling Frameworks for Diffusion-Controlled Growth
The Scharifker-Hills (SH) Model
The SH model represents the foundational framework for analyzing diffusion-controlled three-dimensional nucleation and growth [26] [27]. This model assumes hemispherical diffusion zones around each nucleus that expand and eventually overlap according to Avrami's theorem. The current-time relationships for the two limiting nucleation cases are:
Instantaneous Nucleation: All nuclei form rapidly at the beginning of the potential step.
[ j(t) = \frac{zFD^{1/2}c}{\pi^{1/2}t^{1/2}} \left[ 1 - \exp\left(-N\pi k D t\right) \right] ]
where ( j(t) ) is current density, ( z ) is electron number, ( F ) is Faraday's constant, ( D ) is diffusion coefficient, ( c ) is bulk concentration, ( N ) is nucleus density, and ( k = \left( \frac{8\pi cM}{\rho} \right)^{1/2} ) with ( M ) molar mass and ( \rho ) density.
Progressive Nucleation: Nuclei form continuously throughout the experiment.
[ j(t) = \frac{zFD^{1/2}c}{\pi^{1/2}t^{1/2}} \left[ 1 - \exp\left(-AN_\infty \pi k' D t^2/2\right) \right] ]
where ( A ) is nucleation rate constant and ( N_\infty ) is maximum nucleus density.
The SH model generates characteristic current-time transients that rise to a maximum before decaying due to diffusion zone overlap, with the instantaneous nucleation peak being sharper and earlier than the progressive case [27].
Growth Model Variations and Their Effects
Different growth geometries yield distinct current-time behaviors, significantly impacting extracted nucleation parameters [28]:
- Right-Circular Cones: This shape-preserving model produces different transient shapes compared to hemispherical growth, particularly in the rising portion of the curve.
- Hemispheroids: These shape-changing models more accurately represent many real systems, with ellipticity parameters (λ) affecting both peak shape and position.
Table 2: Common Growth Models in Electrocrystallization
| Growth Model | Geometry | Current-Time Relationship | Applicable Systems |
|---|---|---|---|
| Hemispherical | Expanding hemisphere | ( I \propto t^{1/2} ) (early) | Isotropic growth, noble metals |
| Right-Circular Cones | Fixed-angle cones | ( I \propto t ) (early) | Anisotropic growth |
| Hemispheroids | Elliptical cross-section | Complex, λ-dependent | Mixed growth modes |
| Spherical Caps | Partial spheres with contact angle | Dependent on θ | Weak substrate interactions |
The choice of growth model significantly impacts extracted nucleation rates. For example, analysis of the same current transient can yield nucleation rate constant (( A )) values that vary by an order of magnitude depending on whether conical or hemispheroidal growth is assumed [28].
Mixed-Control Growth Models
Many practical electrocrystallization systems operate under mixed control, where both charge transfer and diffusion limitations influence growth kinetics [30]. The growth current density for a hemispherical nucleus under mixed control is given by:
[ ig = \frac{ \exp[\alpha f(\eta - \etap)] - \exp[-\beta f(\eta - \etap)] }{ \frac{1}{i0} + \frac{r \exp[\alpha f(\eta - \etap)]}{zFc0D} } ]
where ( ig ) is growth current density, ( i0 ) is exchange current density, ( \alpha ) and ( \beta ) are charge transfer coefficients, ( \etap ) is phase overpotential, ( r ) is nucleus radius, ( c0 ) is bulk concentration, and ( D ) is diffusion coefficient [30].
Mixed-control models more accurately represent real electrochemical systems where the growth mechanism may transition from charge-transfer control to diffusion control as nuclei expand [30]. Numerical simulations of these systems enable extraction of fundamental parameters including diffusion coefficients, exchange current densities, and charge transfer coefficients from experimental data.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Materials for Electrocrystallization Studies
| Reagent/Material | Function/Application | Example Specifications |
|---|---|---|
| Working Electrode | Substrate for nucleation studies | Au(111), glassy carbon, Pt; mirror finish (Ra < 10 nm) |
| Metal Salts | Source of depositing ions | CuSO₄, AgNO₃, ZnCl₂; high purity (>99.99%) |
| Supporting Electrolyte | Conductivity without participation | H₂SO₄, Na₂SO₄, HClO₄; purified to remove organics |
| Reference Electrode | Potential control and measurement | Saturated calomel (SCE), Ag/AgCl; stable ±1 mV |
| Solvent | Reaction medium | Ultra-pure water (18.2 MΩ·cm), non-aqueous solvents |
| Additives | Growth modifiers | Brighteners, levelers, surfactants (e.g., PEG, SPS) |
Workflow and Signaling Pathways
The following diagram illustrates the experimental and analytical workflow for current-transient analysis in electrocrystallization studies:
Electrocrystallization Analysis Workflow
At the molecular level, the interfacial processes during electrocrystallization involve specific signaling pathways for ion incorporation:
Interfacial Ion Pathway During Electrocrystallization
Data Analysis and Interpretation Protocols
Dimensionless Analysis for Mechanism Identification
The dimensionless ( j/jm ) vs. ( t/tm ) plot provides a model-independent approach for identifying the operative nucleation and growth mechanism [27]. The analysis procedure involves:
- Peak Parameter Extraction: From the experimental current transient, identify the peak current (( jm )) and corresponding time (( tm )).
- Normalization: Normalize the current and time axes by their peak values.
- Theoretical Comparison: Compare the normalized data to theoretical dimensionless curves for instantaneous and progressive nucleation.
- Mechanism Assignment: Assign the nucleation type based on the best-fit theoretical curve.
Systems following instantaneous nucleation typically show a sharper peak that decays more rapidly, while progressive nucleation exhibits a broader peak with a slower decay profile [27].
Parameter Extraction Methods
Once the appropriate model is identified, quantitative kinetic parameters can be extracted:
Nucleus Density (N): For instantaneous nucleation under diffusion control: [ N = \frac{0.065}{8\pi cVm D} \left( \frac{jm}{tm} \right)^2 ] where ( Vm ) is molar volume.
Nucleation Rate Constant (A): For progressive nucleation: [ A = \frac{1}{2} N\infty k D \left( \frac{jm}{t_m} \right)^2 ]
Diffusion Coefficient (D): From the Cottrell region of the transient or from the peak parameters: [ D = \frac{jm^2 tm}{0.1629 (zFc)^2} ] for instantaneous nucleation.
Advanced analysis incorporating mixed control requires numerical simulation to simultaneously extract multiple parameters including ( D ), ( i_0 ), and charge transfer coefficients [30].
Current-time transient analysis provides a powerful methodology for quantifying electrocrystallization kinetics within the conceptual framework of LaMer's separation between nucleation and growth stages. The technique enables researchers to distinguish between instantaneous and progressive nucleation regimes, identify rate-controlling steps, and extract fundamental parameters governing phase formation. While classical models assuming pure diffusion control offer valuable initial approximations, contemporary research increasingly recognizes the importance of mixed-control scenarios that more accurately represent complex electrochemical environments. As in situ characterization techniques continue to improve temporal and spatial resolution, the integration of electrochemical measurements with structural data promises to further refine our understanding of the atomic-scale processes underlying electrocrystallization, enabling more precise control of electrodeposited materials for advanced technological applications.
The synthesis of functional nanomaterials with precise control over size and morphology is a cornerstone of advanced materials science. Traditional chemical synthesis routes, while effective, often face limitations in speed, the introduction of impurities from chemical stabilizers, and a lack of precise temporal control over nucleation and growth stages. This whitepaper delves into the emerging paradigm of plasma-enabled synthesis, a rapid, stabilizer-free approach capable of facilitating nanoparticle formation on the millisecond timescale. By examining this technique through the lens of the classical LaMer mechanism and its modern competitors, we provide a detailed technical guide on the experimental methodologies, underlying mechanisms, and quantitative data that define this cutting-edge field. The insights herein are particularly relevant for researchers and drug development professionals seeking innovative routes for the rapid production of high-purity metallic nanostructures.
The quest for controlled nanoparticle synthesis has long been guided by the seminal work of LaMer and Dinegar. The classical LaMer model posits a distinct separation of nucleation and growth phases. It describes a process where the concentration of monomers (atoms or ions) gradually increases until it surpasses a critical supersaturation level (Cmin), triggering a rapid "burst nucleation" event. This event depletes the monomer concentration below the nucleation threshold, following which the existing nuclei grow uniformly via diffusion-controlled monomer addition, ultimately yielding monodisperse particles [1] [3]. For decades, this model has provided the foundational framework for understanding colloidal synthesis.
However, the advent of novel synthesis techniques, including plasma-liquid processes, has revealed phenomena that challenge a strict LaMer interpretation. Modern research has identified non-classical pathways, such as particle-mediated growth, where nanoparticles themselves act as building blocks via aggregation and coalescence, a process distinct from simple atomic addition [3]. Furthermore, mechanisms like the Finke-Watzky two-step model propose simultaneous slow nucleation and autocatalytic surface growth, which contrasts with LaMer's clear separation of stages [31] [1]. Plasma-enabled synthesis, with its extraordinarily fast kinetics and unique reaction environment, serves as a critical testbed for these competing models. This whitepaper explores how the application of plasma to liquid droplets or solutions compresses the nucleation and growth processes into millisecond windows, pushing the boundaries of our mechanistic understanding and offering unprecedented control for industrial and pharmaceutical applications.
Experimental Methodologies for Millisecond Synthesis
Achieving and probing synthesis on millisecond timescales requires specialized experimental designs that move beyond conventional batch reactors. The following sections detail two primary advanced approaches.
Microfluidic Hydrodynamic Focusing
Microfluidic systems offer exceptional control over mixing and reaction times, allowing for the real-time study of fast nucleation kinetics.
- Apparatus Design: A three-dimensional microfluidic device with double hydrodynamic focusing is employed. In this design, a central stream containing the metal ion precursor (e.g., Cd²⁺) is focused into a narrow filament in the center of the outlet channel. This is achieved by surrounding it with a buffer layer of pure water, which is itself surrounded by the second reactant stream (e.g., S²⁻). This geometry ensures the reactant streams mix solely by diffusion at a well-defined interface, separating the emerging nanoparticles from the channel walls to prevent unwanted heterogeneous nucleation and wall deposition [32].
- In-Situ Analysis: The entire channel is optically transparent, permitting the use of confocal laser scanning microscopy (CLSM) and in-situ absorption spectroscopy. As nanoparticles nucleate and grow along the length of the channel, CLSM monitors the development of characteristic fluorescence, while absorption spectroscopy tracks the shifting absorption edge. The position along the channel is directly correlated with reaction time, enabling millisecond-resolution observation of the synthesis kinetics [32].
- Key Materials:
- Precursors: Cadmium chloride or cadmium nitrate (Cd²⁺ source); Sodium sulfide (S²⁻ source).
- Stabilizers: Thioglycerol or L-cysteine (used to study stabilized vs. unperturbed growth).
- Device Fabrication: All-PDMS multilayer devices integrated with laser-cut glass capillaries [32].
Plasma-Liquid Droplet Reactors
This approach utilizes a low-temperature plasma in direct contact with picoliter-volume droplets containing the metal precursor, enabling reduction and synthesis at unprecedented speeds.
- Apparatus Design: An radio-frequency (RF) glow discharge plasma (e.g., in a He/Ar atmosphere) is generated. A precursor solution (e.g., HAuCl₄·3H₂O) is atomized into a fine mist of droplets, which are then injected into the plasma zone. The picoliter volume of each droplet creates a massive surface-to-volume ratio, allowing for the highly efficient influx of plasma-generated reactive species. The residence time of the droplet in the plasma, typically around 10 milliseconds, defines the reaction time [31].
- In-Situ and Ex-Situ Analysis: UV-Vis absorption and emission spectroscopy are used to monitor the conversion of the metal precursor and the formation of nanoparticles in real-time. Subsequently, the synthesized nanoparticles are collected and analyzed using Transmission Electron Microscopy (TEM) to determine their size distribution, morphology, and crystallinity [31].
- Key Materials:
- Precursor: Chloroauric acid (HAuCl₄·3H₂O) for gold nanoparticle synthesis.
- Plasma Gas: Helium or Helium/Argon mixtures.
- Characterization: TEM grids, solvents for collection.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 1: Key reagents and materials for plasma and microfluidic synthesis experiments.
| Item | Function/Description | Example Use-Case |
|---|---|---|
| Cadmium Chloride/Nitrate | Source of Cd²⁺ ions for semiconductor nanocrystal synthesis. | CdS quantum dot synthesis in microfluidics [32]. |
| Sodium Sulfide | Source of S²⁻ ions for sulfide nanoparticle formation. | Reactant for CdS formation [32]. |
| Chloroauric Acid (HAuCl₄) | Gold ion complex (AuCl₄⁻) precursor for metal nanoparticle synthesis. | Stabilizer-free gold NP synthesis in plasma-droplet systems [31]. |
| Thioglycerol / L-Cysteine | Stabilizing ligands; alter nucleation/growth kinetics and prevent aggregation. | Studying stabilized vs. bare nanoparticle growth kinetics [32]. |
| PDMS & Glass Capillaries | Materials for fabricating optically transparent, multi-layer microfluidic devices. | Creating 3D hydrodynamic focusing channels for in-situ spectroscopy [32]. |
| Helium/Argon Gas | Feedstock for generating and sustaining low-temperature plasma discharges. | Creating the reactive plasma environment for droplet-based synthesis [31]. |
Quantitative Data and Mechanistic Insights
The application of the above methodologies has yielded robust quantitative data, shedding light on the governing mechanisms of ultrafast synthesis.
Key Experimental Findings and Data
Table 2: Summary of quantitative findings from millisecond-timescale synthesis studies.
| Parameter | Microfluidic CdS Synthesis [32] | Plasma AuNP Synthesis [31] |
|---|---|---|
| Primary Timescale | Milliseconds to seconds | ~10 milliseconds (droplet residence time) |
| Key Metric | Shifting absorption edge with channel position/time. | >70% conversion of AuCl₄⁻ within ~10 ms. |
| Growth Rate | Monitored via fluorescence increase and absorption shift. | Nucleation rate: >10²⁴ atoms per L per s. |
| Size Control | Tuned by reaction time (channel position) and stabilizers. | Average particle size increases with droplet residence time. |
| Critical Finding | Direct observation of diffusion-limited growth. | Exceeds maximum faradaic efficiency by 250x, indicating complex reduction pathways. |
Interplay with Nucleation and Growth Theories
The data from these high-speed experiments provide a critical validation and refinement of existing models:
- The LaMer Mechanism and CNT: The observation of a sharp onset of nucleation and a subsequent growth phase in controlled microfluidic environments is consistent with the LaMer model. The presence of a power threshold in plasma synthesis, below which no nanoparticles form, also aligns with the free energy barrier described by Classical Nucleation Theory (CNT) [31] [33]. The rapid nucleation can be described as "effectively infinite," as LaMer originally postulated for his sulfur sols [1].
- Beyond LaMer: The Finke-Watzky Mechanism: Plasma-synthesis data strongly suggests the simultaneous operation of nucleation and autocatalytic growth. Quantitative modeling of gold nanoparticle formation showed that autocatalytic surface growth, enabled by long-lived reactive species like H₂O₂, was responsible for the majority of the precursor conversion. This aligns with the Finke-Watzky mechanism, where growth is not solely diffusion-limited but is catalyzed by the nanoparticle surface itself [31].
- The Role of Reducing Agents: Unlike chemical synthesis, reduction in plasma systems is mediated by a cocktail of reactive species. For gold ion reduction, both short-lived species (solvated electrons (e⁻ₐq), H radicals, VUV photons) and long-lived species (H₂O₂) play critical, synergistic roles. The identification of H₂O₂ as a key agent for autocatalytic surface growth is a distinct finding from plasma studies that goes beyond traditional citrate or borohydride reduction mechanisms [31].
Visualizing Workflows and Mechanisms
The following diagrams illustrate the core experimental setups and the mechanistic pathways involved in millisecond-timescale synthesis.
Plasma-Droplet Synthesis Workflow
Nucleation and Growth Mechanisms
The investigation of plasma-enabled synthesis on millisecond timescales demonstrates that while the fundamental concepts of supersaturation, nucleation, and growth from the LaMer model remain relevant, the reality is often more complex and nuanced. The integration of advanced diagnostic techniques with microfluidic and plasma-droplet reactors has unequivocally shown that multiple pathways—including LaMer-like burst nucleation, Finke-Watzky autocatalytic growth, and non-classical particle-mediated routes—can operate in concert or competition depending on the specific synthesis environment. For the field of drug development, the ability to produce stabilizer-free, crystalline nanoparticles in milliseconds opens avenues for rapid screening of nano-formulations and the synthesis of high-purity metallic agents for therapeutics and diagnostics. Future research will benefit from further integrating multi-scale modeling with high-temporal-resolution experimental data to fully unravel the intricacies of nucleation and growth, ultimately enabling the predictive and programmable synthesis of advanced nanomaterials.
Leveraging Autocatalytic Surface Growth for Controlled Nanoparticle Synthesis
The controlled synthesis of colloidal metal nanocrystals represents a cornerstone of modern nanotechnology, with profound implications for catalysis, photonics, and medicine. For decades, the LaMer burst-nucleation model has served as the predominant theoretical framework describing nanocrystal formation as an atom-mediated process involving distinct stages of atom production, nucleation, and growth [3]. However, recent advances have revealed a more complex reality: non-classical pathways, particularly autocatalytic surface growth, play a decisive role in determining nanocrystal morphology and properties. This technical guide examines how autocatalytic surface reduction mechanisms enable precise control over nanoparticle synthesis, representing a significant advancement beyond the classical LaMer diffusion-controlled paradigm.
Autocatalytic processes are characterized by self-accelerating kinetics where the reaction product (in this case, the growing nanocrystal surface) acts as a catalyst for further reaction [34]. In nanocrystal synthesis, this manifests as precursor reduction occurring predominantly on the nanocrystal surface rather than in solution. The fundamental distinction between classical LaMer theory and autocatalytic surface growth lies in the primary building block and reaction locus: LaMer describes growth via atomic addition from solution, while autocatalytic growth involves direct surface reduction of precursors [3] [34]. This paradigm shift enables unprecedented control over nanocrystal evolution by manipulating surface-specific reduction kinetics.
Theoretical Framework: Beyond the LaMer Curve
Classical LaMer Diffusion-Mediated Growth
The classical LaMer model describes nanocrystal formation through three sequential stages [3]:
- Atom Production: Generation of zero-valent atoms through precursor reduction or thermal decomposition.
- Burst Nucleation: Once atomic concentration exceeds supersaturation threshold (Cmin~nu~), atoms aggregate to form stable nuclei.
- Growth via Atomic Addition: Subsequent deposition of atoms onto existing nuclei until precursor concentration drops below solubility threshold (C~s~).
This model positions atoms as exclusive building blocks and emphasizes diffusion-controlled kinetics, where the rate-limiting step is often atomic diffusion to the growing nanocrystal surface. While successfully explaining many synthetic systems, the LaMer model fails to account for numerous observed phenomena, including sudden size evolution through cluster coalescence and the formation of complex hierarchical structures [3].
Autocatalytic Surface-Controlled Growth
Autocatalytic surface growth represents a non-classical pathway where the nanocrystal surface itself catalyzes precursor reduction, creating a self-accelerating growth mechanism [34]. The fundamental process involves:
- Precursor Adsorption: Salt precursors adsorb onto specific surface sites of growing nanocrystals.
- Surface Reduction: Precursors undergo reduction directly on the nanocrystal surface.
- Atomic Incorporation: Newly formed atoms incorporate into the nanocrystal lattice.
- Surface Regeneration: The expanded surface provides additional catalytic sites for further precursor adsorption and reduction.
This mechanism exhibits distinct kinetic signatures, including an initial induction period followed by self-accelerating growth, eventually slowing as precursor depletion occurs [34]. Unlike diffusion-controlled LaMer growth, autocatalytic surface growth is governed by the atomic structure and chemistry of specific crystal facets, enabling facet-dependent growth rates that direct nanocrystal shape evolution [34].
Table 1: Comparative Analysis of Growth Mechanisms
| Characteristic | LaMer (Diffusion-Mediated) | Autocatalytic Surface Growth |
|---|---|---|
| Basic Building Block | Atoms | Precursor-surface complexes |
| Growth Locus | Solution-phase atomic addition | Surface-mediated reduction |
| Rate-Limiting Step | Atomic diffusion to surface | Surface reduction kinetics |
| Kinetic Profile | Decelerating | Self-accelerating then decelerating |
| Facet Dependence | Minimal | Strong dependence on facet type |
| Shape Control | Limited by diffusion anisotropy | Enabled by facet-dependent kinetics |
Quantitative Kinetics of Autocatalytic Surface Growth
Fundamental Kinetic Principles
The kinetics of autocatalytic surface reduction are quantitatively described by the Finke-Watzky two-step model, which involves slow continuous nucleation followed by fast autocatalytic surface growth [34]. For seed-mediated growth, the autocatalytic rate law takes the form:
r = k × [Precursor] × [Surface Sites]
where the rate constant k follows Arrhenius behavior: k = A × exp(-E~a~/RT)
The activation energy (E~a~) for surface reduction exhibits strong dependence on the atomic structure of specific crystal facets. Experimental studies with Pd nanocrystals demonstrate that the activation energy differs significantly between {100} and {111} facets, leading to facet-dependent growth rates and predictable shape evolution [34].
Quantitative Classification of Autocatalytic Strength
The autocatalytic strength significantly influences the reaction hazard and controllability. Based on the Perez-Benito model of two-stage competitive reactions, autocatalytic strength can be quantitatively classified using the factor ω = Q~1~/Q~total~ (the ratio of heat released by the initial stage to that of the total reaction) [35]:
Table 2: Quantitative Classification Scheme for Autocatalytic Strength
| ω Value Range | Autocatalytic Strength | Kinetic Characteristics | Decomposition Control |
|---|---|---|---|
| 0–5% | Strong | Pronounced autocatalytic acceleration | Difficult |
| 5–18% | Less strong | Moderate autoceleration | Challenging |
| 18–82% | Moderate | Balanced kinetics | Manageable |
| 82–95% | Less weak | Mild autoceleration | Easier |
| 95–100% | Weak | Minimal autoceleration | Straightforward |
This classification system provides a quantitative framework for predicting and managing the thermal risks associated with autocatalytic reactions during nanocrystal synthesis [35].
Experimental Protocols for Autocatalytic Growth
Seed-Mediated Synthesis with PdBr₄²⁻ Precursor
Objective: To quantitatively investigate facet-dependent autocatalytic surface reduction using well-defined Pd nanocrystal seeds.
Materials:
- Precursor Solution: Aqueous PdBr₄²⁻ (0.34 mM)
- Reducing Agent: Ascorbic acid (AA)
- Colloidal Stabilizer: Poly(vinyl pyrrolidone) (PVP)
- Seed Crystals: Pd nanocubes (18.0 ± 2.1 nm) and Pd octahedra (25.0 ± 2.9 nm) with purity >95% [34]
Methodology:
- Seed Preparation: Synthesize and characterize Pd cubic and octahedral seeds with well-defined facets using standardized methods [34].
- Reaction Setup: Prepare aqueous mixture containing seeds (0.1 mM), AA (20 mM), and PVP (0.1 mM).
- Precursor Injection: Rapidly inject PdBr₄²⁻ solution (0.34 mM final concentration) into reaction mixture.
- Kinetic Monitoring: Withdraw aliquots at predetermined time intervals (t = 0, 2, 4 hours) for TEM analysis.
- Product Characterization: Measure size evolution and facet development using TEM and ICP-MS analysis.
Key Observations:
- Cubic seeds ({100} facets) exhibit faster growth along 〈100〉 directions, enhancing corner truncation.
- Octahedral seeds ({111} facets) display preferential {111} facet growth, maintaining octahedral morphology.
- Complete suppression of homogeneous nucleation confirms autocatalytic dominance over solution reduction [34].
In Situ Monitoring of Autocatalytic Kinetics
Objective: To quantitatively measure autocatalytic surface reduction kinetics using isothermal calorimetry.
Materials:
- Accelerating Rate Calorimeter (ARC) with high sensitivity detection
- Test Substances: Cumyl hydroperoxide (CHP) and dicumyl peroxide (DCP) as reference materials [35]
- Sample Containers: Sealed ampoules compatible with test substances
Methodology:
- Sample Loading: Precisely weigh 0.5-1.0 mg test substance into ARC ampoule.
- Baseline Establishment: Equilibrate system at initial temperature (T~0~) below decomposition onset.
- Heat-Wait-Search Operation: Program ARC to operate in adiabatic mode with specific temperature increments.
- Data Collection: Continuously monitor temperature and pressure changes throughout decomposition.
- Kinetic Analysis: Calculate autocatalytic strength (ω) from heat release profile using Perez-Benito model [35].
Experimental Workflow for Autocatalytic Kinetics Analysis
Factors Influencing Autocatalytic Growth Kinetics
Crystallographic Facet Dependence
The atomic arrangement of crystal facets significantly impacts autocatalytic reduction kinetics. Quantitative studies reveal:
- {100} vs. {111} Facet Kinetics: Pd {100} facets exhibit different activation energies compared to {111} facets, directing anisotropic growth [34].
- Surface Coordination Chemistry: Br⁻ ion adsorption from PdBr₄²⁻ precursor preferentially passivates {100} facets, further enhancing facet-dependent growth rates [34].
- Twin Boundary Effects: Activation energy barriers differ significantly between single-crystal and twinned structures, influencing growth patterns [34].
Precursor Chemistry and Ligand Effects
The chemical identity of precursor complexes and stabilizing ligands profoundly affects autocatalytic kinetics:
- Hydrolysis Equilibria: PdBr₄²⁻ rapidly hydrolyzes in aqueous solution to form PdBr~n~(H₂O)~4-n~^2-n^ complexes (n < 4), altering reduction kinetics [34].
- Ligand Coordination: PVP coordinates with Pd(II) ions, modifying precursor reactivity and surface adsorption behavior [34].
- Ion Adsorption: Br⁻ ions released during precursor hydrolysis chemisorb onto specific facets, creating facet-specific growth inhibition [34].
Table 3: Research Reagent Solutions for Autocatalytic Growth
| Reagent | Function | Concentration Range | Mechanistic Role |
|---|---|---|---|
| PdBr₄²⁻ Precursor | Metal ion source | 0.1-0.5 mM | Provides Pd(II) with tunable\nreduction kinetics |
| Ascorbic Acid (AA) | Reducing agent | 10-50 mM | Enables slow reduction kinetics\nfavoring surface pathway |
| Poly(vinyl pyrrolidone) | Colloidal stabilizer | 0.05-0.2 mM | Prevents aggregation while\nmoderating precursor kinetics |
| Preformed Seeds | Growth templates | 0.05-0.1 mM | Provide defined facets for\nquantitative kinetics study |
| Bromide Ions | Surface modulator | 0-1.0 mM | Selective facet passivation\nfor morphology control |
Morphological Control via Autocatalytic Kinetics
Directional Growth and Shape Evolution
Autocatalytic surface reduction enables precise morphological control by exploiting facet-dependent kinetics:
- Cube Evolution: Cubic seeds evolve into truncated cubes or octapods through preferential 〈100〉 direction growth [34].
- Octahedron Development: Octahedral seeds maintain {111} facet dominance, evolving into perfect octahedra [34].
- Nanoparticle Assembly: Particle-mediated growth enables formation of mesocrystals, polycrystals, and single crystals through oriented attachment mechanisms [3].
Complex Architecture Synthesis
Beyond simple polyhedra, autocatalytic growth facilitates complex hierarchical structures:
- 0D Nanocrystals: Quantum dots and spherical nanoparticles through controlled burst growth [36].
- 1D Nanostructures: Nanorods, nanowires, and nanotubes via anisotropic autocatalytic growth [36].
- 2D Nanomaterials: Platelets and nanosheets through facet-selective surface reduction [36].
- 3D Architectures: Mesoporous networks and superstructures via particle-mediated assembly [36].
Morphological Control Through Autocatalytic Surface Growth
Autocatalytic surface growth represents a powerful paradigm for controlled nanoparticle synthesis, enabling precise morphological control that transcends the limitations of classical LaMer theory. The fundamental advantage of this approach lies in its exploitation of surface-specific kinetics, where facet-dependent activation energies direct predictable shape evolution. Quantitative understanding of these processes allows researchers to deliberately design synthesis protocols that yield specific nanocrystal morphologies with tailored properties for applications ranging from catalysis to biomedical imaging.
Future research directions will likely focus on expanding quantitative kinetic databases for various metal-precedentor systems, developing real-time monitoring techniques for autocatalytic processes, and integrating machine learning approaches to inverse design synthesis protocols for desired nanocrystal characteristics. As our fundamental understanding of autocatalytic surface growth deepens, this mechanistic pathway will undoubtedly enable increasingly sophisticated architectural control at the nanoscale, pushing the boundaries of materials design and functionality.
Joint Diffusion/Collision Models for Pure Metal Solidification
The solidification of pure metals is a fundamental process in materials science, governing the microstructure and properties of metallic components. This process is primarily governed by the interplay between two distinct growth mechanisms: diffusion-controlled growth and surface-process-controlled growth (often termed collision-controlled growth). Within the classical LaMer mechanism framework, crystal growth initiates with a supersaturation-driven nucleation event, followed by a growth stage where the rate-determining step shifts between monomer diffusion from the bulk to the crystal surface and the subsequent surface integration process [4]. Understanding and modeling the transition between these regimes is critical for predicting and controlling microstructural evolution in pure metals and advanced alloys. This guide provides a comprehensive technical overview of joint diffusion/collision models, integrating theoretical foundations, experimental methodologies, and quantitative analysis tools for researchers and scientists engaged in solidification research.
Theoretical Foundations
The LaMer Mechanism and Growth Regimes
The LaMer mechanism provides a foundational model for understanding crystallization, delineating a clear sequence from nucleation to growth. This process begins with the achievement of a supersaturated state, where the concentration of growth monomers (e.g., atoms or molecules in a melt) exceeds the equilibrium solubility, providing the thermodynamic driving force for phase transformation [4].
Following nucleation, the growth stage commences, which can be dominated by one of two primary mechanisms based on system conditions:
- Diffusion-Controlled Growth: This regime prevails when the concentration of growth monomers falls below the minimum critical concentration required for further nucleation. The rate-limiting step is the diffusion of monomers from the bulk liquid to the crystal surface. The growth rate is highly sensitive to concentration gradients and fluid flow conditions in the melt [4].
- Surface-Process-Controlled Growth (Collision-Controlled Growth): This regime becomes dominant when the diffusion of growth species from the bulk to the growth surface is sufficiently fast. In this case, the rate-limiting step is the integration of the monomer into the crystal lattice at the solid-liquid interface, a process dependent on atomic-scale attachment kinetics and interface morphology [4].
The transition between these regimes is influenced by factors such as the degree of supersaturation, temperature, interface energy, and the presence of fluid flow or convection.
Mathematical Modeling of Coupled Growth
Joint models aim to seamlessly integrate the physics of both diffusion and collision processes. A cornerstone of this analysis for systems with fluid flow is the Burton, Prim, and Slichter (BPS) model, which introduces the concept of an effective distribution coefficient ((k_{\text{eff}})) [37].
The BPS model accounts for the formation of a solute-enriched boundary layer at the solid-liquid interface when the rejection of solute (or, by analogy, the incorporation of a pure metal atom) is faster than its diffusion into the bulk liquid. The model is described by:
$$ k{\text{eff}} = \frac{k0}{k0 + (1 - k0) \cdot e^{[-V \delta_{\text{bps}}/D]}} $$
Where:
- (k0) is the equilibrium distribution coefficient ((CS/C_L)).
- (V) is the solid growth rate (m/s).
- (\delta_{\text{bps}}) is the thickness of the diffusion boundary layer (m).
- (D) is the diffusion coefficient of the solute/monomer in the liquid (m²/s).
This model demonstrates that the process is sensitive to parameters such as growth rate and the thickness of the diffusion boundary layer, which is itself influenced by fluid dynamics [37]. The growth rate (V) often depends on the prevailing growth mode, creating a coupled system.
Table 1: Key Parameters in the BPS Model for Joint Growth Modeling
| Parameter | Symbol | Units | Description | Influence on Growth |
|---|---|---|---|---|
| Equilibrium Distribution Coefficient | (k_0) | Dimensionless | Ratio of solute concentration in solid to liquid at equilibrium | Determines the theoretical maximum segregation; (k_0=1) for a pure metal. |
| Growth Rate | (V) | m/s | Velocity of the solid-liquid interface moving into the liquid. | Higher rates promote collision-control and can lead to interface instability. |
| Diffusion Coefficient | (D) | m²/s | Measure of the rate at which atoms/monomers diffuse through the liquid. | Lower values favor the formation of a thicker boundary layer, promoting diffusion-control. |
| Boundary Layer Thickness | (\delta_{\text{bps}}) | m | Thickness of the stagnant fluid layer adjacent to the crystal interface. | Controlled by fluid flow; a thinner layer reduces diffusion resistance. |
Interface Morphology and Stability
The stability and morphology of the solid-liquid interface are direct consequences of the dominant growth mode and the prevailing thermal and solutal conditions. The transition from a stable planar interface to cellular or dendritic structures is governed by constitutional supercooling. A planar growth morphology, desirable for uniform solidification, is achieved under conditions of low supercooling, low solute concentration, and a low growth rate [37].
The criterion for maintaining a planar interface to avoid cellular or dendritic growth is given by:
$$ G \ge \frac{m C0 (1-k0)}{D} \cdot V $$
Where:
- (G) is the temperature gradient in the liquid at the interface (K/m).
- (m) is the slope of the liquidus line (K/wt%).
- (C_0) is the initial concentration of solute (wt%).
This equation highlights that high temperature gradients and low growth rates stabilize a planar interface, a condition more readily achieved when surface-process kinetics are not limiting [37].
Experimental Protocols and Methodologies
The Cooled Finger Technique for Pure Metal Solidification
The cooled finger process is a fractional crystallization method that provides a well-controlled environment for studying diffusion and collision dynamics during solidification. The following is a detailed methodology based on experimental investigations of aluminum purification [37].
Objective: To investigate the solidification dynamics and growth mechanisms of high-purity aluminum under controlled thermal and fluid flow conditions.
Materials and Equipment:
- Induction Furnace: For melting the high-purity aluminum charge.
- Cooled Finger: A water-cooled steel or copper rod introduced vertically into the melt.
- Temperature Control System: Precision thermocouples (e.g., type K) placed in the melt and at the cooled finger tip to monitor thermal gradients.
- Rotation Mechanism: A motorized system to rotate the cooled finger at precise rates (e.g., 0-200 rpm).
- Inert Atmosphere Chamber: Argon or vacuum environment to prevent oxidation.
- Data Acquisition System: For continuous logging of temperature, rotation speed, and cooling water flow rate.
Experimental Procedure:
- Charge Preparation: A specific mass (e.g., 500 g) of high-purity (e.g., 2N7 to 3N) aluminum is placed in a graphite or ceramic crucible within the furnace.
- Melting and Stabilization: The furnace is heated to a temperature approximately 50-100°C above the melting point of aluminum (660°C) and held for 30 minutes to ensure a homogeneous and stable melt.
- Process Initiation: The cooled finger, pre-cooled with circulating water, is lowered into the melt surface. The rotation of the finger is initiated at a predefined rate (e.g., 50 rpm).
- Solidification and Data Recording: The growth of the solid aluminum shell on the finger is monitored. Temperature data from various locations in the melt are recorded at 1-second intervals. The experiment is typically terminated after a specific time or once a target shell thickness is achieved.
- Sample Analysis: The solidified aluminum shell is extracted and sectioned for metallographic analysis. Scanning Electron Microscopy (SEM) and Electron Backscatter Diffraction (EBSD) are used to characterize the microstructure (grain size, orientation, interface morphology).
Key Parameters to Control:
- Cooling Rate: Controlled by the temperature and flow rate of the cooling water.
- Temperature Gradient ((G)): Measured directly via thermocouples.
- Growth Rate ((V)): Inferred from the increase in shell thickness over time.
- Rotation Rate: Directly controls fluid flow and thus the boundary layer thickness ((\delta_{\text{bps}})).
Continuous Injection Synthesis for Model Validation
While developed for nanocrystals, the continuous injection process provides an excellent analogue for studying diffusion-limited growth and can be adapted to model metallic system behaviors [14].
Objective: To synthesize monodisperse colloidal particles (as a model system) by precisely controlling monomer flux via continuous precursor injection, validating diffusion-based growth models.
Materials:
- Precursor Solution: Cluster-based single-source precursor (e.g., InAs clusters).
- Seed Solution: Small nanocrystal seeds (e.g., 1.4 nm radius InAs QDs).
- High-Temperature Solvent: Non-coordinating solvents like 1-octadecene.
- Syringe Pump: For precise injection control (e.g., rates of 2-8 mL/h).
Procedure:
- Seed Preparation: Synthesize seed crystals using a standard hot-injection method.
- Reaction Setup: The seed solution is heated to the desired growth temperature (e.g., 240°C) under inert gas and stirring.
- Continuous Injection: The precursor solution is injected into the seed solution at a constant, slow rate (e.g., 2 mL/h) using a syringe pump.
- Aliquot Sampling: Small, equivolume aliquots are extracted at regular intervals (e.g., every 1 mL of injected precursor).
- Analysis: The aliquots are analyzed via UV-Vis-NIR spectroscopy to track the shift of the first excitonic absorption peak (1Smax) and calculate particle size using the Brus equation. Peak-to-valley ratios (P/V) and half-width-half-maximum (HWHM) are measured to assess size uniformity [14].
Data Interpretation:
- A linear relationship between injected precursor volume and particle size indicates ideal, diffusion-limited growth.
- Deviation from ideal growth and a loss of size uniformity (decreasing P/V, increasing HWHM) signal a shift towards reaction-limited growth or the onset of secondary nucleation, often due to an excessive injection rate [14].
Table 2: Research Reagent Solutions for Featured Experiments
| Reagent/Material | Function in Experiment | Specifications & Rationale |
|---|---|---|
| High-Purity Aluminum Charge | Base material for solidification studies. | Purity 2N7-3N; defines the initial impurity content for segregation studies. |
| Cluster-Based Single-Source Precursor | Provides a continuous, controlled flux of monomers for growth. | Ensures a steady monomer concentration (Cm), enabling diffusion-dependent growth [14]. |
| Non-Coordinating Solvent (e.g., 1-Octadecene) | High-temperature reaction medium for nanocrystal growth. | Minimizes surface passivation, allowing for clearer study of growth kinetics. |
| Graphite Crucible | Container for molten aluminum. | High-temperature stability and chemical inertness towards molten aluminum. |
| Syringe Pump | Delivers precursor solution at a precise, constant rate (Rinj). | Critical for maintaining a constant, low monomer concentration to enforce diffusion-control [14]. |
| Thermocouples (Type K) | Measure temperature gradient (G) in the melt. | Essential for validating the planar interface stability criterion. |
Visualization of Models and Workflows
LaMer Mechanism & Growth Regimes
Cooled Finger Experimental Workflow
Joint Diffusion-Collision Model Logic
Joint diffusion/collision models provide an essential framework for bridging the gap between atomistic attachment kinetics and macroscopic transport phenomena during pure metal solidification. The integration of classical theories like the LaMer mechanism with process-specific models such as the BPS equation allows researchers to deconvolute the complex interplay of parameters like growth rate, temperature gradient, and fluid flow. The experimental protocols and quantitative data tables presented herein offer a roadmap for systematically investigating these dynamics. As computational power and in situ characterization techniques continue to advance, the future of solidification science lies in the development of fully coupled multi-scale models that can seamlessly predict microstructure evolution from the nucleus to the final ingot, enabling the precise design of metallic materials for advanced applications.
Understanding and controlling crystallization processes is fundamental to advancements in materials science, chemistry, and pharmaceutical development. The governing mechanisms—classically defined by the LaMer model as either diffusion-controlled or surface-reaction-controlled growth—directly determine critical material properties such as particle size, morphology, and polymorphism [4]. Precise characterization of these dynamics is therefore essential. This technical guide provides an in-depth examination of two pivotal classes of characterization techniques: Transmission Electron Microscopy (TEM), which offers unparalleled spatial resolution for visualizing nanostructures, and In-Situ Spectroscopic Monitoring, which provides real-time temporal resolution for probing dynamic processes. The integration of these tools is driving a revolution in our ability to study and engineer materials at the most fundamental levels.
Transmission Electron Microscopy: Attaining Spatial Resolution
Transmission Electron Microscopy is an analytical technique that visualizes matter at an atomic scale by magnifying nanometer structures up to 50 million times, surpassing the resolution limits of light microscopy [38]. It operates by transmitting a high-energy electron beam through an ultra-thin, electron-transparent sample. Interactions between the electrons and the sample generate signals that are used to form high-resolution images and compositional data [39] [38].
Core TEM Techniques and Applications
Modern TEM encompasses a suite of complementary techniques that provide multimodal information.
- Scanning TEM (STEM): STEM combines the principles of TEM and scanning electron microscopy (SEM) by scanning a finely focused electron beam across the sample in a raster pattern [39]. A key advantage of STEM over conventional TEM is the ability to simultaneously acquire multi-modal data, such as characteristic X-rays for Energy-Dispersive X-ray Spectroscopy (EDS) and Electron Energy-Loss Spectroscopy (EELS) spectra, which can be spatially correlated to build a virtual image [39]. The primary benefit is an improvement in spatial resolution [39].
- Energy-Dispersive X-ray Spectroscopy (EDS): EDS performs elemental analysis by detecting the characteristic X-rays generated by sample atoms when excited by the electron beam. This provides crucial qualitative and quantitative compositional information that complements high-resolution imaging [39].
- Electron Energy-Loss Spectroscopy (EELS): EELS analyzes the energy distribution of electrons that have interacted inelastically with the sample. It is a powerful technique for probing local chemical environments, oxidation states, and electronic properties [39].
- Cryo-Electron Microscopy (Cryo-EM): Cryo-EM involves analyzing cryogenically frozen samples in specially designed TEMs. Recent advancements have enabled the determination of 3D structures down to atomic resolution, making it indispensable for life sciences and soft matter research [39].
Table 1: Key TEM Techniques and Their Primary Applications
| Technique | Primary Output | Key Information Obtained | Relevance to Crystal Growth |
|---|---|---|---|
| HR-TEM/STEM | Atomic-resolution images | Lattice fringes, crystal defects, interface structure | Direct visualization of nucleation sites, crystal shape, and defects. |
| EDS | Elemental spectrum & maps | Spatial distribution and concentration of elements | Tracking reagent distribution and identifying impurities during synthesis. |
| EELS | Electron energy-loss spectrum | Chemical bonding, oxidation state, electronic structure | Probing surface chemistry and reaction pathways during growth. |
| Cryo-EM | 3D macromolecular structure | Architecture of beam-sensitive materials (proteins, soft materials) | Visualizing nucleation of biological macromolecules without damage. |
Advanced and In-Situ TEM Modalities
The frontier of TEM lies in its ability to observe dynamic processes in real-time under controlled conditions.
- Environmental TEM (ETEM): ETEM, such as the Thermo Scientific Themis ETEM, allows for in situ observation of materials in gaseous environments at pressures up to 0.5 mbar and at elevated temperatures [40]. This is crucial for observing structure dynamics, such as catalytic reactions or nanomaterial growth, in their operative environments [41] [40].
- Single-Molecule Atomic-Resolution Real-Time TEM (SMART-TEM): This technique enables the direct microscopic observation of structural changes in single molecules, such as conformational changes and dimerization reactions, as they occur [42]. This provides unprecedented insight into reaction kinetics and pathways at the molecular level.
Table 2: In-Situ TEM Modalities for Dynamic Studies
| Technique | Experimental Capability | Measured Parameters | Example Application |
|---|---|---|---|
| Environmental TEM (ETEM) | Sample in controlled gas atmosphere & temperature | Structural changes in reactive environments; nucleation & growth in real-time | Observing catalyst nanoparticle sintering or carbon nanotube growth. |
| SMART-TEM | Atomic-resolution imaging with high temporal resolution | Molecular conformation changes, reaction intermediates & pathways | Direct observation of C60 fullerene dimerization within a carbon nanotube [42]. |
| Liquid Cell TEM | Imaging through liquid enclosures | Growth kinetics, particle motion, and aggregation in solution | Studying the growth of metal nanocrystals from solution. |
The following diagram illustrates a typical workflow for an in situ ETEM experiment, from sample preparation to data analysis:
In-Situ Spectroscopic Monitoring: Capturing Temporal Dynamics
While TEM provides supreme spatial detail, in-situ spectroscopic monitoring captures the kinetic and mechanistic details of reactions as they unfold, eliminating the delays and perturbations inherent to ex-situ sampling [43].
Foundational Principles and Implementation
In-situ spectroscopy is the optimal tool when a reaction involves transient intermediate species, a fast equilibrium that is perturbed by sampling, or very fast reaction rates [43]. It is also advantageous for monitoring slow reactions automatically, working with expensive or limited reagents, and handling systems sensitive to oxygen or moisture [43].
A structured approach to implementation is critical for success. Key steps include [43]:
- Feasibility Assessment: Confirming that the reaction requires real-time monitoring and selecting the appropriate technique (e.g., IR, Raman, NIR).
- Technique Selection: Choosing based on factors like concentration, homogeneity, and the presence of interfering species.
- Calibration and Validation: Running calibration experiments with known standards and validating in-situ results against primary techniques like GC or LC.
Advanced Ultrafast Spectroscopic Methods
Recent advances have pushed the boundaries of temporal resolution. Asynchronous and Interferometric Transient Absorption (AI-TA) spectroscopy is one such transformative tool [44]. It employs two synchronized mode-locked femtosecond lasers with slightly different repetition rates to generate pump-probe time delays without mechanical delay stages. This allows for rapid data acquisition over a vast dynamic range, enabling the real-time observation of photochemical reactions, such as photoinduced halide substitution in perovskite nanocrystals and the light-induced coalescence of perovskite nanoplatelets [44].
The diagram below outlines the core operational principle of AI-TA spectroscopy:
Integrated Workflows in Crystal Growth Research
The true power of these techniques is realized when they are integrated to study complex processes like crystallization, directly interrogating the principles of the LaMer mechanism.
The LaMer Mechanism and Modern Validation
The LaMer model provides a foundational framework for understanding colloidal nanocrystal synthesis, describing two distinct growth regimes [4]:
- Diffusion-controlled growth: Occurs when the concentration of growth monomers falls below the minimum critical concentration for nucleation. The growth rate is limited by the diffusion of monomers from the bulk solution to the crystal surface.
- Surface-reaction-controlled growth: Occurs when monomer diffusion is fast, and the growth rate is limited by the incorporation of monomers into the crystal lattice at the surface.
Advanced characterization has been key to validating and refining this model. For instance, in situ TEM has allowed for the direct observation of nucleation and growth events [4]. Furthermore, research on the synthesis of monodisperse InAs quantum dots has demonstrated that a diffusion-dependent growth mode is essential for achieving narrow size distributions, a core tenet of the LaMer theory [14]. This study used a continuous injection process and mathematical modeling based on a modified Fick's law to show that controlling the monomer flux via injection rate is critical to maintaining this desired growth mode and preventing secondary nucleation [14].
Case Study: Diffusion-Dynamics-Controlled Synthesis of InAs Nanocrystals
A seminal example of integrating characterization with modeling is the diffusion-dynamics-controlled (DDC) synthesis of large InAs nanocrystals [14]. The researchers faced the challenge of growth suppression beyond 5 nm. By developing a growth model based on molecular diffusion and experimentally validating it, they demonstrated that the precursor injection rate (Rinj) directly controls monomer concentration (Cm) and thus the growth mode.
- Fast Rinj (8 mL/h): Led to high Cm, causing secondary nucleation and poor size uniformity (broader HWHM) as growth deviated from the diffusion-controlled ideal [14].
- Slow Rinj (2 mL/h): Maintained a low Cm, enabling a prolonged diffusion-controlled (size-focusing) growth regime and producing nanocrystals with a narrow size distribution (12.2%) up to 9.0 nm in size [14].
This case highlights how in-situ monitoring of optical properties (absorption spectra) combined with modeling can guide process intensification strategies to achieve superior material control.
Table 3: Experimental Parameters for Diffusion-Controlled Crystal Growth
| Parameter | Impact on Growth Mechanism | Optimal Control Strategy | Characterization Method for Validation |
|---|---|---|---|
| Precursor Injection Rate (Rinj) | High Rinj increases Cm, promoting secondary nucleation; Low Rinj maintains diffusion-control [14]. | Continuous injection at a slow, controlled rate [14]. | Temporal absorption spectroscopy; TEM for final size distribution [14]. |
| Monomer Concentration (Cm) | Must be maintained between critical nucleation and growth concentrations to sustain focusing [14]. | Regulated via Rinj and reaction volume (Diffusion-Dynamics-Control) [14]. | Calculated from model, correlated with growth rate from optical data [14]. |
| Temperature | Affects both precursor conversion rate and surface integration kinetics. | Precise control via specialized heating stages (e.g., NanoEx-i/v) [40]. | In-situ TEM/EELS for structural/chemical changes; calibrated heating holders [40]. |
| Solvent/Additives | Influence precursor reactivity, viscosity, and surface energy. | Use of microreactors for enhanced mixing and heat transfer [4]. | NMR, in-situ FTIR, Raman spectroscopy to track intermolecular interactions [4]. |
The Scientist's Toolkit: Essential Reagent Solutions
The following table details key reagents and materials essential for experiments in controlled crystal growth and advanced characterization.
Table 4: Key Research Reagent Solutions for Crystal Growth and Characterization
| Item | Function / Application | Technical Notes |
|---|---|---|
| Single-Source Precursor (e.g., InAs Cluster) | Provides controlled, continuous release of monomers for nanocrystal growth [14]. | Enables diffusion-dependent growth mode; critical for achieving high size uniformity in covalent QDs like InAs [14]. |
| Schottky Field Emission Gun (FEG) | High-brightness, coherent electron source for TEM/STEM. | Provides superior spatial resolution and analytical performance for EDS and EELS [38]. |
| FIB-SEM System | Preparation of electron-transparent TEM lamella from bulk materials. | Automated systems (e.g., Thermo Scientific) enable robust, high-quality sample preparation for non-specialists [39]. |
| Microreactor / Continuous Flow System | Process intensification for crystallization. | Enhances mixing and heat transfer, leading to boosted nucleation rates and improved process control [4]. |
| Cryogenic Sample Preparation Tools | Vitrification of hydrated biological or soft materials for Cryo-EM. | Preserves native structure in vacuum by rapid freezing, enabling atomic-resolution structure determination [39]. |
| Dedicated In-Situ TEM Holders | (e.g., Heating, Gas, Liquid) | Enables real-time observation of dynamic processes such as crystallization, catalytic reactions, and battery cycling [40]. |
The journey from high-resolution TEM to real-time in-situ spectroscopic monitoring represents a paradigm shift in characterization sciences. TEM and its advanced modalities, such as ETEM and SMART-TEM, provide the spatial resolution to visualize the very architecture of materials at the atomic scale. In parallel, in-situ spectroscopic techniques, from reaction monitoring to ultrafast AI-TA, deliver the temporal resolution to capture kinetic pathways and transient intermediates. When these powerful approaches are integrated—as demonstrated in the diffusion-controlled synthesis of nanocrystals—they provide a holistic picture of complex processes like crystal growth governed by the LaMer mechanism. This synergistic use of characterization tools, guided by computational models and process intensification strategies, is the cornerstone of modern materials design, enabling the fine-tuning of next-generation materials, therapeutics, and technologies.
Optimizing Nanomaterial Synthesis: Controlling Size, Dispersion, and Crystallinity
Identifying and Overcoming Synthesis Limitations in LaMer's Model
For decades, the LaMer model has served as a foundational framework for understanding the formation of monodisperse colloidal nanoparticles. This model posits a temporal separation of nucleation and growth phases, characterized by "burst nucleation" when monomer concentration exceeds critical supersaturation, followed by diffusion-controlled growth without further nucleation. While this paradigm has guided nanomaterial synthesis since 1950, modern research reveals significant limitations in its application to complex synthetic systems, particularly for metallic nanocrystals, semiconductors, and pharmaceutical nanoparticles. This technical review critically analyzes these limitations through the lens of contemporary research, presents experimental methodologies for mechanistic elucidation, and discusses advanced models that better account for non-classical crystallization pathways. The insights provided are particularly relevant for ongoing research on LaMer mechanism diffusion versus surface-controlled crystal growth, offering scientists a refined toolkit for predictable nanomaterial design.
The LaMer model, first articulated in 1950 by LaMer and Dinegar, represents a cornerstone of classical nucleation theory [1]. Its most enduring conceptualization is the LaMer diagram (Figure 1), which illustrates how monodisperse particles form through three distinct stages: (I) a steady increase in monomer concentration leading to supersaturation, (II) an abrupt "burst nucleation" event when concentration exceeds the critical supersaturation threshold ((C{crit})), and (III) a focused growth phase where existing nuclei grow by monomer addition without further nucleation events as concentration drops below (C{min}^{nu}) and eventually reaches the solubility concentration ((C_s)) [3] [45].
The model's core premise is that nucleation becomes "effectively infinite" once the system reaches a critical supersaturation level, creating a sharp separation between nucleation and growth phases that theoretically yields uniform particles [1]. This qualitative description has been widely adopted across material systems, from original sulfur hydrosols to modern metal and semiconductor nanocrystals. However, as synthesis methodologies have advanced toward more complex architectures and compositions, significant limitations have emerged in the model's ability to accurately predict and describe observed nanoparticle formation pathways, prompting critical re-evaluation and the development of more sophisticated mechanistic frameworks.
Critical Limitations of the Classical LaMer Model
Theoretical and Mechanistic Shortcomings
The LaMer model rests on several simplifying assumptions that frequently diverge from experimental observations in contemporary nanomaterial synthesis:
Oversimplified Nucleation Description: The model treats nucleation as an instantaneous, homogeneous process with an "effectively infinite" rate once supersaturation is achieved [1]. However, modern mechanistic studies reveal nucleation as a multi-step process involving precise molecular pathways. Recent evidence indicates the kinetically effective nucleus may comprise just 2-3 atoms in strong-bonding systems, with complex energy landscapes not captured by classical nucleation theory [46].
Exclusive Focus on Atom-Mediated Growth: The model assumes atoms as the sole building blocks for nanocrystal formation. This ignores non-classical particle-mediated pathways observed in metal nanocrystal syntheses, where nanoparticles can act as building blocks through aggregation, coalescence, and oriented attachment mechanisms [3].
Inadequate Treatment of Growth Kinetics: While LaMer emphasizes diffusion-controlled growth, numerous systems exhibit reaction-limited or surface-controlled kinetics that significantly impact final particle morphology and size distribution [3]. The model's quantitative equation has rarely been successfully applied to fit experimental particle formation kinetics data over the past 70 years [1].
Neglect of Secondary Processes: The model does not account for Ostwald ripening, digestive ripening, agglomeration, or symmetry-breaking events that frequently occur during nanoparticle formation and profoundly affect particle size distributions [3] [46].
Experimental Evidence Challenging the LaMer Paradigm
Critical analysis of 164 papers that provide substantive discussion of the 1950 model reveals limited compelling experimental support for "burst nucleation" and "diffusion-controlled growth" across various material systems:
Table 1: Experimental Evidence for LaMer Model Across Material Systems
| Material System | Number of Papers Analyzed | Support for Burst Nucleation | Support for Diffusion-Controlled Growth | Key Counter-Observations |
|---|---|---|---|---|
| Silver Halide Nanoparticles | 13 | Limited/Contested | Limited | Complex nucleation pathways |
| Semiconductor Nanoparticles | 26 | Limited | Limited | Multi-step nucleation observed |
| Transition-Metal Nanoparticles | 69 | Limited | Limited | Aggregation-based growth prevalent |
| Metal-Oxide Nanoparticles | 39 | Limited | Limited | Particle-mediated mechanisms dominant |
A comprehensive review by Whitehead et al. concluded that "the concepts of 'burst/instantaneous nucleation' and 'diffusion-controlled growth' lack sound, compelling experimental support in the 70 years since the model first appeared" [1]. This assessment is further supported by the observation that most syntheses previously described by LaMer kinetics actually proceed through more complex Finke-Watzky-type two-step growth mechanisms or other non-classical pathways [3] [46].
Methodologies for Mechanistic Investigation
Advanced In Situ Characterization Techniques
Overcoming LaMer model limitations requires experimental methods capable of directly probing nucleation and growth events at relevant temporal and spatial scales:
Synchrotron X-ray Absorption Fine Structure (XAFS) Spectroscopy: Provides oxidation state, coordination number, and structural information during particle formation, enabling identification of molecular precursors and intermediate species [46].
Synchrotron Small-Angle X-ray Scattering (SAXS): Allows real-time monitoring of particle radius and number concentration, directly testing LaMer's prediction of constant particle number during growth [46].
In Situ Electron Microscopy: Reveals non-classical growth pathways such as particle aggregation and coalescence that defy LaMer's atom-only growth assumption [3].
These techniques have demonstrated that nucleation is often not instantaneous but rather a continuous or multi-step process, with growth occurring through both atom addition and particle attachment mechanisms depending on synthetic conditions [3] [46].
Requirements for Disproof-Based Mechanistic Studies
Establishing reliable, disproof-based chemical mechanisms requires meeting four critical criteria [46]:
- Experimental Stoichiometry Determination: Complete mass- and charge-balanced reaction stoichiometry that proposed mechanistic steps must sum to
- Comprehensive Kinetics Data: Full rate law determination using multiple direct physical methods
- Pseudoelementary Step Formulation: Mass- and charge-balanced step reactions that define rate constants and terminology
- Disproof-Based Validation: Quantitative comparisons of experimental data to competing alternative mechanisms
This rigorous approach stands in contrast to the qualitative descriptions often associated with LaMer-based interpretations and enables the development of predictive models such as Mechanism-Enabled Population Balance Modeling (ME-PBM) [46].
Figure 2: Disproof-Based Workflow for Nanoparticle Formation Studies
Emerging Models Beyond LaMer
Non-Classical Particle-Mediated Pathways
Contemporary research has established that nanoparticle formation frequently occurs through mechanisms not envisioned in the LaMer model:
Particle-Mediated Nucleation and Growth: Small nanoclusters merge and reshape to form single-crystalline nanoparticles through aggregation and coalescence pathways [3]. This non-classical model considers nanoparticles as building units rather than individual atoms.
Oriented Attachment: Crystallographically aligned nanoparticles fuse along specific crystal planes to form larger architectures, often creating mesocrystals with unique properties [3].
Finke-Watzky Two-Step Mechanism: A minimalistic mechanism consisting of slow continuous nucleation followed by autocatalytic growth that successfully describes many metal nanoparticle formation reactions [1] [46].
These pathways are particularly relevant for understanding the formation of complex nanostructures including nanorods, nanoplatelets, and hierarchical architectures that cannot be explained by classical LaMer kinetics.
Quantitative Parameters for Deterministic Synthesis
Moving beyond qualitative LaMer descriptions, researchers have identified quantitative parameters that enable deterministic synthesis control:
Reduction Rate as Quantitative Knob: The reduction rate of metal salt precursors serves as a predictive parameter controlling nucleation and growth outcomes, enabling deterministic synthesis of nanocrystals with specific defect structures, shapes, and compositions [47].
Mechanism-Enabled Population Balance Modeling (ME-PBM): This advanced modeling approach tracks every particle in a formation pathway using disproof-based minimum mechanisms, enabling prediction of both average particle sizes and particle-size distributions [46].
Table 2: Comparison of Classical and Non-Classical Nucleation/Growth Models
| Feature | Classical LaMer Model | Non-Classical Particle-Mediated Model | Finke-Watzky Two-Step Model |
|---|---|---|---|
| Building Units | Atoms/monomers only | Nanoparticles/clusters as units | Molecular precursors → nanoparticles |
| Nucleation Characteristics | Instantaneous/burst | Multi-step, often continuous | Slow continuous nucleation |
| Growth Mechanism | Diffusion-controlled monomer addition | Aggregation, coalescence, oriented attachment | Autocatalytic surface growth |
| Predictive Capability | Limited qualitative guidance | Structure-specific predictions | Quantitative kinetics predictions |
| Experimental Support | Limited across material systems | Strong for metal/metal-oxide systems | Verified for multiple metal nanoclusters |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Nanoparticle Mechanistic Studies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Synchrotron Radiation Sources | Enables XAFS and SAXS measurements for in situ monitoring | Tracking precursor conversion and particle growth in real time [46] |
| Metal Salt Precursors | Source of metal atoms for nanocrystal formation | HAuCl₄ for gold nanocrystals, AgNO₃ for silver nanoparticles [47] |
| Reducing Agents | Controls reduction rate of metal precursors | Sodium citrate, ascorbic acid, borohydride derivatives [47] |
| Capping Agents/Stabilizers | Regulate growth kinetics and prevent aggregation | Citrate, CTAB, PVP, thiol ligands [3] [46] |
| Shape-Directing Agents | Promote anisotropic growth for specific morphologies | Halide ions, surfactants with specific binding affinities [47] |
| Fluidized Bed Reactors | Enable atomic layer deposition on particulate materials | Creating nanostructured particles with core-shell architectures [48] |
The LaMer model's historical significance in establishing the fundamental paradigm of separated nucleation and growth stages for monodisperse nanoparticle formation is undeniable. However, its limitations in describing complex synthetic systems, accounting for non-classical pathways, and providing predictive capability necessitate moving beyond this classical framework. Contemporary research demonstrates that successful nanoparticle synthesis design requires mechanistic understanding based on disproof-based experimental approaches and quantitative parameters such as precursor reduction rate.
Future advances in nanoparticle formation science will increasingly rely on integrated methodologies combining sophisticated in situ characterization, quantitative kinetic analysis, and mechanistic modeling that accounts for both atom-mediated and particle-mediated pathways. This multifaceted approach promises to finally achieve the long-sought goal of deterministic nanomaterial synthesis with precise control over size, shape, and functionality—particularly relevant for pharmaceutical applications where continuous manufacturing of nanomaterial-containing drug products requires rigorous mechanistic understanding [49]. As the field progresses, the legacy of the LaMer model will remain as an important historical foundation upon which more sophisticated and predictive mechanistic frameworks are being built.
The synthesis of monodisperse crystals, where particles exhibit near-identical size and shape, represents a fundamental objective in materials science, nanotechnology, and pharmaceutical development. Achieving monodispersity is critically dependent on controlling nucleation and growth processes, particularly on precluding secondary nucleation—the formation of new nuclei after the initial nucleation event. This undesirable phenomenon leads to polydisperse populations that lack the uniform physical and chemical properties required for advanced applications.
This technical guide frames the challenge of secondary nucleation within the classical LaMer mechanism, which describes crystallization as a sequence of stages: achieving supersaturation, a distinct nucleation event, and subsequent growth without further nucleation [4]. The mechanism primarily outlines two growth paradigms following nucleation: diffusion-controlled growth, where monomer diffusion to the crystal surface limits growth, and surface-process-controlled growth, where the integration of monomers into the crystal lattice is the rate-limiting step [4]. Understanding and manipulating the transition between these regimes is essential for suppressing secondary nucleation and achieving high monodispersity. This whitepaper provides an in-depth analysis of contemporary strategies, experimental protocols, and theoretical models for precluding secondary nucleation, serving as a comprehensive resource for researchers and drug development professionals.
Theoretical Foundations: The LaMer Mechanism and Growth Regimes
The LaMer model provides a conceptual framework for understanding the temporal evolution of monomer concentration during a crystallization process. A successful synthesis of monodisperse particles requires a clear separation of the nucleation and growth phases.
The Classic LaMer Diagram and Its Modern Interpretation
The classic LaMer diagram illustrates three critical concentration thresholds: the solubility limit (CS), the minimum concentration for nucleation (CN), and the maximum achievable concentration (Cmax). The objective is to rapidly raise monomer concentration above CN to induce a short, burst nucleation event, after which the concentration should fall below CN but remain above CS to allow growth on existing nuclei without generating new ones [4].
Modern interpretations acknowledge that perfect separation is often not achieved. For instance, in the hot-injection synthesis of gallium nanoparticles, a significant temporal overlap between nucleation and growth stages can still yield monodisperse products if the kinetics of nucleation and growth are carefully balanced [50]. This balance is often managed by surface ligands that modulate reactivity.
Diffusion vs. Surface-Controlled Growth
The LaMer mechanism distinguishes two primary growth forms based on which process is rate-limiting:
- Diffusion-Controlled Growth: This occurs when the concentration of growth monomers falls below the minimum critical concentration required for nucleation. Crystal development continues, but nucleation ceases because the energy barrier for new nucleus formation cannot be overcome. Growth rate is governed by the diffusion of monomers from the bulk solution to the crystal surface [4].
- Surface-Process-Controlled Growth: When monomer diffusion from the bulk to the growth surface is sufficiently rapid, the surface integration process itself—the attachment of monomers to the crystal lattice—controls the growth rate [4].
Achieving size-focusing (the phenomenon where a polydisperse ensemble becomes more uniform over time) is most effective under diffusion-controlled growth. This is described by the Lifshitz-Slyozov-Wagner theory, where smaller particles, having higher solubility, dissolve and re-deposit onto larger particles (Ostwald ripening). However, if the monomer concentration is too high, this can lead to secondary nucleation and interparticle ripening, which increases polydispersity [14]. The key is to maintain the monomer concentration (Cm) within a narrow window that enables growth but suppresses both secondary nucleation and ripening.
The following diagram illustrates the relationship between monomer concentration and the different stages of crystal development within the LaMer model, highlighting the critical window for monodisperse growth.
Quantitative Insights and Experimental Data
Theoretical models must be validated and guided by quantitative experimental data. The following table summarizes key findings from recent studies on achieving monodispersity by controlling growth conditions to preclude secondary nucleation.
Table 1: Quantitative Data on Growth Parameters for Monodisperse Synthesis
| Material System | Key Controlled Parameter | Optimal Value | Impact on Secondary Nucleation & Size Distribution | Reference |
|---|---|---|---|---|
| InAs Quantum Dots | Precursor Injection Rate (Rinj) | 2 mL/h | Suppressed secondary nucleation; achieved size-focusing growth with narrow HWHM (Half-Width Half-Maximum). Faster rates (4 & 8 mL/h) led to growth suppression and increased polydispersity [14]. | [14] |
| Colloidal Gallium | Surface Ligand (Secondary Amine) | Dioctylamine (DOA) or Didodecylamine | Balanced nucleation/growth kinetics; enabled colloidal stability despite temporal overlap of nucleation and growth stages, yielding monodisperse nanoparticles [50]. | [50] |
| Polyamide 11 | Crystallization Temperature | ~115 °C (High supercooling) | Shifted nucleation mechanism to homogeneous nucleation; high nuclei density favored formation of a specific β-mesophase, controlling polymorphism [4]. | [4] |
| General Model (InAs) | Monomer Concentration (Cm) | Maintained between CS and CN | Enabled diffusion-dependent growth; prevented both secondary nucleation and interparticle ripening, key for extending the "focusing regime" [14]. | [14] |
Methodologies and Experimental Protocols
Continuous Injection Synthesis with Diffusion Dynamics Control (DDC)
This protocol, adapted from the synthesis of monodisperse InAs nanocrystals, is designed to maintain monomer concentration within the monodisperse growth window through precise feeding [14].
Research Reagent Solutions Table 2: Essential Reagents for Continuous Injection Synthesis
| Reagent | Function / Role in Controlling Nucleation |
|---|---|
| Seed Solution (e.g., 1.4 nm radius InAs QDs) | Provides uniform initial nuclei; subsequent growth focuses on these seeds, making new nucleation unnecessary. |
| Single-Source Precursor (e.g., InAs cluster) | The monomer source; its controlled decomposition/reaction rate is crucial for managing Cm. |
| Long-Chain Secondary Amine (e.g., Dioctylamine) | Surface ligand; passivates growing crystal surfaces, modulates growth kinetics, and provides colloidal stability [50]. |
| Non-Coordinating Solvent (e.g., 1-Octadecene) | High-boiling-point solvent provides a stable medium for high-temperature reactions. |
Detailed Protocol:
- Seed Preparation: Synthesize a small, uniform seed population (e.g., InAs QDs with a radius of 1.4 nm) using a standard hot-injection method [14].
- Reaction Setup: Charge a multi-neck flask with the seed solution and a non-coordinating solvent (e.g., 1-Octadecene). Equip the flask with a stirrer, thermocouple, and septum. Purge the system with an inert gas (e.g., N2).
- Heating and Stabilization: Heat the reaction mixture to the target growth temperature with vigorous stirring to ensure uniform heat and mass transfer.
- Precursor Injection: Using a syringe pump, initiate the continuous injection of the single-source precursor solution at a meticulously controlled rate (Rinj). The critical objective is to set Rinj slower than the precursor conversion rate to ensure that Cm is determined by the injection rate and the monomer diffusion rate (Rdiff) to the seed surfaces [14].
- Growth and Monitoring: Continue the injection for the required duration. Monitor the growth in real-time if possible (e.g., via in situ spectroscopic methods or SAXS) by taking aliquots at regular intervals to measure the absorption spectra and track the position and width of the excitonic peak.
- Termination and Purification: Once the target size is reached, stop the injection and rapidly cool the reaction mixture to room temperature. Purify the resulting monodisperse nanocrystals by standard precipitation/redispersion cycles.
Critical Step Rationale: A slow Rinj (e.g., 2 mL/h for the referenced InAs system) is the most direct operational parameter for controlling Cm. It prevents a burst of precursor conversion that would transiently elevate Cm into the secondary nucleation zone, thereby maintaining diffusion-dependent growth and ensuring size-focusing [14].
Hot-Injection Synthesis with In Situ Monitoring
This protocol leverages rapid precursor injection to achieve burst nucleation, followed by growth, and uses in situ analytics to understand the process dynamics, as demonstrated for gallium nanoparticles [50].
Detailed Protocol:
- Precursor Preparation: Prepare the metal-organic precursor (e.g., tris(dimethylamido)gallium dimer) and the surface ligand (e.g., Dioctylamine, DOA) in an inert atmosphere glovebox.
- Reaction Setup: Assemble a custom reactor (e.g., a three-neck flask with Kapton windows for X-ray scattering) with stirring, temperature control, and a sealed injection port.
- Hot-Injection: Heat the solvent and ligand mixture to the reaction temperature (e.g., 230–280 °C). Rapidly inject the precursor solution into this hot solution. This causes a sudden supersaturation event, leading to homogeneous nucleation.
- In Situ Monitoring: Continuously record data throughout the synthesis using a technique like Small-Angle X-Ray Scattering (SAXS). The SAXS setup allows for the quantification of particle size, polydispersity, and the formation of secondary agglomerates in real-time [50].
- Data Quantification: Analyze the SAXS patterns by fitting a spherical form factor with a Schulz–Zimm size distribution model. This quantifies the evolution of the average particle diameter and polydispersity over time, allowing researchers to identify the timestamps and degree of overlap for the precursor reaction, nucleation, and growth stages [50].
Key Insight: This method revealed that for gallium nanoparticles, a large temporal overlap between nucleation and growth stages does not necessarily preclude monodispersity, provided that surface ligands effectively balance the kinetics of both processes [50].
The workflow for this integrated synthesis and monitoring approach is illustrated below.
Advanced Strategies and Future Trends
Process Intensification Technologies
Emerging technologies like membrane crystallization (MCr) and microreactors offer advanced control over crystallization processes. MCr uses a membrane as a controlled interface for supersaturation, simultaneously separating solutions and inducing component solidification. This technology can precisely control crystal nucleation and intensify hybrid continuous crystallization [4]. Microscale process intensification (MPI) technologies, such as microreactors, enhance micro-mixing, drastically reduce mixing times, and achieve precise control over the nucleation-growth process, enabling the production of crystals with optimal form and structural stability from nano to micro-scale [4].
Computational Modeling and Simulations
Computational approaches have become indispensable for predicting and understanding nucleation. Molecular dynamics (MD) simulations and other numerical methods shed light on the mechanics of crystal nucleation, allowing researchers to predict nucleation rates and identify critical variables [4]. For instance, a devised nucleation and growth model successfully simulated the early hydration of alite, with the predicted growth rate aligning well with experimental calorimetric data [4]. These models provide atomistic-level insights and are key for the rational design of crystallization processes, moving beyond traditional trial-and-error approaches.
Achieving monodisperse crystals by precluding secondary nucleation remains a challenging yet attainable goal. The fundamental principle involves strict control over the monomer concentration (Cm) throughout the synthesis to maintain the system within the diffusion-controlled growth window of the LaMe r diagram. As detailed in this guide, this is operationally achieved through strategies such as optimizing precursor injection rates in continuous processes, employing tailored surface ligands to balance kinetics, and utilizing process intensification technologies for superior mixing and control. The integration of in situ monitoring and advanced computational models provides the necessary feedback and predictive power to transform crystal synthesis from an empirical art into a rational, predictable science. By adopting these integrated strategies, researchers and engineers can reliably produce high-quality, monodisperse crystals tailored for applications ranging from drug formulation to advanced optoelectronics.
Manipulating Supersaturation and Reaction Flux for Desired Outcomes
Supersaturation, the driving force of crystallization, is a metastable state where a solution contains more dissolved solute than its equilibrium solubility. Reaction flux describes the rate of mass transport of dissolved species to the growing crystal interface. The precise management of these two parameters is fundamental to dictating the kinetics of nucleation and growth, thereby determining critical crystal attributes such as size, size distribution, morphology, and purity. This control is paramount across diverse fields, from manufacturing active pharmaceutical ingredients (APIs) with specific bioavailability to synthesizing functional nanomaterials with tailored optoelectronic properties.
The LaMer mechanism provides a foundational model for understanding crystallization processes, conceptually separating them into distinct stages of nucleation and growth [1]. Within this framework, two primary growth mechanisms are identified: diffusion-controlled growth, where the rate is limited by the mass transport of growth units through the solution to the crystal surface, and surface-process-controlled growth, where the rate is limited by the integration of these units into the crystal lattice at the surface [4]. This whitepaper examines advanced strategies for manipulating supersaturation and reaction flux, contextualizing them within the ongoing research into these competing growth mechanisms to achieve desired crystallization outcomes.
Theoretical Foundations: Supersaturation and Crystal Growth Mechanisms
The LaMer Model and Modern Evolutions
The classical LaMer model postulates a scenario of "burst" or "instantaneous" nucleation, where the rate of nucleation becomes effectively infinite once a critical supersaturation is reached, followed by a growth stage where nucleation is suppressed [1]. The model further distinguishes the growth stage based on the prevailing concentration of growth monomers. If the concentration falls below the critical level for nucleation but remains above the solubility limit, crystal development continues via diffusion-controlled growth. Conversely, if the diffusion of growth species from the bulk to the surface is sufficiently rapid, the slower surface integration process becomes the rate-controlling step [4].
However, modern research, enabled by advanced in-situ characterization techniques, has revealed that the strict temporal separation of nucleation and growth proposed by the classical model is not universal. For instance, recent single-particle fluorescence imaging studies of perovskite nanocrystal crystallization have demonstrated a coupled nucleation-and-growth process, where both phenomena occur simultaneously yet can still produce nanocrystals with a narrow size distribution [51]. This indicates that narrow dispersity can also be achieved through continuous nucleation processes where growth free energy dominates, providing an alternative model to the LaMer mechanism [51].
The Metastable Zone and Supersaturation Control
The metastable zone is the region of supersaturation between the saturation curve and the concentration where spontaneous nucleation occurs. Operating within this zone allows for crystal growth without the formation of new nuclei. The ability to control and maintain a specific supersaturation level within this zone is a powerful tool for process control [52]. For example, in Membrane Distillation Crystallisation (MDC), the membrane area is used as a parameter to adjust supersaturation, thereby modifying kinetics without altering mass and heat transfer within the boundary layer. An increase in concentration rate was shown to shorten induction time and raise supersaturation at induction, which broadened the metastable zone width and favored a homogeneous primary nucleation pathway [52].
Table 1: Key Crystallization Growth Mechanisms and Their Characteristics.
| Growth Mechanism | Rate-Limiting Step | Typical Dependence | Common Experimental Observations |
|---|---|---|---|
| Diffusion-Controlled Growth | Mass transport of growth units from the bulk solution to the crystal surface. | Linear dependence on supersaturation. | Growth rate is sensitive to agitation/stirring speed. |
| Surface-Process-Controlled Growth | Integration of growth units into the crystal lattice (e.g., spiral growth, 2D nucleation). | Parabolic or power-law dependence on supersaturation (e.g., R ∝ σⁿ). | AFM/SEM reveals growth spirals or 2D islands; growth rate is less sensitive to fluid dynamics. |
| Coupling of Nucleation and Growth | Overlapping of nucleation and growth stages, driven by free energy fluctuations. | Described by models like Avrami. | "S"-shaped kinetic curve for ensemble crystals; "T"-shaped curve for individual crystals [51]. |
Experimental Control Strategies and Protocols
Supersaturation Control in Membrane Distillation Crystallisation (MDC)
Objective: To regulate primary nucleation and crystal growth mechanisms by using membrane area to precisely control supersaturation, thereby achieving high yield and superior product quality [52].
Detailed Protocol:
- Setup: Configure a membrane distillation crystallizer equipped with a hydrophobic microporous membrane. The membrane separates the heated feed solution (e.g., concentrated brine) from a cooler permeate stream.
- Supersaturation Generation: Initiate the process by heating the feed solution. Volatile solvent (e.g., water) evaporates, diffuses through the membrane pores, and condenses on the permeate side. This removes solvent from the feed, progressively concentrating it and generating supersaturation.
- Induction and Seeding: Monitor the solution concentration until the metastable zone limit is approached. For seeded crystallization, introduce seed crystals at a predetermined supersaturation level. For unseeded crystallization, allow the system to surpass the metastable limit for spontaneous nucleation.
- Supersaturation Modulation: Adjust the membrane area exposed to the feed solution or the temperature gradient across the membrane to control the rate of solvent removal. A larger active area or steeper temperature gradient increases the supersaturation rate.
- Crystal Retention and Scaling Mitigation: Implement an in-line filtration unit to retain crystals within the crystallizer bulk, reducing deposition and scaling on the membrane surface. This segregation of the crystal phase into the bulk solution creates two discrete regions of supersaturation, allowing for closer control over crystal growth and improving habit, shape, and purity [52].
- Monitoring and Analysis: Track induction time and crystal growth in real-time. Use population balance modeling to confirm the reduction in nucleation rate with longer hold-up times, which results from solvent desaturation due to crystal growth and leads to larger crystal sizes [52].
Investigating Growth History on KDP Crystal Faces
Objective: To determine the influence of the method of changing solution supersaturation (increasing vs. decreasing) on the growth rates and mechanisms of {100} potassium dihydrogen phosphate (KDP) crystal faces [53].
Detailed Protocol:
- Solution Preparation: Prepare an aqueous KDP solution from analytical grade substance (99% purity) and deionized water. Maintain the solution at a saturation temperature of (31.0 ± 0.1) °C and decant to remove impurities.
- Nucleation and Initial Growth: Place the solution in a thermostated growth cell (~15 mL capacity). Temporarily stop the solution flow and introduce air bubbles via a needle to induce spontaneous nucleation. Grow the resulting crystal seeds at 26.0 °C for approximately 2 hours.
- Creating Growth Markers: Partially dissolve the crystals to create clear markers for subsequent growth rate measurements. Achieve this by slowly heating the solution to (34.0 ± 0.1) °C at a rate of 0.5 °C/min and maintaining this temperature for ~30 minutes, reducing crystal sizes in the observed directions by at least 20%.
- Growth Rate Experiments:
- For Decreasing Supersaturation: After dissolution and refaceting, rapidly reduce the solution temperature to 28.0 °C. After a 15-minute stabilization period, measure face displacement. Subsequently, increase the temperature in 1.0 °C steps to 24.0 °C, measuring the growth rate at each temperature (σ₅ to σ₁).
- For Increasing Supersaturation: After preparation, set the initial temperature to 24.0 °C. After measurement, decrease the temperature in 1.0 °C steps to 28.0 °C, measuring the growth rate at each step (σ₁ to σ₅).
- Data Collection: Use an optical microscope with a digital camera to record crystal images at each supersaturation stage. Measure the displacement of the {100} faces with an accuracy of ~±5 μm.
- Analysis: Plot face displacement against time for each constant supersaturation. Determine the linear growth rates by applying the least-squares method. Analyze the growth rate dependence on supersaturation, R(σ), using parabolic and power-law models. Perform surface analysis via SEM and AFM to correlate surface roughness with supersaturation level [53].
Table 2: Experimental Parameters and Key Findings from KDP Crystal Growth Study [53].
| Parameter | Values / Findings |
|---|---|
| Growth Temperature Range | 24.0 °C to 28.0 °C |
| Relative Supersaturation (σ) | 6.2% to 14.7% |
| Observed Crystal Faces | {100} prismatic faces |
| Key Finding on Growth Rate | Higher growth rates were associated with decreasing supersaturation pathways compared to increasing supersaturation. |
| Surface Analysis | Higher supersaturation resulted in greater surface roughness. |
| Proposed Growth Mechanism | The dependence R(σ) was best described by parabolic and power-law models, indicating spiral growth. A significant number of faces showed exponents n > 2, suggesting relevance of multiple nucleation models. |
Reaction-Diffusion Framework for MOF Synthesis
Objective: To control the particle size and morphology of Metal-Organic Framework-199 (MOF-199) single crystals by leveraging a reaction-diffusion framework (RDF) to establish a tunable supersaturation gradient [54].
Detailed Protocol:
- Gel Preparation: Prepare a hydrogel medium (e.g., agarose or silica gel) containing the organic linker, 1,3,5-benzenetricarboxylic acid (BTC), as the inner electrolyte.
- Diffusion Setup: Place the gel in a container and carefully introduce a solution of copper ions (Cu²⁺, outer electrolyte) on top of or adjacent to the gel. The interface between the two phases defines the start of the diffusion path.
- Crystallization: Allow the Cu²⁺ ions to diffuse into the gel medium containing the BTC linker. The reaction between the diffusing metal ions and the static linker occurs at a specific front, creating a supersaturation gradient along the diffusion flux.
- Parameter Tuning: Control the crystal size, distribution, and morphology by adjusting experimental parameters such as temperature, concentrations of the inner and outer electrolytes, and the nature/density of the gel matrix. A steeper concentration gradient typically leads to higher supersaturation, favoring smaller crystals.
- Harvesting and Characterization: After a predetermined time, stop the reaction. Extract the crystals from the gel matrix and characterize them using techniques like scanning electron microscopy (SEM) and X-ray diffraction (XRD).
Visualization of Experimental Concepts and Workflows
LaMer Model and Growth Control Strategies
Membrane Distillation Crystallization Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions and Materials for Crystallization Studies.
| Reagent / Material | Function and Application in Crystallization |
|---|---|
| Potassium Dihydrogen Phosphate (KDP) | A model inorganic crystal system for studying growth kinetics and mechanisms due to its well-defined habit and solubility [53]. |
| Metal-Organic Framework Precursors (e.g., Cu²⁺, BTC linker) | Used in the synthesis of porous crystalline materials via reaction-diffusion frameworks to control particle size and morphology [54]. |
| Perovskite Precursors (e.g., FAPbBr₃) | Enables study of rapid crystallization kinetics; often used with polymeric matrices (e.g., PVDF) for in-situ imaging [51]. |
| Hydrogel Matrix (e.g., Agarose, Silica) | Creates a diffusion-controlled environment for crystallization, used in reaction-diffusion frameworks to establish supersaturation gradients [54]. |
| Microporous Hydrophobic Membrane | The core component in Membrane Distillation Crystallisation (MDC) for controlled solvent removal and supersaturation generation [52]. |
| Polymer Matrix (e.g., Polyvinylidene Difluoride - PVDF) | Provides a confined environment for nanocrystal growth, slowing mass diffusion and enabling single-particle fluorescence studies [51]. |
| In-line Filtration Unit | Used in continuous crystallizers for crystal retention, reducing membrane scaling and promoting controlled growth in the bulk solution [52]. |
The precise manipulation of supersaturation and reaction flux remains a cornerstone of advanced crystallization control. While the LaMer model provides a valuable conceptual framework for understanding diffusion-controlled versus surface-process-controlled growth, contemporary research underscores the complexity and diversity of crystallization pathways. The emergence of techniques like membrane distillation crystallization and reaction-diffusion frameworks, coupled with powerful in-situ characterization tools, provides scientists with an unprecedented ability to design crystallization processes. By moving beyond empirical approaches to a mechanistic-based strategy, researchers can more reliably produce crystalline materials with target properties, accelerating innovation in drug development, materials science, and beyond.
The Role of Stabilizers, Additives, and Surface Modifiers
The pursuit of precise control over nanocrystal morphology is a cornerstone of advanced materials science and pharmaceutical development. For decades, the LaMer mechanism has served as a foundational model for understanding crystallization, describing a process where a rapid burst of nucleation is followed by diffusion-controlled or surface-process-controlled growth [3] [1]. Within this framework, stabilizers, additives, and surface modifiers emerge not as passive spectators but as directive agents that profoundly influence kinetic pathways and thermodynamic outcomes. These compounds enable researchers to navigate beyond classical growth models into the realm of non-classical pathways, such as particle-mediated growth, thereby achieving unprecedented command over crystal size, shape, polymorphism, and stability [3]. This technical guide examines the mechanisms by which these chemical agents dictate crystallization processes, providing detailed methodologies and data frameworks essential for their application in cutting-edge research, particularly in pharmaceutical sciences.
Theoretical Framework: Crystal Growth Within and Beyond the LaMer Model
The LaMer Mechanism and Its Evolution
The classical LaMer model provides a qualitative schematic for achieving monodisperse colloids by temporally separating nucleation and growth. Its cornerstone is the concept of supersaturation as the driving force for crystallization [1]. The model posits three consecutive stages:
- Stage I (Precursor Formation): The initial generation of atomic or molecular precursors, leading to an increase in monomer concentration.
- Stage II (Nucleation Burst): Once the monomer concentration exceeds a critical supersaturation threshold (
C_min_nu), a burst of nucleation occurs, depleting the monomer reservoir and forming stable nuclei. - Stage III (Growth Phase): The monomer concentration falls below the nucleation threshold but remains above the solubility concentration (
C_s), allowing for growth of the existing nuclei without the formation of new ones [3].
The growth in Stage III can be governed by two primary kinetics, as defined by the LaMer mechanism:
- Diffusion-Controlled Growth: The rate of crystal growth is limited by the diffusion of growth species from the bulk solution to the crystal surface. This occurs when the surface integration process is fast relative to monomer supply [3] [4].
- Surface-Process-Controlled Growth: The rate is limited by the integration of monomers into the crystal lattice at the surface, which becomes dominant when diffusion from the bulk is sufficiently rapid [4].
Non-Classical Pathways and the Role of Modifiers
While the atom-mediated LaMer model is foundational, a "non-classical" particle-mediated growth pathway is increasingly recognized. In this model, nanoparticles or clusters act as building blocks for larger crystals via mechanisms like oriented attachment (OA) and coalescence [3]. Stabilizers and modifiers are critical in these pathways; they influence not only atomic addition but also the aggregation and reorganization of primary particles, leading to complex hierarchical structures such as mesocrystals, polycrystals, and unique morphologies unattainable through classical growth alone [3].
The following diagram illustrates the core decision points between classical and non-classical growth pathways, highlighting where modifiers exert their influence.
Mechanisms of Action: How Modifiers Interface with Crystal Growth
Adsorption and Surface Energy Modulation
The primary mechanism of action for most modifiers is selective adsorption onto specific crystal faces. Different crystal facets possess varying surface energies and atomic arrangements. Modifiers preferentially bind to higher-energy facets through electrostatic, steric, or coordination interactions, effectively poisoning their growth. This forces crystal expansion to occur predominantly on lower-energy, less-blocked facets, enabling morphological control [55]. For instance, a polymer additive can adsorb onto a growing calcite lattice, impeding directional growth and promoting the formation of crystals with abnormal shapes and sizes [55].
Kinetically Limited Growth and Supersaturation Control
Modifiers can intentionally create kinetically limited growth conditions, shifting the process from diffusion-control to surface-process-control. By binding to surface sites, they create a significant energy barrier for the integration of new monomers. This decelerates the growth rate and provides a longer time window for atomic rearrangement, which is crucial for producing crystals with high crystallographic perfection and for accessing metastable polymorphs [56]. Furthermore, some additives, like potent chelating agents (e.g., citric acid, EDTA), function by controlling the effective supersaturation of metal ions in solution, yielding purer and more uniform nanoparticles [57].
Influencing Non-Classical Particle-Mediated Pathways
In non-classical crystallization, the role of modifiers expands. Surface ligands on nanoparticles determine the interparticle forces that govern aggregation and oriented attachment. Carefully designed stabilizers can prevent irreversible, random aggregation, allowing for the rotational freedom needed for nanoparticles to find crystallographic alignment before coalescing into a single crystal [3]. Recent in-situ studies have revealed that modifiers can influence the dynamic processes of particle rotation, alignment, and interface elimination that are characteristic of these pathways [3].
A Guide to Key Modifier Classes and Their Applications
The following table summarizes the primary classes of stabilizers, additives, and surface modifiers, along with their distinct functions and applications.
Table 1: Classification of Stabilizers, Additives, and Surface Modifiers
| Modifier Class | Primary Function | Mechanistic Role in Crystal Growth | Exemplary Applications |
|---|---|---|---|
| Surfactants (e.g., CTAB, SDS) | Modulate surface energy and interfacial tension; can form templates. | Selective face blocking; promotes anisotropic growth (e.g., nanorods, nanowires); enhances surface area [57]. | Semiconductor nanocrystals; metal oxide nanoparticles; porous materials. |
| Polymers (e.g., PVP, PEG, Chitosan) | Provide steric stabilization; act as crystal habit modifiers. | Inhibits crystal growth & Ostwald ripening in bulk glasses; effective for polymorph control [57] [56]. | Pharmaceutical API stabilization; inhibition of amorphous phase crystallization; membrane crystallization [56] [4]. |
| Chelating Agents (e.g., Citrate, EDTA) | Control free ion concentration; cap nanoparticle surfaces. | Modulates supersaturation; leads to purer, more uniform nanoparticles [57]. | Synthesis of monodisperse metal & hydroxyapatite nanoparticles. |
| Small-Molecule Additives (e.g., tailor-made inhibitors) | Mimic lattice ions; block specific molecular recognition sites. | Induces highly specific morphological changes by targeting distinct crystal faces. | Specialty chemicals; high-aspect-ratio crystals; pigment industry. |
The Scientist's Toolkit: Essential Research Reagent Solutions
For researchers designing experiments, selecting the appropriate reagent is critical. The following table details key materials used in the field.
Table 2: Research Reagent Solutions for Crystal Engineering
| Reagent / Material | Function in Experimentation | Key Utility & Rationale |
|---|---|---|
| Polyvinylpyrrolidone (PVP) | A versatile steric stabilizer and crystal growth modifier. | Widely used for morphology control of metal nanocrystals; inhibits crystallization from amorphous phases (e.g., in nifedipine) [56]. |
| Cetyltrimethylammonium Bromide (CTAB) | A cationic surfactant and structure-directing agent. | Essential for producing anisotropic gold nanorods; forms micellar templates and selectively blocks specific crystal facets [57]. |
| Citric Acid / Citrate | A multifunctional chelating agent and capping ligand. | Controls ion supersaturation for uniform nucleation; passifies nanoparticle surfaces to prevent aggregation [57]. |
| INITIA 585 Polymer | A dedicated crystal habit modifier. | Adsorbs onto growing crystal lattices (e.g., calcite) to impede directional growth, resulting in modified crystal habits for scale control [55]. |
| Polyethylene Glycol (PEG) | A steric stabilizer and pore-forming agent. | Used in nano-HAp synthesis to adjust pore size and particle dimensions; improves biocompatibility [57]. |
Quantitative Data and Experimental Protocols
Quantifying Modifier Efficacy
The impact of modifiers can be quantified through key performance metrics, as derived from recent literature.
Table 3: Quantitative Data on Modifier Efficacy from Recent Studies
| System | Modifier | Impact on Crystal Growth & Properties | Experimental Conditions |
|---|---|---|---|
| Amorphous Nifedipine [56] | Polyvinylpyrrolidone (PVP) | Inhibited crystal growth in the bulk glass; the VP dimer was far less effective than the polymer. | Study of crystal growth rates below Tg (glass transition temperature). |
| Nano-Hydroxyapatite (HAp) [57] | Citric Acid (Chelator) | Yielded purer, more uniform nanoparticles. | Wet chemical precipitation. |
| Nano-Hydroxyapatite (HAp) [57] | CTAB (Surfactant) | Enhanced surface area of the resulting nanoparticles. | Microwave-assisted synthesis. |
| Lithium-Ion Battery Cathodes [58] | Proprietary Crystal Modifiers | Improved energy density by up to 20%. | Applied in electrode material synthesis. |
| Semiconductor Fabrication [58] | Specific Crystal Modifiers | Improved device performance by 10-15% through reduced defect rates. | Ultra-pure silicon crystal production. |
Detailed Experimental Protocol: Investigating Modifier Effects on Crystal Growth
This protocol outlines a model experiment to study the effect of a polymer additive on the crystallization of an active pharmaceutical ingredient (API), using insights from studies on amorphous solid stability [56].
Objective: To determine the effect of Polyvinylpyrrolidone (PVP K30) on the crystal growth kinetics and polymorphic form of a model drug (e.g., Nifedipine) from its amorphous state.
Materials:
- Model drug (e.g., Nifedipine)
- Polymer additive (e.g., PVP K30)
- Solvent (e.g., Methylene Chloride or Acetone)
- Hot Stage with Temperature Controller
- Polarized Light Microscope (PLM) with digital camera
- Differential Scanning Calorimetry (DSC)
- X-Ray Powder Diffractometer (XRPD)
Methodology:
- Sample Preparation (Thin Film Creation):
- Prepare homogeneous solutions of the pure drug and drug-polymer mixtures (e.g., at 95:5 and 90:10 drug-to-polymer ratios) in a volatile solvent.
- Deposit a small, measured volume of each solution onto a clean, pre-heated microscope coverslip mounted on a hot stage.
- Rapidly evaporate the solvent under a controlled nitrogen flow to create a thin, homogeneous amorphous film. Confirm the amorphous nature of the initial film using in-situ XRPD or the broad halo pattern.
In-Situ Crystal Growth Monitoring:
- Set the hot stage to a pre-determined isothermal temperature below the drug's Tg (e.g., Tg - 10 K).
- Observe the films under PLM. Upon the first appearance of spherulites, begin recording time-lapse images at regular intervals.
- For each spherulite, measure the radius (r) over time (t) from the image sequences. Perform measurements in triplicate for multiple spherulites per sample.
Polymorph Identification:
- At the end of the isothermal growth period, rapidly cool the sample to room temperature.
- Carefully transfer the coverslip to an XRPD stage for analysis to determine the polymorphic form of the crystallized material.
- Compare the diffraction patterns to known forms of the drug.
Data Analysis:
- Plot radius (r) vs. time (t) for each sample. The linear growth rate (u) is the slope of the linear region of this plot.
- Compare the growth rates (u) for pure amorphous drug versus formulations with PVP.
- Correlate the measured growth rates with the polymorphic outcome identified by XRPD.
The workflow for this protocol is systematized in the following diagram.
Expected Outcomes: PVP is expected to significantly reduce the linear crystal growth rate (u) compared to the pure amorphous drug and may also suppress the formation of certain less-stable polymorphs, demonstrating its dual role as a kinetic inhibitor and a polymorph director.
Stabilizers, additives, and surface modifiers are indispensable tools for modern crystal engineering. Their strategic use allows scientists to transcend the limitations of classical LaMer-type growth, enabling sophisticated control over crystallization processes. By understanding their mechanisms—from simple face-specific adsorption to the complex mediation of non-classical particle-attachment pathways—researchers can rationally design crystalline materials with tailored properties. As the field advances, the integration of in-situ characterization techniques, computational modeling, and novel modifier chemistries will continue to unlock new possibilities in the synthesis of advanced materials and the development of more effective pharmaceutical products.
Tuning Crystallization Temperature and Timescales for Lamellar Thickness Control
The control of lamellar thickness in semicrystalline polymers is a fundamental aspect of materials science with significant implications for mechanical properties, thermal stability, and material performance. This parameter exists in a state of non-equilibrium, predominantly determined by the complex interplay between crystallization temperature ((Tc)) and timescales of associated molecular processes [59]. Within the broader context of crystallization mechanisms, this phenomenon sits at the intersection of LaMer-like diffusion-controlled growth and surface-controlled crystal growth theories. The LaMer model, originally describing nanoparticle formation, emphasizes a burst of nucleation followed by diffusion-controlled growth when monomer concentration exceeds a critical supersaturation threshold. In polymer crystallization, this translates to competition between the diffusion of chains to the crystal growth front (a process influenced by the undercooling, (Tm^0 - T_c)) and the subsequent surface integration process, where chains attach to the crystal lattice, often with chain folding [60] [59].
The classical kinetic theory posits that lamellar thickness is selected to maximize the growth rate, resulting in an initial thickness ((lg^*)) close to that of the critical nucleus. However, experimental evidence consistently shows that mature lamellae are significantly thicker, indicating substantial post-growth thickening [60]. This thickening is governed by the competition between the timescale of crystal growth ((\tau{lc})) and the timescale of intracrystalline chain diffusion (ICD, (\tau{stem})) [59]. Polymers are often categorized as "crystal-fixed" (negligible ICD) or "crystal-mobile" (significant ICD), with the latter exhibiting thicker lamellae, higher crystallinity, and a stronger dependence of final lamellar thickness on (Tc) [59]. This technical guide provides a comprehensive resource for researchers and scientists seeking to understand and manipulate these parameters to achieve precise morphological control in semicrystalline polymers.
Theoretical Framework: Mechanisms of Thickness Selection
Foundational Models and the Role of Crystallization Temperature
The fundamental relationship between crystallization temperature and lamellar thickness is rooted in the Gibbs-Thomson equation, which describes the melting point depression of finite-sized crystals:
$$ Tm = Tm^0 \left(1 - \frac{2\sigmae}{\Delta Hf \cdot l}\right) $$
Here, (Tm) is the observed melting point, (Tm^0) is the equilibrium melting point, (\sigmae) is the fold surface free energy, (\Delta Hf) is the bulk heat of fusion, and (l) is the lamellar thickness. This equation establishes the direct correlation between measured melting point and crystal thickness [60]. During isothermal crystallization at temperature (Tc), the initial lamellar thickness is kinetically selected. Theory suggests it approximates the thickness of the critical nucleus for the maximum growth rate, (lg^*) [60]. A crystal of this dimension would be metastable at (Tc), melting only slightly above it. The observation that polyethylene melts approximately midway between (Tc) and (T_m^0) provides definitive evidence that substantial thickening occurs after the initial crystal formation [60].
The Critical Competition: Growth vs. Reorganization
The ultimate lamellar thickness is determined by the competition between two characteristic timescales, a concept crucial for framing within the LaMer mechanism dichotomy:
Layer Crystallization Time ((\tau{lc})): This is the time required for a crystal to grow by one molecular layer, a timescale representative of the surface integration process. It can be calculated from the crystal growth velocity ((\mu)) and an intermolecular distance (approximately 5 Å): (\tau{lc} = 5 \text{ Å} / \mu) [59]. This timescale is highly dependent on (T_c), decreasing with greater undercooling.
Intracrystalline Chain Diffusion Time ((\tau{stem})): This is the time required for a polymer stem to diffuse a distance equal to the crystal thickness via successive helical jumps mediated by the αc-relaxation process [59]. The associated jump correlation time, (\langle \tauc \rangle), follows an Arrhenius temperature dependence: (\langle \tauc \rangle = \tau0 \cdot \exp(E_a / RT)) [59].
The ratio (\tau{stem} / \tau{lc}) dictates the extent of lamellar thickening. If (\tau{stem} \ll \tau{lc}) (fast ICD, as in PEO), significant thickening can occur concurrently with growth. If (\tau{stem} \gg \tau{lc}) (slow ICD, as in PCL), the initial, kinetically-selected thickness is largely preserved [59]. This competition effectively bridges diffusion-controlled (governed by (\tau{stem})) and surface-controlled (governed by (\tau{lc})) growth models.
Diagram 1: Competitive Pathways in Lamellar Thickness Determination.
Quantitative Data and Material-Specific Trends
The following tables consolidate key experimental data from representative polymers, illustrating the quantitative relationship between crystallization conditions, characteristic timescales, and the resulting lamellar morphology.
Table 1: Characteristic Timescales and Lamellar Thickness for Various Polymers [59]
| Polymer | Crystallization Temp, (T_c) (°C) | Growth Velocity, (\mu) (µm/s) | Layer Crystallization Time, (\tau_{lc}) (s) | ICD Jump Time, (\langle \tau_c \rangle) (s) | Final Lamellar Thickness, (l) (nm) | Polymer Class |
|---|---|---|---|---|---|---|
| PEO | 60 | 2.5 | 2.0 × 10(^{-4}) | ~10(^{-8}) | 30.5 | Crystal-Mobile |
| POM130 | 150 | 0.5 | 1.0 × 10(^{-3}) | ~10(^{-1}) | 16.5 | Crystal-Mobile |
| PCL | 50 | 0.1 | 5.0 × 10(^{-3}) | ~10(^{3}) | 7.5 | Crystal-Fixed |
| PBS | 90 | N/A | N/A | Effectively Infinite | ~5.0 | Crystal-Fixed [61] |
Table 2: Isothermal Thickening of Linear Polyethylene (Marlex 50) [60]
| Crystallization Temp, (T_c) (°C) | Crystallization Time (min) | Observed Melting Point, (T_m) (°C) | Calculated Lamellar Thickness, (l) (nm) |
|---|---|---|---|
| 125.0 | 125 | 133.0 | ~12.5 |
| 125.0 | 485 | 133.8 | ~13.2 |
| 130.0 | 300 | 135.2 | ~14.1 |
The data in Table 1 demonstrates the spectrum of polymer behavior. PEO, with extremely fast ICD, develops thick lamellae. PCL and PBS, with effectively no ICD, form thin lamellae. POM occupies an intermediate position, with measurable but slow ICD resulting in a moderate thickness. Table 2 provides a classic example of isothermal thickening in polyethylene, where the lamellar thickness increases logarithmically with time at a constant (T_c), leading to a corresponding increase in melting temperature [60].
Experimental Protocols for Lamellar Thickness Analysis
Dilatometric Crystallization and Melting Point Analysis
This classic protocol is ideal for studying crystallization kinetics and lamellar thickness evolution in materials like polyethylene [60].
Materials and Equipment:
- Polymer sample (e.g., linear polyethylene, ~3 g)
- Mercury-filled dilatometer with spherical bulb (~5 ml) and precision capillary
- Three thermostatically-controlled silicone oil baths
- High-precision thermometer/thermocouple
Procedure:
- Sample Preparation: Load the polymer into the dilatometer bulb. Seal the system and fill it with mercury, ensuring no air bubbles are trapped.
- Initial Melting: Transfer the dilatometer to a bath at 177 °C for 10 minutes to erase all thermal history and remove pre-existing crystallinity.
- Isothermal Crystallization: Rapidly transfer the dilatometer to a second bath held at the desired crystallization temperature ((T_c)). Monitor the descent of the mercury meniscus in the capillary over time to track the volume change associated with crystallization.
- Direct Melting Procedure (to minimize recrystallization): After a predetermined crystallization time, directly transfer the dilatometer to a third bath at a temperature high enough to induce melting but low enough to prevent significant new crystal growth (e.g., 135 °C for PE). The rate of meniscus rise is monitored. This "direct transfer" method is critical to avoid the artifacts of recrystallization that occur during slow heating [60].
- Data Analysis: The observed melting temperature ((Tm)) is taken as the temperature at which the sample reaches a static volume in the melting bath. The lamellar thickness ((l)) can be calculated from (Tm) using the Gibbs-Thomson equation.
Key Consideration: Heat transfer is slow in polymers. Allow sufficient time (e.g., >1 hour) for the sample to reach thermal equilibrium in the melting bath to obtain an accurate (T_m) [60].
Correlating Lamellar Thickness with ICD Timescales
This multi-technique protocol is used to directly test the competition hypothesis by measuring the relevant timescales and the resulting morphology [59].
Materials and Equipment:
- Polymer samples of different molecular weights (e.g., POM130, POM212)
- Polarized Optical Microscope with hot stage
- Solid-State NMR spectrometer (e.g., for (^{13})C MAS CODEX experiments)
- Small-Angle X-Ray Scattering (SAXS) instrument
- Differential Scanning Calorimeter (DSC)
Procedure:
- Isothermal Crystallization for SAXS: Prepare thin films of the polymer. Isothermally crystallize them at a range of temperatures ((T_c)) in a DSC or hot stage. Quench the samples after complete crystallization.
- SAXS Measurement and Analysis: Perform SAXS on the crystallized samples. Use standard analysis methods (e.g., correlation function analysis) to determine the long period. Combine with DSC-derived crystallinity to calculate the average crystalline lamellar thickness, (d_c).
- Crystal Growth Rate Measurement ((\mu)): Using polarized optical microscopy, observe the radial growth of spherulites at different (T_c). Measure the growth rate (\mu).
- Calculation of (\tau{lc}): Calculate the layer crystallization time as (\tau{lc} = (5 \text{ Å}) / \mu) [59].
- NMR Measurement of ICD Timescale ((\langle \tauc \rangle)): Use the (^{13})C MAS CODEX NMR technique on the isothermally crystallized samples. Acquire spectra at a series of temperatures to determine the average jump correlation time (\langle \tauc \rangle) and its activation energy (E_a) [59].
- Data Correlation: Correlate the measured lamellar thickness (dc) with the ratio of the characteristic timescales, (\tau{stem} / \tau_{lc}), to establish the governing relationship.
Diagram 2: Multi-Technique Workflow for Lamellar Thickness and Timescale Analysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Lamellar Crystallization Studies
| Item | Function/Application | Technical Notes |
|---|---|---|
| Linear Polyethylene (e.g., Marlex 50) | Model polymer for foundational studies of lamellar thickening. | Unfractionated material can be used, but fractionated samples reduce heterogeneity in melting behavior [60]. |
| Mercury | Filling fluid for traditional dilatometers to track volumetric changes during crystallization/melting. | Provides high density and non-wetting properties. Note: Significant health and safety hazards require strict handling protocols and containment [60]. |
| Silicone Oil Baths | Thermostatic fluid for precise temperature control during crystallization and melting. | Multiple baths are needed for the direct melting procedure to avoid recrystallization [60]. |
| Dilatometer | Glass apparatus with a spherical bulb and precision capillary to measure volume changes. | Typical configuration: 3 g polymer in a 5 ml bulb with a 2 mm diameter capillary [60]. |
| Poly(oxymethylene) (POM) | Model "crystal-mobile" polymer with intermediate ICD timescale. | Allows study of the competition between growth and diffusion. Molecular weight variants (e.g., POM130, POM212) probe entanglement effects [59]. |
| Poly(ϵ-caprolactone) (PCL) | Model "crystal-fixed" polymer with negligible ICD. | Serves as a baseline for systems where kinetic thickness selection dominates [59]. |
| Poly(butylene succinate) (PBS) | "Crystal-fixed" polymer exhibiting absence of lamellar thickening. | Useful for contrasting against polymers that undergo significant post-growth thickening [61]. |
The control of lamellar thickness is unequivocally governed by the competition between the timescale of crystal growth ((\tau{lc})) and the timescale of intracrystalline chain diffusion ((\tau{stem})). This framework successfully integrates elements of both diffusion-controlled and surface-controlled growth theories. For "crystal-fixed" polymers like PCL and PBS, the initial kinetically-selected thickness is largely preserved, aligning with classical surface-controlled growth models. In contrast, for "crystal-mobile" polymers like PEO and POM, the ability of chains to diffuse and reorganize after attachment allows for substantial thickening, a process more analogous to a post-attachment diffusion-controlled mechanism.
Future research directions will likely focus on a more quantitative mapping of the (\tau{stem} / \tau{lc}) ratio against final lamellar thickness across a wider library of polymers. Furthermore, the application of ultra-fast calorimetry to study the initial stages of thickening, and the development of multi-scale models that integrate chain dynamics, entanglement constraints, and crystallization kinetics, will be crucial for achieving predictive control over polymer morphology. This will ultimately enable the rational design of polymers with tailored lamellar architectures for specific applications in drug delivery, specialty packaging, and high-performance materials.
Evidence and Alternatives: Validating LaMer Against Modern Mechanisms
Critical Experimental Support for 'Instantaneous Nucleation' and 'Diffusion-Limited Growth'
Within the framework of crystallization research, the LaMer mechanism provides a foundational model for understanding the separation of nucleation and growth stages in nanomaterial synthesis. A critical aspect of this model involves distinguishing between instantaneous nucleation, where all nuclei form simultaneously at the onset, and progressive nucleation, where nuclei continue to form over time. The growth stage is further governed by the rate-limiting step, which can be either diffusion-limited growth, controlled by the mass transport of monomers to the crystal surface, or reaction-limited growth, controlled by the surface integration process itself. This whitepaper synthesizes recent, high-quality experimental evidence that validates and refines these classical concepts, providing researchers and drug development professionals with validated protocols and analytical frameworks for controlling nanocrystal properties.
Experimental Case Studies and Quantitative Data
Advanced in situ characterization techniques and electrochemical analysis have yielded direct evidence for instantaneous nucleation and diffusion-limited growth across diverse material systems. The following case studies present key experimental findings and their quantitative interpretation.
Case Study 1: Copper Electrodeposition on HOPG
Experimental Protocol: High-Speed Atomic Force Microscopy (HS-AFM) was used to observe Cu electrodeposition in real-time from a solution of 1.0 mM CuSO₄·5H₂O and 50 mM H₂SO₄ on a Highly Oriented Pyrolytic Graphite (HOPG) substrate. The potentiostatic experiments were conducted at various overpotentials, and the resulting current-time transients were analyzed [62].
Key Findings: HS-AFM videos provided direct visual evidence of a rapid nucleation event, where a fixed number of nuclei appeared suddenly on the HOPG surface. Following this initial burst, the particle density remained constant, and the existing nuclei grew in size without the formation of new ones—a hallmark of instantaneous nucleation. The analysis showed that at more negative overpotentials (e.g., -0.45 V), the nucleation rate was so high that the process was dominated by the initial formation of a large number of nuclei. As the potential was made less negative, the behavior shifted towards growth-dominated kinetics, where the expansion of existing nuclei was the primary process [62].
Quantitative Analysis of Growth: The subsequent growth of the copper particles was analyzed and found to be consistent with diffusion-limited growth. The growth rate was governed by the diffusion of Cu²⁺ ions from the bulk solution to the reducing crystal surface, a process controlled by Fick's law [62].
Case Study 2: Molybdenum Disulfide (MoS₂) Electrodeposition
Experimental Protocol: The nucleation and growth of MoS₂ on a copper substrate from an aqueous solution of sodium molybdate dihydrate and sodium sulphide were investigated using chronoamperometry. Current-time transients were recorded at various applied potentials (-0.9 V to -1.2 V) and compared to the theoretical models of instantaneous and progressive nucleation developed by Scharifker and Hills [63].
Key Findings: The study demonstrated that the nucleation mechanism for MoS₂ is potential-dependent. At an optimal potential of -1.1 V, the experimental data for the initial stages of growth aligned most closely with the model for progressive nucleation, where new nuclei continue to form over time. However, as the deposition progressed, the mechanism transitioned towards instantaneous nucleation [63]. This highlights that nucleation type is not always static and can evolve during the synthesis.
Quantitative Nucleation Parameters: The chronoamperometric data allowed for the calculation of key nucleation parameters, which are summarized in the table below.
Table 1: Nucleation parameters for MoS₂ electrodeposition at different potentials, adapted from [63].
| Applied Potential (V) | Nucleation Rate Constant (A, s⁻¹) | Nucleation Density (N₀, cm⁻²) |
|---|---|---|
| -0.9 | 1.31 × 10⁷ | 2.62 × 10⁷ |
| -1.0 | 5.75 × 10¹⁴ | 1.86 × 10¹⁵ |
| -1.1 | 3.55 × 10¹⁵ | 4.12 × 10¹⁵ |
| -1.2 | 3.19 × 10¹⁵ | 4.32 × 10¹⁵ |
The data shows a dramatic increase in both the nucleation rate and site density between -0.9 V and -1.0 V, indicating a strong dependence on overpotential. The formation of highly monodisperse MoS₂ nanoparticles (5–65 nm) at -1.1 V was attributed to this controlled, progressive nucleation process [63].
Case Study 3: Polymer Crystallization in Ultrathin Films
Experimental Protocol: The crystallization of a fluoropolymer (FK-800) was studied using in situ hot-stage Atomic Force Microscopy (AFM). Ultrathin films (~50 nm) were spin-casted onto silicon and annealed at temperatures between the glass transition (T𝑔 ≈ 31°C) and melting point (T𝑚 ≈ 110°C). AFM was used to track nucleation densities, growth rates, and fractional coverage in real-time [64].
Key Findings: The AFM movies revealed a clear transition in nucleation mechanism from homogeneous at lower temperatures to heterogeneous at higher temperatures, observed near 45°C. Furthermore, a critical transition in the growth mechanism was identified. Below 60°C, growth was primarily reaction-limited, governed by the kinetics of chain attachment to the crystal lattice. Above 60°C, the process became diffusion-limited, where the mobility of polymer chains to the growth front became the rate-controlling step [64]. This temperature-dependent transition underscores the complex interplay between thermodynamic drivers and kinetic limitations in polymer crystallization.
The Scientist's Toolkit: Essential Reagents and Materials
Successful experimental investigation of nucleation and growth requires specific reagents and advanced instrumentation.
Table 2: Key research reagents and materials for nucleation and growth studies.
| Item Name | Function/Application | Example Use Case |
|---|---|---|
| Highly Oriented Pyrolytic Graphite (HOPG) | An atomically flat, chemically inert substrate for fundamental nucleation studies. | Provides a well-defined surface for observing early-stage metal electrodeposition [62]. |
| Potentiostat/Galvanostat | Instrument for applying controlled potentials/currents and measuring electrochemical responses. | Essential for performing chronoamperometry and cyclic voltammetry in electrodeposition studies [63]. |
| Hot-Stage AFM | An atomic force microscope with a integrated temperature-controlled stage. | Enables in situ visualization of temperature-dependent nucleation and growth in polymers and other materials [64]. |
| Scharifker-Hills Model | A theoretical framework for analyzing current-time transients. | Used to discriminate between instantaneous and progressive nucleation mechanisms in electrodeposition [63]. |
| Sodium Molybdate & Sulfide | Precursor ions for the electrodeposition of MoS₂. | Used in aqueous electrolytes to synthesize MoS₂ nanostructures for catalytic and electronic applications [63]. |
Conceptual Workflow and Signaling Pathways
The following diagram synthesizes the experimental evidence into a unified conceptual workflow for identifying nucleation and growth types.
The pathway leading to "Instantaneous Nucleation" followed by "Diffusion-Limited Growth" is often the target for achieving highly monodisperse nanoparticles, as it prevents continued nucleation while ensuring smaller particles "catch up" to larger ones due to the dependence of diffusion flux on particle curvature [14].
The integration of advanced techniques like HS-AFM, hot-stage AFM, and chronoamperometry has provided robust experimental support for the classical models of instantaneous nucleation and diffusion-limited growth. The evidence confirms that these mechanisms are not merely theoretical constructs but are observable and controllable phenomena. The nucleation type (instantaneous vs. progressive) is highly dependent on experimental conditions such as overpotential and temperature, while the growth mode (diffusion vs. reaction-limited) is governed by the relative rates of mass transport and surface integration. For drug development professionals, mastering these concepts and the associated experimental protocols is crucial for the rational design of crystalline drug products, enabling precise control over critical quality attributes like particle size, size distribution, and polymorphic form, which directly impact drug efficacy, stability, and bioavailability.
The Finke-Watzky (F-W) two-step mechanism represents a fundamental shift from classical nucleation theories that have dominated particle formation science for decades. Within the broader context of LaMer mechanism diffusion versus surface-controlled crystal growth research, the F-W mechanism provides an alternative mathematical framework that better explains sigmoidal kinetic data observed in diverse systems ranging from transition-metal nanoparticles to pathological amyloid formations. While the LaMer model postulates "instantaneous nucleation" followed primarily by diffusion-controlled growth, the F-W mechanism introduces the concept of slow continuous nucleation coupled with autocatalytic surface growth [1]. This paradigm shift has proven particularly valuable in explaining kinetic behaviors in protein aggregation and nanoparticle synthesis where the LaMer model fails to account for observed induction periods and subsequent rapid sigmoidal consumption of precursors [65] [66].
The significance of the F-W mechanism lies in its minimalistic "Ockham's razor" approach—reducing complex phase-change processes to two pseudoelementary steps that can quantitatively describe kinetic data from diverse chemical systems [66]. This review comprehensively examines the theoretical foundations, experimental validation, and practical applications of the F-W mechanism while contrasting it with classical nucleation theories.
Theoretical Framework and Key Concepts
Fundamental Mathematical Formulation
The Finke-Watzky mechanism reduces nucleation and growth processes to two essential chemical steps:
- Slow continuous nucleation: A → B (rate constant k₁)
- Autocatalytic surface growth: A + B → 2B (rate constant k₂)
Where A represents the precursor species and B represents the nucleated product [66]. The consumption rate of precursor A is described by the differential equation:
-d[A]/dt = k₁[A] + k₂[A][B] [67]
The integrated form of this rate law provides the concentration of precursor A as a function of time:
[A] = [A]₀ × (1 - exp(-k₁t)) / (1 + (k₂/k₁) × exp(-k₁t)) [67]
Where [A]₀ represents the initial concentration of precursor A. This equation generates the characteristic sigmoidal curve observed in many nucleation-growth processes, with an initial induction period followed by rapid acceleration and eventual deceleration as the precursor is consumed.
Comparative Analysis with Classical Nucleation Theories
Table 1: Comparison of Nucleation-Growth Models
| Feature | LaMer Model | Turkevich "Organizer" Model | Finke-Watzky Model |
|---|---|---|---|
| Year Proposed | 1950 [1] | 1951 [68] | 1997 [66] |
| Nucleation Type | Instantaneous/burst [1] | Inductive organizing process | Slow continuous [66] |
| Growth Mechanism | Diffusion-controlled [1] | Not quantitatively defined | Autocatalytic surface growth [66] |
| Mathematical Formulation | Limited quantitative application [1] | Descriptive without quantitative parameters [67] | Well-defined kinetic equations [66] [67] |
| Applicability to Proteins | Limited | Limited | Excellent for amyloid formation [69] [66] |
Figure 1: Evolution of Nucleation-Growth Models. The Finke-Watzky mechanism represents a significant departure from earlier instantaneous nucleation models.
Experimental Validation and Methodologies
Application to Protein Aggregation Systems
The F-W mechanism has demonstrated remarkable utility in quantifying amyloid formation kinetics, particularly for neurological proteins associated with diseases like Alzheimer's and Parkinson's. In one key study, the mechanism was applied to S100A9 amyloid formation under physiological conditions (pH 7.4, 37°C and 42°C) [69]. The analysis revealed that initial misfolding and β-sheet formation—defined as the "nucleation" step—occurred spontaneously within individual S100A9 molecules at higher rates than subsequent fibrillar growth [69].
Experimental Protocol for Protein Aggregation Kinetics:
- Sample Preparation: Purified S100A9 protein dissolved in appropriate buffer (pH 7.4) at varying concentrations
- Kinetic Monitoring: Incubation at controlled temperatures (37°C and 42°C) with continuous monitoring via:
- Thioflavin T (ThT) fluorescence for β-sheet formation
- Static light scattering for aggregate mass
- Transmission electron microscopy (TEM) for morphological validation
- Data Fitting: Application of F-W kinetic equations to determine k₁ (nucleation) and k₂ (growth) rate constants
- Validation: Comparison with Beker-Döring model for fibril length distribution analysis [69]
The study demonstrated that the autocatalytic growth phase would only proceed if misfolded amyloid-prone S100A9 was populated on a macroscopic timescale, with short fibril lengths consistent with the F-W model prediction that slow growth dominates over fragmentation or secondary pathways [69].
Application to Transition-Metal Nanoparticle Synthesis
The F-W mechanism has been extensively applied to the synthesis of transition-metal nanoparticles, where it better explains the characteristic induction period and sigmoidal precursor consumption compared to the LaMer model [65] [68].
Experimental Protocol for Nanoparticle Synthesis:
- Precursor Solution: Transition-metal salts (e.g., Au, Ag, Pt, Pd) in aqueous or organic solvents
- Reduction System: Appropriate reducing agents (e.g., citrate, borohydride) under controlled conditions
- Stabilizers: Capping agents (e.g., polymers, surfactants) to control growth and prevent aggregation
- Kinetic Monitoring:
- UV-Vis spectroscopy for plasmon resonance changes
- Transmission electron microscopy (TEM) for size distribution
- Dynamic light scattering (DLS) for hydrodynamic radius
- Data Analysis: Nonlinear fitting of precursor decay to F-W equations
Table 2: Quantitative Kinetic Parameters from F-W Analysis of Various Systems
| System | Temperature | k₁ (nucleation) | k₂ (growth) | Data Source |
|---|---|---|---|---|
| S100A9 Amyloid Formation | 37°C | 2.5 × 10⁻³ min⁻¹ | 1.8 × 10² M⁻¹min⁻¹ | [69] |
| S100A9 Amyloid Formation | 42°C | 4.1 × 10⁻³ min⁻¹ | 2.3 × 10² M⁻¹min⁻¹ | [69] |
| Neurological Protein Aggregation | Varies | 10⁻⁴-10⁻² min⁻¹ | 10¹-10³ M⁻¹min⁻¹ | [66] |
| Transition-Metal Nanoparticles | Varies | System-dependent | System-dependent | [65] [68] |
A critical evaluation of the F-W mechanism for nanoparticle synthesis revealed that while it effectively describes precursor consumption, it may produce highly polydisperse particles due to continued nucleation throughout the growth phase [65]. This limitation has led to proposed modifications, including a short nucleation window with delayed onset and rapid suppression [65].
Figure 2: Finke-Watzky Two-Step Mechanism. The mechanism comprises slow continuous nucleation followed by autocatalytic surface growth.
Modifications and Current Developments
Addressing Limitations Through Mechanism Expansion
While the minimal F-W mechanism successfully describes many systems, several modifications have been proposed to address its limitations:
- Bimolecular Agglomeration: Adding a third step to account for particle-particle agglomeration
- Autocatalytic Agglomeration: Including a fourth step for surface-mediated aggregation processes
- Reversible Nucleation: Accounting for the natural reversibility of nucleation steps, particularly important when reactions do not proceed to completion [67]
Recent work has focused on developing a modified F-W mechanism with reversible pseudo-first-order nucleation, creating a kinetic model that more accurately reflects physical reality [67]. This is particularly important for silver nanoparticle synthesis using common reducing agents like sodium borohydride in the presence of capping agents such as polyvinylpyrrolidone (PVP) [67].
Comparative Diagram of Nucleation-Growth Pathways
Figure 3: Comparative Pathways: LaMer vs. Finke-Watzky Mechanisms. The F-W mechanism allows ongoing nucleation during growth, potentially leading to broader size distributions.
Essential Research Reagents and Materials
Table 3: Research Reagent Solutions for F-W Mechanism Studies
| Reagent/Material | Function/Application | Example Systems |
|---|---|---|
| S100A9 Protein | Amyloid-forming protein for neurological studies | Amyloid formation kinetics [69] |
| Thioflavin T (ThT) | Fluorescent dye for β-sheet detection | Quantifying amyloid formation [69] |
| Transition Metal Salts | Precursors for nanoparticle synthesis | Au, Ag, Pt, Pd nanoparticles [65] [68] |
| Sodium Borohydride (NaBH₄) | Strong reducing agent | Silver nanoparticle synthesis [67] |
| Citrate Ions | Reducing agent/stabilizer | Gold nanoparticle synthesis [68] |
| Polyvinylpyrrolidone (PVP) | Capping agent/stabilizer | Controlling nanoparticle growth [67] |
| Buffer Systems (pH 7.4) | Physiological conditions | Protein aggregation studies [69] |
The Finke-Watzky two-step mechanism provides a versatile, minimalistic framework for analyzing nucleation and growth processes across diverse systems from pathological protein aggregation to transition-metal nanoparticle synthesis. Its mathematical formulation successfully deconvolutes the nucleation (k₁) and growth (k₂) rate constants from kinetic data, enabling quantitative comparisons between systems and conditions [66].
When framed within the broader context of LaMer mechanism research, the F-W model addresses significant limitations of classical instantaneous nucleation theories, particularly for systems exhibiting continuous nucleation behavior throughout the growth phase [65] [1]. The mechanism's successful application to neurological protein aggregation datasets—achieving R² values ≥0.98 across 14 datasets from nine different laboratories—demonstrates its robustness and general applicability [66].
Future research directions include further mechanism refinement to address systems with reversible nucleation and size-distribution control, particularly for monodisperse nanoparticle synthesis where the classical LaMer model has traditionally been applied despite contradictory evidence [65] [67]. The ongoing development of modified F-W mechanisms promises enhanced quantitative understanding and control of nucleation-growth processes across chemical, materials, and biological sciences.
The 1950 model proposed by LaMer and Dinegar has served as a foundational framework for understanding the formation of monodisperse nanoparticles for over seven decades [1]. This model postulates a two-stage mechanism: an initial stage of effectively infinite nucleation once precursor concentration exceeds a critical supersaturation level (C_crit), followed by a subsequent stage of diffusion-controlled growth as the concentration drops below the minimum for further nucleation [1] [45]. This process, when perfectly executed, theoretically yields particles with narrow size distributions. The iconic "LaMer curve" schematically represents the temporal evolution of monomer concentration, defining three key stages: (I) initial increase in monomer concentration, (II) burst nucleation when C > C_crit, and (III) growth via monomer diffusion to existing nuclei once C < C_min_nu [3].
This technical guide examines the applicability and limitations of the LaMer mechanism across four critical material classes—silver halide, semiconductors, metals, and metal-oxides—framed within ongoing research on diffusion-controlled versus surface-process-controlled crystal growth. We synthesize evidence from seminal and contemporary studies, providing structured quantitative data and experimental protocols to inform current research and development efforts, particularly in domains requiring precise nanomaterial control such as drug development and diagnostic applications.
Classical Foundations and Competing Theories
The Original LaMer Mechanism
The LaMer model was originally developed to explain the formation of monodispersed sulfur hydrosols [1]. Its core premise is the spatial and temporal separation of the nucleation and growth stages. The model's mathematical basis in Classical Nucleation Theory (CNT) describes nucleation energy barriers and critical radii [1]. A key quantitative expression from the model describes the growth of a particle of radius r at time t by diffusion of monomers from the bulk solution, a relationship that has been critically evaluated in modern contexts [1].
Competing and Contemporary Growth Models
While the LaMer model remains a cornerstone, several other mechanistic pathways have been identified, as illustrated in the diagram below.
The Finke-Watzky (F-W) mechanism presents a significant alternative, proposing that slow, continuous nucleation and autocatalytic surface growth can occur simultaneously rather than as separated stages [70]. Further challenging the classical view is non-classical particle-mediated growth, where nanoparticles themselves act as building blocks, forming larger crystals through aggregation, oriented attachment, and coalescence [3]. This pathway can lead to complex morphologies like mesocrystals and hierarchical structures difficult to achieve via atom-by-atom addition [3].
Critical Analysis of LaMer Model Applications
Silver Halide Nanoparticles
Silver halides represent the historical system considered to provide the strongest evidence for the LaMer model [1].
- Experimental Evidence for LaMer Mechanism: Early work demonstrated that monodisperse AgBr sols could be prepared by rapidly mixing concentrated AgNO₃ and KBr solutions, resulting in a short nucleation burst followed by diffusion-controlled growth [1]. The number of particles remained constant after the initial nucleation period, consistent with the model's predictions.
- Key Experimental Protocol:
- Reagents: AgNO₃, KBr (or other alkali halides), and a stabilizing agent like gelatin.
- Procedure: Rapidly mix equimolar, concentrated solutions of AgNO₃ and KBr under vigorous stirring at a controlled temperature (e.g., 30-60°C). The gelatin acts as a colloidal stabilizer to prevent aggregation.
- Critical Parameters: Supersaturation level is the primary factor controlling nucleation burst and final particle size. Maintaining a concentration between the minimum for nucleation (
C_min_nu) and the saturation concentration (C_s) is crucial for focusing growth without secondary nucleation [45].
- Conflicting Analyses: A critical review of 13 papers on silver halides suggests that even in this "best case" system, the experimental support for "effectively infinite nucleation" and pure "diffusion-controlled growth" is not as robust as often cited. The quantitative equation from the original 1950 LaMer paper is rarely used to fit kinetic data [1].
Semiconductor Nanocrystals (Quantum Dots)
The synthesis of II-VI (e.g., CdSe) and III-V (e.g., InAs) semiconductor quantum dots often employs the hot-injection method, designed explicitly to mimic LaMer conditions [1] [14].
- Hot-Injection Workflow: The following diagram illustrates the standard procedure for achieving a LaMer-like growth regime in quantum dot synthesis.
- Case Study: InAs Quantum Dots: The synthesis of monodisperse InAs QDs highlights both the application and limitations of the diffusion-controlled model. A continuous injection process can maintain a monomer concentration (
C_m) in the "size-focusing regime" [14]. A Diffusion-Dynamics-Controlled (DDC) model, based on a modified Fick's law, shows that growth suppression for larger sizes (>7 nm) is due to reduced monomer flux. By tuning the reaction volume, precursor concentration, and injection rate, monomer flux can be managed to synthesize large InAs QDs up to 9.0 nm (absorption peak at 1600 nm) with a narrow size distribution (12.2%) [14]. - Quantitative Data from InAs QD Synthesis:
| Precursor Injection Rate (mL/h) | Final QD Radius (nm) | 1S Absorption Peak (nm) | Half-Width-Half-Maximum (HWHM) | Growth Regime |
|---|---|---|---|---|
| 8 | ~2.8 | 1060 | Broadened | Defocusing |
| 4 | ~3.2 | 1152 | Broadened | Defocusing |
| 2 | ~4.5 | 1300 | Narrowed | Size-Focusing |
Source: Adapted from [14]
Metal Nanoparticles (Gold and Silver)
The synthesis of noble metal nanoparticles, such as gold, provides a clear view of the competition between classical and non-classical models.
- Turkevich Method (Citrate Reduction): This classic method for synthesizing ~20 nm gold colloids is often interpreted through a LaMer lens [70]. The reduction of AuCl₄⁻ by citrate is thought to cause a short nucleation burst, followed by diffusion-controlled growth.
- Modern Plasma-Driven Synthesis: A recent, rapid synthesis of stabilizer-free gold nanoparticles using a plasma-microdroplet reactor achieved over 70% conversion of gold ions in ~10 milliseconds [70]. Quantitative modeling of this process revealed a hybrid mechanism: nucleation was initiated by short-lived reducing species (e.g., solvated electrons), consistent with a fast, LaMer-like burst. However, the majority of the precursor conversion and particle growth was attributed to an autocatalytic surface growth mechanism mediated by long-lived species like H₂O₂, aligning more closely with the Finke-Watzky model [70].
- Key Experimental Protocol (Plasma Synthesis):
- Reagents: Aqueous solution of HAuCl₄·3H₂O (gold precursor).
- Apparatus: RF glow discharge plasma reactor (e.g., He/Ar/H₂O gas mixture).
- Procedure: Precursor solution is atomized into picoliter-volume droplets, which are completely immersed in the plasma for a controlled residence time (~10 ms). The high surface-to-volume ratio and intense flux of reactive species enable ultra-fast reduction.
- Critical Parameters: Droplet residence time in plasma directly controls final particle size. A power threshold effect for nucleation is observed, supporting CNT [70].
Metal-Oxide Nanoparticles
Metal-oxide systems frequently deviate from the classical LaMer pathway, exhibiting non-classical growth mechanisms.
- Silica (SiO₂) Systems: The base-catalyzed hydrolysis and condensation of tetraalkoxysilanes (e.g., TEOS) can produce highly monodisperse silica spheres [1]. While often framed within the LaMer model, detailed kinetic studies suggest an aggregative growth model where sub-critical nuclei or small particles aggregate to form the final monodisperse particles, rather than growing purely by monomer addition [1] [71].
- Iron Oxides: The formation of uniform iron oxide particles from forced hydrolysis of ferric chloride solutions has been extensively studied. Research indicates that the primary particles form first, which then aggregate and reorganize into the final uniform crystals. This process is highly dependent on the anion concentration (e.g., chloride, perchlorate), which influences the aggregation step, again pointing to a non-classical, particle-mediated pathway [71].
- Titania (TiO₂): Hydrothermal synthesis of titania nanoparticles can involve both Ostwald ripening (a classical process) and oriented attachment (a non-classical process), where nanocrystals align and fuse along specific crystallographic planes to form larger single crystals or mesocrystals [71].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for Nanoparticle Synthesis
| Reagent/Material | Exemplary System | Function in Synthesis |
|---|---|---|
| Tris(trimethylsilyl)arsine ((TMS)₃As) | InAs Quantum Dots | Arsenic precursor for III-V QD synthesis via dehalosilylation [14]. |
| Gold(III) Chloride Trihydrate (HAuCl₄·3H₂O) | Gold Nanoparticles | Oxidized gold metal precursor for both chemical (Turkevich) and plasma synthesis [70]. |
| Trisodium Citrate Dihydrate | Gold Nanoparticles | Reducing agent and colloidal stabilizer (via citrate capping) in the Turkevich method [70]. |
| Tetraethyl Orthosilicate (TEOS) | Silica Nanoparticles | Silicon alkoxide precursor for hydrolysis and condensation polymerization [1]. |
| Cadmium Oxide (CdO) / Dimethylcadmium | CdSe Quantum Dots | Cadmium metal precursor for II-VI QD synthesis [1]. |
| Tri-n-octylphosphine Oxide (TOPO) | Semiconductor QDs | High-boiling-point coordinating solvent that controls growth and stabilizes nanocrystals [1]. |
| Silver Nitrate (AgNO₃) & Potassium Bromide (KBr) | Silver Halide Nanoparticles | Ionic precursors for precipitation synthesis of AgBr crystals [1]. |
| Hydrogen Peroxide (H₂O₂) | Gold Nanoparticles (Plasma) | Long-lived reactive species that enables autocatalytic surface growth on existing nanoparticles [70]. |
The LaMer model remains a profoundly useful conceptual framework for designing syntheses of monodisperse nanoparticles. Its core principle—separating nucleation from growth—is a key strategy for achieving size uniformity. However, a critical analysis of experimental data across material classes reveals that its idealized postulates of "instantaneous nucleation" and purely "diffusion-controlled growth" are seldom fully realized. The empirical evidence often points to more complex realities involving competing mechanisms.
The future of nanoparticle synthesis lies in moving beyond a one-model-fits-all approach. Advanced in situ characterization techniques and computational modeling are revealing intricate pathways, including Finke-Watzky autocatalytic growth and non-classical particle-mediated coalescence. Success in applications from drug delivery to quantum technologies will depend on a nuanced understanding of these mechanisms, enabling the rational design of nanomaterials with precisely controlled properties. The choice between a classical LaMer approach or a non-classical pathway becomes a powerful tool in the materials scientist' arsenal, allowing for the tailored creation of everything from ultra-pure quantum dots to complex hierarchical nanostructures.
The LaMer model has long served as the foundational framework for understanding crystallization processes in solution, elegantly diagramming the journey from monomer supersaturation to nucleation and subsequent crystal growth. [3] [25] This classical theory posits two distinct growth regimes: diffusion-controlled growth, where the diffusion of monomers from the bulk solution to the crystal surface is the rate-limiting step, and surface-reaction-controlled growth, where the incorporation of monomers into the crystal lattice at the surface is slower. [4] In an idealized diffusion-controlled system, the high monomer flux to the surface maintains a constant, high monomer concentration at the interface, ensuring that the crystal's growth rate is governed solely by mass transport, which often leads to uniform growth and superior size distribution.
However, numerous synthetic studies and advanced in-situ characterization techniques have revealed that real-world systems frequently deviate from this idealized picture. Diffusion control can fail when surface reactions become comparably slow or when other processes, such as particle-mediated growth, interfere. This creates a regime known as mixed control, where both diffusion and surface kinetics jointly determine the overall growth rate. [72] [3] [14] Understanding the conditions that lead to this failure and the evidence for mixed-controlled processes is crucial for researchers and drug development professionals seeking precise control over particle size, morphology, and polymorphism—critical factors in applications ranging from drug formulation to nanocrystal-based technologies.
Theoretical Foundations: From Classical to Non-Classical Pathways
The Classical LaMer Mechanism
The classical atom-mediated growth mechanism, as described by LaMer, can be summarized in three key stages [3] [25]:
- Monomer Production: Precursors decompose or are reduced, leading to a gradual increase in the concentration of atomic/molecular monomers in solution.
- Nucleation Burst: Once the monomer concentration exceeds the minimum supersaturation threshold ((C_{min}^{nu})), a rapid nucleation event occurs, forming stable nuclei.
- Crystal Growth: The monomer concentration drops below the nucleation threshold but remains above the solubility limit ((C_s)), allowing the existing nuclei to grow via the sequential addition of atoms. During this stage, if the monomer diffusion rate to the surface is much slower than the rate of atom incorporation, diffusion-controlled growth dominates. Conversely, if the surface incorporation step is slower, interface-controlled growth occurs.
Non-Classical Particle-Mediated Pathways
Beyond the LaMer curve, a non-classical model has gained significant support, in which nanoparticles or clusters act as fundamental building blocks instead of individual atoms. [3] This particle-mediated growth involves:
- Aggregation: The direct physical contact between nanoparticles.
- Coalescence: The merging of nanoparticle domains with extensive atomic-level interactions at their junctions.
- Oriented Attachment (OA): A specific coalescence process where crystals fuse along shared crystallographic orientations to form larger, single-crystalline particles. [3]
The occurrence of these pathways represents a fundamental failure of pure diffusion control and introduces a distinct set of interfacial kinetics and energetic considerations.
Quantitative Evidence and Diagnostic Signatures
The transition from diffusion control to mixed or interfacial control manifests in specific, measurable deviations from classical predictions. The following table summarizes key diagnostic signatures observed in experimental systems.
Table 1: Experimental Evidence for the Failure of Diffusion Control
| Observed Phenomenon | Deviation from Diffusion Control | System | Key Reference |
|---|---|---|---|
| Growth Saturation | Particle growth ceases despite continued monomer/precursor supply, indicating a suppressed monomer flux and a shift to a surface-reaction-limited regime. [14] | Colloidal InAs Quantum Dots | [14] |
| Formation of Mesocrystals & Polycrystalline Structures | Appearance of complex structures via oriented attachment and coalescence of primary nanoparticles, a hallmark of non-classical, particle-mediated growth. [3] | Various Metal Nanocrystals | [3] |
| Negative-Going Delta Plots | In conflict tasks (e.g., Simon task), the congruency effect collapses over time, indicating dynamic, non-monotonic shifts in processing control from automatic to controlled channels. [73] | Psychological Conflict Tasks (as an analog for process dynamics) | [73] |
| Nonlinear Adsorption with Saturation | Adsorbed mass does not scale linearly with liquid saturation due to flow channeling and incomplete mixing at the pore scale, highlighting the role of local interfacial accessibility over bulk diffusion. [72] | Surfactant Adsorption in Unsaturated Porous Media | [72] |
Experimental Protocols for Discriminating Control Mechanisms
Protocol: Quantifying Growth Kinetics to Identify Rate-Limiting Steps
Objective: To determine whether crystal growth is limited by diffusion, surface reaction, or a combination of both. Materials:
- High-purity precursors and solvents
- Thermostated reaction vessel with precise temperature control (±0.1 °C)
- In-situ monitoring tool (e.g., spectrophotometer for UV-Vis, turbidity probe)
- Quenching solvent for rapid sampling
- Electron Microscopy (TEM/SEM) for ex-situ size analysis
Methodology:
- Induce Nucleation: Using a standard hot-injection method, create a sudden supersaturation to generate a nearly monodisperse population of nuclei. [14] [25]
- Monitor Growth: Track the evolution of the average particle size over time ((dr/dt)) using in-situ spectroscopy (e.g., tracking the excitonic peak shift for semiconductors) or by taking aliquots at defined intervals for TEM analysis.
- Vary Agitation Rate: Under constant temperature and initial concentration, repeat the experiment at different stirring speeds. In a diffusion-controlled process, the growth rate will increase with higher agitation due to reduced diffusion layer thickness. In an interface-controlled process, the growth rate remains unaffected by agitation. [74]
- Analyze Temperature Dependence: Measure the growth rate at different temperatures. The apparent activation energy ((Ea)) can be derived from an Arrhenius plot.
- Low (Ea) (< 20 kJ/mol) suggests a diffusion-controlled process.
- High (E_a) (> 40 kJ/mol) suggests an interface-controlled process, as chemical bond formation has a high energy barrier. [25]
Protocol: Investigating Particle-Mediated Growth via In-Situ Techniques
Objective: To directly observe non-classical pathways such as oriented attachment and coalescence. Materials:
- Liquid cell for in-situ Transmission Electron Microscopy (TEM)
- High-resolution TEM with direct electron detectors
- Stable nanoparticle suspension
Methodology:
- Sample Preparation: Load a dilute suspension of the pre-formed primary nanocrystals into a liquid cell holder. [3]
- Real-Time Imaging: Acquire video-rate TEM images under low electron dose conditions to minimize beam effects while observing the dynamic interactions between particles.
- Event Analysis: Identify and analyze sequences involving particle collision, rotation, alignment along congruent crystal planes, and final coalescence. The observation of such "jump-to-contact" events and subsequent reshaping provides direct evidence for the failure of atom-by-atom diffusion control. [3]
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Studying Growth Mechanisms
| Reagent/Material | Function in Experimentation | Example Use-Case |
|---|---|---|
| Architecture-Directing Agents (ADAs) / Surfactants | Modulate surface energy and kinetics; selectively bind to specific crystal facets to control morphology and growth mode. [25] | Poly(vinyl pyrrolidone) to control the shape of metal nanocrystals by promoting or inhibiting atom addition on specific facets. |
| Single-Source Precursors | Provide a continuous, controlled release of monomers to maintain a low supersaturation, favoring diffusion-controlled growth and suppressing secondary nucleation. [14] | InAs cluster precursors for the continuous synthesis of large, monodisperse quantum dots. |
| Chaperone Solvents | A solvent miscible with two immiscible poor solvents for a polymer; its cross-interface diffusion can force the assembly of interfacially non-active polymers, illustrating a complex diffusion-and-assembly process. [75] | Dimethyl sulfoxide (DMSO) to assemble polyacrylonitrile at a water-toluene interface. |
| In-Situ Liquid Cells | Enable direct, real-time observation of nucleation and growth processes within a transmission electron microscope (TEM). [3] [25] | Observing the oriented attachment of gold nanocrystals in an aqueous environment. |
Visualizing Process Dynamics and Transitions
The following diagrams map the logical relationships and transitions between different growth control mechanisms, highlighting the points of failure for pure diffusion control.
The failure of diffusion control is not an anomaly but a common occurrence in complex synthetic environments, leading to interfacial or mixed-controlled processes. Evidence from diverse fields—from nanocrystal synthesis to adsorption studies in porous media—confirms that real systems often operate outside the idealized LaMer regime. The diagnostic signatures, such as growth saturation, the formation of complex superstructures via particle attachment, and nonlinear scaling of interfacial reactions, provide clear markers for this transition.
For researchers, particularly in drug development where crystal form dictates critical product properties, recognizing these signs is paramount. The experimental protocols and toolkit outlined here offer a pathway to diagnose the operative growth mechanism actively. Embracing the complexity of mixed-controlled and non-classical pathways enables a more sophisticated approach to material design, turning the "failure" of simple models into an opportunity for achieving superior control over particle size, morphology, and ultimately, product performance.
Crystallization is a cornerstone process in pharmaceutical development, dictating the critical quality attributes of active pharmaceutical ingredients (APIs), including their solubility, stability, and bioavailability [76]. At the heart of developing robust crystallization processes lies the quantitative modeling of nucleation and growth. For decades, Classical Nucleation Theory (CNT) has provided the foundational framework for understanding the initial stages of particle formation. Its principles underpin the iconic LaMer model, which schematically describes the generation of monodisperse colloids through a process of rapid ("burst") nucleation followed by diffusion-controlled growth [1] [45]. However, over 70 years of research have revealed limitations in this classical view, spurring the development of more nuanced and emerging frameworks that seek to provide a more accurate and predictive description of crystallization phenomena, particularly within the critical context of pharmaceutical development [1]. This whitepaper provides an in-depth technical comparison of these quantitative models, equipping researchers and scientists with the knowledge to select and apply the appropriate framework for their specific challenges in drug development.
Theoretical Foundations of Nucleation and Growth
Classical Nucleation Theory (CNT) and the LaMer Model
Classical Nucleation Theory (CNT) describes the formation of a new, stable solid phase from a supersaturated solution or melt. It posits that this process is governed by a competition between the bulk free energy gain of forming a solid and the surface free energy cost of creating a new interface [1]. The core quantitative output of CNT is the nucleation rate, ( J ), which represents the number of stable nuclei formed per unit volume per unit time.
The Becher–Döring formulation of CNT expresses this rate as: [ J = \frac{3D\phi^2}{2d^2v0(\pi kT\psi^3)^{1/2}} \exp\left(-\frac{\Delta G^*}{kT}\right) ] where ( D ) is the solute diffusion coefficient, ( \phi ) is the supersaturation parameter (( kT \ln S ), with ( S ) being the supersaturation ratio), ( d ) is the monomer diameter, ( v0 ) is the solvent molecular volume, ( \psi ) is the surface energy per monomer unit, ( k ) is the Boltzmann constant, ( T ) is temperature, and ( \Delta G^* ) is the nucleation energy barrier [45].
The energy barrier, ( \Delta G^* ), is a central concept in CNT and is given by: [ \Delta G^* = \frac{4\psi^3}{27\phi^2} ] This equation highlights the inverse square relationship between the supersaturation and the activation barrier; higher supersaturation dramatically lowers the barrier and increases the nucleation rate [45].
The LaMer model, introduced in 1950, provides a qualitative and schematic framework that aligns with CNT principles for the formation of monodisperse particles [1] [45]. It visualizes the crystallization process in three distinct stages, as illustrated in the diagram below.
Diagram: The LaMer Model for Monodisperse Particle Formation. Stage I: Solute concentration increases, surpassing solubility (Cs) and entering a metastable zone. Stage II: At a critical concentration (Ccrit), rapid "burst nucleation" occurs, depleting the solute. Stage III: In the absence of new nucleation, existing nuclei grow by diffusion of solute, leading to uniform particles [45].
Critical Analysis and Emerging Frameworks
While CNT and the LaMer model have been profoundly influential, a critical analysis of the literature over the past 70 years reveals significant challenges and has catalyzed the development of alternative models [1].
A primary critique of the LaMer model is that its core tenets—"effectively infinite nucleation" and "diffusion-controlled growth"—often lack sound, compelling experimental support when applied to modern nanoparticle and API synthesis [1]. The model's quantitative equation is rarely used to fit experimental kinetic data, and its assumptions can be overly simplistic for complex molecular systems. For instance, the formation of different polymorphs (distinct crystal structures of the same API) is a phenomenon of critical importance in pharmaceuticals that is not accounted for in the basic LaMer diagram [76] [77]. The appearance of a new, more stable polymorph can have catastrophic consequences for a drug's bioavailability and efficacy, underscoring the need for models that can predict and control such phase selections [77].
These limitations have driven the proposal of several emerging frameworks and mechanistic insights:
Revised Nucleation Formulations: Newer nucleation theories have been developed to address perceived flaws in CNT. One such formulation corrects assumptions about monomer detachment and deposition rates, yielding a simplified expression for the nucleation rate [45]: [ J = kd C\infty v0 \left( \frac{4v1^2}{3\pi} \right)^{1/3} \frac{\psi}{kT} \exp\left(-\frac{\Delta G^* + \phi}{kT}\right) ] where ( kd ) is the overall deposition rate constant and ( v1 ) is the monomer molecular volume. A key characteristic is that the supersaturation parameter, ( \phi ), appears only in the exponent, simplifying the mathematical application to nucleation event analysis [45].
Integrated Nucleation-and-Growth Models: Modern theories explicitly account for the correlation between nucleation and the growth of generated nuclei during the nucleation stage. These models are built on mass balance, where the supply rate of solute is balanced by its consumption for both nucleation and growth, providing a more holistic framework for quantitative size control of uniform particles [45].
Pathway Complexity and Non-Classical Nucleation: Evidence suggests that nucleation, particularly for complex organic molecules like APIs, may not always follow a simple, single-step classical pathway. The interplay between thermodynamic and kinetic factors can lead to the initial formation of metastable polymorphs or the involvement of intermediate phases, which are not described by CNT [77].
Quantitative Model Comparison
The table below provides a structured, quantitative comparison of the core characteristics of Classical Nucleation Theory against the features of emerging, integrated frameworks.
Table 1: Quantitative Comparison of Classical and Emerging Nucleation Models
| Feature | Classical Nucleation Theory (CNT) | Emerging/Integrated Frameworks |
|---|---|---|
| Core Model | Becher-Döring equation: ( J \propto \exp(-\Delta G^*/kT) ) [45] | Revised equations with simplified supersaturation dependence, e.g., ( J \propto \exp(-(\Delta G^* + \phi)/kT) ) [45] |
| Nucleation Barrier | ( \Delta G^* = \frac{4\psi^3}{27\phi^2} ) (Inverse square dependence on supersaturation) [45] | Often adopts CNT's barrier or modifies the surface energy term based on specific system interactions. |
| View of Process | Two-stage: Sequential nucleation followed by growth [1]. | Integrated: Concurrent nucleation and growth during the nucleation stage, linked by mass balance [45]. |
| Handling of Growth | Often assumed to be diffusion-controlled after nucleation is complete [1]. | Explicitly models growth kinetics (diffusion or surface-controlled) during nucleation to determine final particle number [45]. |
| Predictive Capability | Qualitative and schematic for monodispersity; quantitative use of its full equation is rare [1]. | Aims for quantitative prediction of final particle number and size based on controllable parameters like solute supply rate [45]. |
| Applicability to APIs | Limited in directly predicting or controlling polymorphism and crystal habit [77]. | Provides a more adaptable structure for modeling complex systems, including solvent-mediated polymorph transformation. |
A critical experimental distinction between growth mechanisms is whether the process is diffusion-controlled or surface-integration-controlled (often referred to as surface-controlled). This distinction is vital for designing a crystallization process, as it determines the rate-limiting step and how operators can influence crystal size and habit.
Table 2: Key Experimental Differentiators Between Growth Mechanisms
| Experimental Observation | Diffusion-Controlled Growth | Surface-Controlled Growth |
|---|---|---|
| Dependence on Agitation | Growth rate is highly dependent on agitation intensity. | Growth rate is largely independent of agitation intensity. |
| Apparent Activation Energy | Typically low (~4-20 kJ/mol), characteristic of a mass transfer process. | Typically higher (>40 kJ/mol), characteristic of a chemical reaction or integration process. |
| Dependence on Particle Size | Growth rate is often size-dependent. | Growth rate is often size-independent. |
| Impact on Crystal Morphology | Often results in rounded or dendritic crystals, as growth is limited by solute arrival. | Can produce well-faceted crystals with distinct habits, as integration at different crystal faces dictates shape. |
Experimental Protocols for Mechanism Elucidation
Elucidating the mechanism of crystallization, particularly for a novel API, requires a multi-faceted experimental approach. The following workflow provides a detailed methodology for distinguishing between nucleation and growth mechanisms.
Diagram: Experimental Workflow for Crystallization Mechanism Studies
Detailed Methodologies
Generate Supersaturation:
- Cooling Crystallization: Dissolve the API in a suitable solvent at elevated temperature to create a saturated solution. Induce crystallization by applying a controlled cooling profile. The rate of cooling can be linked to the achieved supersaturation [76].
- Anti-solvent Crystallization: Feed an anti-solvent (a solvent in which the API has poor solubility) into a saturated API solution at a controlled rate. This method allows for precise control over the supersaturation generation rate (( Q_0 ) in integrated models) [76] [45].
- Evaporation Crystallization: Slowly evaporate the solvent from a saturated solution at constant temperature. This method is suitable for compounds sensitive to thermal degradation [76].
In-situ Monitoring:
- Focused Beam Reflectance Measurement (FBRM): Use FBRM to track the real-time chord length distribution of the particle population in a slurry. A sudden spike in fine counts indicates a nucleation event, while a gradual shift in the distribution indicates growth [76].
- Particle Vision Measurement (PVM): Use PVM to capture real-time images of crystals, providing direct visual evidence of nucleation, growth, and changes in crystal habit.
- Attenuated Total Reflectance Fourier-Transform Infrared (ATR-FTIR) Spectroscopy: Use ATR-FTIR to monitor solute concentration in the solution phase in real-time. This allows for the direct construction of a supersaturation profile, similar to the LaMer diagram [76].
Ex-situ Particle Analysis:
- Scanning Electron Microscopy (SEM): Analyze the final particle morphology, size, and shape. Aggregated nanoparticles or specific crystal habits can provide clues about the growth mechanism [45].
- Particle Size Distribution (PSD) Analysis: Use laser diffraction or other techniques to determine the volume-weighted PSD. A narrow, monomodal distribution is consistent with a short nucleation burst and controlled growth [76] [45].
- X-ray Diffraction (XRD): Determine the polymorphic form of the crystallized solid. This is critical for confirming that the desired crystal form has been obtained and for detecting any phase transformations during growth [77].
- Single Crystal X-ray Diffraction (SCXRD): For definitive absolute structure and configuration determination of a new API or polymorph, grow a high-quality single crystal and determine its structure using SCXRD or MicroED for micro/nanocrystals [78].
Growth Kinetics Analysis:
- Agitation Dependence: Conduct identical crystallization experiments at varying agitation speeds. If the crystal growth rate increases with agitation, it suggests diffusion-controlled growth. If it is independent, the growth is likely surface-controlled.
- Temperature Dependence: Conduct isothermal crystallization experiments at different temperatures to determine the apparent activation energy of the growth process. A low activation energy is indicative of diffusion control, while a high one suggests surface integration control.
Data Modeling and Validation:
- Fit the experimental data (e.g., supersaturation profile, final particle number) to the quantitative equations from CNT and integrated models. The model that provides the best fit with the fewest adjustable parameters and most accurately predicts outcomes under new conditions is the most appropriate for that system.
The Scientist's Toolkit: Research Reagent Solutions
The following table details key materials, reagents, and instrumentation essential for conducting rigorous crystallization mechanism studies.
Table 3: Essential Research Tools for Crystallization Studies
| Item | Function/Application | Examples / Notes |
|---|---|---|
| Crystallization Reactors | Provides controlled environment for crystallization (temp, agitation, dosing). | Jacketed lab-scale reactors (e.g., Orb Jacketed Reactor); systems for continuous crystallization [76]. |
| In-situ Analytical Probes | Real-time monitoring of particle and solution properties. | FBRM (particle count/size), PVM (images), ATR-FTIR (concentration) [76]. |
| Solvents & Anti-solvents | Medium for dissolution and generation of supersaturation. | High-purity solvents selected based on API solubility. Anti-solvents must be miscible with the primary solvent. |
| Co-formers | Molecules used to form pharmaceutical co-crystals to modify API properties. | Food-grade acids (e.g., citric, tartaric), amides (e.g., nicotinamide) [77]. |
| Single Crystal Growth Tools | Growing crystals for definitive structure determination. | Vapor diffusion apparatus, slow evaporation setups, temperature-controlled incubators [78]. |
| X-ray Diffractometers | Determining crystal structure and polymorphic form. | Single Crystal X-ray Diffractometer (SCXRD) for large crystals; MicroED for nanocrystals [78]. |
| Thermal Analysis | Characterizing thermal stability and polymorphic relationships. | Differential Scanning Calorimetry (DSC), Thermogravimetric Analysis (TGA) [77]. |
The evolution of quantitative modeling for crystallization, from the foundational Classical Nucleation Theory and the schematic LaMer model to more sophisticated integrated frameworks, reflects the growing complexity and demands of modern pharmaceutical development. While CNT remains a vital conceptual tool, its limitations in predicting and controlling the crystallization of complex APIs are now clear. The emergence of models that dynamically couple nucleation with growth and that can account for pathway complexity offers a more powerful and predictive approach. For researchers and drug development professionals, the path forward lies in employing a robust experimental workflow that combines advanced in-situ analytics with rigorous data modeling. By critically applying these comparative frameworks, scientists can transcend qualitative heuristics and achieve the precise, quantitative control required to ensure the quality, efficacy, and safety of crystalline pharmaceutical products.
Conclusion
The LaMer mechanism remains a pivotal, though not universally applicable, framework for understanding nanoparticle formation. The critical distinction between diffusion-controlled and surface-reaction-controlled growth is paramount for predicting and tailoring nanomaterial properties. While burst nucleation and diffusion-limited growth are validated in specific systems, modern research reveals a richer landscape of simultaneous nucleation and autocatalytic growth, as exemplified by the Finke-Watzky mechanism. Future directions point toward quantitative, multi-parameter models that can seamlessly integrate both nucleation and growth types. For biomedical and clinical research, this refined understanding enables the precise engineering of nanoparticles for drug delivery, imaging, and diagnostics, where control over size, morphology, and surface chemistry directly impacts biological performance, stability, and therapeutic efficacy.
References
- 1. LaMer's 1950 model of particle formation: a review and critical ... [https://pubs.rsc.org/en/content/articlehtml/2021/ma/d0ma00439a]
- 2. [PDF] LaMer's 1950 Model for Particle Formation of ... [https://www.semanticscholar.org/paper/LaMer%E2%80%99s-1950-Model-for-Particle-Formation-of-and-A-Whitehead-%C3%96zkar/71035569a272a4fe67e3ea6af1c3b568f1ca49d7]
- 3. Particle-mediated nucleation and growth of solution ... [https://www.sciencedirect.com/science/article/abs/pii/S1748013215300888]
- 4. Nucleation and Crystal Growth: Recent Advances and ... [https://www.mdpi.com/2673-4591/56/1/22]
- 5. Kinetics of nanoparticle nucleation, growth, coalescence ... [https://www.sciencedirect.com/science/article/abs/pii/S0301010423001842]
- 6. Kinetics for coarsening co-controlled by diffusion and a ... [https://www.sciencedirect.com/science/article/abs/pii/S1359645406005994]
- 7. Diffusion-Controlled Reactions: An Overview - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10674959/]
- 8. Simulation of perovskite thin layer crystallization with varying ... [https://pubs.rsc.org/en/content/articlehtml/2025/mh/d4mh00957f]
- 9. LaMer's 1950 model of particle formation [https://pubs.rsc.org/en/content/articlelanding/2021/ma/d0ma00439a]
- 10. Nucleation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2995260/]
- 11. Classical Nucleation Theory - an overview [https://www.sciencedirect.com/topics/engineering/classical-nucleation-theory]
- 12. Classical nucleation theory [https://en.wikipedia.org/wiki/Classical_nucleation_theory]
- 13. Quantitative studies of crystal nucleation at constant ... [https://pubs.rsc.org/en/content/articlehtml/2014/ce/c4ce00344f]
- 14. Diffusion dynamics controlled colloidal synthesis of highly ... [https://www.nature.com/articles/s41467-021-23259-w]
- 15. Surface control vs. diffusion control during calcite dissolution [https://www.degruyterbrill.com/document/doi/10.2138/am-2002-1002/html?lang=en&srsltid=AfmBOoqk4L5c4naaYr3F61f8bvD2S19gdHO1Dgutucr5HX0ERg-zWPKq]
- 16. Diffusion-controlled reaction [https://en.wikipedia.org/wiki/Diffusion-controlled_reaction]
- 17. Uncovering crystal growth and regeneration mechanism to ... [https://www.sciencedirect.com/science/article/abs/pii/S1383586624013169]
- 18. Using crystals and light, scientists unlock new ways to grow ... [https://natsci.msu.edu/news/2025/25-10-scientists-unlock-new-ways-to-grow-materials-on-demand.aspx]
- 19. Perfecting the art of growing crystals | College of Chemistry [https://chemistry.berkeley.edu/news/perfecting-art-growing-crystals]
- 20. Gold Nanoparticle Formation Kinetics and Mechanism [https://pmc.ncbi.nlm.nih.gov/articles/PMC6641265/]
- 21. Insights into formation kinetics of gold nanoparticles using ... [https://www.sciencedirect.com/science/article/abs/pii/S030101041400192X]
- 22. 3D conformation and crystal interaction insights into drug ... [https://www.nature.com/articles/s42004-025-01618-8]
- 23. A Critical Review on Crystal Growth Techniques for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7663244/]
- 24. Review Current trends and advancements in crystallization ... [https://www.sciencedirect.com/science/article/abs/pii/S0010854524003813]
- 25. Insight into nanocrystal synthesis: from precursor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9425161/]
- 26. The Establishment of Current Transient of Nucleation and ... [https://www.mdpi.com/2079-6412/14/1/62]
- 27. Current Transition of Nucleation and Growth under ... [https://www.mdpi.com/2079-6412/12/8/1195]
- 28. Extracting nucleation rates from current–time transients: Part I [https://www.sciencedirect.com/science/article/abs/pii/S0022072802009725]
- 29. Real–time observation of interfacial ions during ... [https://www.nature.com/articles/s41598-017-01048-0]
- 30. Simulation of 3D Electrochemical Phase Formation: Mixed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8585326/]
- 31. Mechanisms of controlled stabilizer-free synthesis of gold ... [https://pubs.rsc.org/en/content/articlehtml/2024/sc/d4sc01192a]
- 32. Millisecond CdS nanocrystal nucleation and growth ... [https://www.sciencedirect.com/science/article/abs/pii/S0927775718315103]
- 33. Nucleation [https://en.wikipedia.org/wiki/Nucleation]
- 34. Autocatalytic surface reduction and its role in controlling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5748196/]
- 35. Quantitative classification of autocatalytic strength for ... [https://link.springer.com/article/10.1007/s10973-020-10418-2]
- 36. and Nanostructure Nanoparticle and Controlled Synthesis ... Growth [https://pmc.ncbi.nlm.nih.gov/articles/PMC9503496/]
- 37. Experimental and Numerical Investigation of a ... [https://link.springer.com/article/10.1007/s11661-023-07147-0]
- 38. Transmission Electron Microscopy [https://www.nanoscience.com/techniques/transmission-electron-microscopy/]
- 39. Transmission Electron Microscopy Techniques [https://www.thermofisher.com/us/en/home/electron-microscopy/products/transmission-electron-microscopes/techniques.html]
- 40. STEM Microscope | Themis Environmental TEM [https://www.thermofisher.com/us/en/home/electron-microscopy/products/transmission-electron-microscopes/themis-etem.html]
- 41. Review Real-time observations with electron microscopy [https://www.sciencedirect.com/science/article/pii/S1369702109700070]
- 42. Single-molecule atomic-resolution real-time TEM imaging [https://www.jeol.com/solutions/applications/details/EM058.php]
- 43. Seven Essential Steps for In Situ Reaction Monitoring [https://www.spectroscopyonline.com/view/seven-essential-steps-situ-reaction-monitoring]
- 44. In situ and real-time ultrafast spectroscopy of photoinduced ... [https://www.nature.com/articles/s41467-025-60313-3]
- 45. Underlying mechanisms in size control of uniform ... [https://www.sciencedirect.com/science/article/abs/pii/S002197970700080X]
- 46. Particle formation mechanisms supported by in situ ... [https://pubs.rsc.org/en/content/articlehtml/2021/ma/d1ma00222h]
- 47. Reduction rate as a quantitative knob for achieving ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5643889/]
- 48. Emerging applications and trends in atomic layer ... [https://link.springer.com/article/10.1007/s11998-025-01154-z]
- 49. Learning from the future: towards continuous manufacturing of ... [https://aapsopen.springeropen.com/articles/10.1186/s41120-025-00111-9]
- 50. Rational Design for Monodisperse Gallium Nanoparticles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11987021/]
- 51. Single particle fluorescence imaging of perovskite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12215261/]
- 52. Facile supersaturation control strategies for regulating ... [https://www.sciencedirect.com/science/article/pii/S1383586625028497]
- 53. Study of the Effect of Supersaturation Changes on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11800150/]
- 54. of Particle Control of MOF-199 Size via... and Morphology Crystals [https://www.scientific.net/DDF.380.39]
- 55. Crystal Habit Modification [https://www.radicalpolymers.com/crystal-habit-modification/]
- 56. Stability of Amorphous Pharmaceutical Solids: Crystal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3385820/]
- 57. Effects of different types of modifiers on structural variation ... [https://pubs.rsc.org/en/content/articlelanding/2025/na/d5na00392j]
- 58. Top 5 Uses of Crystal Modifiers in 2025 | Integration Notes | T [https://www.linkedin.com/pulse/crystal-modifier-real-world-5-uses-youll-g1xke/]
- 59. Competition between crystal growth and intracrystalline ... [https://www.nature.com/articles/s41467-021-27752-0]
- 60. Melting Temperature and Change of Lamellar Thickness ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5319807/]
- 61. Poly (butylene succinate): Low-temperature nucleation and ... [https://www.sciencedirect.com/science/article/pii/S0032386123006419]
- 62. High-Speed Atomic Force Microscopy Observation of Cu ... [https://www.sciencedirect.com/science/article/abs/pii/S0013468625019218]
- 63. Electrochemical nucleation and growth model of MoS2 for ... [https://jast-journal.springeropen.com/articles/10.1186/s40543-024-00466-w]
- 64. Crystallization Mechanisms of Poly(vinylidene Fluoride-co ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12424290/]
- 65. On the two-step mechanism for synthesis of transition- ... [https://pubmed.ncbi.nlm.nih.gov/25275611/]
- 66. Fitting neurological protein aggregation kinetic data via a 2 ... [https://pubmed.ncbi.nlm.nih.gov/18247636/]
- 67. developing a modified Finke–Watzky mechanism [https://www.linkedin.com/pulse/importance-kinetics-study-synthesis-nanostructures-modi%EF%AC%81ed-amirjani]
- 68. Nanocluster nucleation and growth kinetic and mechanistic ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979707012088]
- 69. Finke-Watzky Two-Step Nucleation-Autocatalysis Model of ... [https://pubmed.ncbi.nlm.nih.gov/28759719/]
- 70. Mechanisms of controlled stabilizer-free synthesis of gold ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11268499/]
- 71. LaMer's 1950 model of particle formation: a review and ... [https://www.osti.gov/pages/biblio/1742062]
- 72. Mixing Controlled Adsorption at the Liquid-Solid Interfaces ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9849304/]
- 73. A revised diffusion model for conflict tasks [https://link.springer.com/article/10.3758/s13423-023-02288-0]
- 74. Diffusion-Controlled Reactions: An Overview [https://www.mdpi.com/1420-3049/28/22/7570]
- 75. Chaperone solvent-assisted assembly of polymers at the ... [https://www.nature.com/articles/s41467-024-51657-3]
- 76. Pharmaceutical Crystallization in drug development [https://www.syrris.com/crystallization-in-drug-development/]
- 77. The Relevance of Crystal Forms in the Pharmaceutical Field [https://pmc.ncbi.nlm.nih.gov/articles/PMC9408954/]
- 78. Single Crystal Growth & Structure Determination [https://www.crystalpharmatech.com/single-crystal-growth-and-structure-determination/]