In Situ Microscopy for Inorganic Crystal Nucleation: Real-Time Insights for Pharmaceutical Development
This article explores the transformative role of in situ microscopy in observing and controlling inorganic crystal nucleation, a critical process in pharmaceutical development.
In Situ Microscopy for Inorganic Crystal Nucleation: Real-Time Insights for Pharmaceutical Development
Abstract
This article explores the transformative role of in situ microscopy in observing and controlling inorganic crystal nucleation, a critical process in pharmaceutical development. It provides a comprehensive overview for researchers and scientists, covering foundational nucleation theories, advanced methodological approaches using electron and optical microscopy, practical troubleshooting for experimental challenges, and validation through comparative analysis with traditional techniques. By synthesizing current research and applications, this review aims to demonstrate how real-time observation at micro- and nanoscales enables precise engineering of crystalline materials for enhanced drug formulation and performance.
Understanding Crystal Birth: Fundamental Principles of Inorganic Nucleation
Classical vs. Non-Classical Nucleation Pathways in Inorganic Systems
Crystal nucleation, the initial step in the formation of solid materials from liquid or vapor phases, represents a critical process in materials science, pharmaceuticals, and chemical engineering. For over a century, Classical Nucleation Theory (CNT) has served as the predominant framework for understanding this phenomenon, positing that nuclei form directly from solution through the stochastic addition of monomers, leading to equilibrium-shaped clusters with macroscopic crystal structure [1]. However, advanced computational and experimental techniques have increasingly revealed the limitations of this classical view, particularly for complex inorganic systems. The emergence of non-classical pathways, characterized by the formation of metastable intermediate phases, has fundamentally challenged our understanding of crystallization processes at the atomic level [1].
This application note examines the competing nucleation pathways in inorganic systems within the context of in situ microscopy observation research. We provide a comprehensive comparison of classical and non-classical mechanisms, detailed experimental protocols for their investigation, and structured data presentation to facilitate research in inorganic crystal nucleation. The insights gained from these advanced methodologies are reshaping fundamental understanding and enabling precise control over crystalline materials across numerous technological applications.
Theoretical Framework: Competing Nucleation Pathways
Classical Nucleation Theory (CNT)
Classical Nucleation Theory describes crystal formation as a single-step process where solutes (atoms, ions, or molecules) directly assemble into critical nuclei with the same atomic arrangement as the final crystal. According to CNT, the formation of a crystal nucleus is governed by a balance between the volume free energy gain and the surface free energy penalty [2]. The critical nucleus size ((r^)) represents the point where the free energy barrier ((\Delta G)) is maximized—clusters smaller than (r^) tend to dissolve, while those larger than (r^*) are likely to continue growing [2].
The mathematical formulation for the free energy change in CNT for a single-crystal TMD with triangular geometry is expressed as:
[ \Delta G = \frac{\sqrt{3}}{4}L^{2}\Delta G_{V} + 3L\sigma ]
Where (L) represents the crystal size (length of triangle edge), (\sigma) is the surface energy per unit length, and (\Delta G{V}) denotes the difference in free energy between solid and liquid/vapor states ((G{S} - G_{X})) [2].
Non-Classical Nucleation Pathways
Non-classical nucleation mechanisms deviate from the direct single-step process of CNT and typically involve the formation of metastable intermediate phases that precede the appearance of the stable crystalline phase [1]. Several distinct non-classical pathways have been identified:
Two-Step Nucleation: This mechanism involves initial formation of a dense liquid droplet or amorphous precursor, within which crystalline nuclei subsequently form [2]. This pathway has been observed in diverse systems including proteins, colloidal particles, and inorganic materials like tungsten disulfide (WS₂) [2].
Precursor-Mediated Pathways: Some inorganic systems form specific intermediate phases prior to crystallization. For instance, sodium halides (NaBr and NaI) exhibit a liquid crystal phase composed of contact ion pairs before nucleating into anhydrous or hydrous single crystals [3].
Polymorphic Competition: Multiple crystal structures may compete during nucleation, as demonstrated in zinc oxide nanoparticles where Wurtzite (WRZ) and body-centered tetragonal (BCT) phases emerge through different pathways depending on supercooling conditions [4].
Table 1: Fundamental Characteristics of Classical vs. Non-Classical Nucleation Pathways
| Characteristic | Classical Nucleation | Non-Classical Nucleation |
|---|---|---|
| Intermediate Phases | None | Metastable clusters, amorphous precursors, or liquid crystalline phases |
| Critical Nucleus Size | Typically nanoscale (1-10 nm) | Can be much larger (up to micrometers) |
| Structural Evolution | Direct to stable crystal | Multiple steps with structural transitions |
| Pathway Dependency | Primarily driven by supersaturation | Influenced by multiple factors (temperature, interfaces, concentration) |
| Representative Systems | Simple inorganic salts (e.g., NaCl) | Complex oxides, TMDs, proteins [2] [3] |
Application Notes: Nucleation in Inorganic Systems
Case Study 1: Zinc Oxide Nanoparticles
ZnO nanoparticle formation exemplifies polymorphic competition between different crystal structures. Through advanced machine-learning force fields (including long-range interactions with PLIP+Q methodology), researchers have demonstrated that nucleation pathways depend critically on the degree of supercooling [4].
At high supercooling, a multi-step process emerges where metastable phases precede the formation of stable crystals. In contrast, under moderate supercooling, nucleation follows a more classical pathway directly to the stable phase. The competition specifically occurs between the wurtzite (WRZ) structure (most stable in bulk) and the body-centered tetragonal (BCT) phase (more stable at small nanoparticle sizes) [4].
This system highlights the importance of advanced simulation approaches for capturing nucleation complexities, including machine-learning interaction potentials that accurately model both bulk and surface effects, particularly for polar surfaces where simpler potentials fail [4].
Case Study 2: Sodium Halides (NaCl, NaBr, NaI)
Microdroplet studies of sodium halides reveal how subtle compositional changes dramatically alter nucleation pathways. While NaCl follows a classical nucleation pathway without detectable intermediates, both NaBr and NaI exhibit clear non-classical behavior with the formation of a liquid crystal intermediate phase composed of contact ion pairs prior to crystal formation [3].
These observations establish a new theoretical framework for crystal nucleation and growth of ionic salts, demonstrating that non-classical pathways are not limited to complex organic molecules but occur even in simple inorganic systems [3]. The findings further suggest opportunities for controlling nucleation pathways to achieve desired crystal structures regardless of specific environmental conditions.
Case Study 3: Calcium Silicate Hydrate (C-S-H)
The nucleation of C-S-H, the most important hydrate of cement, follows a distinct two-step non-classical process. During synthesis, discrete globules initially appear as metastable precursors that subsequently transform into foil-like C-S-H, accompanied by changes in crystallinity and structure with heat release [5].
This transformation from amorphous precursors to crystalline material exemplifies how non-classical pathways dominate in complex inorganic systems with significant practical implications. Understanding these mechanisms benefits functionalization and applications of cement-based materials [5].
Table 2: Experimental Observations of Nucleation Pathways in Selected Inorganic Systems
| System | Observed Pathway | Intermediate Phase | Critical Nucleus Size | Experimental Method |
|---|---|---|---|---|
| Zinc Oxide | Temperature-dependent competition | Metastable crystal phases | Nanoscale (atomistic) | ML-MD simulations [4] |
| WS₂ | Two-step nucleation | Liquid precursor droplets | ~38.7 µm | In situ monitoring CVD [2] |
| NaCl | Classical | None | Not specified | Microdroplet evaporation [3] |
| NaBr/NaI | Non-classical | Liquid crystal phase | Not specified | Microdroplet evaporation [3] |
| C-S-H | Two-step nucleation | Discrete globules | Not specified | TEM, XRD, FT-IR, NMR [5] |
Experimental Protocols
1In SituMonitoring Chemical Vapor Deposition for TMDs
Purpose: Direct visualization of the phase transition from liquid precursors to solid transition metal dichalcogenides (TMDs) to observe critical nuclei and nucleation dynamics [2].
Materials and Equipment:
- Chemical vapor deposition system with optical imaging capability
- Transition metal oxide source powder
- Alkali metal salts
- Substrate
- Automated image analysis system
Procedure:
- Precursor Preparation: Mix transition metal oxide powder with alkali metal salts to enhance vaporization through lowered melting and boiling points [2].
- System Setup: Configure in situ monitoring CVD to capture images at a rate of 1 frame per second throughout the experiment [2].
- Growth Process: Conduct CVD under controlled temperature and gas flow conditions to promote vapor-liquid-solid growth.
- Image Analysis: Apply automated image analysis with predefined HSV color index thresholds to extract monolayer and multilayer regions from optical images [2].
- Data Extraction: Determine incubation time and growth speed from area plots of monolayer coverage versus time.
- Nucleation Analysis: Identify critical nucleation events by tracking cluster size changes and collision events between precursor particles [2].
Key Measurements:
- Incubation time for metastable cluster formation
- Critical nucleus size determination
- Growth velocity calculations
- Transition points from slow to rapid growth phases
Microdroplet Evaporation for Sodium Halides
Purpose: Investigation of crystallization pathways of sodium halides under homogeneous nucleation conditions across a range of supersaturations [3].
Materials and Equipment:
- Aqueous solutions of sodium halides
- Microfluidic or droplet generation apparatus
- Optical microscopy with polarization capability
- Computational resources for data analysis
Procedure:
- Solution Preparation: Prepare aqueous solutions of NaCl, NaBr, and NaI at various concentrations.
- Droplet Generation: Create microdroplets of consistent size using appropriate techniques.
- Controlled Evaporation: Subject droplets to controlled evaporation conditions while monitoring optically.
- Birefringence Monitoring: Use polarized light to detect liquid crystal phase formation through birefringence.
- Pathway Characterization: Document the sequence of phase transitions for each halide compound.
- Computational Analysis: Perform complementary computational analysis to identify structural characteristics of intermediate phases [3].
Key Measurements:
- Identification of intermediate phases
- Transition timing between phases
- Supersaturation thresholds for different pathways
- Structural characterization of liquid crystal phases
Machine-Learning Molecular Dynamics for Oxide Nanoparticles
Purpose: Study polymorphic competition during crystal nucleation using advanced computational approaches that overcome traditional force field limitations [4].
Materials and Equipment:
- High-performance computing resources
- Training datasets from DFT calculations
- Machine-learning interaction potential code
- Structural analysis tools
Procedure:
- Potential Development: Construct machine-learning interaction potentials incorporating long-range interactions using approaches like PLIP+Q [4].
- Validation: Validate the potential against DFT calculations for various polymorphs, phonon density of states, and surface energies.
- Simulation Setup: Prepare liquid nano-droplets of the target material at different temperatures.
- Sampling Approach: Combine brute-force molecular dynamics with rare-event sampling techniques to capture nucleation events [4].
- Trajectory Analysis: Apply data-driven clustering methods based on Gaussian-mixture models to characterize local ordering.
- Pathway Identification: Identify competing nucleation pathways by analyzing structural evolution under different conditions.
Key Measurements:
- Free energy landscapes
- Structural fingerprints of intermediate states
- Nucleation rates for different polymorphs
- Temperature-dependent pathway preferences
Visualization of Nucleation Pathways
This diagram illustrates the fundamental differences between classical and non-classical nucleation pathways. The classical route proceeds directly from solution to crystalline nucleus, while non-classical pathways involve metastable intermediate phases that precede crystal formation, as observed in systems such as sodium halides and tungsten disulfide [2] [3].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Materials for Investigating Inorganic Nucleation
| Reagent/Material | Function | Example Applications |
|---|---|---|
| Alkali Metal Salts | Catalyst to decrease energy barrier and increase surface reaction rate | Salt-assisted growth of TMDs [2] |
| Transition Metal Oxides | Vapor-phase source of metal atoms | Precursors for TMD synthesis [2] |
| Machine-Learning Interaction Potentials | Accurate modeling of atomic interactions including long-range forces | ZnO nanoparticle simulations [4] |
| Microdroplet Platforms | Controlled environment for homogeneous nucleation studies | Sodium halide crystallization [3] |
| In Situ Optical Monitoring | Real-time visualization of nucleation events | WS₂ growth dynamics [2] |
| Aqueous-Organic Solvent Mixtures | Tune supersaturation through antisolvent effects | Potassium chloride/sulfate crystallization [6] |
The investigation of nucleation pathways in inorganic systems has evolved significantly beyond the classical framework, revealing a complex landscape of competing mechanisms that include two-step nucleation, intermediate phase formation, and polymorphic competition. Advanced characterization techniques, particularly in situ monitoring and computational approaches with machine-learning force fields, have been instrumental in uncovering these non-classical pathways [4] [2].
Understanding these diverse nucleation mechanisms provides critical insights for controlling crystal structure and properties in applications ranging from semiconductor manufacturing to pharmaceutical development. The experimental protocols and analytical methods outlined in this application note offer researchers comprehensive tools for investigating nucleation phenomena in diverse inorganic systems, ultimately enabling more precise control over material synthesis and properties.
Nucleation, the initial formation of a new thermodynamic phase, is a critical process in the crystallization of inorganic materials, determining the number, size, perfection, and polymorphic characteristics of the resulting crystals [7]. For researchers in drug development and materials science, controlling nucleation is essential for tailoring product properties, from pharmaceutical bioavailability to catalytic performance [7] [8]. Classical Nucleation Theory (CNT) provides the principal theoretical framework describing this process as a thermally activated event where supersaturation, temperature, and interfacial energies collectively determine the nucleation rate [9] [7]. Within the context of in situ microscopy observation, particularly using advanced techniques like in situ transmission electron microscopy (TEM), researchers can now directly visualize and quantify these parameters at the atomic scale, transforming our understanding of nucleation mechanisms [10] [11]. This application note delineates the key parameters governing nucleation and provides detailed protocols for their investigation within inorganic crystal nucleation research.
Theoretical Foundations of Nucleation
Classical Nucleation Theory (CNT)
Classical Nucleation Theory models nucleation as a process of overcoming a free energy barrier. The formation of a crystalline nucleus from a supersaturated solution involves a balance between the energy gain from forming a new volume and the energy cost of creating a new interface [7]. The Gibbs free energy change (ΔG) for a spherical nucleus is expressed as:
[ \Delta G = -\frac{4}{3}\pi r^3 \Delta g_v + 4\pi r^2 \gamma ]
where (r) is the nucleus radius, (\Delta g_v) is the free energy change per unit volume (driving the phase transition), and (\gamma) is the interfacial energy [9]. This relationship yields a critical nucleus size ((r^)) and the activation barrier for nucleation ((\Delta G^)):
[ r^* = \frac{2\gamma}{\Delta gv} \quad \text{and} \quad \Delta G^* = \frac{16\pi \gamma^3}{3\Delta gv^2} ]
The nucleation rate (J), defined as the number of nuclei formed per unit volume per unit time, follows an Arrhenius-type relationship [9] [7]:
[ J = A \exp\left(-\frac{\Delta G^*}{k_B T}\right) ]
Here, (A) is the pre-exponential factor, (kB) is Boltzmann's constant, and (T) is temperature. The pre-exponential factor incorporates kinetic factors, including the rate of molecular attachment and solution viscosity, and can be modeled as (A \propto C0 / \eta) for interface-transfer controlled nucleation, where (C_0) is the initial solute concentration and (\eta) is the solution viscosity [12].
The Role of Key Parameters
- Supersaturation ((S)): Defined as (S = C0 / C{eq}), where (C0) is the initial concentration and (C{eq}) is the equilibrium solubility, supersaturation provides the thermodynamic driving force for nucleation [12]. It directly influences (\Delta g_v), thereby dramatically affecting the nucleation barrier (\Delta G^*) and rate (J). Higher supersaturation significantly reduces the critical nucleus size and the energy barrier, facilitating faster nucleation [7].
- Temperature: Temperature exerts a complex, dual influence. It directly affects solubility ((C_{eq})), thereby changing supersaturation at a given concentration. Additionally, temperature influences molecular mobility (diffusion coefficient) and the interfacial energy, impacting both the kinetic (pre-exponential factor) and thermodynamic ((\Delta G^*)) components of the nucleation rate [12] [9].
- Interfaces (Interfacial Energy, (\gamma)): The interfacial energy (\gamma) is the energy required to create a unit area of new solid-liquid interface [12]. It is a pivotal property determined by the specific chemical interactions between the solute and solvent. A lower interfacial energy linearly reduces the critical nucleus radius and cubically diminishes the nucleation barrier, profoundly enhancing the nucleation rate [12] [7]. Heterogeneous nucleation on foreign substrates or impurities occurs because these surfaces effectively lower the interfacial energy barrier compared to homogeneous nucleation in the bulk solution [9].
Table 1: Fundamental Equations in Classical Nucleation Theory
| Parameter | Symbol | Equation | Relationship to Nucleation |
|---|---|---|---|
| Supersaturation | (S) | (S = C0 / C{eq}) | Primary thermodynamic driving force |
| Critical Radius | (r^*) | (r^* = \frac{2\gamma}{\Delta g_v}) | Minimum stable nucleus size |
| Nucleation Barrier | (\Delta G^*) | (\Delta G^* = \frac{16\pi \gamma^3}{3\Delta g_v^2}) | Free energy hurdle for nucleation |
| Nucleation Rate | (J) | (J = A \exp\left(-\frac{\Delta G^*}{k_B T}\right)) | Number of nuclei formed per unit time and volume |
Quantitative Data on Nucleation Parameters
Experimental investigations across different systems provide quantitative insights into how these parameters govern nucleation behavior. The interplay between supersaturation, temperature, and solvent-dependent interfacial energy is evident in measured induction times and calculated nucleation parameters.
Supersaturation and Induction Time
The induction time ((ti)), defined as the time required from the creation of supersaturation to the appearance of detectable nuclei, is inversely related to the nucleation rate ((J \propto ti^{-1})) [12]. Studies on model systems like phenacetin in different solvents demonstrate a strong inverse correlation between supersaturation and induction time. Higher supersaturation leads to shorter induction times due to a lower nucleation barrier and increased nucleation rate [12].
Table 2: Experimentally Determined Nucleation Parameters for Phenacetin in Various Solvents at 308 K [12]
| Solvent | Supersaturation Ratio (S) | Average Induction Time (s) | Interfacial Energy, (\gamma) (mJ/m²) | Pre-exponential Factor, (A) |
|---|---|---|---|---|
| Ethanol (ET) | 1.65 | 78 | 2.58 | (1.21 \times 10^{11}) |
| 1.89 | 43 | |||
| 2.14 | 25 | |||
| Methanol (ME) | 1.79 | 162 | 2.63 | (1.02 \times 10^{11}) |
| 2.07 | 46 | |||
| 2.34 | 21 | |||
| Ethyl Acetate (EA) | 1.82 | 384 | 2.79 | (2.21 \times 10^{10}) |
| 2.10 | 128 | |||
| 2.38 | 55 | |||
| Acetonitrile (ACN) | 1.90 | 629 | 2.90 | (4.87 \times 10^9) |
| 2.19 | 175 | |||
| 2.48 | 81 |
Interfacial Energy and Solvent Effects
The solvent environment critically influences nucleation through the interfacial energy ((\gamma)). As shown in Table 2, phenacetin exhibits different interfacial energies in different solvents, with values ranging from 2.58 mJ/m² in ethanol to 2.90 mJ/m² in acetonitrile [12]. This variation arises from specific solute-solvent interactions, such as polarity and hydrogen bonding potential. A lower (\gamma), as seen in ethanol, correlates with a higher pre-exponential factor and faster nucleation (shorter induction times), demonstrating how solvent selection can be used to control nucleation kinetics [12].
Experimental Protocols for In Situ Observation
The following protocols outline methodologies for investigating nucleation parameters using in situ TEM, a powerful technique that enables real-time observation of nucleation and growth dynamics at the atomic scale [10] [8].
Protocol: In Situ TEM for Nucleation in Liquid Environments
This protocol is adapted from studies on Cu-based nanocatalysts and nanomaterial synthesis, focusing on observing nucleation from solution [10] [8].
1. Research Reagent Solutions and Materials Table 3: Essential Materials for In Situ Liquid Cell TEM
| Item | Function/Description |
|---|---|
| Polymer Electrochemical Liquid Cell | A specialized TEM holder that seals a liquid sample between electron-transparent membranes, allowing for high-resolution imaging in liquid [8]. |
| Precursor Solution | A solution containing the solute of interest (e.g., metal salt, inorganic precursor) dissolved in a suitable solvent. The concentration should be chosen to achieve the desired supersaturation. |
| Solvent | High-purity solvent (e.g., water, ethanol, acetonitrile). The choice dictates solubility, interfacial energy, and viscosity [12]. |
| In Situ TEM Holder | A sample stage that allows for electrical biasing, heating, or fluid injection during TEM observation [10] [8]. |
| Aberration-corrected TEM | A microscope capable of atomic-resolution imaging, equipped with fast cameras for capturing dynamic processes [10]. |
2. Experimental Procedure
- Step 1: Liquid Cell Assembly. Load the precursor solution into the polymer electrochemical liquid cell using a micro-syringe under an inert atmosphere if necessary. Assemble the cell according to the manufacturer's instructions, ensuring the silicon nitride windows are clean and intact to avoid imaging artifacts [8].
- Step 2: TEM Insertion and Setup. Insert the loaded liquid cell holder into the TEM column. Allow the system to stabilize to minimize drift. Navigate the electron beam to a region of interest with a thin, uniform liquid layer.
- Step 3: Nucleation Initiation. Induce nucleation by one of several methods:
- Solvent Evaporation: Gently heat the liquid cell using the in-situ holder to increase supersaturation by evaporating solvent [10].
- Solution Mixing: Use a multi-channel liquid cell to mix two precursor solutions directly within the TEM, rapidly creating supersaturation [10].
- Electrochemical Bias: For electrocrystallization, apply a controlled potential/current to the working electrode in the cell to generate supersaturation or cause electrodeposition [8].
- Step 4: Data Acquisition. Begin acquiring data simultaneously upon initiating nucleation.
- Imaging: Record real-time movies at a high frame rate (e.g., 200 fps) to capture nucleation events. Use a low electron dose to minimize beam effects while maintaining sufficient signal [8].
- Spectroscopy: Concurrently acquire Energy Dispersive X-ray Spectroscopy (EDS) for elemental analysis or Electron Energy Loss Spectroscopy (EELS) for chemical state information [10].
- Step 5: Data Analysis. Analyze the recorded videos to quantify nucleation metrics: induction time, nucleation rate, crystal growth speed, and crystal morphology. Correlate these with the applied experimental conditions (supersaturation, temperature, applied potential) [12] [8].
Protocol: Investigating Heterogeneous Ice Nucleation via Cryo-TEM
This protocol, based on the work by Li et al. (2025), details the observation of heterogeneous ice nucleation, a quintessential model for studying the role of interfaces [11].
1. Research Reagent Solutions and Materials
- Translucent Graphene Substrates: Serve as a well-defined substrate for heterogeneous nucleation [11].
- High-Purity Water: Water vapor source for deposition.
- Cryo-TEM Holder: A sample holder capable of cooling to cryogenic temperatures (e.g., 102 K).
- In-Situ Cryo-TEM: A TEM equipped with a cryogenic stage and capabilities for vapor deposition.
2. Experimental Procedure
- Step 1: Substrate Preparation. Mount a pristine graphene substrate onto the cryo-TEM holder.
- Step 2: Cryo-Cooling. Insert the holder into the TEM and cool the substrate to the target temperature (e.g., 102 K) [11].
- Step 3: Vapor Deposition. Introduce water vapor into the TEM column at a controlled pressure (e.g., (10^{-6}) Pa) directed toward the cryogenic substrate. This simulates deposition freezing conditions.
- Step 4: Real-Time Observation. Use high-resolution real-time imaging to monitor the sequence of events:
- Amorphous Ice Adsorption: Observe the formation of an amorphous solid water layer on the substrate [11].
- Spontaneous Nucleation: Capture the emergence of crystalline ice nuclei (Ice Ih and Ice Ic) within the amorphous layer [11].
- Nuclei Evolution: Track subsequent processes like Ostwald ripening (larger nuclei growing at the expense of smaller ones), oriented aggregation, and crystal faceting toward an equilibrium shape [11].
- Step 5: Structural Analysis. Perform Fast Fourier Transform (FFT) on acquired images to identify crystal structures and orientations of the nucleated ice. Map the spatial configuration of different ice polymorphs.
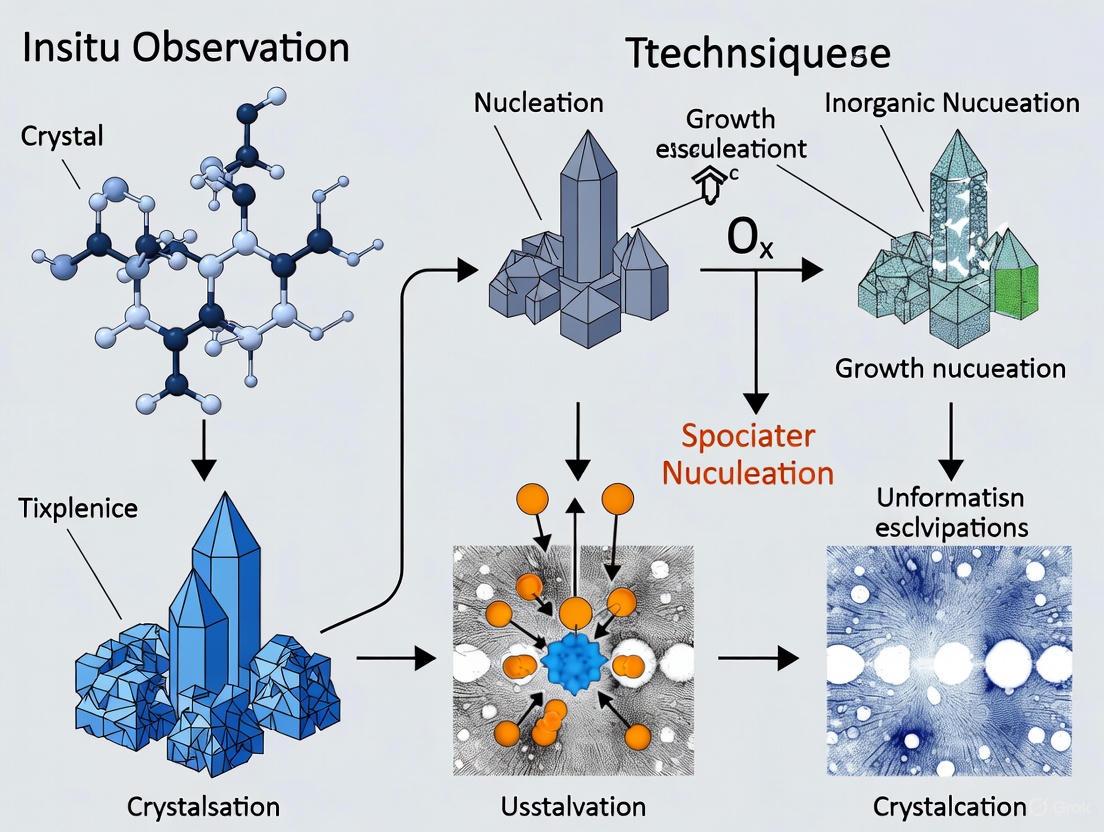
Visualization of Nucleation Pathways
The integration of in situ microscopy with computational models has revealed complex, non-classical nucleation pathways. The following diagram synthesizes the key steps in a heterogeneous nucleation pathway as observed in ice formation studies [11].
This pathway highlights a non-classical, multi-step mechanism where an initial metastable phase (amorphous adsorption layer) precedes the appearance of stable crystalline nuclei [7] [11]. The progression is governed by interfacial free energy minimization, driving the system from a far-from-equilibrium state toward a thermodynamically stable crystalline product with a characteristic equilibrium shape (Wulff construction) [11].
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions and Materials for In Situ Nucleation Studies
| Category/Item | Specific Examples | Function in Experiment |
|---|---|---|
| In Situ TEM Holders & Cells | Polymer Electrochemical Liquid Cell [8], Heating Chip [10], Gas-Phase Cell [10] | Creates controlled microenvironment (liquid, gas, heated) within the TEM for observing dynamic processes. |
| Model Solute Systems | Phenacetin [12], Copper/Silver Nanowires [8], Water Vapor [11] | Well-characterized materials for studying fundamental nucleation parameters and kinetics. |
| Solvents & Precursors | Methanol, Ethanol, Acetonitrile, Ethyl Acetate [12], Metal Salt Solutions [8] | Medium for crystallization; solvent choice directly impacts solubility, supersaturation, and interfacial energy. |
| Substrates for Heterogeneous Nucleation | Graphene Films [11], Functionalized Membranes [13] | Provides a defined surface to study and control heterogeneous nucleation mechanisms and rates. |
| Analytical Software | Fast Fourier Transform (FFT) Analysis [11], Molecular Dynamics Simulation Codes [11] | For analyzing crystal structure from TEM images and simulating molecular-scale nucleation pathways. |
The Critical Role of Prenucleation Clusters and Intermediate Phases
The understanding of crystal nucleation has been fundamentally advanced by the identification of non-classical pathways, in which crystallization does not proceed by the simple addition of single ions or molecules but through the assembly of complex precursor species. These pathways frequently involve the formation of prenucleation clusters and metastable intermediate phases that dictate the structure, phase, and properties of the final crystalline material [13] [5]. The study of these phenomena has been revolutionized by the application of in situ microscopy techniques, particularly advanced transmission electron microscopy (TEM) methods, which allow for the direct, real-time observation of nucleation events at the atomic or near-atomic scale [14] [15].
The critical importance of these precursor states is exemplified by research on calcium silicate hydrate (C-S-H), the most important hydrate in cement. In situ observations have confirmed that its homogeneous nucleation is a two-step, non-classical process [5]. This process begins with the appearance of discrete globules that act as a metastable precursor, which then transform into foil-like C-S-H crystals. This transformation is accompanied by changes in crystallinity, structure, and heat release, underscoring the dynamic role of intermediate species in directing the final material's properties [5].
Similarly, seminal work on ultrathin amorphous nanosheets has provided unprecedented detail on the structural evolution during nucleation. In situ aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) has revealed that nucleation from amorphous structures can be a multi-stage process involving: (1) the aggregation of atoms, (2) crystallization to form lattice-expanded nanocrystals, and (3) relaxation of these nanocrystals to form the final stable product [15]. This pathway has been leveraged for phase engineering, enabling the synthesis of nanomaterials with unconventional crystalline phases, such as face-centered-cubic (fcc) Ru and hexagonal-close-packed (hcp) Rh, which are not typically stable under standard conditions [15].
Table 1: Key Stages in Non-Classical Nucleation Pathways
| Nucleation Stage | Description | Experimental Evidence |
|---|---|---|
| Pre-nucleation Cluster Formation | Formation of thermodynamically stable molecular aggregates in supersaturated solutions prior to nucleation [13]. | Computational modeling and in situ spectroscopy [13]. |
| Amorphous Intermediate Phase | A metastable, disordered aggregate that acts as a precursor to the crystalline phase [15] [5]. | Observed via in situ TEM in C-S-H (discrete globules) [5] and metal nanosheets [15]. |
| Crystallization & Phase Transformation | The solid-state transition of the amorphous intermediate into a crystalline material, often involving lattice strain [15]. | In situ HAADF-STEM showing lattice-expanded intermediates [15]. |
| Final Structural Relaxation | Expulsion of impurities and relaxation of lattice strain to achieve the final stable crystal structure [15]. | In situ EELS analysis showing carbon migration during metal nanosheet nucleation [15]. |
Quantitative Data on Nucleation Phenomena
Quantitative studies of nucleation at constant supersaturation provide crucial data on the kinetics and stochastic nature of the process. The primary measurable is the nucleation time, which is the waiting time before a stable crystal nucleus appears in a supersaturated system [16]. This is distinct from the observation time, which is when a crystal has grown large enough to be detected optically. Isothermal experiments on small droplets are a clean method for obtaining this data, as they ensure constant supersaturation and allow for the monitoring of a large number of identical volumes [16].
The data is best represented by the cumulative probability ( P(t) ) that nucleation has not occurred by time ( t ). For a system with a constant, time-independent nucleation rate ( k ), ( P(t) ) follows a simple exponential decay: ( P(t) = \exp(-kt) ) [16]. The effective nucleation rate ( h(t) ) can be defined from the relationship ( h(t) = -d[\ln P(t)]/dt ). Deviations from a simple exponential decay indicate more complex nucleation mechanisms, such as a time-dependent nucleation rate caused by evolving heterogeneous nucleation sites [16].
Table 2: Quantitative Models and Parameters in Nucleation Studies
| Parameter / Model | Description | Application and Significance |
|---|---|---|
| Nucleation Time (( t_N )) | The time for a nucleus to reach a stable, non-dissolvable size [16]. | The fundamental waiting time of the process; often measured in computer simulations [16]. |
| Observation Time (( t_{OBS} )) | The time when a crystal is first observed (e.g., via optical microscopy) [16]. | The experimental approximation for ( tN ); valid if growth time ( tG \ll t_N ) [16]. |
| Cumulative Probability ( P(t) ) | The fraction of systems (e.g., droplets) that have not nucleated by time ( t ) [16]. | The preferred method for plotting and analyzing isothermal nucleation data [16]. |
| Classical Nucleation Theory (CNT) | A model where nuclei form via the atom-by-atom addition of monomers [13]. | Provides a baseline theory; often fails to predict rates and polymorph selection for complex systems [13]. |
| Non-Classical / Two-Step Nucleation | A model involving the formation of a dense liquid phase or amorphous precursor as an intermediate step [13] [5]. | Explains crystallization in systems like proteins, biominerals, and C-S-H [13] [5]. |
| Hazard Function ( h(t) ) | The instantaneous nucleation rate at time ( t ) for the systems that are still liquid [16]. | Also known as the failure rate in survival analysis; reveals if the nucleation probability changes over time [16]. |
Experimental Protocols forIn SituMicroscopy
Protocol A:In SituTEM of Nucleation in Amorphous Nanosheets
This protocol enables the real-time atomic-scale observation of crystal nucleation from an amorphous precursor, specifically designed for studying phase engineering of unconventional nanocrystals [15].
Sample Preparation:
- Synthesize ultrathin amorphous nanosheets of the target material (e.g., Ru, Rh). The amorphous structure is often stabilized by the incorporation of dopants, such as carbon [15].
- Deposit the nanosheets onto a standard TEM grid suitable for in situ heating.
In Situ Heating Setup:
- Load the sample grid into a commercial TEM heating holder.
- Ensure the holder has double-tilt capabilities to achieve optimal crystallographic orientations for high-resolution imaging [14].
Microscopy and Data Acquisition:
- Use an aberration-corrected High-Angle Annular Dark-Field Scanning TEM (HAADF-STEM) for atomic-number (Z-) contrast imaging [15].
- Begin the in situ heating experiment, gradually increasing the temperature to provide the activation energy for nucleation and growth.
- Record a real-time image series or video at a frame rate sufficient to capture the dynamics of structural evolution.
- Simultaneously, perform in situ Electron Energy-Loss Spectroscopy (EELS) to monitor chemical changes, such as the migration of dopant elements (e.g., carbon) during the process [15].
Data Analysis:
- Analyze the image series to identify the distinct stages of nucleation: atomic aggregation, crystallization into lattice-expanded intermediates, and final lattice relaxation [15].
- Correlate structural changes with the chemical data from EELS.
- Use Density Functional Theory (DFT) calculations to support the experimental findings, for instance, to verify the role of a dopant in stabilizing an unconventional crystal phase [15].
Protocol B: Correlative Light and Electron Microscopy (CLEM) for Extracellular Vesicles and Nanoparticles
This protocol outlines a rapid, cost-effective CLEM method to confirm the vesicular nature of nanoscopic structures, which can be readily adapted to study other biological and synthetic nanoparticles, including those involved in crystallization processes [17].
Sample Isolation and Staining:
- Isolate the nanoparticles of interest (e.g., Extracellular Vesicles from a fungal culture filtrate) via differential centrifugation and ultracentrifugation [17].
- Fluorescent Labeling: Stain the membrane structures of the particles with a lipophilic dye (e.g., FM1-43). This dye fluoresces only upon intercalating into a lipid bilayer [17].
- Fiducial Markers: Mix the sample with fluorescent microspheres to serve as landmarks for correlating images between different microscopes.
Laser Scanning Confocal Microscopy (LSCM) Imaging:
- First, image the sample using LSCM to locate the green fluorescence signal from the stained nanoparticles [17].
- Capture and save high-resolution confocal images of regions of interest, noting the coordinates of the fiducial markers.
Sample Processing for TEM:
- Negative Staining: Subject the same sample to negative staining. This can be done using uranyl acetate solution or, to minimize sample disturbance and artifacts, with osmium tetroxide (OsO(_4)) vapors [17].
Transmission Electron Microscopy (TEM) Imaging:
- Transfer the sample to the TEM and relocate the exact same regions previously imaged with LSCM using the fiducial markers as a guide [17].
- Acquire high-resolution TEM images of the nanoparticles to reveal their detailed morphology and membranous features.
Correlative Analysis:
- Overlay the LSCM fluorescence images with the TEM micrographs.
- Confirm that the dispersed green fluorescence signals correspond to the vesicle-like structures with membranes observed under TEM, thereby validating the presence and nature of the nanoparticles [17].
Visualization of Workflows and Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Nucleation and In Situ Microscopy Studies
| Reagent / Material | Function and Application |
|---|---|
| FM1-43 Dye | A lipophilic styryl dye used for fluorescently labeling lipid membranes in CLEM studies. It fluoresces only upon incorporation into a lipid bilayer, making it ideal for identifying membranous structures like extracellular vesicles [17]. |
| Osmium Tetroxide (OsO4) | Used as a vapor or solution for negative (or positive) staining in TEM. It provides high contrast for biological specimens and membranous structures while minimizing the introduction of artifact-causing particles from liquid stains [17]. |
| Fiducial Markers (Fluorescent Microspheres) | Beads that are both fluorescent and electron-dense. They are added to samples to serve as landmarks, enabling the precise correlation of the same region between light (LSCM) and electron (TEM) microscopes [17]. |
| Aberration-Corrected TEM | A high-resolution transmission electron microscope equipped with correctors for lens aberrations. It enables direct atomic-scale imaging, making it possible to observe the atomic aggregation and lattice transformations during nucleation [15]. |
| In Situ TEM Heating Holder | A specimen holder that allows for controlled heating of the sample inside the TEM. This is essential for providing the activation energy needed to initiate and observe nucleation and growth processes in real-time [14] [15]. |
| Micro-Electromechanical Systems (MEMS) | Devices fabricated using Si technology that act as miniaturized actuators and force sensors. They are used for quantitative in situ TEM deformation studies and stress-strain measurements of nanomaterials [14]. |
The discovery of gallium sulfur iodide (GaSI) represents a significant breakthrough in solid-state chemistry, marking the arrival of a rare inorganic crystal with an atomic-scale helical structure. While helical motifs are common in biological and organic systems, they are exceptionally rare in dense, extended inorganic lattices. GaSI crystallizes in the non-centrosymmetric tetragonal space group P4̅ and belongs to the family of exfoliable one-dimensional (1D) van der Waals (vdW) crystals [18].
Its structure consists of periodic helical chains constructed from corner-sharing GaS3I quasi-tetrahedral building blocks, propagating along the crystal's c-axis. Each helix is composed of three concentric atomic tetrahelices of gallium, sulfur, and iodine atoms. A remarkable feature of GaSI is its manifestation of a "squircular" helical cross-section, a mathematical hybrid between a square and a circle, observed for the first time at the atomic scale in any naturally occurring or engineered crystal [18].
This combination of helicity, non-centrosymmetry, and exfoliability makes GaSI a promising candidate for applications in nonlinear optics and potentially in areas leveraging chiral-induced spin selectivity (CISS) [18].
Experimental Observation & Characterization Protocols
The precise characterization of unconventional crystal structures like GaSI requires a multifaceted approach, combining advanced synthesis with state-of-the-art analytical techniques to elucidate both atomic structure and functional properties.
Synthesis and Structural Analysis
Experimental Protocol: Single Crystal Growth via Melt Method [18]
- Objective: To obtain high-quality, single-phase GaSI crystals for structural and property characterization.
- Materials:
- Gallium (Ga), metal, high purity (≥99.99%)
- Sulfur (S), powder, high purity (≥99.99%)
- Iodine (I), crystal, high purity (≥99.99%)
- Procedure:
- Weigh the constituent elements (Ga, S, I) in a strict 1:1:1 molar ratio.
- Seal the mixture in an evacuated quartz ampoule under vacuum.
- Place the ampoule in a furnace and heat to 500°C.
- Maintain at 500°C for a designated period (e.g., 24-48 hours) to form a homogeneous melt.
- Cool the melt slowly to room temperature, either by turning off the furnace (programmed cooling is preferable) or by using a specified cooling rate.
- Recover the resulting colorless, fiber-like crystals.
- Characterization Techniques:
- Single-Crystal X-ray Diffraction (SC-XRD): Used to determine the refined unit cell, atomic coordinates, and confirm the non-centrosymmetric P4̅ space group.
- High-Resolution Transmission Electron Microscopy (HRTEM): Performed on exfoliated crystals to confirm the retention of crystalline order and directly image the atomic-scale helical motif along the [110] zone axis.
- Energy Dispersive X-ray Spectroscopy (EDS): Confirms the 1:1:1 stoichiometric ratio and uniform distribution of Ga, S, and I.
- Raman Spectroscopy: Assesses the crystal's stability under air exposure.
Table 1: Key Structural Parameters of GaSI and Related Helical Crystal GaSeI [18]
| Parameter | GaSI | GaSeI | Remarks |
|---|---|---|---|
| Space Group | P4̅ (No. 81) | P4̅ | Non-centrosymmetric, tetragonal |
| Helix Type | Distorted Boerdjik–Coxeter (B–C) | Approximate B–C Helix | B–C helix has an irrational twist angle of ~131.81° |
| Helix Cross-Section | "Squircular" | Not squircular | First atomic-scale squircle; squaredness factor ~0.69 (Ga), ~0.65 (I) |
| Noncrystallographic Screw Axis | ~4115 | 41 | |
| Twist Angle (θt) | 132(2)° | ~131.81° | Between consecutive Ga atoms |
| Bandgap | 3.69 eV | From diffuse reflectance spectroscopy |
The Role ofIn SituMicroscopy in Nucleation Research
Understanding how such complex structures form requires techniques that can probe the dynamics of crystal nucleation and growth in real-time. In situ Transmission Electron Microscopy (TEM) has emerged as a transformative tool for this purpose, allowing researchers to observe these processes at the atomic scale under various microenvironmental conditions [10].
Application Note: Utilizing In Situ TEM for Crystal Nucleation Studies [10] [19]
- Objective: To directly observe and analyze the dynamic processes of crystal nucleation, growth, and structural evolution in real-time.
- Methodologies:
- In Situ Heating: Using MEMS-based heater chips to study nucleation and phase transformations at elevated temperatures.
- Gas-Phase Cells: Enabling the introduction of reactive gases (e.g., O₂) to study oxidation or chemical vapor deposition processes.
- Liquid-Phase Cells: Encapsulating a liquid precursor or electrolyte between electron-transparent windows (e.g., SiNₓ) to observe nucleation from solution or electrochemical processes.
- Graphene Liquid Cells: Utilizing single-atom-thick graphene windows to achieve superior imaging resolution for liquid-phase studies, though with limitations for electrochemical experiments.
- Capabilities:
- Real-time imaging: Track atomic migration, interfacial evolution, and defect dynamics.
- Spectroscopic integration: Combine with EDS and EELS for simultaneous chemical analysis.
- Manipulation: Apply external stimuli such as heat, electrical bias, or liquid/gas environments to study responses.
The diagram below illustrates a generalized workflow for conducting in situ TEM studies on crystal nucleation, integrating the various methodologies and analytical capabilities.
Computational Prediction of Crystal Structures
The discovery of novel materials is increasingly aided by computational crystal structure prediction (CSP) methods. These tools are crucial for identifying potential polymorphs—different crystal structures of the same compound—which can have profound implications for the stability and performance of materials, particularly in the pharmaceutical industry [20].
A robust CSP method was recently validated on a large set of 66 diverse molecules, successfully reproducing 137 known polymorphic forms. This method employs a hierarchical ranking approach to efficiently and accurately predict stable crystal structures [20].
Computational Protocol: Hierarchical Crystal Structure Prediction [20]
- Objective: To computationally predict all low-energy polymorphs of a given molecule to complement experimental screening and de-risk the appearance of late-stage, disruptive polymorphs.
- Workflow:
- Systematic Packing Search: A novel algorithm divides the crystal packing parameter space into subspaces based on space group symmetries and searches them consecutively.
- Initial Energy Ranking (FF): Candidate structures are initially ranked using molecular dynamics (MD) simulations with a classical force field (FF).
- Re-ranking with Machine Learning (MLFF): The top candidates are optimized and re-ranked using a machine learning force field (MLFF) that incorporates long-range electrostatic and dispersion interactions for improved accuracy.
- Final Quantum Mechanics Ranking (DFT): A shortlist of structures undergoes final ranking using periodic density functional theory (DFT) calculations, such as with the r2SCAN-D3 functional.
- Clustering Analysis: Similar structures (with an RMSD15 < 1.2 Å) are clustered to remove non-trivial duplicates and provide a cleaner polymorphic landscape.
- Key Insight: For the test set of 66 molecules, the method successfully sampled and ranked the known experimental structure within the top 10 candidates in all cases, and within the top 2 for 26 molecules [20].
Another approach, DeepCSP, leverages artificial intelligence for rapid prediction. This pure machine learning framework uses a coupled generative adversarial network (GAN) and a graph convolutional network (GCN) to generate trial crystal structures and predict their density, respectively. It can predict organic crystal structures in minutes, achieving an accuracy exceeding 80% in marketed drug validations [21].
The relationship between different computational sampling and prediction methods is outlined below.
The Scientist's Toolkit: Essential Research Reagents & Materials
The experimental and computational research into unconventional crystal structures relies on a suite of specialized reagents, materials, and software tools.
Table 2: Key Research Reagent Solutions for Crystal Synthesis and Characterization
| Item / Solution | Function / Application | Specific Examples / Notes |
|---|---|---|
| High-Purity Elements | Starting materials for crystal growth via melt or vapor transport methods. | Ga, S, I for GaSI synthesis; purity ≥99.99% is typically required [18]. |
| Evacuated Quartz Ampoules | Contain reaction mixtures at high temperatures under vacuum or inert atmosphere. | Prevents oxidation and contamination during synthesis [18]. |
| MEMS-based TEM Chips | Platform for in situ TEM experiments, providing heating, electrical biasing, or liquid/gas cell containment. | E-chip heaters, liquid cell holders with SiNₓ windows [10] [19]. |
| Electron-Transparent Windows | Encapsulate samples in in situ TEM while allowing electron beam penetration. | Silicon Nitride (SiNₓ) films, graphene sheets [10] [19]. |
| Machine Learning Force Fields (MLFF) | Accelerate and improve the accuracy of energy evaluations in computational CSP. | Charge recursive neural network (QRNN) [20]. |
| Crystallographic Databases | Source of experimental data for method validation and training AI models. | Cambridge Structural Database (CSD) [21] [20]. |
The discovery of the helical GaSI crystal underscores the fact that novel and complex inorganic structures with potentially disruptive properties await discovery. Its unique "squircular" helix and non-centrosymmetric character were elucidated through a powerful synergy of advanced synthesis, meticulous ex situ characterization, and first-principles calculations. Furthermore, the ongoing development of in situ TEM techniques provides an unprecedented window into the dynamic processes of nucleation and growth that give rise to such structures. Coupled with the rising power of AI-driven and hierarchical computational prediction methods, researchers are now equipped with a robust toolkit to intentionally design, discover, and characterize the next generation of functional crystalline materials.
Bridging Theoretical Models with Experimental Observation Needs
Inorganic crystal nucleation and growth are fundamental processes in materials science, governing the properties of nanomaterials used in catalysis, energy storage, and electronics. Theoretical models of nucleation, including classical and non-classical pathways, provide frameworks for understanding these processes. However, a significant challenge persists in directly validating these models with experimental data, as traditional ex situ characterization techniques only capture initial and final states, missing the transient intermediate stages [10].
In situ transmission electron microscopy (TEM) overcomes this limitation by enabling real-time observation of dynamic processes at the atomic scale [22] [10]. This Application Note details protocols for using in situ TEM to bridge theoretical nucleation models with experimental observation, providing researchers with methodologies to visualize and quantify nanocrystal evolution under various microenvironmental conditions.
Key Capabilities ofIn SituTEM for Nucleation Research
In situ TEM provides a platform for applying external stimuli to a sample while simultaneously observing the resulting dynamic changes with high spatial and temporal resolution [23]. This allows for direct investigation of nucleation and growth phenomena that were previously inferred only from theoretical models.
The table below summarizes the core capabilities of this approach for studying crystal nucleation and growth.
Table 1: Core Capabilities of In Situ TEM for Nucleation Studies
| Capability | Experimental Significance | Theoretical Insight |
|---|---|---|
| Real-time Observation | Enables direct visualization of dynamic processes such as nucleation events, growth pathways, and phase transformations [10] [23]. | Allows for the validation and refinement of theoretical models concerning atomic migration dynamics, interfacial evolution, and structural transformation pathways [10]. |
| Stimulus Control | Permits the application of various external stimuli, including heat, electrical bias, liquid, or gas environments, to replicate synthesis conditions [22] [23]. | Facilitates the study of nucleation mechanisms under controlled conditions, helping to quantify the influence of environmental factors like temperature and pressure predicted by theory [10]. |
| High-Resolution Imaging & Spectroscopy | Combines atomic-scale imaging with techniques like EDS and EELS for simultaneous structural, compositional, and electronic analysis [10] [23]. | Provides multimodal data to correlate structural changes with chemical and electronic states, offering a comprehensive picture that can challenge or confirm non-classical nucleation theories [10]. |
3In SituTEM Methodologies for Nanomaterial Synthesis
In situ TEM experimentation relies on specialized sample holders and cells that introduce controlled environmental conditions into the high vacuum of the microscope column. The choice of methodology is dictated by the material system and the nucleation phenomenon under investigation [10].
The primary classifications relevant to inorganic crystal nucleation research are:
- In Situ Heating Chips: Utilize microfabricated MEMS devices to heat samples, allowing for the study of phase transformations, annealing behavior, and diffusion phenomena at temperatures up to 1200°C [22] [10].
- Liquid Cells: Comprise silicon-based or graphene-sealed chips that encapsulate liquid solutions between electron-transparent windows. These are essential for observing crystal nucleation and growth from solution, mimicking wet-chemical synthesis [10].
- Gas-Phase Cells / Environmental TEM (ETEM): Enable the introduction of gaseous environments around the sample, used to study chemical reactions, such as in catalysis, and oxidation processes in real-time [10].
Detailed Experimental Protocols
Protocol: Investigating Nucleation & Growth in a Liquid Cell
This protocol outlines the procedure for directly observing the nucleation and growth of inorganic nanocrystals from a precursor solution.
1. Experimental Workflow
The following diagram illustrates the key stages of a liquid cell experiment, from preparation to data analysis.
2. Key Research Reagent Solutions
Table 2: Essential Materials for Liquid Cell Experiments
| Item | Function / Explanation |
|---|---|
| Silicon Nitrace Liquid Cell | A MEMS-based device with electron-transparent windows that encapsulate the liquid precursor solution, enabling TEM observation [10]. |
| Metallic Salt Precursors | Source of ions (e.g., HAuCl₄, AgNO₃) for nanocrystal formation. Concentration and composition control nucleation kinetics and crystal phase [10]. |
| Reducing Agent Solutions | Chemicals (e.g., NaBH₄, ascorbic acid) introduced to initiate the reduction of metal ions and trigger nucleation from the solution [10]. |
| In Situ TEM Holder (Liquid) | Specialized TEM holder that interfaces with the liquid cell, providing electrical contacts and fluidic channels for solution delivery [10] [23]. |
3. Methodology Details
- Sample Preparation: Prepare an aqueous precursor solution containing the metal salt (e.g., 1 mM HAuCl₄) and a reducing agent. Using a syringe, load the solution into the liquid cell assembly, ensuring no air bubbles are trapped [10].
- Data Acquisition: Insert the assembled holder into the TEM. Locate a thin, electron-transparent area of the liquid layer using a low electron dose rate to minimize beam effects. Initiate the reaction, often by applying a specific temperature profile via the integrated μHeater [22] [10]. Acquire data using a direct electron detection camera (e.g., Gatan K3 IS) at frame rates of 5 fps or higher to capture rapid nucleation events [23].
- Data Analysis: Analyze the video dataset to extract quantitative metrics. This includes measuring the nucleation rate (number of new crystals per unit time and volume), growth rates of individual crystals, and characterizing the evolution of crystal morphology and phase [10] [23].
Protocol: Studying Phase Evolution Under Thermal Treatment
This protocol describes using a heating holder to investigate temperature-induced phase transformations in pre-synthesized or nascent crystals.
1. Experimental Workflow
The workflow for a heating experiment, from sample loading to phase analysis, is summarized below.
2. Key Research Reagent Solutions
Table 3: Essential Materials for In Situ Heating Experiments
| Item | Function / Explanation |
|---|---|
| MEMS-Based Heater Chip | A microfabricated device (e.g., Thermo Scientific μHeater Holder) that allows for precise temperature control and high-resolution characterization up to 1200°C [22]. |
| In Situ TEM Holder (Heating) | A specialized holder that provides electrical connections and thermal isolation for the MEMS heater chip [22] [23]. |
| Nanomaterial Powder | The sample of interest (e.g., precursor crystals or nanoparticles) dispersed onto the heater chip to observe its thermal transformation. |
3. Methodology Details
- Sample Preparation: Dry-disperse a small amount of the nanomaterial powder onto the MEMS heater chip. Alternatively, a focused ion beam (FIB) can be used to lift-out a site-specific thin section from a bulk material [22].
- Data Acquisition: After inserting the holder, acquire baseline bright-field (BF) and dark-field (DF) images, as well as Selected Area Electron Diffraction (SAED) patterns. Program a thermal ramp (e.g., 10°C/min) or isothermal hold in the control software. Simultaneously record live imaging and diffraction patterns. For fast transformations, use high-speed cameras to capture dynamics with sub-millisecond resolution [23].
- Data Analysis: Monitor the SAED patterns in real-time to identify the emergence of new diffraction rings or spots, indicating a phase change. Correlate the onset temperature of phase transformation with the observed microstructural evolution, such as grain growth or coalescence, measured from the image series [10] [24].
Data Management and Analysis
The large datasets generated by in situ TEM, particularly from video-rate acquisition, require robust management and analysis strategies.
- Data Capture: Modern direct detection cameras stream original, quantitative data directly to disk. This allows each frame to be treated as an individual image for post-processing or played back as a video [23].
- Post-Processing: Apply algorithms for frame summing, drift correction, and binning to enhance signal-to-noise ratio and correct for sample movement [23].
- Advanced Analytics: The integration of machine learning and artificial intelligence is set to enhance data analysis, enabling automated identification and classification of complex structural transformations from large datasets [10].
A Practical Guide to In Situ Microscopy Techniques for Crystal Growth Analysis
Inorganic crystal nucleation, a fundamental process in materials science, has long been a challenge to observe directly at the atomic scale. Traditional characterization techniques provide only static snapshots, limiting insight into dynamic nucleation mechanisms. In situ Transmission Electron Microscopy (TEM) bridges this gap by enabling real-time observation of crystal formation and evolution within controlled gas and liquid environments [19] [10]. This application note details the methodologies and protocols for employing in situ TEM to advance inorganic crystal nucleation research, providing researchers with practical frameworks for implementing these advanced techniques.
Technical Foundations of In Situ TEM Cells
Gas Cell Design Principles
Gas-phase in situ TEM enables atomic-scale observation of nucleation and growth dynamics in reactive gaseous environments. Two primary technical designs facilitate these studies:
Differential Pumping Systems: This design employs multi-stage small apertures placed above and below the sample within the objective pole-piece, enabling direct gas introduction while maintaining high vacuum near the electron gun. This open architecture preserves superior image resolution and supports analytical techniques like electron energy-loss spectroscopy (EELS) but limits maximum pressure to approximately 20 Torr [19] [25].
Thin Window Cells (MEMS-based): These sealed systems confine gas between two electron-transparent membranes (typically amorphous carbon or SiNx), allowing pressures up to one atmosphere without microscope modification. Recent advancements incorporating microelectromechanical system (MEMS)-based heaters enable operation at 800-1000°C, making them suitable for high-temperature nucleation studies [19] [25].
Liquid Cell Design Principles
Liquid-cell TEM allows direct observation of crystallization processes in solution, overcoming the challenges of electron scattering in liquid media:
Silicon Nitride Window Cells: Standard commercial cells use SiNx membranes (typically tens of nanometers thick) to encapsulate liquid samples, with integrated electrodes for electrochemical control. These systems enable real-time observation of electrochemical reactions, metal dissolution, passivation, and oxide formation under applied potentials [19] [26].
Graphene Liquid Cells: Featuring single-atom-thick graphene windows, these cells significantly reduce electron scattering and improve imaging resolution compared to conventional SiNx designs. However, they lack fluid flow capabilities and integrated electrodes, limiting their versatility for certain experiments [19].
Table 1: Comparison of In Situ TEM Cell Configurations
| Parameter | Differential Pumping Gas Cell | Windowed Gas Cell | Standard Liquid Cell | Graphene Liquid Cell |
|---|---|---|---|---|
| Maximum Pressure | ~20 Torr [19] | ~1 atmosphere [19] | Limited by window strength | Limited by graphene seal |
| Temperature Range | Up to 1000°C+ [19] | 800-1000°C [19] | Typically < 400°C | Limited heating capability |
| Resolution | Atomic [19] | Near-atomic [19] | Nanometer | Near-atomic [19] |
| Analytical Compatibility | EELS, EDS [19] | Limited EDS/EELS [19] | Limited | Limited |
| Key Advantage | Superior resolution and analytical capabilities | High-pressure capability | Integrated electrodes for electrochemistry | Minimal electron scattering |
Experimental Protocols
Protocol for Gas-Phase Nucleation Studies
Objective: To observe the initial stages of oxide nucleation on metal nanoparticles under controlled gas environment.
Materials and Equipment:
- Atmosphere AX in situ TEM holder (Protochips) or equivalent MEMS-based gas cell system [27]
- Aberration-corrected TEM with STEM capability
- High-purity oxygen gas (99.999%)
- Metal nanoparticle samples (e.g., Cu, Ni, or Fe nanoparticles supported on SiNx membranes)
Procedure:
- Sample Loading: Deposit metal nanoparticles onto the MEMS chip following standard TEM sample preparation methods. Load the chip into the gas cell holder according to manufacturer specifications.
System Calibration:
- Establish base vacuum in the TEM column (better than 10⁻⁶ Torr).
- Calibrate the heating element using the manufacturer's temperature calibration protocol.
- Verify gas manifold integrity with leak testing.
Experimental Parameters:
- Set initial temperature to 200°C with 5°C/min ramp rate.
- Introduce oxygen gas with gradual pressure increase to target value (typically 1-760 Torr).
- Use electron dose rate of 10-100 e⁻/Ųs to balance signal-to-noise with beam effects.
Data Acquisition:
- Acquire time-resolved HRTEM or HAADF-STEM images at 1-5 frame/second.
- Simultaneously collect EELS spectra when possible to monitor chemical changes.
- Continue acquisition until complete oxide layer formation is observed.
Beam Effects Mitigation:
Protocol for Liquid-Phase Nucleation Studies
Objective: To visualize nucleation and early crystal growth of inorganic salts from solution.
Materials and Equipment:
- Poseidon AX in situ TEM holder (Protochips) or equivalent liquid cell system [27]
- Silicon nitride liquid cells with appropriate spacer thickness (150-500 nm)
- Supersaturated solution of target salt (e.g., sodium chloride, calcium phosphate)
- Syringe pump for precise fluid control
Procedure:
- Cell Assembly:
- Clean silicon nitride chips following manufacturer protocol.
- Pre-wet chips with solvent to eliminate air bubbles.
- Precisely pipette 0.5-1.0 µL of sample solution onto the bottom chip.
- Carefully assemble the liquid cell with specified spacer thickness to control liquid layer height (<1 µm optimal).
Holder Preparation:
- Load assembled liquid cell into the holder.
- Connect fluidic lines and ensure proper sealing.
- Flush cell with clean solution to remove contaminants.
Imaging Parameters:
- Use acceleration voltage of 200-300 kV.
- Employ low-dose imaging techniques (dose rate: 10-50 e⁻/Ųs).
- Set frame rate to 10-20 frames/second for nucleation events.
- Use bright-field TEM or STEM mode depending on resolution requirements.
Nucleation Initiation:
- For radiolysis-induced nucleation: Maintain constant beam exposure.
- For concentration-induced nucleation: Use mixing cells or flow systems to introduce supersaturated solutions.
- For temperature-induced nucleation: Use integrated heaters to control supersaturation.
Data Collection:
- Record continuous video during nucleation phase.
- Capture high-resolution images at critical stages (initial cluster formation, critical nucleus formation, crystal growth).
- Implement machine learning detection for automated event identification where available [28].
Artifact Control:
Diagram 1: Experimental workflow for in situ TEM nucleation studies showing parallel paths for gas-phase and liquid-phase methodologies.
Data Analysis and Interpretation
Quantitative Analysis of Nucleation Dynamics
In situ TEM generates rich datasets requiring specialized analysis approaches. Key quantitative parameters and their analytical methods include:
Nucleation Rate Calculation:
- Direct counting of nucleation events per unit area per unit time
- Machine learning-assisted detection for improved accuracy and efficiency [28]
- Statistical analysis across multiple experiments to account for heterogeneity
Growth Kinetics Analysis:
- Time-resolved particle size measurement from sequential images
- Determination of growth laws (diffusion-limited vs. interface-controlled)
- Activation energy calculation from temperature-dependent studies
Table 2: Key Parameters for Quantitative Analysis of Nucleation Events
| Parameter | Measurement Method | Significance | Typical Values |
|---|---|---|---|
| Critical Nucleus Size | HRTEM image analysis of smallest stable particles | Determines nucleation barrier | 1-5 nm for most inorganic systems |
| Nucleation Rate | Event counting per unit area/time [28] | Quantifies nucleation probability | 10⁶-10¹² events/cm³s |
| Growth Rate | Particle size vs. time tracking | Reveals growth mechanism | 0.1-10 nm/s |
| Activation Energy | Temperature-dependent rate measurement | Identifies rate-limiting steps | 0.1-1 eV for diffusion processes |
| Induction Time | Time from supersaturation to first detection | Measures nucleation difficulty | Milliseconds to hours |
Machine Learning Enhancement
Recent advances integrate machine learning for improved nucleation event detection:
- YOLOv5 Algorithm Implementation: Enables real-time detection of nucleation events at frame rates >10 fps [28]
- Automated Size Tracking: Machine learning models can track particle growth with minimal human intervention
- Early Detection System: Identifies nucleation events earlier than human observation in many cases [28]
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for In Situ TEM Nucleation Studies
| Reagent/Material | Function | Application Example | Considerations |
|---|---|---|---|
| Silicon Nitride MEMS Chips | Electron-transparent windows for environmental cells | Gas-phase nucleation studies at high pressure [19] | Thickness (50-100 nm) critical for resolution |
| Graphene Liquid Cells | Ultra-thin encapsulation for liquid experiments | High-resolution observation of solution nucleation [19] | Limited fluid flow capability |
| Microfabricated Heaters | Precise temperature control during observation | High-temperature nucleation studies [27] | Compatible with gas and liquid cells |
| Supersaturated Salt Solutions | Nucleation precursors for crystallization studies | Sodium chloride crystallization from acetone solutions [28] | Concentration controls supersaturation level |
| High-Purity Gases (O₂, H₂) | Reactive environments for gas-solid interactions | Metal oxide nucleation on nanoparticle surfaces [25] | Gas purity >99.999% recommended |
| Electrochemical Solutions | Electrolytes for bias-induced nucleation studies | Electrodeposition of metals | Concentration affects conductivity |
Technical Challenges and Solutions
Electron Beam Effects
The electron beam can significantly influence observed phenomena through several mechanisms:
- Radiolysis in Liquid Cells: Ionization of solution components creates reactive species that alter nucleation pathways [26] [28]
- Beam-Induced Heating: Local temperature increases may accelerate kinetics or create artificial nucleation events
- Knock-on Damage: Direct atomic displacement in samples, particularly relevant for lighter elements
Mitigation Strategies:
- Dose-rate studies to identify threshold below which artifacts are minimized
- Low-dose imaging techniques and advanced detectors
- Validation with complementary techniques (XRD, light scattering)
Resolution Limitations
Multiple factors challenge atomic-resolution imaging in environmental TEM:
- Electron Scattering: Gas molecules or liquid layers scatter electrons, reducing signal-to-noise
- Window Effects: Supporting membranes add background noise and reduce contrast
- Sample Drift: Thermal and mechanical instabilities in complex cell designs
Advanced Solutions:
- Aberration correction for improved resolution in thick media [19]
- Graphene-based windows for minimal scattering [19]
- Stable MEMS-based designs with integrated thermal management
Diagram 2: Technical challenges in in situ TEM nucleation studies and corresponding mitigation strategies.
In situ TEM with specialized gas and liquid cells provides unprecedented capability to observe inorganic crystal nucleation at atomic resolution. The protocols and methodologies detailed in this application note equip researchers to implement these advanced techniques, accelerating understanding of nucleation mechanisms across materials science applications. As technology advances, particularly in machine learning integration and resolution enhancement, in situ TEM promises to reveal even deeper insights into the fundamental processes of materials formation and transformation.
In Situ Scanning Electron Microscopy (SEM) with Environmental Control
In Situ Scanning Electron Microscopy (SEM) with Environmental Control, specifically using Environmental SEM (ESEM) and related techniques, enables the direct, real-time observation of dynamic processes by allowing samples to be studied under controlled gas environments and, in some cases, while fully hydrated. This capability is a significant advancement over conventional SEM, which requires high vacuum and extensive, often disruptive, sample preparation. Within inorganic crystal nucleation research, this technology provides an unprecedented window into the transient stages and active sites governing crystal formation and growth, moving beyond static snapshots to reveal fundamental kinetic and mechanistic insights [29] [30].
Key Applications in Materials and Crystal Growth
The application of in situ SEM with environmental control has led to critical discoveries in materials science, particularly in understanding the growth dynamics and redox behavior of inorganic crystalline structures.
Real-Time Observation of Nanowhisker Growth
A seminal application is the in situ study of tungsten suboxide (W₁₈O₄₉) nanowhisker growth. Using a specialized microreactor (μReactor) within an SEM chamber, researchers could directly observe the growth process from γ-WO₃/a-SiO₂ nanofibers under a hydrogen atmosphere at 100 Pa [31]. This experiment provided two key findings: first, it offered fundamental insight into the anisotropic growth mode of the crystal shear planes; second, it quantitatively demonstrated that electron beam irradiation markedly slows the growth kinetics of the nanowhiskers [31]. This highlights a critical experimental consideration for in situ studies and underscores the method's ability to quantify dynamic parameters previously inferred from post-mortem analysis.
Spatio-Temporal Redox Dynamics in Metal Catalysts
Beyond crystal growth, in situ SEM is instrumental in visualizing the dynamic behavior of catalysts under reactive conditions. A study on the hydrogen oxidation reaction over copper revealed complex oscillatory redox dynamics near a phase boundary [30].
The competing action of hydrogen and oxygen at 700 °C induced a cyclic sequence of phases on the copper surface, characterized by distinct morphologies. The table below summarizes the observed stages and their characteristics:
Table 1: Observed Stages in Copper Redox Dynamics at 700°C
| Stage | Observed Morphology | Key Characteristics |
|---|---|---|
| Surface Faceting | Heavily facetted structure | Initial state of the metallic surface prior to transformation [30]. |
| Surface Flattening | Microscopically smooth state | Transition induced by propagating waves; precursor to oxidation [30]. |
| Oxide Growth | Formation and expansion of copper oxide islands | Autocatalytic growth with anisotropic propagation speeds of several hundred nm/s [30]. |
| Oxide Reduction | Disappearance of oxide islands, re-faceting | Preceded by a morphological change to a lamellar structure; occurs after a 2-3 minute induction period [30]. |
This work demonstrated that catalytic activity emerges from these spatio-temporal dynamics and phase coexistence, challenging the static picture of active sites and providing a mechanism for their constant regeneration [30].
Experimental Protocols and Workflows
A core strength of in situ SEM is the development of robust protocols that stabilize labile samples and enable reproducible observation under environmental conditions.
The Extended Low Temperature Method (ELTM) for Sample Stabilization
The Extended Low Temperature Method (ELTM) is a universal and inexpensive preparation technique for plant samples, which are analogous to delicate organic or inorganic crystalline structures susceptible to preparation artefacts [29]. The protocol is performed in situ within an ESEM equipped with a cooling Peltier stage and does not require chemical fixation, thus preserving native state morphology [29].
Table 2: ELTM Protocol Phases and Parameters
| Protocol Phase | Key Operational Steps | Critical Parameters & Objectives |
|---|---|---|
| Phase 1: Low Temperature Stabilisation | Simultaneous sample cooling and chamber pumping [29]. | Cool to -20°C while reducing chamber pressure to 200 Pa (water vapour). Rate of T/P change is sample-specific. Goal: Sublimate surface water while retaining internal water [29]. |
| Phase 2: Sample Drying & Transfer | After observation, slowly decrease pressure to minimum ESEM mode (~10 Pa), then gradually increase temperature to +20°C [29]. | Prevents sample collapse and condensation during transfer to atmospheric pressure. Results in a dry, stable sample for high-resolution SEM or storage [29]. |
This method enables repetitive observation of the same sample in its fully hydrated state in ESEM, in a dried state in low vacuum ESEM, and, after conductive coating, in high vacuum SEM, maximizing the information obtained from a single specimen [29].
Workflow for In Situ Crystal Growth Observation
The following diagram illustrates a generalized experimental workflow for conducting in situ crystal growth or redox dynamics studies, integrating components like the μReactor.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of in situ SEM experiments relies on a suite of specialized reagents and instrumentation.
Table 3: Essential Research Reagents and Materials for In Situ SEM
| Item Name | Function / Application | Specific Examples / Notes |
|---|---|---|
| Microelectromechanical System (MEMS) Heater / μReactor | A specialized sample holder enabling precise heating and gas flow within the SEM chamber for simulating reaction conditions [31]. | Allows high-temperature reduction under hydrogen gas; can be transferred between SEM and TEM [31]. |
| Precursor Materials | Source materials for the growth of inorganic crystals or nanostructures. | γ-WO₃/a-SiO₂ nanofibers (for W₁₈O₄₉ nanowhiskers) [31]; metallic copper foils (for redox studies) [30]. |
| Reaction Gases | Create the controlled atmosphere for chemical reactions inside the microscope. | Hydrogen (H₂) for reduction processes [31]; Oxygen (O₂) for oxidation studies [30]. |
| ESEM with Peltier Stage | Microscope capable of low-pressure gas environments and sample cooling for the ELTM protocol [29]. | Enables observation of hydrated or sensitive samples without dehydration artefacts [29]. |
| Correlative Analytical Detectors | Provide chemical and structural information complementary to electron imaging. | Energy Dispersive X-ray Spectroscopy (EDS) for elemental analysis [29]; Mass Spectrometry for gas-phase analysis [30]. |
Quantitative Data from Key In Situ Studies
The following table consolidates critical quantitative data from representative studies, providing a reference for experimental parameters and outcomes.
Table 4: Summary of Quantitative Data from In Situ SEM Studies
| Study Focus | Environmental Conditions | Key Quantitative Observations | Instrumentation Used |
|---|---|---|---|
| W₁₈O₄₉ Nanowhisker Growth [31] | H₂ atmosphere at 100 Pa, high temperature. | Electron beam irradiation was found to slow growth kinetics markedly. | In situ SEM with μReactor; subsequent TEM/STEM analysis. |
| Copper Redox Dynamics [30] | 4% O₂ in H₂ at 20-50 Pa, 700°C. | Oscillatory cycles of ~30 minutes; wave propagation at ~hundreds of nm/s; oxide reduction after 2-3 minute induction. | ESEM; on-line mass spectroscopy; NAP-XPS; ESPEM. |
| Plant Sample Stabilization (ELTM) [29] | Water vapour pressure of 200 Pa, cooling to -20°C. | Method enables repetitive imaging in SEM/ESEM and is suitable for energy-dispersive X-ray microanalysis. | Commercial ESEM with cooling Peltier stage. |
Inorganic crystal nucleation research demands precise observational tools that can operate within native environments without disturbing delicate crystallization processes. In situ optical microscopy has emerged as a powerful solution, enabling real-time, contact-free monitoring of dynamic crystallization events directly within reaction suspensions. This approach eliminates statistical uncertainties inherent in conventional ex situ methods that rely on extracted and prepared samples [32]. When combined with the quantitative capabilities of polarized light microscopy (PLM) and modern computational analysis, researchers gain a formidable toolkit for unraveling complex crystallization mechanisms. These advanced optical techniques provide unprecedented access to crystallization kinetics, polymorph differentiation, and morphological evolution, offering critical insights for pharmaceutical development, materials science, and industrial process optimization. This application note details practical methodologies and experimental protocols for implementing these technologies in inorganic crystal nucleation research, providing scientists with robust frameworks for advancing their investigative capabilities.
Optical Fundamentals and Technical Specifications
Core Principles of In Situ Light Microscopy
In situ light microscopy represents a paradigm shift from conventional microscopy by enabling direct observation of crystallization processes within their native environment. Unlike conventional microscopy performed on static, prepared samples in counting chambers, in situ microscopy couples directly to the suspension via a transparent window, providing a continuous stream of image data from within the reacting suspension [32]. This approach captures particles as they flow naturally through the observation zone, accumulating significant statistical data on concentration and morphological features without sampling artifacts or preparation-induced distortions. The method functions as an imaging flow cytometer integrated directly into the process stream, enabling researchers to monitor crystallization phenomena as they unfold in real-time.
The technical implementation requires specialized optical configurations to maintain submicrometer resolution while operating in challenging suspension environments. A key advancement is the development of adjustment-free immersion objectives where the front surface is directly immersed in the suspension, eliminating the need for precise cover slip positioning and associated mechanical adjustments [32]. This innovation significantly enhances operational reliability in industrial settings where maintenance access may be limited, making the technology particularly valuable for long-term crystallization studies in bioreactors and continuous manufacturing systems.
Polarized Light Microscopy for Crystalline Analysis
Polarized Light Microscopy (PLM) leverages the optical anisotropy of crystalline materials to extract detailed structural and chemical information. A standard compound light microscope equipped with polarizing filters, a compensator, Bertrand lens, and rotating stage transforms into a powerful analytical PLM instrument [33]. When crystals interact with polarized light, they produce distinctive birefringence patterns that reveal information about their crystal system, symmetry, and molecular orientation.
The analytical power of PLM stems from measurements of refractive index, which depends directly on electron density and polarizability within the crystalline lattice [33]. This enables researchers to monitor subtle changes in chemistry and structure as materials undergo transformations during crystallization, ligand binding, or catalytic processes. PLM provides a visual map showing where these changes occur within crystal structures, revealing spatial heterogeneities and domain-specific behaviors that ensemble techniques might average out.
Resolution and Performance Considerations
The performance boundaries of optical microscopy are defined by fundamental physics. For conventional light microscopy, the resolution limit (d) follows the Abbe criterion: d = 0.61 × wavelength / numerical aperture (NA) [34]. With violet light (400 nm) and a high-quality objective (NA=1.4), this translates to approximately 175 nm resolution. In situ microscopy implementations achieve submicrometer resolution through high numerical aperture designs (NA=0.75) optimized for specific immersion media [32].
Table 1: Technical Specifications of Optical Techniques for Crystallization Research
| Technique | Spatial Resolution | Temporal Resolution | Key Measurable Parameters | Sample Requirements |
|---|---|---|---|---|
| In Situ Light Microscopy | ~200 nm (lateral) [32] | Real-time, continuous monitoring [32] | Particle concentration, morphological features, size distribution [32] | Flowing suspensions, minimal transparency required |
| Polarized Light Microscopy (PLM) | ~200 nm (lateral) [34] | Seconds to minutes for static analysis [33] | Crystal system, polymorphism, birefringence, optical activity [33] | Solid crystals on glass slide, minimal sample preparation |
| Interferometric Scattering (iSCAT) | 200-300 nm (lateral), 10-100 nm (axial) [35] | Up to 1 μs temporal resolution [35] | Single-particle growth kinetics, morphological evolution [35] | Samples attached to coverslip, label-free |
Advanced implementations like interferometric scattering (iSCAT) microscopy push these boundaries further, achieving 200-300 nm lateral resolution and 10-100 nm axial resolution while maintaining microsecond temporal resolution for monitoring rapid crystallization events [35]. This exceptional performance enables researchers to track individual crystal growth with single-particle precision, capturing heterogeneous behaviors that ensemble methods inevitably miss.
Experimental Protocols
Protocol for Adjustment-Free In Situ Microscopy of Crystallizing Suspensions
Purpose: To monitor inorganic crystal nucleation and growth in situ within flowing suspensions without mechanical adjustments or sample extraction.
Materials:
- Adjustment-free in situ microscope with water immersion objective (NA=0.75, 50× magnification) [32]
- Transparent reaction vessel with flow cell configuration
- Suspension of crystallizing inorganic compounds
- Monochromatic illumination source (660±10 nm)
- High-speed camera system
- Data acquisition computer with image analysis software
Procedure:
- Microscope Configuration: Employ a specially designed immersion objective whose front surface will be directly immersed in the crystallizing suspension. Verify the objective is corrected for applications without a cover slip [32].
- Flow Cell Setup: Couple the microscope objective directly to the transparent window of the flow cell containing your crystallizing inorganic suspension. Ensure the object plane is positioned approximately 15 μm from the objective's front surface to minimize scattering effects while allowing individual crystals to fully enter the observation zone [32].
- Optical Alignment: Adjust only the distance between the objective and camera sensor (image plane). Utilize the geometrical imaging with a magnification factor of 50×, where millimeter precision at the image plane ensures micrometer positioning of the conjugated object plane within the suspension [32].
- Image Acquisition: Initiate continuous image capture as new crystals are transported through the observation zone at a rate proportional to the flow speed and crystal concentration. Maintain consistent illumination intensity throughout the experiment.
- Data Processing: Analyze captured images to extract crystal count, size distribution, and morphological features. Utilize the continuous stream of image data to calculate nucleation rates and growth velocities directly from the native crystallization environment.
Technical Notes: The adjustment-free system eliminates the need for precise distance optimization between objective and optical window, significantly simplifying operation during long-term crystallization experiments. The curved object plane inherent in this optical design is acceptable for crystallization studies as the high numerical aperture defines a small volume within the suspension that is sharply imaged to the flat sensor according to the narrow depth of field [32].
Protocol for Polarized Light Microscopy Analysis of Crystal Polymorphs
Purpose: To identify and characterize polymorphic forms in inorganic crystals through birefringence patterns and optical properties.
Materials:
- Polarized light microscope with two polarizing filters, rotating stage, compensator, and Bertrand lens [33]
- Hot stage for temperature-controlled experiments (range: -196°C to 1500°C) [33]
- Glass slides and coverslips
- Inorganic crystal samples
- Immersion oils with calibrated refractive indices
Procedure:
- Microscope Setup: Orient polarizers to crossed position (90° separation) to produce a dark background. Ensure the light path includes the compensator for retardation measurements.
- Sample Preparation: Disperse inorganic crystals on a clean glass slide. For solution-based crystallization, place a droplet of crystallizing suspension and carefully lower a coverslip to minimize air bubbles.
- Initial Observation: Observe samples under low magnification (10×) to identify areas of interest with well-distributed crystals.
- Crystallographic Analysis:
- Rotate the stage through 360° while observing birefringence intensity changes.
- Note extinction positions (where crystals appear darkest) and maximum birefringence positions.
- Record interference color patterns using the Michel-Lévy chart for retardation estimates.
- Refractive Index Measurement: Employ immersion methods with calibrated oils to determine principal refractive indices. Compare crystal visibility in oils of known refractive indices to estimate crystal refractive indices.
- Temperature-Dependent Studies: For polymorphic transformation studies, program the hot stage to ramp temperature at controlled rates (e.g., 1-10°C/min) while recording birefringence changes that indicate phase transitions [33].
Technical Notes: The anisotropy of optical properties measured by PLM provides insight into the distribution of electron density in different crystallographic planes within the material. This can be correlated to anisotropy of other properties like thermal expansion, hardness, and electrical conductivity [33]. For inorganic crystals, careful attention to dispersion staining colors can rapidly differentiate polymorphic forms with similar morphology but different crystalline structures.
Protocol for Real-Time Crystallization Monitoring Using iSCAT Microscopy
Purpose: To monitor crystal nucleation and growth with high spatiotemporal resolution using label-free interferometric scattering microscopy.
Materials:
- iSCAT microscope setup with laser illumination (637 nm), high-numerical-aperture objective, and camera with microsecond exposure capability [35]
- Glass coverslips
- Sample chamber with fluidic injection system
- Inorganic crystal precursors in solution
Procedure:
- Sample Chamber Preparation: Clean glass coverslips thoroughly. Assemble sample chamber with silicone spacer to create defined reaction volume.
- Microscope Alignment: Align the iSCAT microscope to optimize interference between light scattered from growing crystals and reference light reflected from the glass-solution interface [35].
- Baseline Imaging: Acquire reference images of the field of view before introducing crystal precursors to establish background interference patterns.
- Reaction Initiation: Introduce crystal precursor solution into the sample chamber while continuing image acquisition.
- Real-Time Monitoring: Capture image sequences with appropriate temporal resolution (milliseconds to seconds depending on crystal growth rate). iSCAT signals will appear as Airy discs of concentric dark and light rings caused by interference between reflected and scattered signals [35].
- Image Analysis: Segment individual crystals and track their size evolution over time. Apply kinetic models to extract growth rate constants from trajectory data.
Technical Notes: iSCAT microscopy provides exceptional temporal resolution (up to 1 μs) and can monitor processes over long observation times without photobleaching concerns, as it is a label-free technique [35]. The common-path configuration of iSCAT makes it robust against environmental vibrations compared to conventional interferometry, allowing deployment in less controlled environments like industrial crystallization plants.
Data Analysis and Computational Integration
Deep Learning Analysis of Crystallization Textures
Modern crystallization research increasingly integrates computational methods with optical techniques to extract quantitative information from complex image data. Deep learning approaches, particularly U-Net convolutional neural networks, have demonstrated remarkable efficacy in automating the analysis of crystallization textures captured via polarized light microscopy [36].
The U-Net architecture employs a symmetric encoder-decoder structure with skip connections that preserve spatial information throughout the network. This design enables precise semantic segmentation of microscopy images, automatically identifying and classifying crystalline phases within complex mixtures [36]. Implementation typically involves:
- Data Preparation: Collect paired input images and corresponding segmentation masks where each pixel is labeled according to its crystalline phase.
- Model Training: Train the U-Net model for 15-50 epochs using Adam optimizer (learning rate = 0.0001) with binary cross-entropy as the loss function.
- Probability Mapping: Generate probability maps indicating the likelihood of each pixel belonging to specific crystalline phases.
- Quantification: Binarize probability maps to quantify the degree of crystallization over temperature or time using the equation: D(T) = SCr(T)/S, where SCr is the area of crystal phase and S is the total surface area [36].
This automated approach enables researchers to process large datasets generated during in situ crystallization studies, extracting robust kinetic parameters such as nucleation rates and growth velocities that are essential for modeling and optimizing crystallization processes.
Crystallization Kinetics Analysis
The quantitative data extracted from optical microscopy can be further analyzed using kinetic models to understand fundamental crystallization mechanisms. For 1D crystal growth observed via iSCAT microscopy, length evolution trajectories of individual crystals can be fitted to established kinetic models to extract rate constants [35]. Similarly, for polymorphic transformations monitored through PLM, the temperature-dependent phase evolution can be analyzed using sigmoidal fitting, with the inflection point identifying the temperature of maximum crystallization rate [36].
Advanced analysis may include fitting data to the Ozawa model or other crystallization kinetics models that account for nucleation and growth mechanisms. The combination of high-quality optical data with appropriate kinetic modeling transforms qualitative observations into quantitative parameters that can guide process development and fundamental understanding of crystallization behavior.
Essential Research Reagent Solutions
Table 2: Key Research Reagents and Materials for Crystallization Microscopy
| Reagent/Material | Function/Application | Technical Specifications | Experimental Considerations |
|---|---|---|---|
| Water Immersion Objectives | High-resolution imaging in aqueous crystallization environments [32] | NA=0.75, 50× magnification, working distance 10-20 μm [32] | Requires correction for no cover slip; sealed for pressure resistance |
| Polarizing Filters | Creating polarized light for birefringence analysis [33] | High extinction ratio (>10,000:1), minimal strain | Should be precisely aligned at 90° for crossed polarization |
| Refractive Index Oils | Determining crystal refractive indices [33] | Calibrated RI values at specific wavelengths and temperatures | Must match temperature during measurement due to RI temperature dependence |
| Hot Stage System | Temperature-controlled crystallization studies [33] | Range: -196°C to 1500°C, precision ±0.1°C [33] | Programmable temperature ramps essential for polymorphism studies |
| Block Copolymer Unimers | Model systems for crystallization kinetics studies [35] | PCL-based polymers (e.g., PCL45-b-PDMA348) [35] | Concentration-dependent kinetics; biocompatible options available |
| iSCAT Coverslips | Substrate for interferometric scattering microscopy [35] | High-quality glass with precise thickness specifications | Critical for interference quality; requires meticulous cleaning |
Workflow and System Integration
Optical Crystallization Analysis Workflow
The integrated workflow for optical crystallization analysis begins with precise formulation of the research question, which determines appropriate sample preparation and microscopy selection. Each optical technique provides complementary information: PLM reveals crystalline structure and polymorphism, in situ microscopy captures morphological evolution in native environments, and iSCAT microscopy delivers high-resolution single-particle kinetics [32] [33] [35]. The computational integration through deep learning transforms raw image data into quantitative parameters that feed kinetic models, creating an iterative cycle of hypothesis generation and testing that continuously advances understanding of crystallization mechanisms.
Technical Implementation Diagrams
In Situ Microscope Optical Configuration
The optical configuration for adjustment-free in situ microscopy employs a specialized objective design that eliminates mechanical adjustments through direct immersion in the crystallizing suspension [32]. The optical path incorporates a solid immersion lens combined with an aspheric lens to achieve high numerical aperture (NA=0.75) while maintaining a simple, robust design. Critical technical features include the acceptance of a curved object plane (radius ≈1.4 mm) and the elimination of separate cover slip adjustments, significantly enhancing operational reliability in industrial crystallization environments [32]. This configuration enables real-time monitoring of crystal nucleation and growth directly within flowing suspensions, providing continuous data streams for statistical analysis of crystallization phenomena.
In the field of inorganic crystal nucleation research, understanding the initial stages of formation and growth within a native, hydrated environment is paramount. Traditional single-modality microscopy techniques provide limited insights; differential interference contrast (DIC) microscopy visualizes unstained, living cells and crystals based on density gradients, fluorescence microscopy pinpoints specific molecular components and dynamic processes, and electron microscopy (EM) offers nanoscale structural resolution. Correlative microscopy integrates these techniques into a unified workflow, providing a comprehensive view from dynamic cellular context to atomic-level detail. This protocol details advanced approaches for combining DIC, fluorescence, and electron microscopy, with specific application to studying in situ biomineralization and inorganic crystal nucleation in biological environments, enabling researchers to link dynamic cellular functions with ultrastructural analysis in a near-native state.
Key Research Reagent Solutions
The following reagents are essential for successful implementation of correlative microscopy workflows in crystal nucleation research.
Table 1: Essential Research Reagents for Correlative Microscopy
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| Fluorescent Probes | Hoechst, Mitotracker Red, GFP-fusion proteins (e.g., SP7-nls-GFP) [37], Fluorescein-labeled nanoparticles [38] | Labeling specific cellular compartments (nuclei, mitochondria), tracking osteoblast activity [37], and monitoring nanoparticle uptake and localization [38]. |
| Fiducial Markers | Quantum Dots (QDs) [39], Gold Nanoparticles [39] | Provide visible landmarks in both fluorescence and electron microscopy images, enabling precise image correlation and registration between different modalities [39]. |
| Vitrification Media | 10% Dextran solution [37], Phosphate Buffered Saline (PBS) | Used as a filler medium during high-pressure freezing to support samples and promote the formation of vitreous ice, preserving native hydration and structure [37]. |
| Sample Support | Indexed Gold EM Grids [40], Silicon Microchips with Silicon Nitride Windows [39], FinderTOP HPF Carriers [37] | Provide a stable, electron-transparent substrate for cells and tissues. FinderTOP carriers imprint a navigational pattern onto the sample surface for precise relocation [37]. |
Experimental Protocols
This section provides detailed methodologies for key correlative workflows applicable to crystal nucleation studies.
Protocol: Cryo-Correlative Light and Electron Microscopy (Cryo-CLEM) for Tissue Mineralization
This protocol is adapted for studying mineralization processes in tissues, such as zebrafish scales, using high-pressure freezing and patterned carriers for precise targeting [37].
Table 2: Key Steps in Cryo-CLEM for Tissues
| Step | Procedure | Purpose & Notes |
|---|---|---|
| 1. Sample Preparation & Labeling | 1. Harvest and stain tissue (e.g., zebrafish scales).2. Transfer to a FinderTOP high-pressure freezing (HPF) carrier filled with 10% dextran.3. Vitrify using HPF. | Fluorescent stains label nuclei and mitochondria. Dextran fills volume. HPF preserves native state. The FinderTOP imprints a grid for navigation [37]. |
| 2. Cryo-Fluorescence Microscopy | 1. Mount sample in universal cryo-holder.2. Acquire overview images (reflection & fluorescence).3. Record high-resolution 3D Airyscan confocal images of the ROI. | Locate Region of Interest (ROI). Use grid pattern and fluorescence for coarse-to-fine correlation. Determine ROI depth relative to surface [37]. |
| 3. Image Correlation & Registration | 1. Use correlation software to overlay fluorescence and reflection images.2. Computationally align imaging modalities using the FinderTOP pattern and fluorescent features. | Precisely map the fluorescent ROI onto the sample surface for targeting in the FIB/SEM [37]. |
| 4. 3D Cryo-FIB/SEM Volume Imaging | 1. Transfer sample to FIB/SEM.2. Use correlated data to locate ROI.3. Perform sequential FIB milling and SEM imaging to generate a 3D volume stack. | Reveals the nanoscale structural context of the fluorescently labeled ROI in its native, vitrified state [37]. |
Protocol: Liquid-Phase STEM Correlative Microscopy for Nanoparticle-Cell Interactions
This protocol enables the study of nanoparticle interactions with living cells in liquid, combining fluorescence microscopy and scanning transmission electron microscopy (STEM) without the need for thin sections [39].
- Cell Culture and Labeling: Grow cells (e.g., COS7 fibroblasts) directly on silicon microchips featuring an electron-transparent silicon nitride window. Incubate cells with functionalized nanoparticles, such as Quantum Dots (QDs) bound to Epidermal Growth Factor (EGF) [39].
- Live-Cell Fluorescence & DIC Imaging: Fix cells lightly and image them on the microchip. Acquire both DIC and fluorescence images. The DIC signal visualizes the cellular material, while fluorescence identifies the location of the QD-labeled nanoparticles. Use the window frame as a navigational landmark [39].
- Microfluidic Chamber Assembly: Assemble the imaged microchip into a microfluidic chamber, using spacer microspheres to create a liquid-filled cavity of defined thickness [39].
- Liquid STEM Imaging: Transfer the assembled chamber to the STEM. Image the same cellular regions previously identified with fluorescence. Locate the window and specific cells using low-magnification overviews. In ADF-STEM mode, QDs appear as bright spots against the darker cellular background and liquid layer, allowing their distribution to be resolved at nanometer resolution [39].
- Image Correlation: Correlate the fluorescence and STEM images by aligning the common features (cell edges, window frame) visible in both modalities. This confirms the co-localization of fluorescent signals and electron-dense nanoparticles [39].
Protocol: In Situ pH Titration for Mapping Supramolecular Polymerization
This combinatorial methodology precisely probes the self-assembly of molecules (e.g., peptide amphiphiles) in aqueous environments under thermodynamic equilibrium, which is relevant for understanding bio-inspired mineralization scaffolds [41].
- Initial Depolymerization: Prepare an aqueous stock solution of cationic peptide amphiphiles (PAs). Add hydrochloric acid (HCl) to fully protonate the molecules, maximizing electrostatic repulsion and breaking pre-formed polymers into micellar states. Confirm the random-coil conformation of micelles using Circular Dichroism (CD) spectroscopy [41].
- Controlled Titration and In Situ Monitoring: Use a syringe pump to titrate a sodium hydroxide (NaOH) solution into the PA micellar solution at a precisely controlled, slow flow rate (e.g., 0.5 mL/hour). Monitor the evolution of secondary structures in situ using CD spectroscopy, tracking the shift from random coils to β-sheets [41].
- Ex Situ Validation: At key points in the titration process (e.g., the first and second CD plateau), extract aliquots for ex situ characterization. Use Transmission Electron Microscopy (TEM) and Small-Angle X-Ray Scattering (SAXS) to confirm the morphological transition from spheroidal micelles to filamentous polymers or nanoribbons [41].
- Data Analysis: Normalize the CD titration curves to calculate the extent of polymerization. Plot this as a function of the [NaOH]/[PA] ratio and [PA] to construct a detailed assembly landscape, revealing how conditions influence the equilibrium and the resulting supramolecular architecture [41].
Data Presentation and Analysis
Quantitative data from correlative studies provides critical insights into the relationships between molecular interactions, assembly conditions, and final nanostructures.
Table 3: Correlative Metrics for Assembly and Imaging
| Technique / System | Quantifiable Parameter | Typical Value / Observation | Biological/Material Significance |
|---|---|---|---|
| Liquid STEM [39] | Spatial Resolution on QDs | 3 nm | Enables precise localization of individual protein labels (e.g., EGF receptors) in liquid. |
| Liquid STEM [39] | Liquid Layer Thickness | 6 ± 1 μm | Maintains hydrated state of whole eukaryotic cells during EM imaging. |
| PA Supramolecular Polymerization [41] | Critical Titration Ratio ([NaOH]/[PA]) | Molecule-dependent (e.g., C16V3A3K3) | Indicates the precise condition for micelle-to-polymer transition under equilibrium. |
| Cryo-CLEM on Tissues [37] | FinderTOP Pattern Dimensions | 200 x 200 μm grid, 4 μm high lines | Provides navigational landmarks on otherwise featureless vitrified ice surfaces. |
| In Situ AFM on Mineralization [42] | Nucleation Rate on Collagen | Hours (vs. days in solution) | Quantifies the potent nucleating effect of organic matrices on crystal formation. |
In Situ Microscopy Technologies for Crystal Monitoring
In situ microscopy enables direct, real-time observation of crystallization processes within reactors, providing invaluable insights into nucleation, crystal growth, and morphology without the need for manual sampling. This non-invasive analytical approach aligns with the goals of the Process Analytical Technology (PAT) initiative, enhancing process control and efficiency in both materials science and pharmaceutical development [43]. The following table summarizes the key implementation and outputs of this technology.
Table 1: In Situ Microscopy Implementations for Crystallization Monitoring
| Application Area | Specific Technology | Key Measured Parameters | Reported Benefits and Outputs |
|---|---|---|---|
| Technical Protein Crystallization | In situ microscope with machine learning-based image analysis [44] | Real-time crystal volume, yield, crystallization kinetics, Crystal Size Distribution (CSD), crystal count | Correlates well with offline protein concentration; superior to offline data when amorphous precipitation occurs [44]. |
| MOF Crystallization | Automated optical microscopy with computer vision (Bok Choy Framework) [45] | Crystal morphology, aspect ratio, crystal area, crystallization outcome classification | Analysis efficiency improved by ~35x compared to manual methods; enables rapid screening of synthesis parameters [45]. |
| General Crystallization (e.g., ASS, HEWL) | In-situ microscope (ISM) type III-XTF [43] | Crystal size distribution (CSD), crystal morphology | Allows direct access to important crystallization parameters for process control; distinguishes various crystal forms [43]. |
Advanced image analysis, particularly machine learning (ML), has significantly augmented the power of in situ microscopy. For example, convolutional neural networks (CNNs) can automatically detect protein crystals in microscopic images and identify unwanted crystals in slurry samples, facilitating immediate corrective actions [44]. These technologies provide a scalable foundation for data-driven discovery in catalysis, energy storage, and pharmaceuticals [45].
Detailed Experimental Protocols
Protocol 1: Automated Synthesis and Morphological Screening of Co-MOF-74
This protocol details a high-throughput workflow for synthesizing and characterizing cobalt-based MOF-74, leveraging automation and computer vision to efficiently map synthesis parameters to crystal morphology [45].
Key Research Reagent Solutions:
- Metal Precursor: Cobalt-based salt (e.g., Co(NO₃)₂ or CoCl₂).
- Organic Linker: 2,5-dioxido-1,4-benzenedicarboxylic acid (H₄dobdc).
- Solvent Systems: Dimethylformamide (DMF), ethanol, and deionized water.
- Equipment: Opentrons OT-2 liquid handling robot, high-resolution optical microscope (e.g., EVOS imaging system) with an automated XY stage.
Procedure:
- Robotic Precursor Formulation: Program the liquid-handling robot to automate the pipetting and dispensing of MOF precursor solutions into a multi-well reaction plate (e.g., 96-well plate). This ensures consistent reagent volumes and concentrations, minimizing human error.
- Solvothermal Synthesis: Subject the reaction plates to solvothermal conditions. Systematically vary key parameters such as solvent composition (ratios of DMF/water/ethanol), reaction time, temperature, and precursor stoichiometry to explore the synthesis space.
- High-Throughput Imaging: After synthesis, use an automated optical microscope to rapidly capture images of the solid products from each well. The automated XY stage allows for high-throughput imaging without manual repositioning.
- Computer Vision Analysis: Process the acquired images using the "Bok Choy Framework" or a similar computer vision algorithm. The software automatically:
- Detects and distinguishes between isolated crystals and crystal clusters.
- Extracts quantitative morphological features such as crystal area, perimeter, and aspect ratio.
- Classifies the overall crystallization outcome (e.g., success, failure, amorphous).
- Data Correlation and Analysis: Correlate the extracted morphological data with the corresponding synthesis parameters from each well. This structured dataset enables the identification of conditions that promote desired crystal morphologies and inhibits unwanted growth.
This integrated workflow reduces hands-on labor by approximately one hour per synthesis cycle and improves image analysis efficiency by about 35 times compared to manual methods [45].
Protocol 2: Real-Time Monitoring of Technical Protein Crystallization
This protocol describes the use of in situ microscopy to monitor the batch crystallization kinetics of proteins, such as alcohol dehydrogenase (LbADH) and its mutants, in a stirred-tank crystallizer [44].
Key Research Reagent Solutions:
- Protein Solution: Purified protein (e.g., LbADH wild type or crystal contact mutants like T102E, Q207D).
- Precipitant: Polyethyleneglycol 550 monomethyl ether (PEG 550 MME).
- Buffer: Phosphate buffered saline (PBS) or other suitable buffer.
- Equipment: 1 L stirred-tank crystallizer, in situ microscopy probe (e.g., Mettler Toledo, Sartorius Stedim Biotech), image analysis software with ML capabilities.
Procedure:
- Reactor Setup and Calibration: Install the in situ microscopy probe directly into a standard port of the 1 L stirred crystallizer. Ensure the probe's focal plane is positioned within the main crystallization broth. Calibrate the system according to the manufacturer's instructions.
- Process Initiation: Charge the crystallizer with the protein solution in buffer. Initiate agitation to ensure homogeneous mixing. Begin data acquisition with the in situ microscope to establish a baseline.
- Induction of Crystallization: Slowly add the precipitant solution (e.g., PEG) to the reactor to induce supersaturation. Continue real-time image acquisition throughout this process and for the duration of the crystallization.
- Real-Time Image Analysis: The acquired images are automatically analyzed by machine learning-based software. The software:
- Identifies and counts nascent crystals.
- Tracks the growth of individual crystals over time.
- Calculates real-time estimates of total crystal volume and other kinetic parameters.
- Offline Validation and Correlation: Periodically, take offline samples to measure protein concentration in the solution (e.g., via UV-Vis spectroscopy). Correlate the offline protein concentration data with the online estimated crystal volume from image analysis. This validation confirms the reliability of the in situ monitoring technique, which is particularly advantageous when amorphous precipitation complicates other analytical methods [44].
Protocol 3: In Situ Liquid-Phase TEM of MOF Encapsulation
This advanced protocol utilizes ultra-low electron flux Liquid-Phase Transmission Electron Microscopy (LP-TEM) to visualize the dynamic encapsulation process of nanoparticles (e.g., gold) by MOFs (e.g., ZIF-8) at near-atomic resolution [46].
Key Research Reagent Solutions:
- Metal Nanoparticles (NPs): Colloidal gold nanoparticles (Au NPs).
- MOF Precursors: Zinc nitrate (Zn(NO₃)₂) and 2-Methylimidazole (2-MeIm).
- Surfactant: Cetyltrimethylammonium chloride (CTAC) to promote defined crystalline shapes.
- Equipment: Liquid-Phase TEM holder and chips, Transmission Electron Microscope capable of low-dose imaging.
Procedure:
- Precursor Solution Preparation: Prepare an aqueous solution containing the MOF precursors (e.g., 0.4 M 2-MeIm and 6 mM Zn(NO₃)₂), surfactant (CTAC), and the Au NPs.
- Liquid Cell Loading: Load a small volume of the precursor solution into the liquid cell assembly, typically sandwiched between two silicon nitride windows.
- In Situ TEM Imaging with Ultra-Low Electron Flux: Insert the liquid cell into the TEM. Use an ultra-low electron flux (e.g., ~0.5 e⁻ nm⁻² s⁻¹) to initiate and image the reaction. This low dose is critical to minimize electron beam damage to the beam-sensitive MOF structures.
- Real-Time Visualization and Analysis: Record image sequences or videos to track the nucleation and growth of ZIF-8 on the surface of individual Au NPs in real-time. Key observations include:
- The formation of a low-contrast layer around the NP indicating initial shell growth.
- The evolution of this layer into a mature, cuboidal ZIF-8 shell.
- The impact of precursor concentration on growth kinetics and shell morphology (e.g., single-crystalline vs. polycrystalline shells).
- Post-Reaction Analysis: After the in situ experiment, disassemble the liquid cell and perform post-mortem analysis on the synthesized structures using techniques like Scanning TEM (STEM) and Energy-Dispersive X-ray Spectroscopy (EDX) to confirm composition and structure [46].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of in situ crystallization monitoring relies on a suite of specialized reagents and equipment. The following table catalogs key solutions and their functions.
Table 2: Essential Research Reagent Solutions for In Situ Crystallization Studies
| Reagent/Material | Function in Experiment | Application Context |
|---|---|---|
| Polyethyleneglycol (PEG) | Acts as a precipitating agent to induce protein crystallization by excluding volume and promoting protein-protein interactions. | Technical protein crystallization [44]. |
| Dimethylformamide (DMF) | Common solvent for solvothermal synthesis of MOFs, dissolving organic linkers and metal salts. | MOF synthesis and crystallization [45]. |
| 2-Methylimidazole | Organic linker molecule used for constructing Zeolitic Imidazolate Frameworks (ZIFs) like ZIF-8. | MOF encapsulation studies [46]. |
| Cetyltrimethylammonium chloride (CTAC) | Surfactant used to control MOF crystal morphology, promoting the formation of well-defined, cuboidal crystals for clearer imaging. | In situ TEM of MOF formation [46]. |
| Liquid Handling Robot (e.g., Opentrons OT-2) | Automates precise dispensing of precursor solutions, ensuring reproducibility and enabling high-throughput experimentation. | Automated MOF synthesis [45]. |
| In Situ Microscope (ISM) Probe | Optical probe inserted directly into bioreactors/crystallizers for real-time, non-invasive image acquisition of the crystallization process. | Protein and MOF crystallization monitoring [44] [43]. |
| Liquid-Phase TEM Holder | Specialized TEM holder that encapsulates liquid samples, allowing for real-time observation of dynamic processes in a liquid environment. | Visualizing NP encapsulation by MOFs [46]. |
Workflow and Signaling Pathway Diagrams
Automated Crystallization Development Workflow
This diagram illustrates the integrated, closed-loop workflow for accelerated crystallization development, combining automated synthesis, high-throughput characterization, and data-driven analysis [45].
MOF Encapsulation Pathway and Morphology Control
This diagram details the mechanistic pathway for encapsulating a nanoparticle within a MOF, highlighting how precursor concentration critically determines whether a uniform single-crystalline shell or an irregular polycrystalline shell forms [46].
Overcoming Experimental Hurdles: Optimization and Troubleshooting in Live Imaging
Mitigating Electron Beam Effects on Sensitive Nucleation Processes
In situ electron microscopy has revolutionized the study of crystal nucleation and growth, enabling direct observation of these dynamic processes at unprecedented spatial and temporal resolution. However, the high-energy electron beam necessary for imaging can significantly alter the very phenomena researchers seek to observe. For researchers investigating inorganic crystal nucleation, particularly in sensitive organic and pharmaceutical systems, distinguishing beam-induced artifacts from genuine nucleation pathways presents a major challenge. This application note provides a structured framework for identifying, quantifying, and mitigating electron beam effects on sensitive nucleation processes, with specific protocols developed within the context of advanced materials research.
The fundamental issue stems from the interaction between incident electrons and the sample environment. Ionizing radiation can radiolyze solvents, generate reactive species, cause localized heating, and directly damage developing crystal structures. These effects can artificially accelerate or decelerate nucleation rates, modify polymorph selection, and even create nucleation pathways that would not occur under standard conditions. Understanding and controlling these parameters is therefore not merely a technical consideration but a fundamental prerequisite for obtaining biologically and physically relevant data from in situ experiments.
Quantitative Analysis of Electron Beam Effects
A critical first step in mitigation is understanding the measurable impacts of electron irradiation on materials. Systematic studies across different material classes provide a baseline for anticipating potential beam-specimen interactions.
Table 1: Quantitative Effects of Electron Beam Irradiation on Organic Crystalline Materials
| Material | Irradiation Dose | Observed Physical Change | Measurement Technique | Significance for Nucleation Studies |
|---|---|---|---|---|
| Flufenamic Acid (FFA) [47] | >150 e⁻/Ų/s | Induction of nucleation in undersaturated solutions | Liquid Phase EM | Beam can trigger nucleation, confounding native pathway analysis. |
| Diclofenac (DC) [48] | 25 kGy (standard sterilization) | No significant chemical changes | HPLC, FT-IR | Below-threshold dose for this API; establishes upper stability limit. |
| Diclofenac (DC) [48] | 400 kGy (high dose) | Melting point decreased by 5.0°C (291.0°C to 286.0°C) | Capillary Melting Point | High doses alter thermodynamic properties critical to crystallization. |
| Aceclofenac (AC) [48] | 25 kGy | No change in melting point | Capillary Melting Point | Demonstrates compound-specific stability. |
| Aceclofenac (AC) [48] | 400 kGy (high dose) | Melting point decreased by 2.0°C (153.0°C to 151.0°C) | Capillary Melting Point | Confirms even stable compounds degrade at sufficient doses. |
| Polyacrylonitrile (PAN) Fibers [49] | 200 kGy | Color change from white to pale yellow; radical formation | ESR Spectroscopy, Visual Inspection | Visual and spectroscopic signs of radiolysis. |
| Lysozyme Protein [50] | 3.2 x 10² e⁻/nm²/s | Continuous crystal growth without damage | TEM Growth Rate | Establishes a "safe" imaging dose rate for a biological system. |
| Lysozyme Protein [50] | 2.9 x 10³ e⁻/nm²/s | Crystal dissolution initiated | TEM Observation | Threshold dose where beam damage overwhelms natural process. |
The data in Table 1 reveal several key patterns. Pharmaceutical compounds like diclofenac and aceclofenac show remarkable stability at standard irradiation doses (25 kGy), but exhibit measurable degradation—such as melting point depression and color change—at higher doses [48]. This suggests a dose threshold exists for these materials. Furthermore, the dose-rate dependency is crucial, as demonstrated by the lysozyme study where a tenfold increase in electron flux switched the outcome from controlled growth to dissolution [50]. For the organic molecule FFA, the beam was used to initiate nucleation via radiolysis, illustrating that the effect can be harnessed but must be explicitly accounted for in the experimental design [47].
Diagram 1: Experimental workflow for identifying electron beam artifacts in nucleation studies by comparing low-dose and high-dose observations.
Underlying Mechanisms of Beam-Induced Alterations
The perturbations observed in nucleation studies arise from specific physical and chemical mechanisms initiated by the electron beam.
Radical Formation and Propagation
A primary mechanism of beam damage is the radiolysis of chemical bonds and the subsequent formation of radical species. As demonstrated in polyacrylonitrile (PAN) fibers, electron beam irradiation produces a variety of long-lived chain radicals, including alkyl, allyl, and polyenyl radicals, detectable via Electron Spin Resonance (ESR) spectroscopy [49]. These radicals gradually oxidize in the presence of oxygen to form peroxy radicals, which can then act as intermolecular cross-linking points or initiate successive cyclization reactions [49]. In organic nucleation systems, similar radical chemistry could artificially link molecules, lowering the energy barrier for aggregation and leading to non-physical nucleation kinetics and the formation of metastable phases not observed in bulk crystallization.
Solvent Radiolysis and Precursor Modification
In liquid-phase experiments, the electron beam interacts extensively with the solvent. Radiolysis of solvents like water or ethanol generates reactive species—including hydrated electrons, hydrogen radicals, and hydroxyl radicals—that can rapidly react with solute molecules. A study on flufenamic acid (FFA) in ethanol explicitly hypothesized that the breakup of ethanol molecules and the formation of reactive species altered the local chemical environment, thereby lowering the energy barrier for nucleation [47]. This indicates that the beam may not act directly on the solute but can catalyze nucleation by modifying the solvent or creating intermediate chemical species that facilitate aggregation.
Non-Classical Nucleation Pathways
Beam effects may preferentially influence one nucleation pathway over another. Non-classical crystallization (NCC) pathways, which involve pre-nucleation clusters (PNCs) or dense liquid phases (DLPs) as intermediates, may be particularly susceptible [47]. The beam-induced formation of reactive species could stabilize these otherwise transient amorphous precursors or alter their internal structure and dynamics. For example, in-situ TEM of lysozyme revealed that crystal nucleation did not occur within pre-existing amorphous solid particles (ASPs) but rather within a second type of noncrystalline particle that assembled on the ASP surface, a subtle pathway that could be easily biased by beam effects [50]. Distinguishing between these complex, multi-step pathways and beam-facilitated shortcuts is a central challenge.
Experimental Protocols for Mitigation and Control
The following protocols provide actionable methods for minimizing beam effects or accounting for them in data analysis.
Protocol 1: Establishing a Safe Dose Rate
Purpose: To determine the maximum electron dose rate that allows for observation of nucleation events without inducing significant beam-damage artifacts.
- Preparation: Prepare a series of identical, supersaturated solutions of the target material.
- Calibration: Calibrate the electron flux of the microscope using a Faraday cup.
- Imaging: Image each sample at a different, precisely recorded dose rate (e.g., 10, 50, 100, 500 e⁻/Ų/s). Use the lowest possible dose to achieve adequate signal-to-noise ratio, leveraging modern direct electron detectors.
- Analysis: Compare nucleation kinetics, crystal growth rates, and polymorphic outcomes across the different dose rates.
- Threshold Identification: Identify the critical dose rate where parameters (e.g., growth rate) diverge from those observed in bulk crystallization or at the lowest doses. The "safe" dose rate is the highest value below this divergence point. For lysozyme, this was found to be ~3.2 x 10² e⁻/nm²/s [50].
Protocol 2: Controlled Beam-Induced Nucleation for Pathway Analysis
Purpose: To actively exploit beam effects to study potential nucleation pathways, while clearly delineating them from native processes.
- Preparation: Load a solution at a known, undersaturated concentration into a liquid cell.
- Initial Survey: Perform a rapid, low-dose survey of the entire window to confirm the absence of pre-existing nuclei.
- Beam Application: Focus a high-dose beam (e.g., >150 e⁻/Ų/s [47]) on a small, predefined region for a short pulse to initiate radiolysis and nucleation.
- Observation: Immediately after nucleation is initiated, switch back to a low-dose rate to monitor the subsequent growth and evolution of the crystal.
- Interpretation: Clearly annotate all data derived from this method as "beam-induced" and do not conflate the initiation mechanism with spontaneous nucleation pathways.
Table 2: Research Reagent Solutions for In Situ Nucleation Studies
| Reagent/Material | Function in Experiment | Considerations for Beam Interactions |
|---|---|---|
| Diclofenac/Aceclofenac [48] | Model pharmaceutical crystalline compounds. | High radiation stability at standard 25 kGy dose; useful for establishing baseline stability. |
| Flufenamic Acid (FFA) [47] | Model small organic molecule API for nucleation studies. | Demonstrates beam-induced nucleation in ethanol; a model for radiolysis studies. |
| Lysozyme Protein [50] | Model protein for studying biomolecular crystallization. | Provides a benchmark for "safe" electron flux limits in biological systems. |
| Ethanol Solvent [47] | Common solvent for organic molecules. | Susceptible to radiolysis, generating reactive species that can catalyze nucleation. |
| Aqueous Buffers [50] | Solvent for biological macromolecules. | Radiolysis produces hydrated electrons, H•, and •OH radicals that can damage proteins. |
| Amorphous Solid Particles (ASPs) [50] | Heterogeneous nucleation substrates. | Beam effects may alter their surface chemistry or ability to promote nucleation. |
Diagram 2: Logical map of electron beam effect mechanisms, from primary physical interactions to observable experimental artifacts in nucleation studies.
Concluding Recommendations
Mitigating electron beam effects requires a multi-faceted strategy that combines technical precision with critical data interpretation. The following integrated approach is recommended:
- Employ a Dose-Resolved Study Design: Always begin a new system with a dose-rate series (Protocol 1) to establish a "safe" imaging window. This is the single most important step for generating reliable data.
- Correlate with Offline Validation: Where possible, correlate in situ observations made at low dose with offline techniques such as optical microscopy or X-ray scattering to confirm key findings like growth rates and polymorph identity.
- Document and Report Beam Parameters: Meticulously record and report all imaging parameters, including accelerating voltage, electron flux, total accumulated dose, and frame rate. This is essential for the reproducibility and correct interpretation of results.
- Utilize Reference Materials: Use materials with known radiation stability (e.g., diclofenac [48]) as internal standards to calibrate and validate experimental conditions for new, uncharacterized systems.
The most powerful approach is to combine low-dose observation of native processes with controlled, high-dose probing of beam-induced effects. By systematically mapping the influence of the electron beam, researchers can confidently extract meaningful mechanistic insights into inorganic crystal nucleation while explicitly accounting for the influence of their primary investigative tool.
Strategies for Improving Temporal and Spatial Resolution in Dynamic Experiments
In the field of in situ microscopy for inorganic crystal nucleation research, the interplay between temporal and spatial resolution is a fundamental challenge. Capturing fast, transient dynamics at relevant length scales requires a sophisticated integration of instrumentation, methodology, and data processing. Recent paradigm shifts in (scanning) transmission electron microscopy ((S)TEM) have led to unprecedented improvements, with spatial resolution now extending to the sub-angstrom level and temporal resolution for single-shot imaging reaching the nanosecond timescale [51]. This application note details current strategies and protocols to enhance these capabilities, providing a structured guide for researchers investigating dynamic materials processes.
Core Concepts and Definitions
Spatial Resolution refers to the smallest distinguishable distance between two adjacent points in an image. In dynamic transmission electron microscopy (DTEM), for example, a spatial resolution of <10 nm has been achieved using 15 ns electron pulses [52].
Temporal Resolution is the minimum time interval between consecutive observations needed to resolve a dynamic process. The Rose criterion for signal-to-noise ratio (SNR) defines how many electrons are needed for a given resolution, creating a fundamental trade-off where higher frame rates typically reduce SNR and thus spatial resolution [53].
The core challenge lies in optimizing one without critically compromising the other, a balance governed by instrumental limitations and the physics of electron-sample interactions.
Instrumental and Methodological Approaches
Improving resolution requires a multi-faceted strategy. The following sections outline key technological and methodological pathways.
Advanced Microscope Platforms
Specialized microscope platforms overcome the limitations of conventional TEMs.
- Dynamic TEM (DTEM): The DTEM developed at Lawrence Livermore National Laboratory (LLNL) uses a single-shot approach with nanosecond-duration, high-current electron pulses to capture irreversible material processes. It has demonstrated a spatial resolution of <10 nm with 15-ns temporal resolution [52]. This is ideal for studying rapid, irreversible events like solid-state reactions in NiAl multilayer films or martensitic transformations [52].
- Ultrafast TEM (4D-EM): This approach, often using a stroboscopic (pump-probe) method, synchronizes a drive laser (initiating the process) with an electron probe. It can achieve femtosecond to nanosecond temporal resolution but typically requires the process under study to be perfectly reversible [53].
- Environmental TEM (ETEM): ETEMs feature a modified sample area that can accommodate gas pressures up to 2000 Pa, allowing for the study of gas-solid interactions at elevated temperatures under realistic conditions without the obfuscation of windowed cells [53].
Specialty Sample Holders and Stimuli
The development of micro-electro-mechanical system (MEMS)-based holders allows for the application of various external stimuli during imaging.
- Windowed Cell Holders: These enable the study of solid/liquid and solid/gas interactions. Liquid cells are popular for studying nanoparticle nucleation from solutions and electrochemical processes in battery materials [53].
- Heating and Biasing Holders: Specially designed holders can reproduce solvothermal reaction conditions, such as temperatures up to ~300 °C in an organic solvent, which is necessary for studying the thermal decomposition of precursors for cobalt-based nanoparticles [54].
- Photonic Stimulation: Lasers can be introduced via modified holders or independent microscope ports to initiate photocatalytic reactions, phase transformations, or for local heating [53].
Detectors and Data Acquisition
The recording media often defines the temporal resolution boundary.
- Direct Detection Cameras: These cameras have a high detection quantum efficiency (DQE) and are capable of high-speed imaging, with rates of up to ~1600 frames per second [53]. This represents a significant advance over conventional CCD cameras.
- Data Handling: The large data sets generated by high-speed, high-resolution in situ experiments require robust data acquisition, transfer, and mining procedures to extract meaningful scientific knowledge [53].
Quantitative Comparison of Techniques
The table below summarizes the performance characteristics of different advanced microscopy techniques relevant to dynamic experiments.
Table 1: Performance Comparison of Advanced Microscopy Techniques
| Technique | Typical Spatial Resolution | Typical Temporal Resolution | Key Strengths | Primary Limitations |
|---|---|---|---|---|
| In Situ TEM/STEM with Direct Detection | Atomic (~0.1 nm) [51] | Up to ~1,600 fps [53] | High spatial resolution; versatile in situ capabilities | Speed limited by camera readout and electron dose |
| Dynamic TEM (DTEM) - Single Shot | < 10 nm [52] | ~15 ns [52] | Captures irreversible processes; nanosecond resolution | High current pulses limit spatial resolution; technical complexity |
| Ultrafast TEM (Stroboscopic) | Atomic scale possible [53] | Femtosecond - Nanosecond [53] | Extremely high temporal resolution | Requires perfectly reversible/repeatable processes |
| Environmental TEM (ETEM) | Atomic resolution at low pressure [53] | Video-rate to high-speed | Direct observation in gas environments; high temperature | Resolution degrades with increasing gas pressure [53] |
| Environmental-Cell TEM (EC-TEM) | Sub-nm to a few nm [54] | Video-rate | Observation in liquid environments; solvothermal reactions | Liquid layer and windows scatter electrons, reducing resolution |
Experimental Protocols
Protocol: In Situ EC-TEM for Nanoparticle Nucleation and Growth
This protocol is adapted from studies investigating the confinement effect of carbon nanotubes on the synthesis of cobalt-based nanoparticles via thermal decomposition [54].
- Aim: To visualize the nucleation and growth mechanisms of cobalt-based nanoparticles under solvothermal conditions in real-time.
- Experimental Setup:
- Microscope: TEM equipped with an aberration corrector for high spatial resolution.
- Holder: Commercial environmental cell TEM (EC-TEM) holder, adapted to withstand temperatures up to ~300 °C with an organic solvent.
- Sample: Precursor solution (e.g., cobalt stearate complex and oleic acid in an organic solvent) loaded into a windowed liquid cell. For confinement studies, carbon nanotubes (CNTs) are introduced into the solution [54].
- Procedure:
- Loading: Inject the precursor solution into the liquid cell, ensuring a thin, continuous liquid layer is formed between the electron-transparent windows.
- Initial Characterization: Image the initial state at room temperature to identify areas of interest, such as empty CNT channels.
- In Situ Reaction: Ramp the holder temperature to the precursor decomposition range (typically 280–330 °C) at a controlled rate.
- Data Acquisition:
- Use a high-speed direct detection camera to record the dynamic process.
- For high-resolution snapshots, temporarily pause heating or reduce the frame rate to increase electron dose and SNR.
- Monitor the reaction mixture as it incorporates into CNT channels via capillarity at low temperatures [54].
- Observation: Directly visualize key events:
- For unconfined synthesis (outside CNTs): Observe the formation of vesicle-like structures in the solvent. Nucleation is initiated at the liquid-gas interface of these vesicles, where monomer concentration is higher. Clusters grow to a critical size of 4–5 nm before forming chain-like structures and sintering to ~20 nm [54].
- For confined synthesis (inside CNTs): Observe the formation of a porous micellar liquid structure that densifies at higher temperatures (~300 °C), leading to the formation of separated Co-based nanoparticle entities [54].
- Data Analysis: Perform particle tracking and size analysis on the recorded image series to quantify nucleation rates and growth kinetics.
Protocol: Nanosecond Single-Shot Imaging with DTEM
This protocol is based on the use of the DTEM for studying rapid, irreversible solid-state reactions and phase transformations [52].
- Aim: To capture transient structural changes occurring on nanosecond timescales.
- Experimental Setup:
- Microscope: Modified TEM (e.g., JEOL 2000FX platform) with laser access to the photocathode and specimen.
- Laser Systems: A cathode laser (e.g., 10 ns, 211 nm pulse) for generating electron pulses and a separate pump laser for initiating the material process.
- Procedure:
- Sample Preparation: Prepare a thin sample, such as a NiAl reactive multilayer foil (RMLF) or nanocrystalline Ti.
- Synchronization: Precisely synchronize the pump laser (which initiates the reaction) and the cathode laser (which generates the single electron pulse for imaging) with a defined time delay.
- Single-Shot Data Capture:
- The pump laser pulse rapidly heats the sample, initiating the transient process (e.g., a self-propagating reaction in an RMLF).
- After a preset time delay (e.g., nanoseconds to microseconds), a single, intense pulse of electrons is generated at the photocathode and used to form a TEM image or diffraction pattern in a single shot.
- The jitter between the pump laser and electron pulse in this configuration is typically ~1 ns [52].
- Multi-shot Experiment: Repeat the experiment on a fresh area of the sample with a different time delay to build a "movie" of the transient process.
- Key Considerations:
Visualizing the Strategy Selection Workflow
The following diagram outlines the decision-making process for selecting the appropriate high-resolution strategy based on the characteristics of the dynamic process under investigation.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for In Situ Crystal Nucleation Studies
| Item | Function/Application | Example Use-Case |
|---|---|---|
| Metal-Carboxylate Precursors (e.g., cobalt stearate, cobalt oleate) | Acts as the metal source for nanoparticle synthesis via thermal decomposition. | Formation of cobalt-based nanoparticles (Co, CoO, Co3O4) inside and outside carbon nanotubes [54]. |
| Capping Agents (e.g., oleic acid, citric acid) | Ligands that control nanoparticle growth, size, and morphology by binding to crystal surfaces. | Used in thermal decomposition synthesis to precisely control the properties of the resulting cobalt-based NPs [54]. |
| Carbon Nanotubes (CNTs) | Provide a confined nano-environment for synthesis, influencing NP morphology, stability, and catalytic properties. | Studying the confinement effect on the nucleation and growth mechanisms of cobalt-based NPs [54]. |
| Micro-Electro-Mechanical System (MEMS) Based Holders | Enable in situ application of external stimuli (heating, biasing, liquid/gas environment) during TEM imaging. | Reproducing solvothermal reaction conditions for the real-time visualization of NP synthesis at high temperatures [54] [53]. |
| Reactive Multilayer Foils (RMLFs) | Model systems for studying rapid, exothermic solid-state reactions and transient phase evolution. | Investigating rapid chemical reactions and morphological changes with nanosecond resolution in the DTEM [52]. |
The pursuit of visualizing dynamic processes, such as inorganic crystal nucleation, at the nanoscale requires sample preparation that faithfully preserves the liquid or gas microenvironment in which these events occur. The fundamental challenge lies in bridging the gap between a material's native operational conditions and the high-vacuum environment of a transmission electron microscope (TEM). Cryogenic electron microscopy (cryo-EM) has emerged as a powerful tool for this purpose, enabling the analysis of nanoscale structures by rapidly freezing samples to liquid nitrogen temperatures, which fixes them in a vitrified, near-native state [55]. This approach mitigates two primary issues associated with conventional TEM imaging: structural changes induced by the electron beam and the alteration of solution-state processes due to vacuum exposure [55]. For research on inorganic crystal nucleation, this capability is indispensable, as it allows for "snapshots" of solution-based processes, providing unprecedented structural insights into the early stages of crystal formation and growth [55].
Core Challenges in Creating Representative Microenvironments
Preparing samples for in situ microscopy of dynamic processes presents three interconnected challenges that must be overcome to obtain meaningful data.
Challenge 1: Isolation from High Vacuum. Standard TEM operates under high vacuum, which causes most liquids and solvents to vaporize, thereby destroying the native microenvironment. Liquid cell (LC)-TEM techniques address this by encapsulating the sample in a miniature reactor that isolates it from the vacuum [56]. The core of this challenge lies in designing cells that are sufficiently thin for electron transparency while being robust enough to contain the sample and, in some cases, integrate additional capabilities such as fluid flow or electrical biasing.
Challenge 2: Electron Beam Effects. The interaction between the high-energy electron beam and the sample is a critical consideration. In liquids, the beam causes radiolysis, breaking molecular bonds and generating reactive radical species [56]. These radicals can damage biological samples or trigger unintended chemical reactions in synthetic systems, such as the nucleation of crystals that are artifacts of the imaging process rather than representative of the natural mechanism. Cooling the specimen to cryogenic temperatures near liquid nitrogen significantly reduces this structural damage during imaging [55].
Challenge 3: Verifying Native-State Preservation. A paramount challenge, especially in "live-cell" or dynamic process studies, is confirming that the prepared sample remains in a state representative of its native conditions. This is particularly complex for biological or process-oriented samples. Reliability requires the use of sensitive viability assays for biological components and stringent controls to ensure that the captured nucleation events are genuine [56].
Established Protocols for Liquid and Gas Microenvironment Preparation
The following protocols detail methodologies for creating and analyzing representative microenvironments, with a focus on liquid phase studies relevant to crystal nucleation.
Protocol 1: General Workflow for Cryo-EM Sample Preparation of Solution-Phase Materials
This protocol is adapted from established cryo-EM procedures in materials science for vitrifying solution-phase samples to capture their native-state structure [55].
- Objective: To prepare a thin film of vitrified solution containing the material of interest (e.g., nucleating crystals, nanoparticles) for observation by cryo-TEM.
- Principle: Rapid freezing of a thin liquid film immobilizes the samples in a glassy, non-crystalline ice layer, preserving their native structure and spatial distribution.
- Materials:
- Aqueous or organic solution containing the sample of interest.
- Cryo-EM Grids (e.g., Lacey carbon, Quantifoil).
- Cryogen (typically liquid ethane or a mixture of liquid ethane/propane).
- Plunger apparatus (vitrification robot or manual system).
- Cryo-transfer holder for TEM.
- Blotting paper.
- Method:
- Grid Preparation: Apply a few microliters of the sample solution onto a clean TEM grid.
- Blotting: Use blotting paper to remove excess liquid, leaving a thin, continuous film (typically 10-500 nm thick) spanning the grid holes.
- Vitrification: Rapidly plunge the grid into a cryogen (e.g., liquid ethane) cooled by liquid nitrogen. The speed of this process is critical to prevent the formation of crystalline ice.
- Storage and Transfer: Transfer the vitrified grid under liquid nitrogen to a cryo-box for storage or directly into a cryo-TEM holder.
- Imaging: Insert the holder into the TEM, maintained at cryogenic temperatures, for imaging.
Protocol 2: Assembly of a Silicon Nitride (SiN)-Based Liquid Cell
This protocol outlines the procedure for creating a closed liquid cell for true in situ liquid phase TEM (LP-TEM) observations, allowing for the visualization of dynamic processes like crystal nucleation in real-time [56].
- Objective: To encapsulate a liquid sample between two ultrathin silicon nitride windows for observation in the TEM while isolated from the vacuum.
- Principle: Two SiN chips, each featuring an electron-transparent window, are separated by a spacer to create a sealed chamber for the liquid medium.
- Materials:
- Pair of SiN membrane chips.
- Spacer (e.g., metallic or polymeric shims of defined thickness).
- Liquid sample of interest.
- Gaskets or sealing fixtures (specific to the commercial liquid cell holder).
- Liquid cell TEM holder.
- Method:
- Cell Assembly: Carefully place a spacer on the surface of the bottom SiN chip. The spacer defines the height of the liquid chamber, which is typically between 100 nm and 1 µm.
- Sample Loading: Pipette a small volume (e.g., 0.5-1 µL) of the liquid sample onto the spacer.
- Sealing: Align and lower the top SiN chip onto the assembly, creating a sealed liquid chamber.
- Holder Integration: Secure the assembled liquid cell into the dedicated TEM holder according to the manufacturer's instructions, ensuring a leak-tight seal.
- Insertion and Imaging: Insert the holder into the TEM and begin imaging. The electron beam can pass through the SiN windows and the liquid layer, allowing dynamic processes to be recorded.
Table 1: Comparison of Liquid Microenvironment Preparation Techniques
| Parameter | Cryo-EM (Protocol 1) | Liquid Cell TEM (Protocol 2) |
|---|---|---|
| State of Sample | Vitrified (arrested) | Liquid (dynamic) |
| Temporal Resolution | Snapshot | Real-time video |
| Primary Application | High-resolution structure determination | Imaging dynamic processes (e.g., growth, motion) |
| Electron Dose | Can use higher doses for resolution | Must be minimized to reduce radiolysis |
| Complexity | Moderate | High |
| Risk of Artifacts | From freezing process | From electron beam effects |
Quantitative Data and Performance Metrics
The selection and optimization of a sample preparation method rely on key performance metrics that quantify the quality of the resulting data and the integrity of the microenvironment.
Table 2: Key Metrics for Assessing Microenvironment and Image Quality
| Metric | Description | Relevance to Microenvironment |
|---|---|---|
| Liquid Thickness | Height of the liquid chamber or ice layer. | Thicker layers better mimic bulk conditions but reduce resolution and increase electron scattering. |
| Electron Dose (e⁻/Ų) | Number of electrons incident on the sample per unit area. | Determines the extent of radiolysis damage; must be balanced with the need for signal-to-noise [56]. |
| Resolution (Å or nm) | Smallest discernible detail in the image. | Defines the level of structural detail achievable, from near-atomic for single particles to nanometer scale for cellular structures [55]. |
| Signal-to-Noise Ratio (SNR) | Ratio of the true signal level to the background noise. | Critical for identifying low-contrast features in a thick liquid layer or vitreous ice [57]. |
| Contrast-to-Noise Ratio (CNR) | Measure of the discernibility of a feature from its surroundings [57]. | Determines the visibility of a nucleating crystal against the liquid background. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the aforementioned protocols requires a suite of specialized tools and materials.
Table 3: Research Reagent Solutions for Microenvironment Preparation
| Item | Function/Description |
|---|---|
| SiN Membrane Chips | Form the electron-transparent windows of liquid cells, sealing the liquid sample from the microscope vacuum [56]. |
| Liquid Cell TEM Holder | A specialized TEM holder that houses the sealed liquid cell and provides the necessary fluidic or electrical interfaces. |
| Cryo-EM Grids | Perforated grids that support the thin film of vitreous ice and sample during cryo-EM. |
| Plunger/Vitrification Robot | Apparatus for the rapid and reproducible vitrification of samples to preserve their native state [55]. |
| Radical Scavengers | Chemical additives (e.g., ascorbic acid) introduced to the liquid medium to mitigate damage from radiolysis-generated radicals [56]. |
| Cryogen (Liquid Ethane) | Cryogen of choice for rapid heat transfer, enabling vitrification of water instead of crystalline ice formation. |
Workflow and System Diagrams
The following diagrams illustrate the logical flow of a cryo-EM experiment and the configuration of a key tool, the silicon nitride liquid cell.
Diagram 1: Cryo-EM sample preparation workflow for capturing snapshots of processes like crystal nucleation.
Diagram 2: Configuration of a closed SiN liquid cell, showing the sealed liquid chamber and electron-transparent windows.
Inorganic crystal nucleation and growth research has been transformed by in situ transmission electron microscopy (TEM), which enables direct observation of dynamic processes at the nanoscale. These experiments generate complex time-resolved datasets that present significant data management challenges [58] [23]. This application note details standardized protocols for processing and analyzing these datasets, framed within ongoing inorganic crystal nucleation research. The methodologies outlined enable researchers to extract quantitative insights into nucleation mechanisms, growth kinetics, and structure-property relationships from complex multidimensional data.
Experimental Protocols for In Situ TEM Nucleation Studies
In Situ TEM Setup for Thermal Decomposition Synthesis
Purpose: To directly visualize the nucleation and growth of cobalt-based nanoparticles via thermal decomposition within controlled environments [54].
Materials and Reagents:
- Precursor: Cobalt stearate complex (e.g., 0.1 M in oleic acid)
- Support Material: Carbon nanotubes (CNTs), both non-treated and annealed at 900°C under Ar
- Capping Agent: Oleic acid (e.g., 0.15 M)
- Solvent: High-boiling point organic solvent (e.g., octadecene)
- TEM Grids: Commercially available in situ TEM holders compatible with environmental cells
Procedure:
- Sample Preparation:
- For confined synthesis: Use annealed CNTs to remove oxygen functions from external surfaces
- For comparative external surface synthesis: Use non-treated CNTs retaining external oxygen functions
- Disperse CNTs in precursor solution via ultrasonication for 30 minutes
In Situ TEM Setup:
- Load prepared sample into commercial environmental cell TEM (EC-TEM) holder
- Implement custom adaptations to achieve solvothermal reaction conditions (temperatures up to 300°C)
- Establish stable liquid phase environment within the microscope
Data Acquisition:
- Initiate temperature ramp from room temperature to 300°C at 5-10°C/min
- Begin continuous image acquisition at frame rates of 1-30 fps depending on phenomenon of interest
- Simultaneously acquire complementary data: STEM imaging, EDS, EELS where applicable
- Record temperature and time metadata synchronized with image frames
Termination:
- Cool system rapidly to room temperature upon completion of reaction
- Extract sample for additional ex situ characterization if required
Data Acquisition Parameters for Time-Resolved Studies
Table 1: Representative Data Acquisition Parameters for In Situ TEM Nucleation Experiments
| Parameter | Typical Range | Optimal Value for Nucleation Studies | Notes |
|---|---|---|---|
| Temporal Resolution | 1 ms - 30 s | 1-5 s | Dependent on nucleation kinetics |
| Spatial Resolution | 0.1 - 10 nm | <1 nm | Critical for early nucleus detection |
| Frame Size | 1K×1K - 8K×8K pixels | 4K×4K | Balance between field of view and resolution |
| Acquisition Duration | 1 min - several hours | 30-60 min | Cover complete nucleation and growth phases |
| Complementary Techniques | EDS, EELS, Diffraction | EDS + STEM | Elemental and structural mapping |
| Temperature Control | ±1°C | ±2°C | Critical for thermal decomposition studies |
Processing Workflow for Time-Resolved Image Data
The following diagram illustrates the comprehensive data processing workflow for managing large, complex datasets from in situ TEM nucleation experiments:
Step 1: Pre-processing
- Apply frame alignment and drift correction algorithms to compensate for sample movement
- Implement noise reduction filters (Gaussian, median, or non-local means) while preserving edges
- Normalize intensity across frames to account for beam fluctuations
- Correct for uneven illumination (flat-field correction)
Step 2: Segmentation and Feature Identification
- Apply edge detection algorithms (Canny, Sobel) for initial boundary identification
- Utilize machine learning approaches (U-Net, Random Forest) for robust nanoparticle segmentation
- Implement tracking algorithms to follow individual nuclei across frames
- Manually validate segmentation accuracy across multiple timepoints
Step 3: Quantitative Parameter Extraction
- Measure nanoparticle size (diameter, area), shape parameters (aspect ratio, circularity), and position
- Calculate growth rates from size versus time curves
- Determine nucleation density and spatial distribution
- Quantify size distributions at sequential timepoints
Step 4: Temporal Analysis and Kinetic Modeling
- Fit nucleation kinetics to classical models (LaMer, Ostwald ripening) [54]
- Calculate nucleation rates from cumulative nucleus count versus time
- Determine growth exponents from particle size evolution
- Identify critical nucleus size through statistical analysis of stable versus unstable nuclei
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for In Situ TEM Studies of Inorganic Crystal Nucleation
| Reagent/Material | Function | Example Application | Notes |
|---|---|---|---|
| Cobalt Stearate | Metal-organic precursor | Thermal decomposition synthesis of Co nanoparticles [54] | Decomposes at ~300°C to form metallic Co |
| Carbon Nanotubes (CNTs) | Confinement support | Creating nanoscale environments for controlled nucleation [54] | Diameter affects nanoparticle size and morphology |
| Oleic Acid | Capping agent/ligand | Controls nanoparticle growth and prevents aggregation [54] | Concentration affects final particle size |
| Environmental Cell TEM Holder | Sample environment control | Maintaining liquid or gas environments during TEM imaging [54] [23] | Enables realistic synthesis conditions |
| High-Speed TEM Camera | Data acquisition | Capturing rapid nucleation events with high temporal resolution [23] | Frame rates up to 30 fps standard; specialized systems higher |
| Microscopy Software | Data management and analysis | Processing large time-resolved datasets (e.g., MicroManager, ImageJ) [59] | Custom scripts often required for specific analyses |
Data Management Framework
Experimental Design and Data Acquisition
The experimental setup for in situ TEM nucleation studies generates complex multidimensional data, as illustrated below:
Effective data management begins with experimental design that considers the ultimate data analysis requirements. For in situ TEM nucleation studies, this involves synchronizing multiple data streams [58] [23]:
- Primary imaging data: Time-resolved TEM or STEM images
- Spectroscopic data: EDS or EELS for compositional analysis
- Structural data: Electron diffraction patterns for crystal structure identification
- Metadata: Experimental parameters (temperature, time, beam conditions)
Quantitative Analysis of Nucleation Phenomena
Nucleation Kinetics Analysis:
- Calculate nucleation rates from the appearance frequency of new particles
- Determine critical nucleus size through statistical analysis of stable versus unstable nuclei
- Fit experimental data to nucleation models (classical, non-classical, two-step)
Growth Kinetics Analysis:
- Measure growth rates of individual particles from size versus time curves
- Identify growth mechanisms (Ostwald ripening, coalescence, monomer addition)
- Calculate growth exponents and compare with theoretical models
Morphological Evolution:
- Quantify shape parameters (aspect ratio, faceting, symmetry) over time
- Analyze spatial distribution and organization of nanoparticles
- Correlate morphological evolution with experimental conditions
Table 3: Key Quantitative Parameters in Nanoparticle Nucleation and Growth Studies
| Parameter Category | Specific Metrics | Analysis Method | Information Gained |
|---|---|---|---|
| Nucleation Kinetics | Nucleation rate, Induction time, Critical size | Cumulative nucleus count vs. time, Size stability analysis | Mechanism (homogeneous/heterogeneous), Energy barriers |
| Growth Behavior | Growth rate, Size distribution, Growth exponent | Size vs. time curves, Distribution width analysis | Rate-limiting steps, Growth mechanism |
| Morphological Evolution | Aspect ratio, Facet geometry, Shape uniformity | Geometric analysis, Crystallographic modeling | Surface energy anisotropy, Confinement effects |
| Spatial Distribution | Inter-particle distance, Spatial correlation | Radial distribution function, Nearest-neighbor analysis | Cooperative effects, Diffusion fields |
Applications to Inorganic Crystal Nucleation Research
The protocols described enable detailed investigation of fundamental phenomena in inorganic crystal nucleation:
Confinement Effects: Studies of cobalt-based nanoparticles synthesized inside versus outside carbon nanotubes reveal profound confinement effects. NPs within CNTs exhibit homogeneous size (~50 nm) and octahedral morphology with Co–CoO structure, while external NPs show random morphologies, smaller sizes (~20 nm), and oxidized Co3O4 composition [54].
Nucleation Mechanisms: In situ observations challenge classical nucleation theory, revealing that nucleation frequently occurs at heterogeneous interfaces. For cobalt-based nanoparticles formed outside CNTs, nucleation initiates within vesicle-like structures at liquid-gas interfaces characterized by higher monomer concentration [54].
Growth Pathways: Direct visualization reveals multiple growth pathways including monomer addition, particle coalescence, and Ostwald ripening. The formation of chain-like structures from 4-5 nm primary particles followed by sintering to ~20 nm particles has been documented [54].
Effective management of large, complex time-resolved datasets from in situ TEM experiments requires robust, standardized protocols from acquisition through analysis. The frameworks presented here enable comprehensive investigation of inorganic crystal nucleation mechanisms, growth kinetics, and morphological evolution. By implementing these methodologies, researchers can extract maximal quantitative information from challenging experiments, advancing our fundamental understanding of nanoscale materials synthesis.
The precise observation of inorganic crystal nucleation and growth is pivotal for advancing research in materials science, geology, and pharmaceutical development. In situ microscopy enables researchers to capture these dynamic processes in real time, revealing details that are often lost in ex situ analyses. However, the fidelity of such observations is highly dependent on the careful calibration of experimental protocols. This application note provides a detailed framework for optimizing three critical parameters—temperature, concentration, and triggering conditions—within the context of inorganic crystal nucleation research. By integrating quantitative data from recent studies and providing standardized methodologies, this document aims to enhance the reproducibility and accuracy of in situ crystallization experiments for researchers, scientists, and drug development professionals.
Quantitative Parameter Optimization
Successful in situ observation of crystal nucleation requires precise control over experimental conditions. The following tables consolidate optimal parameters from recent studies on different inorganic systems to guide experimental design.
Table 1: Optimized Temperature and Triggering Parameters for Inorganic Crystal Nucleation
| Crystal System | Optimal Temperature Range | Heating Rate | Triggering Environment | Key Observed Nucleation Phase |
|---|---|---|---|---|
| Al-Cu-Li Alloy [60] | 180°C (aging) | 1°C/s | MEMS-based heating chip, vacuum | T1 precipitation |
| Ice I (deposition mode) [11] | 102 K (-171.15°C) | N/A | Vapor deposition on graphene at 10⁻⁶ Pa | Amorphous ice to ice I transition |
| Calcium Phosphate on TiO₂ [61] | Room temperature (solution) | N/A | Liquid cell in 50 mM Tris-HCl, pH 7.4 | Amorphous CaP to crystalline HAP |
Table 2: Concentration and Sample Preparation Parameters
| Material System | Critical Concentration/Thickness | Preparation Method | Impact on Nucleation & Growth |
|---|---|---|---|
| Al-Cu-Li Alloy [60] | 150–200 nm (sample thickness) | FIB milling with low-energy (3 kV) ion polishing | Balances resolution with bulk-like precipitation kinetics; thinner samples cause abnormal coarsening |
| TiO₂ Nanoparticles [61] | 17.3 nm average nanoparticle size | Ultrasonication (2 h at 50°C) in Tris buffer | Forms aggregation sites for amorphous CaP layer formation |
| Ice Nucleation [11] | Substrate-specific (graphene) | Vapor deposition on cryogenic graphene | Mediates amorphous ice adsorption prior to crystalline nucleation |
Experimental Protocols
Sample Preparation and Transfer for Metallic Alloys
This protocol mitigates gallium (Ga) and platinum (Pt) contamination during Focused Ion Beam (FIB) preparation, which can significantly distort precipitation behavior in aluminum alloys [60].
- Materials: Bulk Al-Cu-Li alloy (2195Al composition), MEMS-based heating chip, FIB system with Ga⁺ ion source.
- Solution Treatment: Subject bulk sample to 510°C for 30 minutes, followed by immediate water quenching [60].
- FIB Milling:
- Initial Thinning: Tilt sample to 52° and mill to approximately 2 µm thickness.
- Transfer: Use a micromanipulator for ex situ transfer to the MEMS chip to minimize Ga⁺ implantation.
- Final Thinning: Employ low-energy ion milling at 3 kV accelerating voltage at 0° tilt to achieve the target thickness of 150–200 nm [60].
- Thickness Verification: Confirm sample thickness using HAADF-STEM imaging. Maintain uniform electron transparency without exceeding 250 nm to avoid resolution loss [60].
Liquid-Phase Mineralization for Biomaterials
This procedure enables real-time observation of calcium phosphate (CaP) mineralization on titanium dioxide (TiO₂) nanoparticles using liquid-phase TEM [61].
- Materials: Anatase TiO₂ nanoparticles (avg. 17.3 nm), 50 mM Tris-HCl buffer (pH 7.4), liquid TEM cell with silicon nitride (SiNx) windows.
- Nanoparticle Dispersion:
- Suspend TiO₂ nanoparticles in Tris buffer.
- Stir continuously for 5 minutes.
- Sonicate in a 50°C ultrasonic bath for 2 hours to disperse aggregates [61].
- Liquid Cell Assembly: Inject prepared suspension into the liquid cell, ensuring nanometer-scale liquid thickness to minimize electron scattering.
- Imaging Parameters: Use low electron flux density (e.g., 17.7 e⁻/nm²/s) to prevent beam-induced nanoparticle aggregation and morphological changes [61].
- Process Monitoring: Record real-time aggregation of TiO₂, formation of amorphous CaP layers, and subsequent crystallization into hydroxyapatite (HAP) [61].
Vapor-Phase Deposition Freezing for Ice Nucleation
This protocol details the setup for observing molecular-scale heterogeneous ice nucleation via deposition freezing on graphene substrates [11].
- Materials: Graphene substrate, in-situ cryogenic TEM system.
- Substrate Cooling: Cool graphene substrate to 102 K (-171.15°C) within the TEM chamber [11].
- Environment Control: Maintain chamber pressure at 10⁻⁶ Pa to ensure vapor deposition conditions.
- Vapor Introduction: Introduce water vapor to initiate the deposition process.
- Imaging and Analysis:
- Use high-resolution real-time imaging to track amorphous solid water cluster formation.
- Monitor subsequent spontaneous nucleation and growth of ice I (hexagonal) and ice Ic (cubic) phases.
- Analyze crystal structure and orientation using Fast Fourier Transform (FFT) of sequential TEM images [11].
Visualization of Experimental Workflows
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions and Materials
| Item | Function/Application | Example Specifications |
|---|---|---|
| MEMS-based Heating Chip [60] | In situ thermal stimulation with precise temperature control | Protochips-type, SiN membrane, embedded microheater, 8-μm observation window |
| Liquid TEM Cell [61] | Real-time observation of solution-phase reactions | SiNx windows (≤50 nm thick), liquid encapsulation capability |
| Gallium FIB Source [60] | Site-specific sample milling and thinning | Ga⁺ ion source, adjustable kV (3 kV for final polish) |
| Tris-HCl Buffer [61] | Physiological pH environment for biomaterial studies | 50 mM concentration, pH 7.4 |
| Graphene Substrates [11] | Atomically smooth surface for deposition nucleation studies | Commercial graphene, cryogenically compatible |
| Silicon Nitride Membranes [62] | Sample support for electron transparency | Various thicknesses, electron-transparent |
Benchmarking Performance: Validating and Comparing In Situ Against Traditional Methods
Understanding the nucleation and growth of inorganic crystals is a fundamental challenge in materials science. The pathway a material takes from its constituent parts to a stable crystalline solid governs its final properties, performance, and functionality. For decades, researchers have relied on ex situ characterization methods, which analyze samples before and after experiments, and bulk techniques, which average information over large volumes. While invaluable, these approaches provide only snapshots or averaged data, missing the transient stages and dynamic mechanisms of crystal formation.
The advent of in situ microscopy has revolutionized this field by enabling real-time observation of materials processes under controlled stimuli. This application note provides a comparative analysis of these characterization paradigms, framed within inorganic crystal nucleation research. It details specific protocols and highlights how in situ methods offer unparalleled insights into dynamic processes, empowering researchers and scientists to advance the design of next-generation materials.
The table below summarizes the core differences between bulk, ex situ, and in situ characterization methodologies.
Table 1: Comparison of Characterization Techniques for Crystal Nucleation Studies
| Feature | Bulk Characterization | Ex Situ Microscopy | In Situ Microscopy |
|---|---|---|---|
| Temporal Resolution | Indirect, averaged over time | "Before and after" snapshots | Real-time, video-rate monitoring of dynamic events [63] |
| Spatial Resolution | Micrometer to millimeter scale | Nanometer to atomic scale (for TEM) | Nanometer to atomic scale (e.g., via aberration-corrected TEM) [58] [10] |
| Representative Data | Averages over large sample volumes (mg to g) | Interrogates specific, pre-selected locations | Direct observation of nascent events (nucleation, growth fronts) [64] [65] |
| Key Advantage | Statistically significant average data | High-resolution structural/chemical analysis | Reveals kinetics and transient mechanisms [63] |
| Primary Limitation | Lacks spatial and temporal detail | May miss key intermediates; potential for sample damage/post-processing changes | Complex setup; potential for electron beam effects on samples [58] [10] |
| Example Techniques | X-ray Diffraction (XRD), Neutron Scattering | Standard SEM, TEM, AFM | In Situ (S)TEM, in situ AFM, Environmental TEM [66] [67] [10] |
Experimental Protocols for In Situ Characterization
This section provides detailed methodologies for key in situ experiments relevant to inorganic crystal nucleation and growth.
Protocol: In Situ TEM Lithiation of Silicon Nanomaterials
Application: Directly observing the structural and phase evolution of anode materials for lithium-ion batteries during electrochemical cycling [64].
Materials & Reagents:
- Porous Silicon Nanoparticles/Nanowires: Synthesized via etching of metallurgical Si (e.g., using Fe(NO3)3/HF or AgNO3/HF etchants) [64].
- Lithium Metal: Serves as the lithium source and counter electrode.
- Solid-State Electrolyte (Li2O): Forms a native layer on the lithium metal tip for the nanobattery setup.
- Copper Wire: Acts as the current collector.
Procedure:
- Nanobattery Fabrication: A single silicon nanoparticle or nanowire is placed on the copper current collector. A tungsten or lithium metal tip coated with a native Li2O layer is precisely maneuvered into contact with the particle using nanomanipulators inside the TEM [64].
- Application of Bias: A DC bias (-2 V to -4 V) is applied to the copper electrode relative to the Li/Li2O tip to drive lithiation.
- Real-Time Data Acquisition:
- Imaging: Acquire bright-field TEM or high-angle annular dark-field (HAADF-STEM) images at high frame rates to monitor volume expansion, crack formation, and lithiation front propagation [64].
- Electron Diffraction: Collect Selected Area Electron Diffraction (SAED) patterns at regular intervals to identify phase transitions (e.g., from crystalline Si to amorphous LixSi to crystalline Li15Si4) [64].
- Spectroscopy: Perform electron energy-loss spectroscopy (EELS) or energy-dispersive X-ray spectroscopy (EDS) to track chemical and electronic state changes.
Key Observations: This protocol revealed that porous Si particles exhibit an "end-to-end" lithiation manner, have a much larger critical fracture diameter (~1.52 μm vs. 150 nm for solid Si), and form an amorphous LixSi phase instead of crystalline Li15Si4 upon full lithiation, highlighting the profound impact of nanostructure on phase stability [64].
Protocol: In Situ SEM Characterization of Intermetallic Compound (IMC) Nucleation
Application: Investigating the early-stage nucleation, growth kinetics, and phase evolution at the interface of bimetallic systems, such as Fe/Al composites [65].
Materials & Reagents:
- Cold-Rolled Fe/Al Composite: Pure Fe and pure Al plates are pickled, brush-polished, and roll-bonded to form a composite interface.
- In Situ Heating Stage: A specialized SEM holder capable of precise temperature control (e.g., MINI-HT1200-SE) [65].
Procedure:
- Sample Preparation: The Fe/Al composite is cut into a specific size (e.g., 460 × 30 × 1 mm) using wire electrical discharge machining (EDM) to ensure a clean, observable interface.
- In Situ Heating: The sample is loaded into the heating stage inside the SEM chamber. The temperature is ramped to the target (e.g., 380°C or 520°C) while a thermocouple maintains accurate temperature control (±2°C) [65].
- Real-Time Data Acquisition:
- Imaging: Continuously record secondary electron images to monitor the nucleation and lateral growth of IMCs at the interface.
- Statistical Analysis: Use image analysis software (e.g., Image Pro Plus) to statistically measure the thickness and lateral dimensions of the IMC layer over time from multiple fields of view.
- Post-Process Correlation: After the in situ experiment, prepare site-specific samples via Focused Ion Beam (FIB) lift-out for ex situ TEM and Electron Backscatter Diffraction (EBSD) analysis to identify the specific crystal phases (e.g., Fe4Al13, Fe2Al5) and their crystallographic relationships with the substrate [65].
Key Observations: This protocol enabled the direct observation that the primary phase nucleating at 380°C is Fe4Al13, which subsequently transforms to the more stable Fe2Al5. It also allowed for the quantification of growth kinetics, identifying a shift from reaction control to diffusion control once the IMC layer reaches a critical thickness of ~4.5 μm [65].
Essential Research Reagent Solutions
The following table catalogues key materials and reagents essential for conducting advanced in situ microscopy experiments.
Table 2: Key Research Reagent Solutions for In Situ Characterization
| Reagent/Material | Function in Experiment | Application Example |
|---|---|---|
| Microfabricated Liquid Cell (SiN windows) | Encapsulates liquid solution between electron-transparent membranes for observation in liquid phase [63] [10]. | Studying nanocrystal nucleation and growth from solution (e.g., CaCO3, proteins) [63]. |
| Photoreactive Organosilane (e.g., Mol-1) | Forms a self-assembled monolayer for photopatterning; enables FFM-based characterization of surface terminations [67]. | Creating and analyzing 2D patterns in surface functionality via photolithography [67]. |
| Porous Silicon Nanostructures | High-capacity anode material with pre-formed pores to accommodate lithiation-induced volume expansion [64]. | Investigating fracture behavior and phase transitions during battery operation via in situ TEM [64]. |
| Graphene Liquid Cell | Provides a ultrathin liquid enclosure for high-resolution TEM of solution-phase processes [10]. | Observing the growth and oriented attachment of nanocrystals with near-atomic resolution [10]. |
| In Situ TEM Holder (Heating/Bias) | Applies external stimuli (heat, electrical bias) to the sample inside the TEM column [58] [10]. | Mimicking operational conditions of devices like batteries or catalysts under the microscope [64] [10]. |
Workflow and Decision Pathway for Characterization
The diagram below outlines a logical workflow for selecting and applying characterization techniques in crystal nucleation research.
The comparative analysis underscores that in situ microscopy is not merely an incremental improvement but a paradigm shift in inorganic crystal nucleation research. By providing direct, real-time observation of dynamic processes at the nanoscale, it uncovers critical mechanisms—such as non-classical growth pathways, transient phases, and reaction kinetics—that are inaccessible to ex situ and bulk methods. While the choice of technique must always be guided by the specific research question, the integration of in situ microscopy into the materials science workflow is indispensable for developing a fundamental, mechanistic understanding that will accelerate the rational design of advanced materials for pharmaceuticals, energy storage, and beyond.
Cross-Validation with Scattering Techniques (SAXS, XRD) and Spectroscopy
Within the framework of advanced research into in situ microscopy observation of inorganic crystal nucleation, a multi-technique analytical approach is paramount for developing a complete mechanistic understanding. The cross-validation of data obtained from Small-Angle X-ray Scattering (SAXS), X-ray Diffraction (XRD), and various spectroscopic methods provides a powerful synergy, coupling nanoscale structural evolution with chemical and atomic-level information. SAXS is exceptionally sensitive to electron density fluctuations, making it an ideal tool for probing the formation of pre-nucleation clusters and the subsequent growth of nanoparticles, typically in the size range of 1 to 100 nm [68]. XRD complements this by identifying the emergence of long-range crystalline order and determining phase composition once nuclei have matured beyond a critical size [69]. Meanwhile, spectroscopy yields crucial data on local electronic structure, oxidation states, and molecular bonding. This application note details the protocols and data integration strategies for employing these techniques in tandem, specifically within the context of observing dynamic crystallization processes in situ.
Experimental Protocols and Workflows
Core Experimental Workflow
The following diagram illustrates the integrated workflow for conducting cross-validated in situ studies of inorganic crystal nucleation, from sample preparation through data synthesis.
Protocol 1: In Situ SAXS for Monitoring Pre-Nucleation and Growth
Objective: To characterize the formation and evolution of precursor species and nanoparticles during the early stages of crystallization.
Detailed Methodology:
- Sample Environment: Utilize a microfluidic or milli-fluidic sample cell [70]. These devices enable precise control over reaction conditions, rapid mixing of reagents, and minimize radiation damage during X-ray exposure. The flow path should be designed to provide an X-ray pathlength of approximately 1 mm for aqueous solutions to optimize signal-to-noise ratio [70].
- Reaction Initiation: Load precursor solutions into syringes and use a syringe pump to initiate the reaction. For studies on temperature-dependent nucleation, use an integrated temperature controller. Commercial stages (e.g., Linkam HFSX350, -196 to 350 °C; Anton Paar DHS1100, up to 1100 °C) or custom Joule/Laser heating setups can be employed [68].
- Data Collection: Perform time-resolved SAXS measurements. The required time resolution depends on the kinetics of the process, ranging from minutes for slow crystallization to milliseconds for fast nanoparticle synthesis using continuous-flow mixers [68]. Collect 2D scattering patterns.
- Data Analysis:
- Radially average the 2D patterns to obtain 1D scattering profiles, I(q), where q is the scattering vector.
- In the Guinier region (at low q, where q*Rg < 1), analyze the data to determine the radius of gyration (Rg) of the scattering particles, which provides information about their overall size [71].
- Analyze the intermediate q-range (Porod region) to infer surface-to-volume ratio and morphology.
- Apply appropriate form factor models (e.g., for spheres, rods, core-shell structures) to extract quantitative parameters like particle size distribution and shape [71] [68].
Protocol 2: Combined SAXS/WAXS for Correlating Nanoscale and Atomic Structure
Objective: To simultaneously track the development of nanoscale precursors (via SAXS) and the emergence of crystalline phases (via Wide-Angle X-ray Scattering, WAXS).
Detailed Methodology:
- Beamline Setup: Employ a combined SAXS/WAXS instrument, typically at a synchrotron source, equipped with two detectors to capture the small-angle and wide-angle signals concurrently [71].
- Sample Preparation: Similar to Protocol 1, a microfluidic or capillary-based cell is ideal for in situ studies of liquid-phase crystallization [70].
- Data Collection: Acquire simultaneous SAXS and WAXS patterns with high temporal resolution. For example, a study on LTA-type zeolite formation used this setup to observe ~10 nm precursors prior to the detection of crystalline Bragg peaks [71].
- Data Analysis:
- Process SAXS data as in Protocol 1 to monitor particle size and morphology.
- Integrate WAXS patterns to generate 1D intensity vs. 2θ profiles. Identify Bragg peaks and match them to known crystal structures using databases (e.g., ICDD PDF-4+). Quantitative phase analysis can be performed via Rietveld refinement [72].
- Correlate the timelines from SAXS (growth of nanoscale particles) and WAXS (appearance of crystalline phases) to establish the sequence of events in the crystallization pathway.
Protocol 3: Integrating X-ray Spectroscopy with Scattering
Objective: To link the evolving structural information from scattering with chemical state analysis.
Detailed Methodology:
- Technique Selection:
- Experimental Setup: For in situ studies, a compatible reaction cell (e.g., a microfluidic device with X-ray transparent windows) must be used. Laboratory-based XAFS spectrometers are now available, making this technique more accessible alongside synchrotron measurements [72].
- Data Collection and Analysis:
- Collect XAFS spectra at the absorption edge of the element of interest (e.g., Fe K-edge for iron oxide nucleation).
- Analyze the XANES region to determine average oxidation state by comparing with reference compounds.
- Fit the EXAFS region to extract bond distances, coordination numbers, and species identification via Linear Combination Fitting (LCF) [72].
- Correlate the temporal evolution of chemical species from XAFS with particle size from SAXS and crystalline phase from XRD/WAXS on a common timeline.
Data Presentation and Key Findings
Quantitative Parameters from Scattering and Spectroscopy
Table 1: Key Quantitative Parameters Accessible via Cross-Validated Techniques
| Analytical Technique | Primary Measurable Parameters | Typical Size/Scale Sensitivity | Key Information on Nucleation |
|---|---|---|---|
| SAXS | Radius of gyration (Rg), particle size distribution, shape, surface area, aggregation state [71] [68] | 1 – 100 nm (can extend to ~200 nm) [71] | Formation of pre-nucleation clusters, precursor particle size and morphology, growth kinetics |
| XRD / WAXS | Crystalline phase identification, lattice parameters, crystallite size, strain, quantitative phase abundance [69] [72] | Atomic / Molecular (Å scale); Crystallite size ~1 nm and above | Onset of long-range order, phase transitions, identification of stable vs. metastable crystalline products |
| XAFS (XANES/EXAFS) | Oxidation state, local coordination chemistry, bond distances, coordination numbers, quantitative species analysis via LCF [72] | Atomic / Short-range (0.1-0.5 nm) | Evolution of oxidation states during nucleation, identification of intermediate chemical species |
Illustrative Data from Literature
Table 2: Exemplary In Situ Findings from Cross-Technique Studies
| Material System | SAXS Findings | XRD/WAXS Findings | Spectroscopy Findings | Integrated Conclusion |
|---|---|---|---|---|
| LTA Zeolite | Formation of homogeneous precursor particles of ~10 nm prior to crystallization [71] | Detection of LTA crystal structure; Final crystal size depends on synthesis temperature [71] | (Not reported in source, but could be used to probe Al/Si coordination) | Crystallization proceeds through the assembly of nanoscale homogeneous precursors |
| CoAlPO-5 | Formation of polydispersed precursor particles with an average size of ~50 nm [71] | Crystallization of CoAlPO-5 structure | Effect of organic structure-directing cations on production (suggests chemical interaction) [71] | Crystallization mechanism and precursor properties are influenced by synthesis temperature and organic additives |
| Iron Oxides (e.g., α-Fe2O3/Fe3O4) | (Can be used to track nanoparticle size during growth) | Quantitative phase analysis via Rietveld refinement [72] | Quantitative species analysis via XAFS-LCF; Oxidation state determination [72] | Lab-XAFS can compete with XRD for quantitative species analysis of elemental composition [72] |
| Ordered Mesoporous Carbons (OMC) | In situ GISAXS shows structure formation during thermopolymerization, not evaporation [68] | (Used to confirm final crystalline structure) | (Not reported in source) | Mesostructure formation is driven by thermally induced self-assembly |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Equipment for In Situ Scattering and Spectroscopy Experiments
| Item / Reagent | Function / Application | Key Considerations |
|---|---|---|
| Microfluidic / Milli-fluidic Cells | Sample environment for in situ reaction control; enables rapid mixing, precise temperature control, and mitigates radiation damage [70] | Channel dimensions (millifluidic for better SNR with aqueous solutions); X-ray transparent windows (e.g., Kapton, diamond) [70] |
| Temperature Control Stages | To study temperature-dependent nucleation kinetics and pathways (e.g., Linkam, Anton Paar stages) [68] | Range (e.g., -196°C to 1600°C); Heating/Cooling rate (up to 1.5·10⁴ K/s with conductive cooling); Atmosphere control [69] [68] |
| Synchrotron Beamline Access | Provides high-flux X-rays for time-resolved SAXS/WAXS/XAFS measurements with millisecond-to-second resolution [71] [70] | Requires competitive proposal; Offers specialized setups for combined techniques (SAXS/WAXS, XAFS/XRD) [71] |
| Laboratory XAFS Spectrometer | Enables element-specific oxidation state and speciation analysis in a home laboratory, complementing lab XRD [72] | Emerging technology; Uses von Hamos geometry with mosaic crystals for efficient dispersion [72] |
| Reference Compounds (High Purity) | Essential for quantitative spectroscopy (XAFS Linear Combination Fitting) and phase identification in XRD [72] | Purity and structural characterization are critical for accurate quantification and model validation |
The cross-validation of scattering and spectroscopic techniques provides an unparalleled, multi-dimensional view of the inorganic crystal nucleation process. By simultaneously probing nanoscale structure, crystalline phase, and chemical state, researchers can move beyond simple observational studies to construct predictive, mechanistic models of crystallization. The protocols and data integration strategies outlined here provide a robust framework for designing in situ experiments that can decode the complex pathways from dissolved ions to mature crystals, with significant implications for materials design, pharmaceutical development, and industrial process optimization.
This application note details how in situ microscopy techniques, particularly liquid-phase transmission electron microscopy (TEM), are revolutionizing our understanding of inorganic crystal nucleation by directly capturing transient intermediates that evade conventional offline methods. We provide a comparative analysis of observational capabilities, detailed protocols for real-time experimentation, and a toolkit for implementing these advanced techniques in materials science and pharmaceutical development research.
Classical nucleation theory posits that crystals form through a simple, two-step process of nucleation and monomer-by-monomer growth. However, this model fails to account for the complex, multi-pathway crystallization processes observed in many inorganic and soft material systems. For decades, our understanding was limited because conventional offline methods—which involve extracting samples at various time points for ex situ analysis—inherently miss short-lived, non-equilibrium states [73].
The advent of in situ microscopy has fundamentally altered this landscape. By allowing researchers to observe dynamic processes in real-time within their native environments, these techniques have uncovered a rich variety of non-classical crystallization pathways, including the formation of pre-nucleation clusters, dense liquid phases, and particle-based attachment mechanisms that were previously undetectable [73]. This case study examines how these transient intermediates are finally being revealed.
Comparative Analysis: Offline vs. Real-Time Observation Capabilities
Table 1: Capabilities of Offline Methods versus Real-Time In Situ Observation for Detecting Transient Crystallization Intermediates
| Feature/Aspect | Conventional Offline Methods | Real-Time In Situ Observation |
|---|---|---|
| Temporal Resolution | Low (snapshots at fixed intervals) | High (milliseconds to seconds) [23] |
| Spatial Resolution | Atomic to microscale (post-process) | Nanoscale to microscale in liquid/real conditions [73] |
| Key Intermediates Detected | Stable crystalline phases | Pre-nucleation clusters, dense liquid phases, amorphous precursors, particle attachment events [73] |
| Primary Techniques | Ex situ TEM, XRD, SEM | Liquid-phase TEM (LP-TEM), in situ spectroscopy [23] |
| Data Type | Static, discontinuous | Dynamic, continuous, time-resolved |
| Information on Kinetics | Inferred | Directly measured |
| Major Limitation | Misses transient, non-equilibrium states | Potential for electron beam effects on sensitive materials [73] |
Table 2: Quantitative Data on Transient Intermediates Revealed by Real-Time Observation
| Transient Intermediate | Example Material System | Key Characteristic | Observation Technique |
|---|---|---|---|
| Pre-nucleation Clusters | Calcium carbonate, organic molecules [73] | Stable ion associations preceding nucleation; size: 1-2 nm [73] | In situ liquid-phase TEM |
| Dense Liquid Phases | Proteins, small organic molecules [73] | Liquid-like droplets acting as nucleation precursors [73] | In situ liquid-phase TEM |
| Amorphous Precursors | Cerium oxalate, calcium phosphate | Metastable solid particles that transform into crystals [73] | In situ microscopy |
| Oriented Attachment | Metal oxides, semiconductors | Crystallographic alignment and fusion of nanoparticles [73] | In situ liquid-phase TEM |
| Mesocrystals | Various organic & inorganic systems | 3D ordered superstructures of crystallographically aligned nanoparticles [73] | In situ microscopy |
Experimental Protocols
Protocol 1: Direct Observation of Non-Classical Crystallization via Liquid-Phase TEM
Principle: Liquid-phase TEM (LP-TEM) enables direct, real-time visualization of crystallization events within a native liquid environment at nanoscale resolution [73] [23].
Materials & Equipment:
- In situ liquid cell holder for TEM
- Microfabricated silicon liquid cell with viewing windows
- Syringe pump for controlled reagent injection
- High-speed, high-sensitivity TEM camera (e.g., Gatan K3 IS camera) [23]
- Aqueous solution of precursor ions/species
Procedure:
- Cell Assembly: Load the liquid cell with the precursor solution, ensuring no air bubbles are trapped in the viewing area.
- Holder Insertion: Carefully insert the sealed liquid cell holder into the TEM column, following manufacturer protocols.
- Imaging Parameter Optimization:
- Data Acquisition: Initiate video recording simultaneously with the onset of reaction, often triggered by mixing reagents or changing environmental conditions (e.g., temperature) within the cell.
- Data Processing: Analyze the recorded video sequence to track the formation, evolution, and disappearance of transient species. Apply post-processing algorithms (e.g., drift correction, frame summing) to enhance signal-to-noise ratio [23].
Protocol 2: In Situ Identification of Pre-Nucleation Clusters
Principle: This protocol focuses on detecting and characterizing the small, stable associations of ions or molecules that exist in solution before the appearance of a stable crystalline nucleus [73].
Materials & Equipment:
- In situ liquid cell TEM holder
- Analytical TEM capable of performing Electron Energy Loss Spectroscopy (EELS) or Energy Dispersive X-ray Spectroscopy (EDS)
- Solution containing pre-nucleation species (e.g., 1-10 mM calcium bicarbonate for CaCO₃ studies)
Procedure:
- Solution Preparation: Prepare a metastable supersaturated solution and allow it to equilibrate.
- Spectral Mapping: Acquire a series of spectrum images (e.g., EELS or EDS) over time at a fixed location within the liquid cell. The Gatan GIF Continuum upgrade, for example, enables such in-situ EELS spectrum imaging with real-time quantification [23].
- Cluster Identification: Analyze the spectral data to identify regions with consistent elemental or bonding signatures that differ from the bulk solution but do not exhibit crystalline order.
- Stability Assessment: Monitor these regions over time to determine their lifetime and track their evolution into amorphous nanoparticles or direct crystalline phases.
Figure 1: Experimental workflow for observing transient crystallization intermediates using in situ liquid-phase TEM.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for In Situ Crystallization Studies
| Item/Reagent | Function/Application | Example Use Case |
|---|---|---|
| Microfabricated Liquid Cell | Encapsulates liquid sample between electron-transparent windows (e.g., SiNₓ) for TEM observation. | Provides a native liquid environment for observing crystal growth in real time [73]. |
| In Situ TEM Holder | Holds the liquid cell and interfaces with the TEM, often allowing for control of temperature, electrical bias, or fluid flow. | Enables the application of external stimuli during observation [23]. |
| Low-Dose Imaging Software | Minimizes the electron dose exposed to the sample to reduce beam-induced artifacts. | Essential for studying beam-sensitive soft materials and organic crystals [73] [23]. |
| High-Speed Direct Electron Detector | Captures high-resolution images at video rates with high signal-to-noise ratio. | Allows for the clear resolution of fast dynamics, such as nanoparticle attachment events [23]. |
| Programmable Syringe Pump | Precisely controls the injection and mixing of reagents within the liquid cell. | Used to initiate crystallization reactions or change solution conditions during observation. |
| Pre-nucleation Cluster Stabilizers | Additives (e.g., certain polymers or ions) that stabilize transient intermediates for longer observation windows. | Facilitates the detailed spectroscopic and structural analysis of pre-nucleation species [73]. |
Data Interpretation and Pathway Analysis
The data gathered from in situ experiments reveals that crystallization often proceeds through complex energy landscapes rather than a single-pathway mechanism. The discovery of multiple, parallel routes to the final crystalline state necessitates a re-evaluation of crystal formation kinetics and control strategies.
Figure 2: Complex crystallization pathways revealed by real-time observation, showing multiple non-classical routes involving transient intermediates (red) that lead to final crystalline products (green). The dashed lines represent less frequently observed pathways [73].
The implementation of real-time observation techniques, chiefly in situ liquid-phase TEM, has unequivocally demonstrated that the pathway from dissolved ions to a mature crystal is far more complex and fascinating than classical models suggested. The consistent detection of transient intermediates like pre-nucleation clusters and amorphous precursors across diverse material systems underscores their critical role in crystallization. For researchers in inorganic chemistry and drug development, embracing these in situ methodologies is no longer optional for cutting-edge research; it is essential for gaining a complete, mechanistic understanding of crystallization that enables precise control over material properties and product outcomes.
The transition from laboratory-scale observation to industrial-scale production presents a significant challenge in materials science, particularly in the field of inorganic crystal nucleation research. This protocol details a structured approach for assessing the predictive power of laboratory data for successful industrial scale-up, with specific application to the synthesis of novel inorganic crystalline materials such as helical GaSI crystals [74] and cobalt-based nanoparticles [54]. We present a comprehensive framework integrating advanced characterization techniques, quantitative data analysis, and scale-up methodologies to bridge the gap between fundamental research and industrial application, enabling researchers and drug development professionals to make informed decisions based on laboratory observations.
Theoretical Framework and Scale-up Methodology
Scale-up Decision Framework
Prospective scaling requires robust methodologies to translate laboratory data into industrial process predictions. Table 1 summarizes the primary scaling methods identified through systematic review of Life Cycle Assessment (LCA) studies [75].
Table 1: Scaling Methods for Prospective Technological Assessment
| Method Category | Specific Methods | Technology Applicability | Data Requirements | Uncertainty Handling |
|---|---|---|---|---|
| Engineering Models | Process simulation, Stoichiometric calculations | Chemical processes, Reactor design | High (detailed process parameters) | Sensitivity analysis |
| Mathematical Scaling | Power law correlations, Dimensional analysis | Particle systems, Fluid processes | Medium (key performance parameters) | Parameter variation |
| Similarity-based | Geometric, kinematic, dynamic similarity | Equipment scale-up | Medium to High (benchmark data) | Comparative analysis |
| Statistical | Regression analysis, Machine learning | Diverse systems with historical data | Varies with model complexity | Confidence intervals, Prediction bounds |
| Hybrid Approaches | Combined engineering and statistical methods | Complex systems with partial mechanistic understanding | Medium to High | Integrated uncertainty propagation |
An Excel-based decision tool has been developed to assist researchers in selecting the most appropriate scaling methodology based on evaluation criteria including complexity, data intensity, duration, and uncertainty [75]. This tool enables customization according to specific project constraints and data availability.
DBTL Cycle for Process Optimization
The Design-Build-Test-Learn (DBTL) framework provides an effective iterative approach for strain engineering and process optimization that can be adapted for crystal nucleation and growth processes [76]. This framework is particularly valuable for managing the complexity of biological systems and can be extended to inorganic crystallization processes:
- Design: Strategies span from rational (defined specific edits) to semi-rational (hypothesis-driven targets) to random approaches (chemical mutagenesis), with selection informed by phenotyping capacity and hypothesis confidence [76].
- Build: Comprehensive process engineering typically requires multiple tools to introduce diverse edit types, with tradeoffs between throughput, cost, edit precision, and genomic accessibility [76].
- Test: Encompasses phenotyping methods and workflows for connecting genotype to associated phenotype.
- Learn: Computational tools analyze collected data to draw conclusions and make predictions about which genetic changes achieve strain improvement goals [76].
Experimental Protocols
In Situ Electron Microscopy for Nucleation Observation
Protocol: Real-time observation of nucleation and growth processes using Environmental Cell TEM (EC-TEM) [54]
This protocol enables direct visualization of nanocrystal formation under solvothermal conditions, providing unprecedented insights into reaction mechanisms and how confinement fundamentally changes thermodynamic and kinetic pathways.
3.1.1 Materials and Equipment
- Transmission Electron Microscope equipped with environmental cell (EC-TEM) capability
- Carbon nanotubes (CNTs) as confinement templates
- Cobalt stearate precursor (or other metal carboxylate)
- Oleic acid (capping agent)
- Organic solvent (e.g., octadecene)
- Heating holder capable of reaching 300°C
- Microfabricated liquid cells with heating capability
3.1.2 Procedure
Sample Preparation
- Prepare reaction mixture: 0.1 M cobalt stearate, 0.2 M oleic acid in octadecene
- For confinement studies: Introduce CNTs to reaction mixture at 1 mg/mL concentration
- Load mixture into microfabricated liquid cell using capillary action
- Seal cell to contain solvent at elevated temperatures
EC-TEM Setup
- Mount liquid cell in heating holder
- Align electron microscope for high-resolution imaging (200 kV)
- Set up video recording capability for time-series documentation
- Calibrate temperature settings for precise control
In Situ Thermal Decomposition
- Ramp temperature from room temperature to 320°C at 10°C/min
- Maintain at 320°C for nanoparticle growth phase
- Record real-time nucleation events at 10 frames/second
- Focus on liquid-gas interfaces and CNT confinement regions
Data Collection
- Document nucleation initiation points and timing
- Measure critical nucleus size (typically 4-5 nm for cobalt-based NPs)
- Track growth rates for confined vs. unconfined nanoparticles
- Characterize final nanoparticle morphology and structure
3.1.3 Key Observations
- Nucleation initiates preferentially at liquid-gas interfaces in vesicle "walls" with higher monomer concentration [54]
- Critical nucleus size of 4-5 nm observed before chain-like structure formation [54]
- Confined NPs within CNTs exhibit homogeneous size (~50 nm) and octahedral morphology [54]
- External NPs show random morphologies and smaller sizes (~20 nm) with oxidized Co3O4 layer [54]
- Anisotropic growth observed along CNT direction during synthesis [54]
Quantitative Analysis of Nucleation Parameters
Protocol: Statistical assessment of predictive laboratory parameters [77]
This protocol provides a standardized approach for comparing quantitative data between experimental conditions to identify parameters with strongest predictive power for industrial performance.
3.2.1 Data Collection
Laboratory-scale Metrics
- Nucleation rate (events/unit time)
- Crystal growth rate (μm/hour)
- Final crystal size distribution
- Purity and yield measurements
- Morphological consistency
Industrial-scale Performance Indicators
- Batch-to-batch consistency
- Scalability factor (performance maintenance at scale)
- Process efficiency (yield/time/energy)
- Product quality specifications
3.2.2 Statistical Analysis
Graphical Comparison
Numerical Summaries
- Compute means and medians for each group
- Calculate differences between group means/medians
- Determine standard deviations and interquartile ranges
Figure 1: Predictive Power Assessment Workflow
Data Presentation and Analysis
Quantitative Comparison of Confined vs. Unconfined Nanoparticle Synthesis
Table 2 presents experimental data comparing the characteristics of cobalt-based nanoparticles synthesized inside carbon nanotubes (confined) versus on their external surfaces (unconfined), demonstrating the significant impact of confinement on final nanoparticle properties [54].
Table 2: Quantitative Comparison of Confined vs. Unconfined Nanoparticle Characteristics [54]
| Parameter | Confined NPs (inside CNTs) | Unconfined NPs (external) | Difference | Industrial Significance |
|---|---|---|---|---|
| Size Distribution | Homogeneous (~50 nm) | Heterogeneous (~20 nm core) | +30 nm | Improved batch uniformity |
| Morphology | Regular octahedral | Random, irregular | Crystalline faceting | Enhanced material properties |
| Crystal Structure | Co-CoO monocrystalline | Co3O4 oxidized layer | Structural purity | Predictable performance |
| Size Control | CNT diameter dependent | Precursor concentration dependent | Anisotropic growth | Design flexibility |
| Sintering Resistance | High (stabilized by walls) | Moderate (sintering observed) | Improved stability | Longer catalyst lifetime |
| Faceting Analysis | Elongated octahedral (anisotropic) | Multifaceted, irregular | Controlled growth direction | Tailored surface properties |
Statistical Data Comparison Methods
When comparing quantitative data between different experimental conditions or scale-up stages, appropriate statistical methods must be employed [77]:
- For two groups: Compute the difference between means and/or medians
- For multiple groups: Calculate differences between one group mean/median (benchmark) and other group means/medians
- Graphical representation: Use back-to-back stemplots (two groups), 2-D dot charts (multiple groups), or boxplots (distribution comparison) [77]
Table 3 illustrates a summary table format for comparing quantitative data across different groups, as demonstrated in gorilla chest-beating research but applicable to crystal nucleation metrics [77].
Table 3: Statistical Summary Format for Group Comparisons (Adapted from [77])
| Group | Mean | Median | Std. Dev. | Sample Size |
|---|---|---|---|---|
| Laboratory-scale | 2.22 | 1.70 | 1.270 | 14 |
| Pilot-scale | 0.91 | 0.85 | 1.131 | 11 |
| Difference | 1.31 | 0.85 | - | - |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for In Situ Crystallization Studies
| Reagent/Material | Function | Application Example | Industrial Relevance |
|---|---|---|---|
| Carbon Nanotubes (CNTs) | Confinement template | Nanoparticle synthesis with controlled morphology [54] | Catalyst support, confined reactors |
| Porous Nucleating Agents | Heterogeneous nucleation inducers | Protein crystallization optimization [78] | Controlled crystal initiation |
| Cobalt Stearate | Metal carboxylate precursor | Thermal decomposition synthesis of Co-based NPs [54] | Metal oxide nanoparticle production |
| Oleic Acid | Capping agent/ligand | Surface stabilization during NP growth [54] | Size and morphology control |
| Environmental Cell TEM Holders | In situ reaction monitoring | Real-time visualization of nucleation [54] | Process optimization and understanding |
| Bioactive Glasses | Nucleation-inducing material | Crystallization facilitation [78] | Controlled material synthesis |
| Molecularly Imprinted Polymers | Template-controlled nucleation | Protein crystallization facilitation [78] | Selective crystallization processes |
Figure 2: Laboratory to Industrial Parameter Relationships
This protocol establishes a comprehensive framework for assessing the predictive power of laboratory observations for industrial process scale-up in inorganic crystal nucleation research. Through the integration of advanced in situ characterization techniques, quantitative data analysis, and systematic scale-up methodologies, researchers can significantly improve the reliability of translating nanoscale observations to industrial-scale production. The application of these protocols to specific case studies involving helical GaSI crystals [74] and confined nanoparticle synthesis [54] demonstrates the critical importance of understanding fundamental nucleation and growth mechanisms for successful technological implementation. Future developments in high-throughput characterization and machine learning-assisted data analysis promise to further enhance our predictive capabilities in materials design and manufacturing optimization.
Quantifying Improvements in Crystal Size Distribution, Polymorph Control, and Purity
Within the field of inorganic crystal nucleation research, achieving precise control over Crystal Size Distribution (CSD), polymorphic form, and purity is paramount for determining the functional properties of crystalline materials. These characteristics influence critical aspects such as drug bioavailability, product stability, filterability, and performance in technological applications like optoelectronics [79] [80]. The inherent challenge of crystallization lies in its multiparametric nature, where factors like supersaturation, mixing, and temperature interact in complex ways, making traditional offline analysis insufficient for capturing transient process dynamics [43] [81].
The adoption of Process Analytical Technology (PAT) represents a paradigm shift, enabling real-time, in-situ monitoring and control. This application note details how integrating advanced PAT tools, specifically in-situ microscopy and spectroscopy, with targeted experimental protocols provides researchers with a quantitative framework to optimize crystallization processes. We demonstrate measurable improvements in key crystallization outcomes, framed within the context of inorganic materials research.
Quantitative Data on Crystallization Improvements
The implementation of in-situ monitoring and controlled crystallization strategies yields significant, quantifiable enhancements. The tables below summarize documented improvements in CSD, polymorph control, and purity across various studies.
Table 1: Documented Improvements in Crystal Size Distribution (CSD)
| System/Method | Key Parameter | Result | Citation |
|---|---|---|---|
| SNP·2H2O / Antisolvent | Target Crystal Size | Achieved microcrystals of (50 ± 10) μm, tailored for photocrystallography. | [80] |
| SNP·2H2O / Slow Evaporation | Crystal Size Range | Produced long, lath-like crystals from 100 to 1500 μm with broad distribution. | [80] |
| LbADH / In-situ Microscopy | Monitoring Capability | Real-time estimation of crystal volume, kinetics, and CSD correlated with offline protein concentration. | [44] |
| General Crystallization | CSD Expansion | Theoretical analysis confirms CSD continues to expand during the crystal growth stage. | [79] |
Table 2: Documented Improvements in Polymorph Control and Purity
| System/Method | Key Parameter | Result | Citation |
|---|---|---|---|
| ROY / Acoustic Cavitation | Polymorph Selectivity (Batch) | Sonication promoted the formation of the stable Y polymorph over other forms. | [81] |
| ROY / Silent Crystallization | Polymorph Selectivity | Outcome influenced by local supersaturation and mixing, leading to mixed polymorphs. | [81] |
| CaCO3 / In-situ Raman | Phase Transformation Monitoring | Real-time tracking of amorphous (ACC) → vaterite → calcite transformation. | [82] |
| LbADH / In-situ Microscopy | Detection Specificity | Superior to offline data in distinguishing crystals from amorphous precipitation. | [44] |
Experimental Protocols
The following protocols provide detailed methodologies for implementing key in-situ techniques to quantify crystallization improvements.
Protocol: In-Situ Microscopy for CSD and Growth Kinetics Analysis
This protocol outlines the use of in-situ microscopy (ISM) for real-time monitoring of crystal size, distribution, and morphology in a stirred crystallizer, adapted for inorganic systems [43] [44].
1. Equipment and Reagents:
- Crystallizer: Jacketed, stirred tank reactor (e.g., 0.5 L to 1 L volume) with temperature control.
- In-Situ Microscope: Sterilizable, focusable probe (e.g., Sartorius Stedim Biotech ISM type III-XTF) mounted in a standard reactor side-port.
- Image Analysis Software: Capable of real-time image processing and feature extraction (e.g., using machine learning for crystal detection).
- Chemicals: Inorganic compound of interest (e.g., Ammonium sulfate, Potassium sulfate); solvents (e.g., deionized water); antisolvents if applicable.
2. Experimental Procedure:
- Step 1: Setup. Install the ISM probe in the crystallizer side-port. Connect the probe to the light source and camera controller. Calibrate the microscope's magnification and focus using a calibration slide.
- Step 2: Solution Preparation. Prepare a supersaturated solution of the target inorganic compound. This can be achieved by dissolution at elevated temperature followed by cooling, or by antisolvent addition.
- Step 3: Process Initiation and Monitoring. Transfer the supersaturated solution to the crystallizer. Initiate stirring at a defined rate (e.g., 300 rpm) and temperature control. Start the ISM system to acquire images at regular intervals (e.g., every 30 seconds) directly from the crystallization slurry.
- Step 4: Image Analysis. Use the integrated software to automatically analyze images in real-time. The algorithm should:
- Identify crystal contours and separate overlapping particles.
- For each crystal, measure key dimensions (e.g., length, width, area).
- Calculate real-time CSD, mean crystal size, and crystal count.
- Step 5: Data Correlation. Correlate the estimated total crystal volume from image analysis with offline measurements (e.g., solute concentration in solution) to track yield and kinetics [44].
3. Key Measurements and Outcomes:
- Quantitative CSD: Obtain temporal evolution of crystal size histograms.
- Growth Kinetics: Determine crystal growth rates by tracking size increase of individual crystals over time.
- Morphology Identification: Distinguish between different crystal habits and detect the presence of amorphous solids.
Protocol: In-Situ Raman Spectroscopy for Polymorph Evolution
This protocol describes the use of in-situ Raman spectroscopy to monitor and control polymorphic transformations in real-time, as demonstrated in CaCO3 precipitation [82].
1. Equipment and Reagents:
- Reactor: Jacketed batch reactor (e.g., 500 mL) with overhead agitation.
- Raman Spectrometer: Equipped with a immersion probe (e.g., Blaze Metrics probe coupled to a Wasatch spectrometer).
- PAT Software: For spectral acquisition and multivariate analysis.
- Chemicals: Calcium chloride (CaCl2), sodium carbonate (Na2CO3) or dissolved CO2 as a carbonate source; deionized water.
2. Experimental Procedure:
- Step 1: System Configuration. Place the Raman immersion probe into the reactor, ensuring it is immersed in the reaction mixture during operation. Set the Raman spectrometer to collect spectra at defined intervals (e.g., every 1 minute).
- Step 2: Baseline Acquisition. Collect Raman spectra of the pure solvent (water) and individual reactant solutions (e.g., CaCl2 solution) to establish a spectral baseline.
- Step 3: Nucleation and Growth. Initiate the crystallization reaction (e.g., by adding the carbonate source to the calcium solution under controlled dosing and mixing). Begin continuous Raman monitoring.
- Step 4: Spectral Identification. Identify characteristic Raman peaks for different polymorphs. For CaCO3, key bands are:
- Vaterite: Peak at ~1090 cm⁻¹ with a characteristic split.
- Calcite: Sharp peak at ~1088 cm⁻¹.
- Amorphous Calcium Carbonate (ACC): Broad, featureless peaks in the same region [82].
- Step 5: Kinetic Profiling. Monitor the intensity of polymorph-specific peaks over time to quantify the phase transformation kinetics (e.g., the decline of vaterite and concurrent rise of calcite).
3. Key Measurements and Outcomes:
- Polymorph Identity: Confirm the presence and sequence of appearing polymorphs.
- Transformation Kinetics: Determine the rate of transformation from metastable to stable phases.
- Process Control: Use the real-time data to trigger process interventions (e.g., temperature change, additive introduction) to isolate a desired metastable polymorph.
Protocol: Controlled Microcrystallization via Antisolvent Addition
This protocol describes a controlled antisolvent method to produce narrow CSDs of microcrystals with a defined habit, optimized for functional inorganic materials like sodium nitroprusside dihydrate (SNP·2H2O) [80].
1. Equipment and Reagents:
- Reaction Vessel: Small scale (e.g., 5-10 mL) vial or flask with magnetic stirring.
- Syringe Pump: For controlled addition of antisolvent.
- Microscope: For offline verification of crystal size and habit.
- Chemicals: Inorganic compound (e.g., SNP·2H2O), solvent (e.g., deionized water), antisolvent (e.g., acetonitrile).
2. Experimental Procedure:
- Step 1: Solution Preparation. Prepare a saturated solution of the inorganic compound in the primary solvent (e.g., water for SNP·2H2O) at room temperature.
- Step 2: Antisolvent Addition. Place a known volume of the saturated solution in the vessel under constant stirring. Using a syringe pump, add the antisolvent (e.g., acetonitrile) at a slow, controlled rate (e.g., 0.1 mL/min).
- Step 3: Nucleation Induction. The controlled addition of antisolvent steadily increases supersaturation, inducing a homogeneous nucleation event. The slow rate prevents excessive nucleation and promotes uniform growth.
- Step 4: Quenching and Isolation. Once the desired antisolvent volume is added, continue stirring for a short period to allow for growth. Then, filter the suspension to isolate the crystals.
- Step 5: Analysis. Analyze the resulting crystals using microscopy to determine the crystal size distribution and habit. Compare with crystals obtained from uncontrolled slow evaporation.
3. Key Measurements and Outcomes:
- Narrow CSD: Achieve a target microcrystal size with low variance (e.g., 50 ± 10 μm).
- Habit Control: Produce a uniform crystal habit (e.g., plates) suitable for downstream applications like serial crystallography.
- Reproducibility: High batch-to-batch reproducibility by controlling the supersaturation profile.
Workflow and Signaling Pathways
The following diagram illustrates the integrated experimental and data analysis workflow for using in-situ microscopy to control crystallization processes, from setup to quantitative outcome.
The Scientist's Toolkit: Research Reagent Solutions
The table below lists key materials and instruments essential for implementing the described in-situ crystallization control strategies.
Table 3: Essential Research Reagents and Equipment for In-Situ Crystallization Studies
| Item | Function / Application | Specific Example |
|---|---|---|
| In-Situ Microscope (ISM) | Non-invasive, real-time imaging of crystals in slurry for CSD and morphology. | Sartorius Stedim Biotech ISM probe [43] [44]. |
| In-Situ Raman Spectrometer | Real-time identification and monitoring of polymorphic forms and phase transformations. | Blaze Metrics probe with Wasatch spectrometer [82]. |
| Stirred Crystallizer Reactor | Provides controlled environment (mixing, temperature) for scalable crystallization studies. | Jacketed glass reactor (e.g., Chemglass) [82] [44]. |
| Acoustic Cavitation Setup | Applies ultrasound to influence nucleation, polymorph selection, and reduce agglomeration. | Ultrasonic transducer with amplifier and signal generator [81]. |
| Syringe Pump | Enables precise, controlled addition of antisolvent or reactants for reproducible supersaturation. | For controlled microcrystallization protocols [80]. |
| Model Compounds | Well-characterized systems for method development and validation. | ROY (polymorphs), SNP·2H2O (microcrystallization), CaCO3 (polymorph transformation) [81] [80] [82]. |
Conclusion
In situ microscopy has fundamentally changed our understanding of inorganic crystal nucleation by providing direct, real-time visualization of processes once considered theoretical. The synergy of advanced TEM, SEM, and optical methods now allows researchers to move from inferring mechanisms to directly observing them, leading to more predictable control over crystal properties critical for pharmaceutical applications. Future progress hinges on overcoming current limitations in temporal resolution and electron beam effects, with emerging trends pointing toward integration with machine learning for automated analysis and the development of more sophisticated multi-modal correlative platforms. For drug development professionals, these advancements promise a new era of rational crystal engineering, enabling the design of materials with tailored bioavailability, stability, and performance characteristics, ultimately accelerating the development of more effective therapeutics.
References
- 1. Crystal nucleation: Rare made common and captured by Raman [https://pmc.ncbi.nlm.nih.gov/articles/PMC9173772/]
- 2. Non-classical nucleation in vapor–liquid–solid growth of ... [https://www.nature.com/articles/s41598-021-01666-9]
- 3. Discovery of a liquid crystal phase of sodium halides via ... [https://www.sciencedirect.com/science/article/pii/S2772834X25000727]
- 4. Competing nucleation pathways in nanocrystal formation [https://www.nature.com/articles/s41524-024-01371-x]
- 5. New insights into the non-classical nucleation of C-S-H [https://www.sciencedirect.com/science/article/abs/pii/S0008884623000479]
- 6. Extension to Inorganic Salts [https://www.sciencedirect.com/science/article/pii/S0263876225005064]
- 7. Nucleation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2995260/]
- 8. In-situ/operando study of Cu-based nanocatalysts for CO 2 ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1525245/full]
- 9. Classical nucleation theory [https://en.wikipedia.org/wiki/Classical_nucleation_theory]
- 10. In situ transmission electron microscopy characterization and ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc01214g]
- 11. Molecularly resolved mapping of heterogeneous ice ... [https://www.nature.com/articles/s41467-025-62900-w]
- 12. Determining the nucleation parameters for the same solute ... [https://www.sciencedirect.com/science/article/abs/pii/S0022024825002052]
- 13. Nucleation and Crystal Growth: Recent Advances and ... [https://www.mdpi.com/2673-4591/56/1/22]
- 14. In situ experimental mechanics of nanomaterials at the ... [https://www.nature.com/articles/am201270]
- 15. In situ Observation of Structural Evolution and Phase ... [https://pubmed.ncbi.nlm.nih.gov/36222376/]
- 16. Quantitative studies of crystal nucleation at constant ... [https://pubs.rsc.org/en/content/articlehtml/2014/ce/c4ce00344f]
- 17. A practical protocol for correlative confocal fluorescence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12211028/]
- 18. GaSI: A Wide-Gap Non-centrosymmetric Helical Crystal [https://pmc.ncbi.nlm.nih.gov/articles/PMC11345776/]
- 19. In situ electron microscopy: atomic-scale dynamics of metal ... [https://www.nature.com/articles/s41529-025-00568-9]
- 20. A robust crystal structure prediction method to support ... [https://www.nature.com/articles/s41467-025-57479-1]
- 21. Organic crystal structure prediction via coupled generative ... [https://www.sciencedirect.com/science/article/pii/S266667582300190X]
- 22. In Situ Electron Microscopy [https://www.thermofisher.com/us/en/home/materials-science/in-situ-experimentation.html]
- 23. In situ [https://www.gatan.com/techniques/situ]
- 24. Ex-situ and in-situ S/TEM study of bainitic ferrite nucleation ... [https://www.sciencedirect.com/science/article/pii/S135964542500672X]
- 25. In Situ TEM Studies of Catalysts Using Windowed Gas Cells [https://www.mdpi.com/2073-4344/10/7/779]
- 26. Exploring Biomineralization Processes Using In Situ Liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11735904/]
- 27. In Situ Electron Microscopy for Materials Science [https://www.protochips.com/solutions/by-applications/materials-science/]
- 28. Early Detection of Nucleation Events From Solution in LC- ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.818230/full]
- 29. In-situ preparation of plant samples in ESEM for energy ... [https://www.nature.com/articles/s41598-019-38835-w]
- 30. In situ observation of oscillatory redox dynamics of copper [https://www.nature.com/articles/s41467-020-17346-7]
- 31. Lessons from In Situ SEM/TEM Growth to Large Scale ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10767701/]
- 32. In situ microscopy using adjustment-free optics [https://www.spiedigitallibrary.org/journals/journal-of-biomedical-optics/volume-20/issue-11/116007/In-situ-microscopy-using-adjustment-free-optics/10.1117/1.JBO.20.11.116007.full]
- 33. Light Microscopy in the Chemistry Laboratory [https://www.americanlaboratory.com/914-Application-Notes/726-Light-Microscopy-in-the-Chemistry-Laboratory/]
- 34. Visualizing Cells through Microscopy [https://open.oregonstate.education/cellbiology/chapter/microscopy/]
- 35. Real-time label-free imaging of living crystallization-driven ... [https://www.nature.com/articles/s41467-025-57776-9]
- 36. Deep Learning Analysis of Crystallization Using Polarized ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12516727/]
- 37. Precise targeting for 3D cryo-correlative light and electron ... [https://www.nature.com/articles/s42003-023-04887-y]
- 38. of C60(C(COOH) 2) 2 interacting with living cells... In situ observation [https://link.springer.com/article/10.1007/s11434-006-1060-1]
- 39. Correlative Fluorescence Microscopy and Scanning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2919632/]
- 40. Correlative Cryo-electron Tomography and Optical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3812453/]
- 41. Mapping in situ the assembly and dynamics in aqueous ... [https://www.nature.com/articles/s41467-025-60138-0]
- 42. Microscopy techniques for investigating the control of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4922639/]
- 43. Adaptation of in-situ microscopy for crystallization processes [https://www.sciencedirect.com/science/article/abs/pii/S0022024809006551]
- 44. In Situ Microscopy with Real-Time Image Analysis Enables ... [https://www.mdpi.com/2073-4352/14/12/1009]
- 45. Controlling metal–organic framework crystallization via ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d5ta03199k]
- 46. Encapsulation of Metal Nanoparticles by Metal–Organic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224958/]
- 47. Non-classical crystallisation pathway directly observed for ... [https://www.nature.com/articles/s41598-020-75937-2]
- 48. Electron Beam Radiation as a Safe Method for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9325154/]
- 49. Comprehensive stabilization mechanism of electron-beam ... [https://www.nature.com/articles/srep27330]
- 50. Two types of amorphous protein particles facilitate crystal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5338535/]
- 51. In-Situ (S)TEM/DTEM: From High Spatial Resolution to High ... [https://materialscience.uoregon.edu/in-situ-stemdtem-from-high-spatial-resolution-to-high-temporal-resolution/]
- 52. Nanosecond time-resolved investigations using the in situ ... [https://www.sciencedirect.com/science/article/abs/pii/S0304399108001794]
- 53. Current status and future directions for in situ transmission ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5100813/]
- 54. 3D and in situ electron microscopy studies of the nucleation ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr01112d]
- 55. Unravelling complex mechanisms in materials processes with ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc05188b]
- 56. Imaging of Hydrated and Living Cells in Transmission ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11984313/]
- 57. Quantitative Statistical Methods for Image Quality Assessment [https://pmc.ncbi.nlm.nih.gov/articles/PMC3840409/]
- 58. Tutorial on In Situ and Operando (Scanning) Transmission ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11697340/]
- 59. Direct Visualization and Quantification of the Actin Nucleation and... [https://bio-protocol.org/en/bpdetail?id=2146&type=0]
- 60. Optimizing Sample Preparation for In situ TEM Heating ... [https://www.sciencedirect.com/science/article/abs/pii/S096843282500109X]
- 61. Real‐Time Visualization of Calcium Phosphate Formation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12632435/]
- 62. Articles - AMT - Copernicus.org [https://amt.copernicus.org/articles/18/5223/2025/]
- 63. Nucleation and phase transformation pathways in ... [https://www.sciencedirect.com/science/article/abs/pii/S1359029417301334]
- 64. In Situ and Ex Situ TEM Study of Lithiation Behaviours ... [https://www.nature.com/articles/srep31334]
- 65. In Situ SEM, TEM, EBSD Characterization of Nucleation ... [https://www.mdpi.com/1996-1944/16/17/6022]
- 66. In operando and in situ characterization tools for advanced ... [https://www.sciencedirect.com/science/article/pii/S0378775325020245]
- 67. Ex situ and in situ characterization of patterned ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4286208/]
- 68. In situ capabilities of Small Angle X-ray Scattering [https://www.degruyterbrill.com/document/doi/10.1515/ntrev-2019-0032/html?lang=en&srsltid=AfmBOop-0YvwEBk-inYQtjDFz92PVHQFzi1PvPOEUV3iIjfsTaspsZHu]
- 69. X-ray Scattering [https://www.mri.psu.edu/materials-characterization-lab/characterization-techniques/x-ray-scattering]
- 70. and milli-fluidic sample environments for in situ X-ray analysis ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d4lc00637b]
- 71. Insights into the formation of microporous materials by in ... [https://www.sciencedirect.com/science/article/abs/pii/S0920586109003149]
- 72. Can laboratory-based XAFS compete with XRD and ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0323678]
- 73. Non-classical crystallization in soft and... | Nature Reviews Materials [https://www.nature.com/articles/s41578-023-00637-y?error=cookies_not_supported&code=6f865b46-6d8f-4224-9724-99c5a35aea61]
- 74. 2025 Seed: Discovery of Helical Inorganic Crystals as ... [https://ccam.uci.edu/2025-seed-discovery-of-helical-inorganic-crystals-as-building-blocks-for-chiral-and-non-reciprocal-excitations/]
- 75. Systematic review of scale-up methods for prospective life ... [https://www.sciencedirect.com/science/article/pii/S0959652624016093]
- 76. Optimizing the strain engineering process for industrial-scale ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10548853/]
- 77. 14 Comparing quantitative data between individuals [https://bookdown.org/pkaldunn/SRM-Textbook/BetweenQuantData.html]
- 78. Porous nucleating agents for protein crystallization | Nature Protocols [https://www.nature.com/articles/nprot.2014.109?error=cookies_not_supported&code=099e9d9a-18a7-45ab-9f30-a077023f1997]
- 79. Theoretical Analysis of the Factors Determining the Crystal ... [https://www.mdpi.com/2073-4352/15/7/653]
- 80. Controlled microcrystallization for in situ photocrystallography [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00367a]
- 81. Altering ROY polymorph crystallization in conventional and ... [https://www.nature.com/articles/s41598-025-97635-7]
- 82. In situ Raman characterization of polymorph evolution ... [https://www.sciencedirect.com/science/article/abs/pii/S0022024825003306]