High-Throughput DFT for Thermodynamic Stability Screening: Accelerating Materials and Drug Discovery
This article provides a comprehensive overview of high-throughput Density Functional Theory (DFT) calculations for thermodynamic stability screening, a transformative approach in materials science and drug development.
High-Throughput DFT for Thermodynamic Stability Screening: Accelerating Materials and Drug Discovery
Abstract
This article provides a comprehensive overview of high-throughput Density Functional Theory (DFT) calculations for thermodynamic stability screening, a transformative approach in materials science and drug development. It covers the foundational principles of thermodynamic stability and its critical role in predicting compound synthesizability and properties. The piece details established high-throughput DFT methodologies, workflow automation tools, and specific applications ranging from perovskite discovery to organic piezoelectric materials. It further addresses key challenges in computational screening, including accuracy versus efficiency trade-offs and bias mitigation strategies, and explores validation protocols that integrate experimental data and machine learning. Designed for researchers and development professionals, this guide serves as a strategic resource for leveraging computational power to accelerate the discovery and optimization of novel materials and pharmaceutical compounds.
The Fundamentals of Thermodynamic Stability and Its Role in Discovery
In high-throughput computational materials science, accurately assessing thermodynamic stability is a critical first step for predicting synthesizable compounds among vast numbers of candidates. This process primarily relies on two interconnected concepts: the formation energy of a compound and the construction of the convex hull within a phase diagram. The formation energy provides a measure of the energy gained when a compound forms from its constituent elements, while the convex hull defines the set of the most thermodynamically stable phases at various compositions. Compounds lying on this hull are considered stable, while those above it are metastable or unstable. The vertical distance from a compound's formation energy to the hull, known as the *energy above hull (Ehull*), quantifies its relative instability and propensity to decompose into more stable neighboring phases [1]. With the advent of extensive materials databases and advanced machine learning, the methodologies for determining these stability metrics are rapidly evolving, enabling the efficient screening of novel inorganic compounds, metal-organic frameworks, and energetic materials [2] [3] [4].
Core Definitions and Quantitative Metrics
Formation Energy (Ef)
The formation energy is the energy change associated with forming a compound from its elemental constituents in their standard reference states. A negative formation energy indicates that the compound is stable with respect to its elements.
Calculation: For a compound ( AxBy ), the formation energy per atom, ( Ef ), is calculated as: [ Ef = \frac{E{total}(AxBy) - x\muA - y\muB}{x+y} ] where ( E{total}(AxBy) ) is the total energy of the compound, and ( \muA ) and ( \muB ) are the chemical potentials of the elemental phases of A and B, respectively [1].
Energy Above Hull (Ehull)
The energy above hull is the energy difference between a compound's calculated formation energy and the corresponding point on the convex hull at the same composition. It represents the decomposition energy—the energy released when the compound decomposes into the most stable phases on the hull.
- Ehull = 0 meV/atom: The compound is thermodynamically stable.
- Ehull > 0 meV/atom: The compound is metastable or unstable. The magnitude indicates the degree of instability; even small positive values (e.g., 20-30 meV/atom) can indicate a metastable compound that may still be synthesizable [1].
Table 1: Key Thermodynamic Stability Metrics
| Metric | Symbol | Definition | Stability Criterion |
|---|---|---|---|
| Formation Energy | ( E_f ) | Energy to form a compound from its elemental references [1]. | ( E_f < 0 ) |
| Energy Above Hull | ( E_{hull} ) | Vertical distance from a compound's ( E_f ) to the convex hull [1]. | ( E_{hull} = 0 ) |
| Decomposition Energy | ( \Delta H_d ) | Energy difference between a compound and its stable decomposition products [2]. | ( \Delta H_d > 0 ) |
Computational Construction of the Convex Hull
The convex hull is a geometrical construction in energy-composition space that represents the set of the most stable phases.
Protocol for Convex Hull Construction:
- Data Collection: Gather the calculated formation energies (( E_f )) for all known and candidate compounds within a given chemical system (e.g., the A-B binary system).
- Plotting: Plot the formation energy per atom against composition for every compound.
- Geometrical Construction: Determine the lower envelope of points—the convex hull. In a binary system, this is the set of straight lines connecting the stable phases such that all other compounds lie on or above these lines.
- Stability Assignment: Any compound whose data point lies on this hull is deemed stable. Any compound above the hull is unstable with respect to decomposition into the stable phases defining the hull at that composition.
- Calculating Ehull: For a compound above the hull, its ( E_{hull} ) is the vertical energy difference to the hull surface. The decomposition path is not always to a single phase but often to a combination of multiple phases in thermodynamic equilibrium [1].
Example: For a quaternary compound ABO*2N, the decomposition reaction might be into a mixture of stable phases, such as: ( \text{ABO}2\text{N} \rightarrow \frac{2}{3} \text{A}4\text{B}2\text{O}9 + \frac{7}{45} \text{A}(\text{BN}2)2 + \frac{8}{45} \text{B}3\text{N}5 ) The ( E_{hull} ) is calculated using the normalized (eV/atom) energies of all involved phases according to the stoichiometry of this specific reaction [1].
Diagram 1: Convex hull in a binary system (A-B). The unstable compound's E_hull is its energy distance to the hull.
Advanced Methods and High-Throughput Protocols
High-Throughput Screening Workflow
The integration of stability assessment into high-throughput screening is crucial for identifying viable materials.
Diagram 2: High-throughput stability screening workflow.
Machine Learning and Enhanced Sampling Methods
Traditional Density Functional Theory (DFT) is computationally expensive. Emerging methods accelerate stability prediction:
- Machine Learning (ML) for Formation Energy: Advanced ML models can predict formation energies directly from composition or structure, bypassing costly DFT. State-of-the-art models include graph neural networks (e.g., ALIGNN) and image-based approaches using voxel representations of crystals, achieving accuracy comparable to DFT at a fraction of the cost [5].
- Ensemble ML for Stability: Ensemble models like ECSG integrate multiple models based on different knowledge domains (elemental properties, graph representations of formulas, and electron configurations) to predict thermodynamic stability with high accuracy (AUC > 0.98) and superior data efficiency [2].
- Enhanced Molecular Dynamics (MD) for Thermal Stability: For applications like energetic materials, an optimized MD protocol using Neural Network Potentials (NNP), nanoparticle models, and low heating rates (e.g., 0.001 K/ps) can reliably predict decomposition temperatures, reducing errors from >400 K to ~80 K compared to experiments [4].
Table 2: Computational Methods for Stability Prediction
| Method | Core Principle | Key Advantage | Example/Performance |
|---|---|---|---|
| DFT + Convex Hull | First-principles energy calculation followed by geometrical construction [1]. | High fidelity; considered the benchmark. | Foundation of Materials Project database. |
| ML Formation Energy | Trains models on DFT databases to predict ( E_f ) from structure or composition [5]. | Speed (orders of magnitude faster than DFT). | Voxel-based CNN achieves MAE competitive with graph models [5]. |
| Ensemble ML (ECSG) | Combines multiple models to reduce bias and improve accuracy [2]. | High accuracy and data efficiency. | AUC = 0.988 for stability classification [2]. |
| NNP-MD Protocol | Uses machine-learned interatomic potentials for realistic simulations of decomposition [4]. | Captures finite-temperature and kinetic effects. | Reduces T_d error to ~80 K for RDX [4]. |
The Scientist's Toolkit: Essential Computational Reagents
Table 3: Key Resources for Thermodynamic Stability Analysis
| Tool / Resource | Type | Function | Access/Reference |
|---|---|---|---|
| Materials Project (MP) | Database | Provides pre-calculated ( Ef ), ( E{hull} ), and decomposition pathways for over 150,000 materials [1]. | https://materialsproject.org |
| AFLOW | Database | A large repository of computed material properties, including formation energies for convex hull construction [5]. | http://aflow.org |
| pymatgen | Software Library | A Python library for materials analysis that includes robust modules for constructing phase diagrams and calculating ( E_{hull} ) [1]. | Open-source |
| Density Functional Tight Binding (DFTB) | Computational Method | A faster, approximate alternative to DFT for calculating formation energies and convex hulls, suitable for large systems [6]. | Integrated with codes like DFTB+ |
| Agentic Automation (DREAMS) | Framework | An LLM-agent framework that automates DFT simulation setup, execution, and error handling for high-fidelity property prediction [7]. | Prototype (academic) |
Application Notes and Protocols
Protocol 1: Calculating Energy Above Hull for a Novel Compound
Objective: To determine the thermodynamic stability of a newly modeled compound, "ABO*2N".
Materials: DFT calculation setup (e.g., VASP, Quantum ESPRESSO), computational resources, pymatgen library, access to the Materials Project API.
Procedure:
- Relaxation and Energy Calculation: Perform a full structural relaxation of ABO*2N using DFT to obtain its ground-state total energy.
- Energy Normalization: Normalize this total energy to eV per atom.
- Reference Element Energies: Obtain the energies per atom of the standard reference states for elements A, B, O, and N (e.g., the crystal structures of pure A, pure B, O_2 gas, and N_2 gas).
- Calculate Formation Energy (Ef): Use the formula in Section 2.1 to compute the formation energy of ABO*2N.
- Retrieve Relevant Phase Diagram: Use the Materials Project API or pymatgen to retrieve all existing compounds in the A-B-O-N chemical system and their formation energies.
- Construct the Convex Hull: Use pymatgen's
PhaseDiagramclass to construct the convex hull from the dataset. - Query Ehull: Use the
PhaseDiagram.get_e_above_hull(compound_entry)method, inputting an entry for your calculated ABO*2N, to directly obtain its ( E_{hull} ) [1].
Notes:
- Ensure consistency in DFT calculation parameters (functional, pseudopotentials, convergence settings) between your new compound and the data in the reference database.
- An ( E_{hull} ) less than 20-30 meV/atom often suggests a potentially synthesizable metastable material.
Protocol 2: High-Throughput Screening with Integrated Stability Metrics
Objective: To screen a large database of hypothetical materials for application-specific properties while ensuring thermodynamic and mechanical stability.
Materials: A database of candidate structures (e.g., hypothetical MOFs), DFT or ML force fields, stability evaluation tools.
Procedure (Inspired by MOF Screening [3]):
- Initial Performance Screening: Screen the database for primary application performance metrics (e.g., CO_2 uptake and CO_2/N_2 selectivity for carbon capture). Shortlist the top performers.
- Assess Thermodynamic Stability: For each shortlisted candidate, evaluate thermodynamic stability. For MOFs, this can be done by calculating its free energy and comparing it to a baseline of known experimental MOFs. Candidates with free energies significantly higher than the baseline are filtered out [3].
- Evaluate Mechanical Stability: Perform molecular dynamics (MD) simulations at relevant temperatures to calculate elastic constants (bulk, shear, Young's moduli). This assesses whether the framework can withstand synthesis and processing conditions [3].
- Predict Thermal and Activation Stability: Use pre-trained machine learning models or additional simulations to predict the thermal decomposition temperature and stability during activation (e.g., solvent removal) [3].
- Final Candidate Identification: Select the final list of candidates that simultaneously exhibit high application performance and pass all stability filters.
Why Stability Screening is a Critical First Step in Materials and Drug Design
Stability screening serves as the foundational gatekeeper in both materials science and pharmaceutical development, determining the ultimate feasibility and commercial viability of new compounds and drug candidates. This application note details the critical role of high-throughput stability assessment, with a specific focus on thermodynamic stability screening via Density Functional Theory (DFT) calculations. We provide comprehensive protocols for implementing these strategies, enabling researchers to identify promising candidates with the highest potential for success while efficiently allocating resources. By establishing stability as a primary filter in discovery pipelines, organizations can significantly reduce late-stage failures and accelerate the development of robust materials and therapeutics.
Stability is a fundamental quality attribute that dictates the practical utility of any engineered substance, be it a catalytic material or a pharmaceutical compound. Stability screening provides the first major checkpoint, ensuring that only candidates with sufficient longevity under relevant environmental conditions progress through costly development stages. In materials design, thermodynamic stability determines whether a synthesized material will maintain its structure and function under operational stresses. Similarly, in pharmaceuticals, chemical stability ensures that drug products maintain their safety, identity, strength, quality, and purity throughout their intended shelf-life [8] [9].
The consequences of inadequate stability screening are severe. For materials, unstable compounds can lead to catastrophic failure in applications such as fuel cells or catalysts. In pharmaceuticals, instability can result in reduced efficacy, harmful degradation products, product recalls, and most critically, potential harm to patients [8]. Comprehensive stability assessment is therefore not merely a regulatory hurdle but a fundamental scientific and economic imperative throughout the product lifecycle.
Theoretical Foundations: Thermodynamic Stability in Materials and Drug Design
Thermodynamic Basis of Stability
At its core, stability is governed by thermodynamics. The Gibbs free energy (ΔG) provides the crucial parameter describing the spontaneity of processes, where negative values indicate favorable, spontaneous reactions [10]. For molecular interactions in drug design, ΔG determines binding affinity, while for materials, it dictates phase stability. The relationship between free energy, enthalpy (ΔH), and entropy (ΔS) is defined by the fundamental equation:
ΔG = ΔH - TΔS [10]
This separation of ΔG into enthalpic and entropic components reveals critical insights into binding modes and stability mechanisms. Similar ΔG values can mask radically different ΔH and ΔS contributions, describing entirely different physical phenomena that have profound implications for further optimization [10].
Computational Assessment of Thermodynamic Stability
In high-throughput materials design, thermodynamic stability is quantitatively assessed through the convex hull distance, which represents the energy difference between a compound and the most stable combination of other phases at that composition [11]. Compounds with convex hull distances ≤ 0 are considered thermodynamically stable, while those with positive values are metastable or unstable [11]. This approach enables rapid computational screening of thousands of candidate materials before any synthetic effort is undertaken.
Table 1: Key Thermodynamic Parameters for Stability Assessment
| Parameter | Definition | Significance in Stability Assessment | Experimental/Calculation Method |
|---|---|---|---|
| Gibbs Free Energy (ΔG) | Energy change determining process spontaneity | Negative values indicate thermodynamically favorable processes; determines binding affinity and phase stability | ITC, van't Hoff analysis, DFT calculations |
| Formation Energy (Hf) | Energy change when a compound forms from its constituent elements | More negative values indicate greater stability of the compound relative to its elements | DFT calculations [11] |
| Convex Hull Distance | Energy difference between a compound and the most stable phases at that composition | Values ≤ 0 indicate thermodynamic stability; positive values indicate instability [11] | DFT calculations with convex hull construction [11] |
| Enthalpy (ΔH) | Heat change during a process at constant pressure | Indicates net bond formation/breakage; negative values suggest stronger bonding | Calorimetry (ITC, DSC) [10] |
| Entropy (ΔS) | Measure of system disorder | Positive values often associated with hydrophobic interactions and desolvation | Calculated from ΔG and ΔH measurements [10] |
High-Throughput Stability Screening Protocols
Protocol 1: High-Throughput DFT Screening for Material Stability
This protocol outlines a computational workflow for assessing thermodynamic stability of materials, specifically ABO3 perovskites, using high-throughput Density Functional Theory (DFT) calculations [11].
Experimental Workflow
Materials and Computational Parameters
Table 2: Research Reagent Solutions for High-Throughput DFT Screening
| Component | Specification | Function/Role |
|---|---|---|
| DFT Software | Vienna Ab initio Simulation Package (VASP) [11] | Performs quantum mechanical calculations to determine electronic structure and total energy |
| Exchange-Correlation Functional | GGA-PBE [11] | Approximates quantum mechanical interactions between electrons |
| Pseudopotential Method | Projector-Augmented Wave (PAW) [11] | Efficiently handles electron-ion interactions |
| DFT+U Corrections | Applied to 3d transition metals and actinides [11] | Accounts for strong electron correlations in specific elements |
| Elemental References | Ground-state structures from OQMD [11] | Provides reference states for calculating formation energies |
| High-Throughput Framework | qmpy python package [11] | Automates workflow management and thermodynamic analysis |
Step-by-Step Procedure
Define Composition Space: Select 73 metallic and semi-metallic elements for A and B site substitution in ABO3 perovskites, generating 5,329 possible compositions [11].
Generate Crystal Structures: Create initial structures for ideal cubic perovskite configuration and three common distortions (rhombohedral, tetragonal, orthorhombic) [11].
Perform DFT Calculations: Execute high-throughput DFT calculations using parameters in Table 2. Apply Hubbard U corrections for 3d transition metals and actinides according to established values [11].
Calculate Formation Energy: For each compound, compute the formation energy HfABO3 using: HfABO3 = E(ABO3) - μA - μB - 3μO where E(ABO3) is the DFT total energy of the perovskite, and μA, μB, and μO are the chemical potentials of the constituent elements [11].
Construct Convex Hull: Build a convex hull using all known phases in the A-B-O ternary phase diagram from the Open Quantum Materials Database (OQMD), which contains approximately 470,000 compounds [11].
Calculate Stability Metric: Compute the convex hull distance HstabABO3: HstabABO3 = HfABO3 - Hfhull where Hfhull is the convex hull energy at the ABO3 composition [11].
Classify Stability: Designate compounds with convex hull distance ≤ 0.025 eV per atom as stable, approximately kT at room temperature [11].
Protocol 2: Automated Solution Stability Assay for Drug Discovery
This protocol describes an automated high-performance liquid chromatography (HPLC)-based method for screening solution stability of drug candidates in discovery phases [12].
Experimental Workflow
Materials and Equipment
Table 3: Research Reagent Solutions for Automated Stability Screening
| Component | Specification | Function/Role |
|---|---|---|
| HPLC System | HPLC with temperature-controlled autosampler [12] | Automated sample preparation, injection, and separation |
| Detection Method | Mass spectrometry (LC-MS) or UV detection [12] | Detects and quantifies parent compound and degradation products |
| Stability Matrices | Buffers at various pH, biological assay media, physiological fluids [12] | Provides relevant environmental conditions for stability assessment |
| Incubation System | Temperature-controlled compartment [12] | Maintains precise temperature during stability study |
| Analytical Columns | Reversed-phase C18 or similar [12] | Separates analytes of interest |
Step-by-Step Procedure
Sample Preparation: Prepare compound solutions in relevant stability matrices including buffers at various pH, biological assay media, or simulated physiological fluids [12].
Automated Setup: Program temperature-controlled autosampler to deliver compounds to stability matrices, mix, and incubate at specified temperatures [12].
Time-Course Sampling: Configure system for automated injections at predetermined time points (e.g., 0, 1, 2, 4, 8, 24 hours) without operator intervention [12].
Chromatographic Analysis: Use HPLC or LC-MS with stability-indicating methods to separate and quantify parent compound and degradation products [12].
Data Analysis: Calculate percentage of parent compound remaining at each time point and determine degradation kinetics.
Stability Ranking: Rank compounds based on degradation half-life under each condition to identify candidates with optimal stability profiles.
Protocol 3: Phase-Appropriate Stability Testing for Pharmaceutical Development
This protocol outlines a phase-appropriate approach to stability testing throughout pharmaceutical development, from pre-clinical stages to commercial marketing [9].
Experimental Workflow
Materials and Equipment
Table 4: Research Reagent Solutions for Pharmaceutical Stability Testing
| Component | Specification | Function/Role |
|---|---|---|
| Stability Chambers | Temperature and humidity-controlled [8] | Maintains specified storage conditions (e.g., 25°C/60% RH, 40°C/75% RH) |
| Container-Closure System | Same as proposed marketed packaging [9] | Evaluates product-package interactions and protective properties |
| Analytical Methods | Stability-indicating HPLC, GC, spectroscopy [9] | Detects and quantifies changes in quality attributes over time |
| Reference Standards | Qualified working standards [13] | Ensures analytical method performance and data integrity |
| Data Loggers | Continuous temperature and humidity monitors [8] | Provides documentation of storage conditions |
Step-by-Step Procedure
Preclinical/Phase 1: Initiate an ongoing stability program using representative samples. Conduct studies under accelerated and long-term storage conditions to support clinical trial duration [9].
Forced Degradation Studies: Perform stress testing on the API under conditions including hydrolytic (acid/base), thermal, oxidative, and photolytic stress to identify potential degradation pathways and validate stability-indicating methods [9].
Phase 2 Studies: Expand stability testing to include formal protocols with defined tests, analytical procedures, acceptance criteria, and test intervals. Use stability data to support formulation development and container-closure selection [9].
Phase 3/Registration: Conduct formal ICH-compliant stability studies on three primary batches of the final formulation manufactured using the commercial process [9]. Implement storage conditions per ICH guidelines:
Testing Intervals: Schedule testing appropriately (e.g., 0, 3, 6, 9, 12, 18, 24, 36 months for long-term studies) [8].
Data Analysis and Shelf-Life Determination: Analyze stability data using statistical methods including regression analysis and Arrhenius modeling to establish recommended storage conditions and expiration dating [8].
Applications and Significance
Materials Design Applications
High-throughput DFT stability screening has enabled rapid discovery of novel materials across multiple domains. In ABO3 perovskites, this approach identified 395 predicted stable compounds, many not yet experimentally reported, opening avenues for applications in solid oxide fuel cells, piezo-, ferro-electricity, and thermochemical water splitting [11]. Similarly, machine learning models trained on DFT-calculated formation energies and convex hull distances have identified novel stable Ag-Pd-F catalyst materials for formate oxidation reaction in direct formate fuel cells [14].
Pharmaceutical Development Applications
Stability screening in pharmaceutical development provides critical data for formulation optimization, container-closure selection, and establishment of shelf-life and storage conditions [9]. Automated solution stability assays enable rapid profiling of compound stability under physiological conditions, guiding structural modifications to improve metabolic stability [12]. The extensive stability data required for regulatory submissions ultimately ensures that marketed drug products maintain quality, safety, and efficacy throughout their shelf-life [8] [9].
Stability screening represents an indispensable first step in both materials and pharmaceutical design pipelines. The protocols detailed herein—from high-throughput DFT calculations for materials to automated solution stability assays and phase-appropriate pharmaceutical testing—provide robust frameworks for implementing these critical assessments. By integrating comprehensive stability screening early in development workflows, researchers can make informed decisions, mitigate downstream risks, and accelerate the development of viable materials and therapeutics. As high-throughput and computational methods continue to advance, stability screening will become increasingly predictive and efficient, further solidifying its role as the critical gatekeeper in materials and drug design.
In the field of high-throughput materials discovery, accurately assessing thermodynamic stability is a critical first step for identifying synthesizable compounds. Density Functional Theory (DFT) calculations enable the efficient screening of vast compositional spaces by computing key thermodynamic parameters. This application note details the core thermodynamic parameters—formation energy (ΔHf), decomposition energy (ΔHd), and stability criteria—within the context of high-throughput DFT screening, providing protocols and guidelines for researchers.
Defining the Key Thermodynamic Parameters
Formation Energy (ΔHf)
The formation energy (ΔHf) quantifies the energy released or absorbed when a compound forms from its constituent elements in their reference states at T=0 K. It is calculated according to Equation 1 [11]:
$$ Hf^{ABO3} = E(ABO3) - \muA - \muB - 3\muO $$
where:
- ( E(ABO_3) ) is the total DFT energy of the compound.
- ( \muA ), ( \muB ), and ( \mu_O ) are the chemical potentials of elements A, B, and oxygen, respectively.
For most elements, the chemical potentials are equal to the DFT total energies of their standard elemental reference states. Corrections are applied for certain elements where the T=0 K DFT ground state is not an adequate reference (e.g., diatomic gases like O₂, room-temperature liquids like Hg, and elements with structural phase transformations between 0 and 298 K) [11]. A negative ΔHf indicates that the compound is stable with respect to its elements.
Table 1: Key Quantitative Data on Formation and Decomposition Energies from High-Throughput Studies
| Property | Typical Value Range | Dataset Context | Significance | Citation |
|---|---|---|---|---|
| Formation Energy (ΔHf) | Mean ± AAD = -1.42 ± 0.95 eV/atom | Materials Project (85,014 compositions) | Quantifies stability relative to elements; wide energy range. | [15] |
| Decomposition Energy (ΔHd) | Mean ± AAD = 0.06 ± 0.12 eV/atom | Materials Project (85,014 compositions) | Subtle but critical for stability; positive = unstable. | [15] |
| Stability Threshold | < 0.025 eV/atom | ABO₃ Perovskites (5,329 compositions) | Approximate kT at room temperature; labels compounds as stable. | [11] |
| Number of Predicted Stable Perovskites | 395 compounds | ABO₃ Perovskites (5,329 compositions) | Demonstrates the power of high-throughput screening for discovery. | [11] |
Decomposition Energy (ΔHd) and Phase Stability
While formation energy measures stability against elements, the decomposition energy (ΔHd), or "energy above the convex hull," determines a compound's thermodynamic stability with respect to all other competing phases in its chemical space. It is defined as [15]:
$$ \Delta Hd = Hf^{compound} - H_f^{hull} $$
where ( Hf^{compound} ) is the formation energy of the compound in question and ( Hf^{hull} ) is the convex hull energy at that composition, derived from the lowest-energy combination of other phases.
The convex hull is constructed in formation energy-composition space, forming the lower envelope that connects the most stable phases. Any phase lying on this hull (ΔHd ≤ 0) is considered thermodynamically stable, while phases above the hull (ΔHd > 0) are unstable or metastable [15] [2]. The magnitude of ΔHd indicates the energy penalty for a compound's decomposition into more stable neighboring phases. As shown in Table 1, ΔHd typically operates on a much smaller energy scale than ΔHf, making it a more sensitive metric [15].
Stability Criteria and the "Stable" Label
In computational high-throughput screening, a compound is typically classified as thermodynamically stable if its ΔHd is negative or zero. In practice, a small positive tolerance is often applied to account for numerical uncertainties and nearly-stable compounds. For instance, a stability criterion of ΔHd < 0.025 eV/atom (approximately kT at room temperature) has been used to label ABO₃ perovskites as stable [11].
High-Throughput Workflow for Stability Assessment
The process for determining these parameters at a high-throughput scale involves a multi-step, automated workflow. The diagram below illustrates the logical sequence from initial setup to final stability classification.
Computational Protocols and Methodologies
DFT Calculation Parameters for Thermodynamic Properties
High-throughput DFT relies on standardized protocols to ensure consistency and accuracy across thousands of calculations. The following parameters are critical [11] [16].
Table 2: Essential DFT Setup Parameters for Thermodynamic Stability Screening
| Parameter | Typical Setting / Consideration | Function/Role in Calculation |
|---|---|---|
| Software | Vienna Ab initio Simulation Package (VASP) | Performs electronic structure calculation and geometry relaxation. |
| Pseudopotential | Projector-Augmented Wave (PAW) method | Represents core electrons and interaction with valence electrons. |
| Exchange-Correlation Functional | GGA-PBE (Perdew-Burke-Ernzerhof) | Approximates the quantum mechanical exchange-correlation energy. |
| DFT+U | Applied for 3d transition metals and actinides (e.g., U=3-4 eV) | Corrects self-interaction error for strongly correlated electrons. |
| k-point Sampling | Automated meshes (e.g., Monkhorst-Pack); protocols exist for precision/efficiency tradeoffs [17] | Samples the Brillouin zone for numerical integration. |
| Energy Cutoff | System-specific; tested for convergence (see Fig. S1 in [16]) | Determines the basis set size (plane-wave kinetic energy cutoff). |
| Energy Convergence Criterion | Tight settings (e.g., 10⁻⁵ eV per atom) | Ensures electronic self-consistency is achieved. |
| Force Convergence Criterion | (e.g., 0.01 eV/Å) | Determines when ionic relaxation is complete. |
Standard solid-state protocols (SSSP) have been developed to optimize the selection of parameters like k-point sampling and smearing, balancing precision and computational efficiency for high-throughput workflows [17]. For charged defect calculations, which are related but distinct, advanced corrections or hybrid functionals are often necessary to address band gap errors [18].
Machine Learning for Accelerated Stability Prediction
Machine learning (ML) has emerged as a powerful tool to predict formation energies and stability, bypassing expensive DFT calculations for initial screening. However, a critical examination reveals that accurate prediction of ΔHf does not guarantee accurate prediction of ΔHd and stability [15]. This is because stability depends on the small energy differences between competing phases, where errors in ΔHf may not cancel out.
Ensemble models that combine different knowledge domains show improved performance. For example, the ECSG framework integrates models based on elemental properties (Magpie), interatomic interactions (Roost), and electron configuration (ECCNN), achieving an Area Under the Curve (AUC) score of 0.988 for stability classification and requiring only one-seventh of the data to match the performance of other models [2]. For composition-based models without structural information, important features for predicting ΔHf include anion electronegativity, electron affinity, and orbital properties [19].
Successful high-throughput screening relies on a suite of computational tools and databases.
Table 3: Key Resources for High-Throughput Thermodynamic Screening
| Tool / Resource | Type | Primary Function | Example/URL |
|---|---|---|---|
| VASP | Software | First-principles DFT code using PAW pseudopotentials. | https://www.vasp.at/ |
| Quantum ESPRESSO | Software | Open-source DFT code using plane waves and pseudopotentials. | https://www.quantum-espresso.org/ |
| AiiDA | Workflow Manager | Automates, manages, and reproduces computational workflows. | https://www.aiida.net/ |
| pymatgen | Materials API | Python library for analyzing, generating, and manipulating structures. | https://pymatgen.org/ |
| OQMD | Database | Open Quantum Materials Database; source of DFT-calculated formation energies and phase data. | http://oqmd.org |
| Materials Project | Database | Provides computed properties for over 85,000 materials, including formation energies. | https://materialsproject.org |
| SSSP | Protocol | Standard Solid-State Protocols for optimized pseudopotentials and calculation parameters. | https://www.sssp.eu |
The rigorous application of formation energy (ΔHf), decomposition energy (ΔHd), and well-defined stability criteria forms the foundation of high-throughput thermodynamic stability screening. While high-throughput DFT provides the primary data, integrated machine-learning models and robust computational protocols are enhancing the efficiency and scope of materials discovery. Adherence to standardized methods ensures reliable identification of stable, synthesizable materials, accelerating the development of new functional compounds for energy and electronic applications.
The Limitation of Traditional Methods and the Need for High-Throughput Approaches
The discovery and development of new functional materials and drug compounds are fundamental to technological and medical progress. Traditionally, this process has been dominated by experimental approaches characterized by sequential, labor-intensive testing of individual candidates. In computational materials science, conventional density functional theory (DFT) calculations, while powerful, are often too resource-intensive for exploring vast chemical spaces. The limitations of these traditional methods have become increasingly apparent, creating a pressing need for high-throughput (HT) approaches that can rapidly screen thousands to millions of compounds efficiently and systematically [20] [21].
Traditional screening methods in pharmaceutical research were limited to 20-50 compounds per week per laboratory, requiring large assay volumes (∼1 ml) and substantial amounts of each compound (5-10 mg) [20]. Similarly, in materials science, conventional DFT studies are often restricted to investigating a manageable subspace of potential materials due to prohibitive computational costs, creating a bottleneck for discovery [22]. This article details the specific limitations of traditional methodologies and presents structured protocols for implementing high-throughput alternatives, with a specific focus on high-throughput DFT for thermodynamic stability screening.
Quantitative Comparison: Traditional vs. High-Throughput Methods
The quantitative advantages of high-throughput screening over traditional methods are substantial, as detailed in Table 1.
Table 1: Performance Comparison of Traditional and High-Throughput Screening Methods
| Screening Parameter | Traditional Methods | High-Throughput Methods |
|---|---|---|
| Throughput (Compounds/Week) | 20-50 [20] | 1,000-100,000+ [20] [23] |
| Assay Volume | ∼1 ml [20] | 1-100 μL [20] [23] |
| Compound Consumption | 5-10 mg [20] | ∼1 μg [20] |
| Automation Level | Manual, single-tube processing [20] | Robotic, array-based (96 to 1586-well plates) [20] [23] |
| Data Integration | Limited, non-standardized | Centralized databases (e.g., OQMD) [11] [21] |
The impact of these limitations is clearly demonstrated in specific research contexts. For example, a high-throughput DFT screening of single-metal and high-entropy MOF-74 structures identified Mn-MOF-74 as optimal for CO₂/N₂ separation and MnFeCoNiMgZn-MOF-74 for H₂ storage, a task impractical through sequential study [24]. In another case, screening 4350 bimetallic alloy structures via HT-DFT identified eight promising Pd-substitute catalysts, with four experimentally confirmed and one (Ni₆₁Pt₃₉) showing a 9.5-fold enhancement in cost-normalized productivity [25].
Limitations of Traditional Workflows
Low Throughput and High Resource Consumption
The most significant limitation of traditional methods is their low throughput. The serial "one-at-a-time" paradigm for testing compounds or materials is inherently slow, creating a critical path in research and development timelines [20]. Furthermore, traditional experimental screening consumes large amounts of reagents and compounds (5-10 mg per test), making it expensive and often impractical when novel compounds are difficult to synthesize in large quantities [20]. In computational settings, conventional DFT calculations, while faster than experimentation, can require "several months... for computing a full energetics picture of even one metallic catalyst system" [25].
Limited Exploration of Chemical Space
The combinatorial nature of chemical and materials spaces is enormous. Relying on intuition or incremental modifications for candidate selection means that vast regions of potentially fruitful chemical space remain unexplored [22] [21]. This often results in suboptimal materials or drug candidates being selected for development. High-throughput computational screening has revealed that promising materials can be exceptionally rare; for instance, among ternary Heusler compounds, only 0.5% (135 of 27,000+) simultaneously met the criteria for high magnetic anisotropy and thermodynamic, dynamic, and magnetic stability [22].
Inefficient Data Management and Knowledge Transfer
Traditional research often lacks standardized data management protocols, leading to "data silos" where information is difficult to aggregate, analyze, and reuse across different projects [21]. The absence of a formal data flow strategy in conventional computational studies hinders the extraction of broader scientific trends and makes it challenging to assess the transferability and accuracy of computational methods across diverse chemical systems [21].
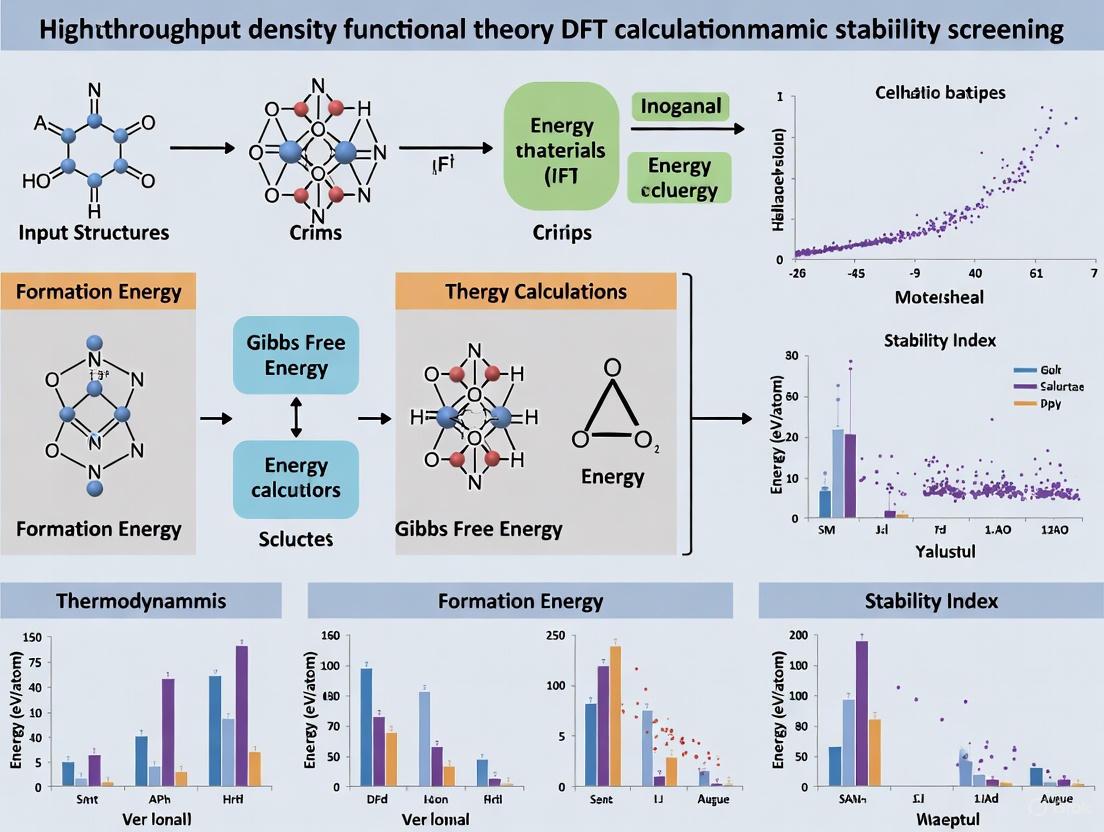
High-Throughput Screening Protocol for Thermodynamic Stability
This protocol outlines a standardized workflow for using high-throughput DFT calculations to screen materials for thermodynamic stability, a critical first step in identifying viable new compounds.
Protocol Workflow
The high-throughput screening process for thermodynamic stability involves a multi-stage workflow, illustrated in Figure 1.
Figure 1: High-Throughput DFT Screening Workflow. The process is a sequential pipeline from target definition to final candidate validation [21].
Detailed Experimental Procedures
Data Selection and Compound Management
- Objective: Define the target chemical space and generate a list of candidate structures for computation.
- Procedure:
- Define Search Space: Systematically enumerate possible compositions, such as all possible ABO₃ perovskites [11] or all conventional quaternary Heusler compounds [22].
- Select Structural Prototypes: For each composition, identify relevant crystal structures (e.g., cubic, tetragonal, rhombohedral for perovskites [11]).
- Generate Input Files: Automate the creation of all necessary computational input files (e.g., for VASP) with standardized parameters [21].
- Note: For stability screening, it is crucial to include not only the target compounds but also all competing phases (elements and other known compounds) to construct accurate phase diagrams [11].
High-Throughput Data Generation (DFT Calculations)
- Objective: Perform automated, consistent first-principles calculations to determine the total energy and optimized geometry for each candidate.
- Computational Parameters (Generalized, based on [24] [11] [21]):
- Software: Vienna Ab initio Simulation Package (VASP).
- Functional: Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA).
- Van der Waals Correction: D3 Becke-Johnson damping [24].
- Basis Set: Plane-wave basis set with projector-augmented wave (PAW) pseudopotentials.
- k-point Mesh: Automated mesh generation ensuring energy convergence (e.g., 500 k-points per reciprocal atom) [21].
- Convergence Criteria: Define strict thresholds for electronic (e.g., 10⁻⁵ eV) and ionic (e.g., 0.01 eV/Å) relaxation [21].
- Automation: Use job-management scripts to submit, monitor, and manage thousands of calculations in parallel on high-performance computing clusters.
Data Storage and Management
- Objective: Store calculated properties in a queryable, persistent database to facilitate analysis and sharing.
- Procedure:
- Database Schema: Design a relational database (e.g., MySQL, PostgreSQL) with tables for compositions, crystal structures, energies, and derived properties [21].
- Automated Parsing: Implement scripts to automatically parse the output of DFT calculations and populate the database.
- Public Databases: Consider contributing to or using public databases like the Open Quantum Materials Database (OQMD), which contains hundreds of thousands of calculated compounds [11].
Data Analysis: Stability and Property Screening
- Objective: Identify the most stable and promising candidates from the calculated dataset.
- Key Calculations:
- Formation Energy (ΔEf): Calculate using
H_f_ABO₃ = E(ABO₃) - μ_A - μ_B - 3μ_O, whereE(ABO₃)is the total energy of the compound andμare the chemical potentials of the constituent elements [11]. - Distance to Convex Hull (ΔH): The energy difference between a compound and the most stable mixture of phases at the same composition. A stable compound has
ΔH = 0; a near-stable compound has a small, positive value (e.g., < 0.025 eV/atom) [11] [22].
- Formation Energy (ΔEf): Calculate using
- Screening: Apply filters to select compounds that are stable or near-stable (
ΔH< threshold) and that possess the target functional properties.
Experimental and Advanced Validation
- Objective: Validate the computational predictions with targeted experiments or higher-fidelity calculations.
- Procedure:
- Synthesis Attempt: Prioritize the top predicted stable candidates for experimental synthesis [11].
- Property Characterization: Experimentally measure the target properties (e.g., gas adsorption, catalytic activity, thermoelectric performance) [24] [25] [26].
- Higher-Level Theory: Validate key findings using more accurate (but computationally expensive) methods like hybrid DFT or quantum Monte Carlo [27].
Successful implementation of high-throughput screening relies on a suite of computational and physical resources, as detailed in Table 2.
Table 2: Key Research Reagent Solutions for High-Throughput Screening
| Tool / Resource | Function / Description | Example Use Case |
|---|---|---|
| VASP (Vienna Ab initio Simulation Package) [24] [11] | A widely used software package for performing DFT calculations. | Computing formation energies and electronic structures in HT screens. |
| Open Quantum Materials Database (OQMD) [11] | A public database containing DFT-calculated data for ~40,000 experimental and ~430,000 hypothetical compounds. | Provides reference energies for stability analysis (convex hull construction). |
| 96/384-Well Microplates [20] [23] | Standardized array formats for miniaturized and parallelized experimental assays. | Conducting thousands of biological or chemical tests in parallel. |
| Machine Learning Interatomic Potentials (MLIPs) [22] | ML models that accelerate structure optimization by orders of magnitude vs. DFT. | Rapidly relaxing crystal structures of thousands of candidates in a HT workflow. |
| Robotic Liquid Handling Systems [20] [23] | Automation workstations for precise, high-speed dispensing of assay reagents. | Enabling the preparation and testing of vast compound libraries. |
| qmpy Python Package [11] | An open-source tool for managing high-throughput calculations and thermodynamic analysis. | Automating workflow and analyzing phase stability via convex hull constructions. |
The limitations of traditional, sequential discovery methods are clear: they are slow, resource-intensive, and ill-suited for exploring the vastness of chemical space. High-throughput approaches, particularly those leveraging high-throughput DFT, represent a paradigm shift. By integrating automated computation, systematic data management, and machine learning, these protocols enable researchers to rapidly identify stable, functional materials with a precision and scale previously unimaginable. As these methodologies continue to mature and integrate with experimental validation, they promise to dramatically accelerate the pace of innovation across materials science and drug discovery.
Implementing High-Throughput DFT Workflows: From Theory to Practice
Core Components of a High-Throughput DFT Framework
High-throughput density functional theory (HT-DFT) has emerged as a transformative paradigm in computational materials science, enabling the rapid screening and discovery of novel materials with targeted properties. This approach leverages automated computational workflows to systematically evaluate thousands of materials, dramatically accelerating the materials design cycle that traditionally relied on resource-intensive trial-and-error experimentation [28]. In the specific context of thermodynamic stability screening—a critical prerequisite for experimental synthesis—HT-DFT provides invaluable predictions of phase stability, formation energies, and decomposition pathways, guiding researchers toward synthesizable materials candidates [29]. The framework's core components work in concert to manage the immense computational scale, ensure data consistency, and facilitate the extraction of scientifically meaningful trends from massive datasets, thereby establishing a robust foundation for data-driven materials innovation.
Core Component Architecture
A functional HT-DFT framework rests on several interconnected pillars, each addressing a distinct challenge in the high-throughput pipeline. The seamless integration of these components is what enables efficient, reliable, and scalable materials screening.
Workflow Automation & Management
This component manages the execution of complex computational tasks, from initial structure generation to final property extraction. It handles error recovery, job scheduling, and dependency management, which is crucial for maintaining throughput across thousands of calculations. Specialized software packages are essential for this automation, with several established options available:
- FireWorks & Atomate: A widely adopted combination for designing, managing, and executing workflows. Atomate provides a library of pre-defined workflows for common materials simulation tasks, built upon the FireWorks workflow engine [30].
- AiiDA: A powerful informatics platform that not only automates workflows but also tracks the provenance of every calculation, ensuring full reproducibility and transparency of the data generation process [30].
- qmpy: This package was used to automate high-throughput calculations and thermodynamic analysis for a comprehensive study on ABO₃ perovskites, demonstrating its application in large-scale stability screening [29].
Computational Infrastructure & DFT Codes
This component encompasses the core quantum mechanics engines that perform the actual energy and property calculations. The choice of DFT code and its configuration dictates the accuracy and computational cost of the simulations.
- DFT Codes: Common plane-wave basis set codes include VASP (Vienna Ab initio Simulation Package) and Quantum ESPRESSO. For instance, a massive HT-DFT study on 5,329 perovskites was conducted using VASP [29], while the nanoHUB resource provides a platform for running high-throughput calculations with Quantum ESPRESSO [31].
- Exchange-Correlation Functional: The approximation for the exchange-correlation functional is a critical choice. While the GGA-PBE functional is widely used in high-throughput studies [32] [29], its tendency to underestimate band gaps is well-documented. For more accurate electronic properties, hybrid functionals like HSE06 or meta-GGAs like HLE17 can be employed, though at a higher computational cost [32].
- Pseudopotentials: The projector-augmented wave (PAW) method is commonly used to describe electron-ion interactions [33] [29]. HT frameworks often integrate pseudopotential libraries to ensure consistent accuracy across the periodic table.
Data Management & Analysis
This component is responsible for storing, retrieving, and analyzing the vast volumes of structured data generated by HT-DFT calculations. Effective data management transforms raw calculation outputs into actionable knowledge.
- Databases and Platforms: Public databases such as the Materials Project and the Open Quantum Materials Database (OQMD) are central repositories for HT-DFT data [32] [31] [29]. They provide curated data and tools for analysis. The OQMD, for instance, was used to assess the thermodynamic stability of perovskites by constructing convex hulls from hundreds of thousands of compounds [29].
- Data Analysis Techniques: Dimensionality reduction techniques like Principal Component Analysis (PCA) can be employed to reveal latent structure in complex data, such as identifying correlations between bulk and surface electronic properties [34]. Furthermore, computed properties are often validated against existing experimental data to ensure predictive fidelity [29].
Validation & Uncertainty Quantification
This final component ensures the reliability and physical meaningfulness of the high-throughput predictions. It involves benchmarking computational results against experimental data or higher-level theories and assessing the impact of methodological choices.
- Stability and Convergence Testing: Protocols are established to test for the convergence of key parameters like plane-wave cutoff energy and k-point sampling [33].
- Functional Benchmarking: Studies systematically compare results from different DFT approximations (e.g., LDA, PBE, PBE+U, HSE06) to evaluate their performance for specific classes of materials, such as finding that PBE+U provided the best agreement with experimental data for zinc-blende CdS and CdSe [33].
- Experimental Cross-Validation: Predicted stable materials are compared against known experimental compounds to validate the computational framework. For example, a HT-DFT study curated a list of 223 experimentally synthesized perovskites to compare against its predictions [29].
Table 1: Key Software Tools for High-Throughput DFT Frameworks
| Tool Name | Primary Function | Application Example |
|---|---|---|
| FireWorks/Atomate [30] | Workflow Automation | Managing complex job dependencies in high-throughput screening. |
| AiiDA [30] | Workflow Automation & Data Provenance | Tracking the entire calculation history for reproducibility. |
| pymatgen [31] [30] | Materials Analysis | Parsing output files, analyzing structures, and generating input files. |
| qmpy [29] | Thermodynamic Analysis | Constructing convex hulls to assess phase stability. |
| VASP [29] | DFT Engine | Large-scale property calculation for thousands of compounds. |
| Quantum ESPRESSO [31] | DFT Engine | High-throughput simulations on platforms like nanoHUB. |
Experimental Protocol for Thermodynamic Stability Screening
The following protocol outlines a standardized methodology for using an HT-DFT framework to screen for thermodynamically stable materials, using the extensive study on ABO₃ perovskites as a benchmark [29].
Workflow Visualization
The logical flow of the high-throughput stability screening process is summarized in the diagram below.
Step-by-Step Methodology
Step 1: Define the Target Chemical Space
- Objective: Systematically define the set of chemical compositions to be screened.
- Procedure: For an ABO₃ perovskite screening, select all possible combinations of 73 metallic and semi-metallic elements on the A and B sites, resulting in 5,329 unique compositions [29]. For other systems, define the relevant structure types and compositional ranges.
Step 2: Generate Initial Crystal Structures
- Objective: Create initial structural models for DFT relaxation.
- Procedure:
- For each composition, create input files for the primary crystal structure of interest (e.g., the ideal cubic perovskite structure).
- Optionally, generate input files for common distorted variants (e.g., rhombohedral, tetragonal) for a subset of compositions to capture lower-energy phases [29].
Step 3: Perform High-Throughput DFT Calculations
- Objective: Compute the total energy of each structure in its ground state.
- Computational Parameters:
- Code: Vienna Ab initio Simulation Package (VASP) [29].
- Functional: GGA-PBE [29].
- HF Exchange: For more accurate electronic properties, a hybrid functional like HSE06 can be used, though it increases computational cost [32].
- DFT+U: Apply a Hubbard U correction for elements with localized d- or f-electrons (e.g., U = 3.1 eV for V, 3.5 eV for Cr) to improve the description of on-site electron correlations [29].
- Pseudopotentials: Use the projector-augmented wave (PAW) method [29].
- Convergence: Ensure convergence of total energy with respect to plane-wave cutoff energy and k-point sampling [33].
Step 4: Calculate Formation Energies
- Objective: Determine the energy of formation for each compound from its constituent elements.
- Procedure: Use the equation:
H_f(ABO₃) = E(ABO₃) - μ_A - μ_B - 3μ_OwhereE(ABO₃)is the total energy from DFT, andμare the chemical potentials of the elements, typically derived from the DFT total energies of their standard elemental phases [29].
Step 5: Construct Convex Hulls for Stability Assessment
- Objective: Identify which compounds are thermodynamically stable.
- Procedure:
- Gather formation energies for all known and calculated phases in the A-B-O ternary chemical space from a reference database (e.g., OQMD) [29].
- Construct the convex hull—the set of stable phases with the lowest formation energy at their composition.
- For each perovskite, calculate its hull distance (Ehull):
E_hull = H_f(Perovskite) - H_f(Convex Hull at ABO₃ composition)A compound is considered theoretically stable if Ehull ≤ 0 eV/atom. A small positive value (e.g., < 0.025 eV/atom, approximately thermal energy at room temperature) may indicate a "nearly stable" compound that might be synthesizable under non-equilibrium conditions [29].
Step 6: Data Storage and Analysis
- Objective: Curate and disseminate results for further use.
- Procedure: Store relaxed structures, formation energies, hull distances, and other computed properties (band gaps, magnetic moments) in a structured, publicly accessible database. This data can be used for direct candidate selection or to train machine learning models [29].
Table 2: Key Calculated Properties for Thermodynamic Stability Screening
| Property | Calculation Method | Significance in Stability Screening |
|---|---|---|
| Formation Energy [29] | H_f = E_total - Σ μ_elements |
Measures thermodynamic stability relative to elemental phases. |
| Hull Distance (E_hull) [29] | E_hull = H_f - H_f,hull |
Definitive metric for thermodynamic stability. E_hull ≤ 0 indicates stability. |
| Oxygen Vacancy Formation Energy [29] | E_vac = E(A₂B₂O₅) + μ_O - 2E(ABO₃) |
Key for assessing application-relevant stability under reducing conditions. |
| Band Gap [32] [29] | From electronic DOS calculation | Important for functional properties; sensitive to DFT functional choice. |
In the context of HT-DFT, "research reagents" refer to the essential software, data, and computational resources required to build and operate the framework.
Table 3: Essential "Reagents" for a High-Throughput DFT Framework
| Reagent / Resource | Type | Function in the HT-DFT Workflow |
|---|---|---|
| Pymatgen [30] | Python Library | Provides core materials analysis capabilities, file input/output, and workflow management. |
| Materials Project API [31] [30] | Database & Tool | Allows query and retrieval of existing crystal structures and properties to define initial screening sets. |
| Standard Solid State Pseudopotentials (SSSP) | Pseudopotential Library | A curated set of high-quality pseudopotentials ensuring consistency and accuracy across calculations. |
| Open Quantum Materials Database (OQMD) [29] | Database | Provides a vast repository of calculated phases essential for constructing convex hulls and assessing stability. |
| High-Performance Computing (HPC) Cluster | Infrastructure | Provides the massive parallel computing resources necessary to execute thousands of DFT calculations. |
Data Analysis and Technical Validation
The final stage of the HT-DFT protocol involves rigorous analysis and validation to ensure the predictive power of the results. This often involves techniques that manage the complexity of the generated data.
Data Analysis Workflow
The process of analyzing results, particularly for establishing structure-property relationships, can be visualized as a pipeline from raw data to predictive models.
A key example of an advanced analysis is mapping the surface density of states (DOS) from bulk properties. This involves:
- Dimensionality Reduction: Using Principal Component Analysis (PCA) to obtain compact, low-dimensional representations of both bulk and surface DOS [34].
- Latent Feature Alignment: Identifying a linear transformation that maps the latent features of the bulk DOS to those of the surface DOS. This transformation is learned from a small set of materials (e.g., CuNbS, CuTaS, CuVS) for which both bulk and surface calculations are available [34].
- Prediction: Applying the learned transformation to the bulk DOS of new, unseen compositions (e.g., CuCrS, CuMoS) to predict their surface DOS, thereby bypassing the need for computationally expensive surface calculations [34].
Technical Validation and Benchmarking
Validation is critical. For a stability screening study, this includes:
- Comparison with Experiment: Cross-referencing predicted stable compounds with known experimental materials. In the perovskite study, 395 compounds were predicted stable, with a significant number representing new predictions awaiting experimental confirmation [29].
- Functional Benchmarking: Systematically evaluating different DFT approximations. For instance, one should note that while PBE is efficient, it can severely underestimate band gaps compared to hybrid functionals like HSE06, which can affect the predicted electronic properties of stable compounds [32].
- Sensitivity Analysis: Testing the robustness of predictions to computational parameters, such as the choice of Hubbard U value or the effect of different structural distortions on the calculated formation energy [33] [29].
This structured approach to analysis and validation ensures that the high-throughput framework produces not just data, but reliable and actionable scientific insights.
High-Throughput Density Functional Theory (HT-DFT) has emerged as a transformative approach in computational materials science, enabling the systematic calculation of properties for thousands of materials in an automated fashion. This paradigm shift from traditional single-structure calculations to large-scale screening has been facilitated by the development of sophisticated software frameworks and extensive databases. These resources allow researchers to predict material properties, assess thermodynamic stability, and identify promising candidates for various applications before undertaking experimental synthesis. The core infrastructure enabling these efforts consists of automation tools like Pymatgen and AFLOW, alongside comprehensive databases such as the Open Quantum Materials Database (OQMD) and the Materials Project (MP). These platforms collectively provide researchers with standardized methods for generating structures, performing calculations, analyzing results, and accessing curated data, thereby accelerating materials discovery across diverse fields including energy storage, catalysis, and electronic materials.
The critical importance of these tools is underscored by their widespread adoption in the materials community. They have been used to screen over 80,000 inorganic compounds for applications ranging from Li-ion batteries to topological insulators [35]. Within the context of thermodynamic stability screening—a fundamental step in materials discovery—these tools provide robust methodologies for determining whether a proposed compound is stable against decomposition into competing phases. This article provides detailed application notes and protocols for leveraging these tools effectively in HT-DFT research, with particular emphasis on thermodynamic stability assessment.
Major HT-DFT Databases: Contents and Specializations
Table 1: Key Features of Major HT-DFT Databases
| Database | Primary Focus | Number of Entries | Key Properties | Access Method |
|---|---|---|---|---|
| OQMD (Open Quantum Materials Database) | DFT formations energies and stability assessment [36] | ~300,000 calculations [36] (ICSD compounds + hypothetical structures) | Formation energies, thermodynamic stability, oxygen vacancy energies [11] | Public download at oqmd.org/download [36] |
| Materials Project | Calculated properties of all known inorganic materials [35] | ~80,000 compounds screened as of 2013 [35] | Crystal structures, band gaps, elastic properties, electrochemical data [37] | RESTful API via Pymatgen [35] |
| AFLOW (Automatic Flow) | High-throughput calculation of crystal structure properties [35] | Not specified in sources | Structural properties, stability, superconductors, topological insulators [35] | Online database access |
The Open Quantum Materials Database (OQMD) specializes in DFT formation energies and thermodynamic stability assessments, containing nearly 300,000 DFT calculations as of 2015 [36]. Its dataset includes both experimentally reported structures from the Inorganic Crystal Structure Database (ICSD) and hypothetical compounds generated through decoration of common crystal prototypes [36]. A notable specialized dataset within OQMD includes 5,329 ABO₃ perovskites with calculated formation energies, stability, and oxygen vacancy formation energies, highly relevant for applications like solid oxide fuel cells and water splitting [11].
The Materials Project aims to compute properties of all known inorganic materials and make this data publicly available [35]. It provides a diverse range of calculated properties accessible through a RESTful API that can be interfaced via Pymatgen [35]. The project has historically used the PBE GGA functional but has more recently transitioned to the r²SCAN meta-GGA functional, which generally provides improved accuracy for magnetic moments and thermodynamic stability [37].
AFLOW (Automatic Flow) provides a software framework for high-throughput calculation of crystal structure properties, with applications including binary alloys, superconductors, and topological insulators [35]. While specific current statistics were not provided in the search results, it represents one of the three major HT-DFT databases alongside OQMD and Materials Project.
Quantitative Comparison of Database Methodologies and Outputs
Table 2: Methodological Comparison and Reproducibility Across Major Databases
| Aspect | OQMD | Materials Project (Legacy) | Materials Project (Newer) |
|---|---|---|---|
| DFT Functional | GGA-PBE with DFT+U for specific elements [11] | PBE (GGA) and GGA+U [37] | r²SCAN meta-GGA [37] |
| Formation Energy Accuracy | MAE: 0.096 eV/atom vs. experiments [36] | Not specified | Improved thermodynamic accuracy [37] |
| Magnetic Properties | Not specified | Good for elemental metals [37] | Overestimates on-site ferromagnetic moments in elemental metals [37] |
| Inter-database Variance | Median Relative Absolute Difference: 6% for formation energy [38] |
A comparative analysis reveals both consistencies and variations across the major databases. Formation energies and volumes generally show higher reproducibility than band gaps and total magnetizations [38]. The median relative absolute difference between databases is approximately 6% for formation energy and 4% for volume, while band gaps show 9% difference, and total magnetization 8% [38]. Notably, these variances are comparable to differences between DFT and experimental measurements, highlighting the importance of understanding methodological choices when utilizing these databases.
Key sources of discrepancy include choices involving pseudopotentials, the DFT+U formalism, and elemental reference states [38]. For instance, the Materials Project's transition from PBE to r²SCAN has improved thermodynamic accuracy but tends to overestimate magnetic moments in transition metals compared to experimental measurements [37]. These differences underscore the benefit of having multiple databases available, as they enable cross-validation and uncertainty quantification for high-throughput calculations.
Experimental Protocols for Thermodynamic Stability Screening
Workflow for Thermodynamic Stability Assessment
The assessment of thermodynamic stability for new materials represents a fundamental application of HT-DFT databases. The following protocol outlines a standardized workflow for determining phase stability using the OQMD and Materials Project frameworks.
Protocol 1: Phase Stability Assessment via Convex Hull Construction
Structure Generation and Input Preparation
- Generate initial crystal structures for compounds of interest. Pymatgen provides powerful tools for structure generation via substitution based on data-mined rules [35].
- For perovskite systems (ABO₃), consider multiple structural distortions (rhombohedral, tetragonal, orthorhombic) beyond the ideal cubic structure, as distortions generally lower the energy [11].
DFT Calculation Parameters
- Perform DFT calculations using consistent parameters across all compounds. The OQMD methodology uses:
- Vienna Ab initio Simulation Package (VASP) [11] [36]
- GGA-PBE exchange-correlation functional [11]
- Projector-augmented wave (PAW) method [11]
- DFT+U for 3d transition metals and actinides with element-specific U-values [11]
- Plane-wave cutoff energy: 520 eV [11]
- k-point density: 6,000 k-points per reciprocal atom (KPPRA) for structural relaxation [11]
- Perform DFT calculations using consistent parameters across all compounds. The OQMD methodology uses:
Formation Energy Calculation
- Calculate the formation energy (H(f^{ABO3})) using: [ Hf^{ABO3} = E(ABO3) - \muA - \muB - 3\muO ] where E(ABO₃) is the total energy of the perovskite, and μ(A), μ(B), and μ(_O) are the chemical potentials of the constituent elements [11].
- Use appropriate reference states for elements (standard states for most elements, with corrections applied for diatomic gases, room temperature liquids, and elements with structural phase transformations) [11].
Stability Assessment via Convex Hull Construction
- Construct the energy convex hull for the A-B-O ternary system using all known competing phases from the database [11].
- Calculate the convex hull distance (H({stab}^{ABO3})) as: [ H{stab}^{ABO3} = Hf^{ABO3} - Hf^{hull} ] where H(f^{hull}) is the convex hull energy at the ABO₃ composition [11].
- Classify compounds with stability below 0.025 eV per atom (approximately kT at room temperature) as stable [11].
Validation and Analysis
Specialized Protocol: Oxygen Vacancy Formation Energy Calculation
For energy materials such as perovskites in fuel cell applications, oxygen vacancy formation energy represents a critical property beyond basic thermodynamic stability. The following protocol details its calculation.
Protocol 2: Oxygen Vacancy Formation Energy in Perovskites
Defect Structure Generation
- Create a 2×2×2 supercell of the perovskite structure (A(8)B(8)O(2)(4)) or use a standardized A(2)B(2)O(_5) 9-atom supercell for high-throughput screening [11].
- Remove one oxygen atom to create the defective structure.
DFT Calculation of Defect System
- Perform DFT relaxation of the defective A(2)B(2)O(_5) structure using consistent calculation parameters with the pristine system.
- Ensure consistent k-point sampling and convergence criteria.
Energy Calculation
- Calculate the oxygen vacancy formation energy (E({vO})) using: [ E{vO} = E(A2B2O5) + \muO - 2E(ABO3) ] where E(A(2)B(2)O(5)) is the total energy of the defective cell, E(ABO(3)) is the total energy of the pristine perovskite, and μ(O) is the chemical potential of oxygen [11].
Analysis and Correlation
- Correlate oxygen vacancy formation energies with stability metrics and application-specific performance.
- For water-splitting applications, use both stability and oxygen vacancy formation energy as screening parameters [11].
Essential Tools and Integration Strategies
The Scientist's Toolkit: Software and Databases
Table 3: Essential Software Tools and Databases for HT-DFT Research
| Tool/Database | Type | Primary Function | Key Applications in Stability Screening |
|---|---|---|---|
| Pymatgen | Python Library | Materials analysis, input generation, and post-processing [35] | Structure manipulation, phase diagram analysis, API access to Materials Project [35] |
| qmpy | Python Framework | High-throughput DFT calculations and database management [11] [36] | OQMD database management, thermodynamic analysis [36] |
| VASP | DFT Code | First-principles electronic structure calculations [11] | Energy calculations, structural relaxation, property prediction [11] |
| OQMD | Database | DFT formation energies and stability data [36] | Convex hull construction, stability assessment, new compound prediction [11] [36] |
| Materials Project API | Web API | Programmatic access to calculated materials data [35] | Retrieving reference data, comparative analysis, structure retrieval [35] |
Integration Workflow for Materials Screening
Effective integration of these tools enables robust materials screening pipelines. The following diagram illustrates the relationships and data flow between key components in a typical HT-DFT workflow for stability screening.
Best Practices and Uncertainty Management
Data Representation and Functional Considerations
When working with HT-DFT databases, researchers should adhere to several best practices to ensure robust and reproducible results:
Structure Representation: Be aware that databases may store structures in non-standard representations. Always convert to primitive or conventional cells using tools like Pymatgen's
to_conventional()orget_primitive_structure()methods for consistent analysis [37].Functional Differences: Understand the implications of different DFT functionals. While PBE (GGA) provides reasonable accuracy, r²SCAN (meta-GGA) generally improves thermodynamic accuracy and magnetic ordering in oxides but may overestimate magnetic moments in elemental metals [37].
Cross-Validation: Leverage multiple databases when possible to quantify uncertainty. The observed variances between databases (6% for formation energy, 9% for band gaps) are significant and should be considered when making stability predictions [38].
Stability Thresholds: Use appropriate energy thresholds for stability classification. The OQMD uses 0.025 eV/atom (approximately kT at room temperature) as a practical threshold for predicting synthesizable materials [11].
Uncertainty Quantification in HT-DFT
Recent research has highlighted the importance of quantifying uncertainty in high-throughput calculations. Key considerations include:
Phonon-Induced Uncertainty: Ab initio molecular dynamics (AIMD) calculations reveal significant uncertainties (standard deviation ≈ 0.16 V in overpotentials) due to phonon-induced energy fluctuations [39].
Experimental Comparison: The mean absolute error between DFT and experimental formation energies is 0.096 eV/atom in OQMD, but a significant fraction of this error may be attributed to experimental uncertainties, which show a mean absolute error of 0.082 eV/atom between different experimental measurements [36].
Methodological Consistency: Maintain consistent calculation parameters (pseudopotentials, k-point sampling, U-values) throughout a screening project to minimize systematic errors and ensure comparable results [38].
These protocols and best practices provide a foundation for robust thermodynamic stability screening using modern HT-DFT tools and databases. By leveraging these standardized approaches, researchers can efficiently identify promising new materials while properly accounting for computational uncertainties and methodological limitations.
The discovery of advanced functional materials is often limited by the vastness of chemical space and the resource-intensive nature of traditional experimental approaches. ABO₃ perovskites, characterized by their versatile oxide structure, exhibit remarkable properties for applications including solid oxide fuel cells, ferroelectrics, and thermochemical water splitting [11]. Their stability with respect to cation substitution makes them particularly promising, with many potential compounds awaiting discovery [11] [40]. This case study details a high-throughput density functional theory (HT-DFT) framework for computationally screening ABO₃ perovskites, focusing on two critical properties: thermodynamic stability and oxygen vacancy formation energy. The protocol demonstrates how integrated computational workflows can accelerate materials discovery by rapidly identifying promising candidates for further experimental investigation.
Key Quantitative Data from High-Throughput Screening
A high-throughput DFT screening of 5,329 ABO₃ perovskites, comprising both cubic and distorted structures, provides a robust dataset for analysis [11] [41]. The key quantitative results are summarized in the following tables.
Table 1: Summary of High-Throughput DFT Screening Results for ABO₃ Perovskites
| Property Category | Metric | Value | Description |
|---|---|---|---|
| Dataset Scope | Total Compounds Calculated | 5,329 | Includes cubic and distorted structures [11] |
| Thermodynamic Stability | Predicted Stable Compounds | 395 | Stability threshold: < 36 meV/atom [11] [42] |
| Predicted Metastable Compounds | 13 | Stability range: 36-70 meV/atom [42] | |
| Structural Analysis | Compositions with Multiple Distortions | 2,162 | Rhombohedral, tetragonal, and orthorhombic [11] |
| Experimental Validation | Experimentally Known Stable Perovskites | 223 | Curated from literature for comparison [11] |
Table 2: Key Property Definitions and Calculated Ranges
| Property | Definition/Calculation | Significance | Representative Range/Notes |
|---|---|---|---|
| Formation Energy ((H_f)) | (Hf^{ABO3} = E(ABO3) - \muA - \muB - 3\muO) [11] | Measures stability of compound from its elements. | Negative values indicate exothermic formation [11]. |
| Energy Above Convex Hull ((E_{hull})) | (H{stab}^{ABO3} = Hf^{ABO3} - H_f^{hull}) [11] | Primary metric for thermodynamic stability. | Stable: (E{hull} < 36) meV/atom; Metastable: (36 \leq E{hull} < 70) meV/atom [42]. |
| Oxygen Vacancy Formation Energy ((E_{VO})) | (E{VO} = E(A2B2O5) + \muO - 2E(ABO3)) [11] | Indicates ease of oxygen loss; crucial for redox applications. | Lower values favor oxygen vacancy formation [11]. |
Experimental and Computational Protocols
High-Throughput DFT Workflow
The screening process follows a structured, automated workflow to ensure consistency and reliability across thousands of calculations [11].
Diagram 1: High-throughput DFT screening workflow for ABO₃ perovskites.
Protocol: Density Functional Theory Calculations
This protocol details the core computational parameters used for the high-throughput screening [11].
- Objective: To perform high-throughput first-principles calculations of ABO₃ perovskites for formation energy, stability, and oxygen vacancy formation energy.
- Software: Vienna Ab initio Simulation Package (VASP) [11].
- Exchange-Correlation Functional: GGA-PBE [11].
- Pseudopotentials: Projector-Augmented Wave (PAW) method [11].
- DFT+U: Applied for 3d transition metals and most actinides to account for strong electron correlations (specific U values are tabulated in the source) [11].
- Spin-Polarization: Calculations were spin-polarized with a ferromagnetic alignment for relevant elements [11].
- Reference Database: Calculations were performed within the framework of the Open Quantum Materials Database (OQMD) [11].
Protocol: Thermodynamic Stability Assessment
- Objective: To determine the thermodynamic stability of each ABO₃ compound relative to all other competing phases in the A-B-O ternary system.
- Stability Metric: Energy above the convex hull ((E_{hull})).
- Method: A convex hull is constructed using the formation energies of all ~470,000 phases in the OQMD, which includes both experimental phases from the ICSD and hypothetical compounds [11].
- Calculation:
- The formation energy of the perovskite, (Hf^{ABO3}), is calculated.
- The convex hull energy, (Hf^{hull}), at the ABO₃ composition is determined from the lowest-energy linear combination of phases.
- The stability is computed as: (H{stab}^{ABO3} = Hf^{ABO3} - Hf^{hull}) [11].
- Stability Classification:
Protocol: Oxygen Vacancy Formation Energy Calculation
- Objective: To compute the energy required to form an oxygen vacancy in the perovskite structure, a key property for redox applications.
- Supercell Model: A 9-atom cubic supercell (A₂B₂O₅) is used, which corresponds to two perovskite unit cells with one oxygen atom removed [11].
- Calculation:
- The energy of the pristine cell, (E(ABO3)), and the defective cell, (E(A2B2O5)), are calculated after full structural relaxation.
- The oxygen chemical potential, (\mu_O), is referenced to the DFT total energy of an O₂ molecule [11].
- The oxygen vacancy formation energy is calculated as: (E{VO} = E(A2B2O5) + \muO - 2E(ABO3)) [11].
Machine Learning for Accelerated Screening
To further enhance screening efficiency, machine learning (ML) models can be trained on the DFT dataset to predict stability without performing full DFT calculations for every new candidate [42] [2].
Table 3: Machine Learning Models for Perovskite Screening
| Model Name | Model Type | Input Features | Reported Performance |
|---|---|---|---|
| Gradient Boosting Decision Tree (GBDT) [42] | Ensemble Tree | 9 primary features (e.g., tolerance factor, octahedral factor, Mendeleev numbers) [42] | 94.6% accuracy for perovskite formability classification [42] |
| ECSG (Proposed Framework) [2] | Ensemble Super Learner | Electron configuration, atomic properties, interatomic interactions [2] | AUC = 0.988 for stability prediction [2] |
Protocol: Machine Learning Screening for Stability
- Objective: To rapidly screen new ABO₃ compositions for thermodynamic stability using a pre-trained ML model.
- Feature Engineering: Compute the following 9 crucial features for each A-B pair [42]:
- Tolerance factor (t)
- Octahedral factor (μ)
- Radius ratio of A to O (rA / rO)
- A-O bond length
- B-O bond length
- Electronegativity difference for A-O multiplied by rA / rO
- Electronegativity difference for B-O multiplied by rB / rO
- Mendeleev number for A
- Mendeleev number for B
- Model Application: Input the feature vector into the trained GBDT model to obtain a prediction of stability ((E_{hull}) class) and the probability of perovskite formability [42].
- Validation: Promising candidates identified by ML should be validated with subsequent DFT calculations.
Table 4: Key Computational Tools and Databases for Perovskite Screening
| Tool/Resource | Type | Function in Screening | Access/Reference |
|---|---|---|---|
| VASP [11] | Software | Primary DFT engine for calculating total energies and electronic properties. | Proprietary (vasp.at) |
| Open Quantum Materials Database (OQMD) [11] | Database | Provides reference phases for convex hull construction and a repository of calculated data. | Public (oqmd.org) |
| qmpy [11] | Python Package | Manages high-throughput workflow, thermodynamic analysis, and data visualization. | Open Source (MIT License) |
| Materials Project [42] | Database | Alternative source of DFT-calculated data for training ML models and validation. | Public (materialsproject.org) |
| scikit-learn [42] | Python Library | Provides ML algorithms (e.g., GBDT) for building classification and regression models. | Open Source (BSD License) |
Data Interpretation and Candidate Selection
The final step involves integrating the calculated properties to identify the most promising candidates. The relationship between stability and oxygen vacancy energy is crucial for targeting specific applications, such as thermochemical water splitting [11].
Diagram 2: Decision logic for candidate selection based on Ehull and EVO.
- Primary Candidates for Synthesis: Compounds located in the green quadrant (low (E{hull}), low (E{VO})) are ideal for applications requiring reversible oxygen exchange, as they are both stable and active for reduction [11].
- Stable Host Materials: Compounds with low (E{hull}) but high (E{VO}) (blue quadrant) are structurally robust but may be less active for processes like fuel cell electrodes or water splitting.
- Secondary Metastable Candidates: Compounds with slightly positive (E{hull}) (e.g., < 70 meV/atom) but desirable (E{VO}) can be considered for experimental synthesis, as metastable materials are often synthesizable under non-equilibrium conditions [42]. The dataset from this study predicted 37 stable and 13 metastable ABO₃ perovskites worthy of further investigation [42].
The need for environmentally sustainable alternatives to lead-based piezoelectrics, such as lead zirconate titanate (PZT), has catalyzed intensive research into lead-free materials [43] [44]. Organic molecular crystals represent a promising class of eco-friendly piezoelectric materials due to their diverse chemistries, biocompatibility, and potential for tailored electromechanical properties through crystal engineering principles [44]. Unlike traditional inorganic piezoelectrics, organic crystals offer significantly lower dielectric constants, which can result in exceptionally high voltage constants ideal for specific sensing and energy harvesting applications [44].
High-throughput computational screening, powered by density functional theory (DFT), has emerged as a powerful paradigm for accelerating the discovery of functional materials [45] [46]. This approach enables researchers to systematically evaluate vast chemical spaces and predict electromechanical properties before undertaking resource-intensive experimental synthesis [44]. The "materials by design" paradigm, supported by initiatives like the Materials Genome Initiative, has transformed materials discovery from traditional trial-and-error approaches to rational, computation-driven strategies [46]. This case study examines the application of high-throughput DFT screening for discovering organic piezoelectric crystals, detailing the computational protocols, key findings, and research infrastructure enabling this innovative approach.
Computational Framework and Workflow
High-Throughput Screening Methodology
The screening of organic piezoelectric materials employs an automated computational workflow based on quantum mechanical modelling (QMM) via DFT calculations [44]. This high-throughput methodology minimizes human intervention through sequential scripts that automate three critical stages of the calculation process: file preparation, calculation submission and maintenance, and output analysis [44]. This automation is essential for handling the hundreds of crystal structures involved in comprehensive screening studies.
The process begins with curating a dataset of noncentrosymmetric organic structures from crystallographic databases, primarily the Crystallographic Open Database (COD) [44]. A crucial initial filter applies symmetry constraints, selecting only crystals belonging to noncentrosymmetric space groups that permit piezoelectric behavior [44]. To maintain computational tractability while maximizing the number of calculable structures, researchers typically set an initial limit of 50 atoms per unit cell [44]. For validation and benchmarking purposes, calculations are also performed on well-studied piezoelectric crystals including zinc oxide, aluminum nitride, α-quartz, and barium titanate [44].
Table 1: Key Screening Criteria for Organic Piezoelectric Crystals
| Screening Parameter | Criteria | Purpose |
|---|---|---|
| Crystal Symmetry | Noncentrosymmetric space groups | Essential for piezoelectric activity |
| System Size | ≤50 atoms per unit cell | Computational tractability |
| Source Database | Crystallographic Open Database (COD) | Reliable structural data |
| Reference Materials | ZnO, AlN, α-quartz, BaTiO₃ | Validation and benchmarking |
Density Functional Theory and Density Functional Perturbation Theory
DFT serves as the fundamental computational engine for predicting the electromechanical properties of candidate materials [44]. For piezoelectric properties specifically, density functional perturbation theory (DFPT) provides an efficient methodology for computing energy derivatives with respect to atomic displacements, strain, and electric fields [46]. DFPT enables calculation of the full piezoelectric tensor, which describes the change in material polarization due to mechanical stress or strain, and conversely, the change in stress or strain due to an external electric field [46].
The accuracy of high-throughput DFT calculations depends critically on controlling numerical parameters and convergence criteria [45]. Recent advances have led to the development of standardized solid-state protocols (SSSP) that provide automated methods for selecting optimized parameters based on different precision and efficiency tradeoffs [45]. These protocols help control errors in total energies, forces, and other properties while consistently managing k-point sampling errors across a wide range of crystalline materials [45].
Experimental Protocols and Workflow Implementation
High-Throughput Workflow for Piezoelectric Property Calculation
The following diagram illustrates the automated computational workflow for high-throughput screening of organic piezoelectric crystals:
High-Throughput Screening Workflow
Validation of Computational Predictions
Rigorous validation against experimental data is essential for establishing the reliability of computational predictions. A comparison of calculated and experimental piezoelectric constants for various bioorganic systems demonstrates strong correlations across distinct tensor components [44]. For example:
- γ-glycine (COD ID: 7128793) exhibits experimental strain coefficients of 5.33 pC/N (d₁₆) and 11.33 pC/N (d₃₃), compared to DFT-predicted values of 5.15 pC/N and 10.72 pC/N, respectively [44].
- l-histidine shows a close match, with reported values of 18 pC/N (d₂₄) against predicted values of 18.49 pC/N and 20.68 pC/N for its two distinct entries (COD IDs 2108877 and 2108883) [44].
- Other materials, including l-aspartate and dl-alanine, display good agreement between literature values and predictions, further reinforcing the accuracy of the computational high-throughput simulation pipeline [44].
Table 2: Validation of DFT Predictions Against Experimental Data
| Material | COD ID | Tensor Component | Experimental Value (pC/N) | DFT Prediction (pC/N) |
|---|---|---|---|---|
| γ-glycine | 7128793 | d₁₆ | 5.33 | 5.15 |
| γ-glycine | 7128793 | d₃₃ | 11.33 | 10.72 |
| l-histidine | 2108877 | d₂₄ | 18.00 | 18.49 |
| l-histidine | 2108883 | d₂₄ | 18.00 | 20.68 |
| l-aspartate | - | d₂₅ | 13.40 | 12.87 |
| dl-alanine | - | d₁₄ | 5.70 | 5.43 |
It is important to note that experimental measurements of piezoelectricity involve multiple methods with inherent variations due to differences in measurement conditions, sample geometry, and technique-specific limitations [44]. These methods include the quasistatic Berlincourt method, resonance-based frequency measurements, and piezoresponse force microscopy (PFM) [44].
Key Research Findings and Database Development
CrystalDFT Database
The high-throughput screening of approximately 600 noncentrosymmetric organic structures has led to the development of CrystalDFT, a comprehensive database of small molecular crystals and their DFT-predicted electromechanical properties [44]. This database highlights the broad range of electromechanical properties among organic crystals, with a particular significance placed on the high number of crystals that possess a naturally occurring (unpoled) longitudinal piezoelectric response (d₁₁/d₂₂/d₃₃) [44]. This longitudinal electromechanical coupling is a prerequisite for several conventional sensing and energy harvesting applications but has been notably rare in the literature on biomolecular crystal piezoelectricity until now [44].
The CrystalDFT database represents a significant step in establishing a large-scale, systematic resource for organic piezoelectrics. Rather than focusing solely on individual high-performing materials, this initiative emphasizes developing a systematic methodology that facilitates broader analysis and comparison of electromechanical properties across diverse chemical spaces [44]. The database is accessible at https://actuatelab.ie/CrystalDFT, providing an essential data infrastructure for scalable data generation and analysis in large-scale projects [44].
Promising Organic Piezoelectric Candidates
The high-throughput screening has identified several organic crystals with promising piezoelectric properties, some exceeding the performance of well-known inorganic piezoelectric materials [44]. The database encompasses a diverse range of bio/organic materials, including:
- l-ornithine hydrochloride, previously reported for its nonlinear optical properties (NLO) [44]
- dl-malic acid (COD ID: 5000188), noted for its piezoelectric contribution [44]
- Derivatives of both malic acid and tartaric acid (COD ID: 200) [44]
The diversity of these materials demonstrates the rich chemical space available for developing sustainable piezoelectric materials through crystal engineering approaches such as cocrystallization [44].
Table 3: Essential Resources for High-Throughput Piezoelectric Screening
| Resource Name | Type | Function | Access |
|---|---|---|---|
| CrystalDFT | Database | Repository of organic crystals and DFT-predicted electromechanical properties | https://actuatelab.ie/CrystalDFT |
| Crystallographic Open Database (COD) | Database | Source of crystal structure data for screening candidates | Public |
| VASP | Software | DFT and DFPT calculations for piezoelectric properties | Proprietary |
| WIEN2K | Software | Full-potential linearized augmented plane wave (FP-LAPW) DFT calculations | - |
| JARVIS-DFT | Database | Contains computed IR, piezoelectric, and dielectric properties for inorganic materials | https://www.ctcms.nist.gov/~knc6/JVASP.html |
| SSSP | Protocol | Standardized solid-state protocols for precision and efficiency in high-throughput DFT | - |
Key Methodological Components
The high-throughput screening infrastructure incorporates several critical methodological components:
- Automated Workflow Management: Specialized scripts streamline and parallelize each stage of the calculation process, including file preparation, calculation submission and maintenance, and output analysis [44].
- Quality Control Protocols: Automated protocols control k-point sampling errors and ensure consistent precision across diverse materials systems [45].
- Machine Learning Integration: Supervised machine learning models based on classification and regression methods can pre-screen high-performance materials without performing additional first-principles calculations [46] [2].
- Thermodynamic Stability Assessment: Formation energies and convex hull distances evaluate compound stability, with compounds having stability below 0.025 eV per atom (approximately kT at room temperature) typically classified as stable [29].
High-throughput computational screening represents a transformative approach for discovering organic piezoelectric crystals as sustainable alternatives to lead-based piezoelectrics. The methodology detailed in this application note demonstrates how DFT and DFPT calculations can be scaled to efficiently screen hundreds of organic crystals, predicting their electromechanical properties with validated accuracy against experimental data.
The development of the CrystalDFT database provides researchers with a systematically curated resource that captures the broad range of electromechanical properties among organic crystals. This infrastructure supports the discovery of materials with promising piezoelectric responses, some of which may exceed the performance of well-known inorganic materials for specific applications.
Future advancements in this field will likely focus on integrating machine learning models more deeply into the screening pipeline, expanding the chemical space of screened materials, and improving the accuracy of property predictions through advanced exchange-correlation functionals. As high-throughput methodologies continue to mature, they offer the promise of significantly accelerating the development of sustainable piezoelectric materials for sensing, actuation, and energy harvesting applications.
Active Pharmaceutical Ingredients (APIs) and excipients are fundamental components of drug development. A significant challenge in modern pharmaceuticals is the poor aqueous solubility of many APIs, which hampers their bioavailability and therapeutic efficacy [47]. Among the various strategies to overcome this, the formation of pharmaceutical co-crystals has emerged as a powerful and innovative approach. Co-crystals are crystalline materials composed of two or more different molecular and/or ionic compounds, typically an API and a co-former, in a stoichiometric ratio within the same crystal lattice [47]. Unlike salts, which involve proton transfer and ionization, the components in a co-crystal remain in a neutral state and interact via non-ionic interactions such as hydrogen bonds, π bonds, and van der Waals forces [47]. This unique structure allows for the modification of key physicochemical properties of an API—including solubility, dissolution rate, stability, and hygroscopicity—without altering its fundamental chemical structure or pharmacological activity [48]. The primary objective of this application note is to provide a detailed protocol for the high-throughput screening of API-excipient co-crystals, with a specific emphasis on leveraging molecular interactions and computational thermodynamics to accelerate discovery.
Background and Significance
The ability to improve the properties of poorly soluble APIs (falling into BCS Class II or IV) through co-crystallization is of paramount importance to the pharmaceutical industry [47] [49]. It is estimated that about 40% of commercial compounds and up to 80% of drug substances in the development pipeline suffer from solubility issues [47]. Co-crystals offer a distinct advantage over other solid forms like amorphous dispersions, which are thermodynamically unstable and prone to recrystallization, potentially leading to variable performance [47]. In contrast, co-crystals are crystalline and generally more stable, providing a more reliable and durable formulation strategy.
The traditional discovery of co-crystals has been a resource-intensive process, requiring the extensive exploration of a complex experimental landscape that includes variations in co-formers, solvents, and stoichiometric ratios [50]. However, the integration of high-throughput experimental techniques and advanced computational modeling has dramatically accelerated this process. High-throughput methods, such as Encapsulated Nanodroplet Crystallisation (ENaCt), allow for the rapid screening of thousands of experimental conditions with minimal sample consumption [50]. Concurrently, computational tools like Density Functional Theory (DFT) and molecular dynamics (MD) simulations provide a theoretical foundation for predicting the thermodynamic stability and interaction strength between APIs and potential co-formers before any wet-lab experiments are initiated [51] [48]. This synergy between computation and experiment is crucial for the efficient design and development of superior pharmaceutical solid forms.
High-Throughput Screening of Co-crystals: A Computational and Experimental Workflow
The following section outlines an integrated protocol for co-crystal screening, from initial computational prediction to experimental validation and characterization.
In-silico Co-former Screening
Objective: To rapidly identify promising co-former candidates for a target API using computational chemistry and thermodynamic modeling.
Protocol:
System Setup:
- API and Co-former Selection: Obtain the 3D molecular structures of the target API and a library of potential co-formers from databases such as the Cambridge Structural Database (CSD) or PubChem. Co-formers are typically selected from the FDA's "Generally Recognized As Safe" (GRAS) list [47].
- Geometry Optimization: Perform geometry optimization on all structures using computational chemistry software (e.g., Gaussian, Materials Studio) with an appropriate functional (e.g., B3LYP) and basis set (e.g., 6-31G*).
Interaction Energy Calculation:
- Dimer Model Construction: Build a simplified model of a 1:1 API/co-former dimer complex.
- DFT Single-Point Energy Calculation: Calculate the single-point energy of the optimized dimer (Ecomplex) and of the individual, optimized monomer units (EAPI and Eco-former).
- Binding Energy Determination: Compute the binding energy (ΔEbind) of the complex using the formula:
ΔE_bind = E_complex - (E_API + E_co-former)A more negative ΔEbind value indicates a more stable complex and a higher probability of co-crystal formation [51].
Thermodynamic Stability Assessment (PC-SAFT):
- Parameterization: Obtain pure-component parameters for the API and co-former for the Perturbed-Chain Statistical Associating Fluid Theory (PC-SAFT) equation of state. These can be fitted to experimental solubility data or estimated using group contribution methods [49].
- Phase Behavior Prediction: Use PC-SAFT to predict the thermodynamic phase behavior and solubility of the API in the presence of the co-former and solvent. The model can identify conditions where the co-crystal is the most stable solid phase [49].
- Activity Coefficient and Solubility: The solubility of a crystalline API in a solvent (or co-former melt) is governed by Solid-Liquid Equilibrium (SLE) [49]:
x_API^L = 1 / (γ_API^L) * exp( -Δh_API^SL / (R * T) * (1 - T / T_API^SL) )wherex_API^Lis the mole fraction solubility,γ_API^Lis the activity coefficient (calculated by PC-SAFT),Δh_API^SLis the melting enthalpy of the API,Tis the temperature, andT_API^SLis the melting temperature of the API.
Key Quantitative Metrics for Co-former Selection: Table 1: Key quantitative metrics for computational screening of co-crystals.
| Metric | Description | Target Value/Indicator | Computational Method |
|---|---|---|---|
| Binding Energy (ΔEbind) | Energy released upon complex formation | More negative values indicate stronger, more favorable interactions [51]. | DFT |
| Interaction Energy (Etotal) | Sum of electrostatic (Ecoul) and Lennard-Jones (ELJ) energies | More negative values suggest better compatibility. Example: NPX-Eudragit L100: -143.38 kJ/mol [51]. | Molecular Dynamics (MD) |
| Hydrogen Bonding | Number and strength of H-bonds between API and co-former | Higher number and stronger bonds improve stability. | DFT/MD |
| Solubility Parameter (δ) | Measure of miscibility (Hansen solubility parameters) | Closer δ values between API and co-former indicate better miscibility [51]. | PC-SAFT/MD |
| Solubility (x_API^L) | Predicted mole fraction solubility of the co-crystal | Higher values indicate improved dissolution potential. | PC-SAFT |
Experimental Validation and Workflow
After in-silico screening, the top candidate pairs must be validated experimentally. A high-throughput workflow is essential for efficiency.
The following DOT code defines the workflow diagram:
Diagram 1: High-throughput co-crystal screening workflow, integrating computational and experimental methods.
Objective: To experimentally produce and confirm the formation of co-crystals predicted by in-silico models.
Protocol:
High-Throughput Co-crystal Synthesis (ENaCt):
- Solution Preparation: Prepare nanodroplet solutions containing the target API and selected co-former in various stoichiometric ratios (e.g., 1:1, 2:1, 1:2) dissolved in a range of organic solvents.
- Encapsulation: Disperse the nanodroplets into an immiscible carrier oil within a 96-well or 3456-well plate format, creating thousands of isolated micro-reactors [50].
- Crystallization: Allow the plates to stand or control the evaporation of the solvent to induce crystallization within the nanodroplets. This method requires minimal sample volume and explores a vast experimental space in parallel [50].
Solid-State Characterization:
- Powder X-ray Diffraction (PXRD): Analyze the solid material from each successful experiment. A PXRD pattern distinct from the patterns of the pure API and co-former indicates the formation of a new crystalline phase [48].
- Differential Scanning Calorimetry (DSC): Measure the thermal behavior of the solids. The appearance of a new, unique melting endotherm that is different from the starting materials suggests co-crystal formation [52].
- Spectroscopic Confirmation:
- Raman Spectroscopy: Use to detect changes in vibrational modes, pinpointing specific interactions (e.g., hydrogen bonding) between the API and co-former molecules [48].
- Solid-State Nuclear Magnetic Resonance (SSNMR): Provide detailed information on molecular conformation and the nature of intermolecular interactions within the crystal lattice, helping to differentiate between co-crystals and salts [48].
Ultimate Confirmation: Single Crystal X-ray Diffraction (SCXRD)
- If a single crystal of suitable size and quality is obtained, perform SCXRD. This technique is considered the "gold standard" for unequivocally confirming co-crystal formation, as it provides a three-dimensional map of the crystal structure, revealing the precise stoichiometry and the specific intermolecular interactions stabilizing the lattice [50].
The Scientist's Toolkit: Essential Research Reagents and Materials
A successful co-crystal screening campaign relies on a suite of computational and experimental tools.
Table 2: Essential research reagents and materials for co-crystal screening.
| Category/Item | Specific Examples | Function in Co-crystal Screening |
|---|---|---|
| Computational Software | Gaussian, Materials Studio, COSMO-RS | Performs DFT/MD calculations for predicting interaction energies, binding affinity, and thermodynamic stability [51] [48]. |
| Thermodynamic Model | PC-SAFT (Perturbed-Chain SAFT) | Predicts solubility and phase behavior of API/excipient/solvent systems, guiding solvent and co-former selection [49]. |
| APIs (Model Compounds) | Mesalazine, Allopurinol, Naproxen (NPX), Omeprazole (OPZ) | Poorly soluble BCS Class II/IV drugs used as model compounds to test and validate co-crystallization approaches [51] [49]. |
| Polymeric Co-formers | HPMC-based polymers (HPMC(P), HPMC(AS)), Eudragit L100, PVPK25 | Commonly used excipients that act as co-formers; their functional groups can form specific interactions with APIs [51] [49]. |
| Solvents | Methanol, Ethanol, Acetonitrile, Ethyl Acetate | Used to dissolve API and co-former for high-throughput screening; solvent choice can dictate the polymorphic outcome. |
| High-Throughput Platform | Encapsulated Nanodroplet Crystallisation (ENaCt) | Enables thousands of parallel crystallization experiments with minimal material consumption, drastically accelerating discovery [50]. |
| Characterization Instruments | PXRD, DSC, Raman Spectrometer, SSNMR, SCXRD | A suite of analytical techniques used to confirm the formation, structure, and stability of the new co-crystalline phase [50] [48] [52]. |
Application Notes and Troubleshooting
- Focus on Hydrogen Bond Donors/Acceptors: During co-former selection, prioritize molecules with complementary hydrogen bond donors and acceptors to the target API. This significantly increases the probability of forming a stable co-crystal lattice [47].
- Handling Hygroscopic Materials: Some co-crystals or starting materials may be hygroscopic. Perform stability testing under controlled humidity (e.g., using dynamic vapor sorption) to ensure the solid form does not change during storage [52].
- Distinguishing Co-crystals from Salts: The extent of proton transfer can be ambiguous. Use a combination of techniques: SCXRD is definitive, but SSNMR and Raman spectroscopy can also differentiate between a co-crystal (neutral components) and a salt (ionized components) by probing the electronic environment of key atoms [47] [48].
- Accelerated Stability Testing: For a rapid assessment of chemical stability, prepare a saturated aqueous slurry of the API-co-former mixture and heat it at an elevated temperature (e.g., 75°C for 72 hours). Analyze the sample for degradation products. This method accelerates degradation kinetics and provides quick feedback on formulation stability [53].
The integration of high-throughput computational screening, particularly using DFT for thermodynamic stability assessment, with advanced experimental platforms like ENaCt, represents a paradigm shift in pharmaceutical co-crystal discovery. This synergistic approach allows researchers to efficiently navigate the vast and complex experimental space of co-crystallization. By leveraging molecular interactions to rationally design co-crystals, it is possible to significantly enhance the physicochemical properties of challenging APIs, paving the way for the development of more effective and reliable drug products. The protocols and tools outlined in this application note provide a robust framework for accelerating this critical process in modern drug design.
Overcoming Computational Challenges and Enhancing Screening Accuracy
The pursuit of predictive computational screening for materials and drug discovery represents a fundamental shift in scientific research, moving from observation-driven to simulation-led discovery. In this paradigm, high-throughput density functional theory (HT-DFT) calculations have emerged as an indispensable tool for evaluating thermodynamic stability and electronic properties across vast chemical spaces. However, the widespread implementation of HT-DFT faces a critical challenge: navigating the intricate balance between computational cost and predictive accuracy when selecting exchange-correlation functionals and basis sets.
The selection of computational parameters is not merely a technical consideration but a strategic decision that directly impacts the reliability and scalability of research outcomes. As noted in benchmark studies, outdated methodological choices can persist despite known limitations, highlighting the need for clear protocols that reflect contemporary understanding and capabilities [54]. This document provides a structured framework for researchers engaged in thermodynamic stability screening, with a particular focus on protocols that maintain an optimal accuracy-efficiency balance for high-throughput applications.
Theoretical Foundation: The Cost-Accuracy Paradigm in DFT
The Fundamental Trade-off
At the core of DFT calculations lies a fundamental trade-off between computational expense and result accuracy. This balance manifests primarily through two methodological choices: the exchange-correlation functional approximation and the atomic orbital basis set. The computational cost of DFT calculations generally scales cubically with system size, but specific functional and basis set choices can significantly alter the prefactor of this relationship [55].
The accuracy of a functional depends critically on its treatment of electron correlation effects. Traditional functionals are organized hierarchically on "Jacob's Ladder," ascending from local density approximations (LDA) to meta-generalized gradient approximations (meta-GGAs) and hybrid functionals, with each rung offering potential accuracy improvements at increased computational cost [56]. For high-throughput applications, this hierarchy provides a structured approach to selecting an appropriate functional based on target accuracy and available computational resources.
Quantifying Thermodynamic Stability
In thermodynamic stability screening, the primary property of interest is the energy above the convex hull (E$__{\text{hull}}$), which quantifies a compound's stability relative to competing phases in a chemical space. Accurate prediction of this property requires functional and basis set combinations capable of reproducing formation energies with uncertainties typically below 20-30 meV/atom to reliably distinguish stable from unstable compounds [57] [2]. This precision threshold establishes the minimum accuracy requirement for methodological selections in high-throughput workflows.
Functional and Basis Set Selection Guidelines
Recommended Method Combinations for High-Throughput Screening
Table 1: Recommended DFT Method Combinations for High-Throughput Thermodynamic Stability Screening
| Application Scope | Recommended Functional | Recommended Basis Set | Accuracy Estimate | Computational Cost | Key Limitations |
|---|---|---|---|---|---|
| Initial screening of large compositional spaces | r$^2$SCAN-3c [54] | def2-mSVP [54] | ~30 meV/atom for formation energies [54] | Low | Limited accuracy for weak dispersion interactions |
| Primary stability prediction for inorganic compounds | PBEsol [21] | Plane-wave (500-600 eV cutoff) | ~20-30 meV/atom for formation energies [21] | Medium | Less accurate for molecular systems |
| High-accuracy validation of candidate materials | Hybrid functionals (e.g., PBE0) [54] | def2-TZVP [54] | < 20 meV/atom for formation energies [54] | High | Prohibitive for systems >100 atoms |
| Molecular/organic systems with dispersion interactions | B97M-V [54] | def2-SVPD [54] | ~15 meV/atom for organic formation energies [54] | Medium-High | Requires robust integration grids |
Basis Set Selection Protocol
Basis set selection requires careful consideration of both accuracy requirements and computational constraints:
- Minimal basis sets should be avoided except for initial structural prototyping due to significant basis set superposition error (BSSE) and poor convergence [54].
- Double-zeta basis sets with polarization functions (e.g., def2-SVP) provide a reasonable compromise for initial geometry optimizations but may insufficient for final energy evaluations [58].
- Triple-zeta basis sets with polarization functions (e.g., def2-TZVP) are recommended for final single-point energy calculations where highest accuracy is required [54].
- Diffuse functions augmentation (e.g., def2-SVPD) becomes important for systems with significant non-covalent interactions or electron-rich atoms [58].
- Basis set superposition error (BSSE) corrections should be applied when using smaller basis sets or for systems dominated by weak interactions [54].
Diagram 1: Basis set selection workflow for high-throughput DFT calculations. The pathway guides researchers through critical decision points based on system type, accuracy requirements, and chemical interactions.
Integrated High-Throughput Screening Protocol
Tiered Screening Workflow
A tiered screening approach maximizes efficiency while maintaining reliability in thermodynamic stability assessment:
Initial Compositional Screening (Tier 1)
- Objective: Rapid evaluation of large compositional spaces (10$^3$-10$^5$ compounds)
- Methods: Machine learning models trained on DFT data or low-cost DFT functionals (r$^2$SCAN-3c)
- Basis set: def2-mSVP or minimal basis sets for initial ranking
- Validation: Compare against known stable compounds from materials databases [2]
Intermediate DFT Screening (Tier 2)
- Objective: Accurate stability assessment for top candidates (10$^2$-10$^3$ compounds)
- Methods: Robust meta-GGA functionals (B97M-V, SCAN) with dispersion corrections
- Basis set: def2-SVP or def2-TZVP for molecular systems; plane-wave 500-600 eV for solids
- Output: E$_{\text{hull}}$ values with uncertainty <30 meV/atom
High-Accuracy Validation (Tier 3)
- Objective: Final validation of most promising candidates (10-100 compounds)
- Methods: Hybrid functionals (PBE0, ωB97X-V) or composite methods
- Basis set: def2-TZVP or larger with diffuse functions if needed
- Validation: Comparison with experimental data where available [54]
Diagram 2: Tiered high-throughput screening protocol for thermodynamic stability assessment. The multi-stage approach efficiently narrows candidate pools while maintaining accuracy through successive refinement.
Machine Learning-Augmented Workflows
Recent advances enable synergistic integration of machine learning with DFT calculations:
- Stability prediction: Ensemble models like ECSG (Electron Configuration models with Stacked Generalization) achieve AUC scores of 0.988 in predicting stability, requiring only one-seventh of the data needed by conventional models to achieve comparable accuracy [2].
- Feature selection: For perovskite stability prediction, feature selection algorithms have identified that approximately 70 key features extracted from elemental properties are sufficient for accurate modeling without overfitting [57].
- Active learning: ML models can prioritize DFT calculations on compositions most likely to be stable, dramatically reducing computational burden [2].
Advanced Methods and Emerging Approaches
Machine-Learned Density Functionals
The emerging paradigm of machine-learned functionals represents a potential breakthrough in computational materials discovery:
- Deep learning approaches: Neural network-based functionals like Microsoft's Skala model learn the exchange-correlation functional directly from high-accuracy reference data, reaching "hybrid-like accuracy" at significantly lower computational cost [55].
- Data requirements: These approaches require generating extensive high-accuracy training data from wavefunction methods, but once trained, can generalize to unseen molecules with experimental-level accuracy [55] [56].
- Nonlocal descriptors: ML functionals can incorporate nonlocal density descriptors that have hitherto been impractical in traditional functional design, potentially overcoming long-standing accuracy limitations [56].
Error Assessment and Validation Protocols
Robust validation remains essential for reliable high-throughput screening:
- Internal consistency checks: Compare formation energies across different rungs of Jacob's Ladder to identify potential functional-driven errors [54].
- Database comparison: Validate predicted stable compounds against experimental databases and high-quality computational repositories like the Materials Project [21] [2].
- Uncertainty quantification: Implement statistical error estimation for predicted E$_{\text{hull}}$ values, with particular attention to compositions near the stability threshold (typically 0-50 meV/atom) [57].
Table 2: Error Ranges for Different Functional Classes in Thermodynamic Property Prediction
| Functional Class | Typical Formation Energy Error (meV/atom) | Typical E$_{\text{hull}}$ Error (meV/atom) | Representative Examples |
|---|---|---|---|
| LDA | 50-150 | 40-100 | SVWN, PZ81 |
| GGA | 30-80 | 25-60 | PBE, PBEsol |
| meta-GGA | 20-50 | 15-40 | SCAN, B97M-V |
| Hybrid | 15-35 | 10-25 | PBE0, HSE06 |
| ML Functionals | 10-25 | 8-20 | Skala [55] |
| Composite Methods | 5-15 | 5-12 | B3LYP-3c, r$^2$SCAN-3c |
Table 3: Key Research Reagent Solutions for High-Throughput DFT Screening
| Resource Category | Specific Tools/Platforms | Primary Function | Application in Stability Screening |
|---|---|---|---|
| DFT Codes | VASP, Quantum ESPRESSO, Gaussian, ORCA | Electronic structure calculation | Core engine for energy and property calculations |
| High-Throughput Frameworks | AFLOW, PyMatGen, AiiDA | Workflow automation | Manages thousands of calculations with consistent parameters |
| Materials Databases | Materials Project, JARVIS-DFT, OQMD | Reference data repository | Provides training data for ML models and validation benchmarks |
| Machine Learning Libraries | Scikit-learn, DeepChem, Roost, Magpie | Predictive modeling | Accelerates initial screening and identifies promising candidates |
| Analysis Tools | Pymatgen (phase diagrams), VESTA (visualization) | Data interpretation | Constructs convex hull diagrams and analyzes structural properties |
| Reference Data | W4-17, GMTKN55 | Benchmark datasets | Validates functional/basis set accuracy for thermochemical properties |
The strategic selection of density functionals and basis sets represents a critical determinant of success in high-throughput DFT screening for thermodynamic stability. By implementing the tiered protocols outlined in this document—beginning with rapid ML-based screening, progressing through medium-level DFT calculations, and culminating in high-accuracy validation—researchers can efficiently navigate vast compositional spaces while maintaining the accuracy necessary for predictive materials discovery. As machine-learned functionals and increasingly sophisticated basis sets continue to evolve, the balance between computational cost and accuracy will progressively shift, enabling ever more ambitious exploration of chemical space for next-generation materials and pharmaceutical compounds.
Addressing Systematic Biases with Ensemble Machine Learning and Stacked Generalization
Systematic biases present a significant challenge in high-throughput Density Functional Theory (DFT) calculations, particularly in thermodynamic stability screening for materials discovery and drug development. These biases can arise from various sources, including limitations in exchange-correlation functionals, insufficient convergence criteria, and the inherent constraints of modeling complex systems. Ensemble machine learning, particularly stacked generalization, offers a powerful framework for mitigating these biases by combining multiple models to produce more robust and reliable predictions. This approach leverages the strengths of diverse algorithms and datasets, effectively reducing both bias and variance that plague individual models [59]. The application of these techniques is especially valuable in computational materials science and pharmaceutical research, where high-throughput screening generates vast datasets requiring accurate and unbiased interpretation for successful experimental validation.
Understanding Systematic Biases in High-Throughput DFT
High-throughput DFT calculations, while powerful, are susceptible to several systematic biases that can compromise the reliability of thermodynamic stability predictions. Key biases include:
Functional-driven biases: Approximations in exchange-correlation functionals (e.g., GGA-PBE) systematically affect formation energy calculations, often overbinding or underbinding specific chemical environments [11].
Configuration sampling biases: Incomplete exploration of configuration space, particularly in complex systems like high-entropy materials, leads to inadequate representation of the true thermodynamic landscape [24].
Numerical convergence biases: Incomplete convergence with respect to k-point sampling, basis set size, or other computational parameters introduces systematic errors in property predictions [11].
Data representation biases: Underrepresentation of certain chemical spaces or structural motifs in training data creates gaps that propagate through the screening pipeline [60].
These biases manifest quantitatively in calculated properties. For example, in perovskite stability screening, formation energy errors can exceed 0.1 eV/atom for certain compositions, significantly impacting stability predictions [11].
Consequences for Materials and Drug Discovery
The impact of these biases extends throughout the discovery pipeline. In materials research, biased stability predictions can lead to failed synthetic attempts or overlooking promising candidates. For instance, in ABO₃ perovskite screening, approximately 8% of predicted stable compounds may be misclassified due to systematic biases [11]. In pharmaceutical applications, biased predictions of protein-ligand interactions or compound stability can misdirect experimental resources and delay drug development timelines [61]. The financial and temporal costs of these inaccuracies underscore the critical need for robust bias mitigation strategies.
Table 1: Quantifying Common Biases in High-Throughput DFT Screening
| Bias Type | Typical Magnitude | Primary Impact | Common Mitigation Approaches |
|---|---|---|---|
| Functional Bias | 0.05-0.15 eV/atom | Formation energies, band gaps | Hybrid functionals, DFT+U, ML corrections |
| Configuration Sampling | 0.02-0.10 eV/atom | Stability rankings | Enhanced sampling, genetic algorithms |
| Numerical Convergence | 0.01-0.05 eV/atom | Absolute energies | Convergence testing, standardized protocols |
| Data Representation | Varies by system | Predictive accuracy | Data augmentation, transfer learning |
Ensemble Learning Fundamentals
Core Principles and Terminology
Ensemble learning operates on the principle that combining multiple models (base learners) produces superior predictive performance compared to any single constituent model. This approach effectively addresses the bias-variance tradeoff fundamental to machine learning [59]. Key terminology includes:
Base learners: Individual models (e.g., regression models, neural networks) that form the ensemble components [59]
Weak vs. strong learners: Weak learners perform slightly better than random guessing, while strong learners achieve high predictive accuracy (≥80% in binary classification) [59]
Bias-variance tradeoff: Bias measures average prediction error on training data, while variance measures prediction differences across model realizations [59]
Ensemble methods are particularly effective because they combine diverse models with different bias-variance characteristics, yielding lower overall error while retaining individual model strengths [59]. Research indicates that greater diversity among combined models generally produces more accurate ensemble predictions [59].
Ensemble Methodologies for Bias Reduction
Three primary ensemble methodologies have demonstrated effectiveness in addressing systematic biases:
Bagging (Bootstrap Aggregating): A parallel ensemble method that creates multiple dataset replicas through bootstrap resampling, trains base learners on these samples, and aggregates predictions through voting or averaging [59]. This approach primarily reduces variance without increasing bias, making it particularly effective for high-variance models like decision trees.
Boosting: A sequential method that iteratively trains learners, with each new model focusing on instances misclassified by previous models [59]. Algorithms like AdaBoost and Gradient Boosting progressively reduce both bias and variance by weighting misclassified samples or using residual errors to guide subsequent learning [59].
Stacked Generalization (Stacking): A heterogeneous parallel method that trains multiple base learners using different algorithms, then combines their predictions through a meta-learner [59]. This approach exemplifies meta-learning and often achieves superior performance by leveraging the complementary strengths of diverse algorithms [62].
Table 2: Comparative Analysis of Ensemble Methods for Bias Mitigation
| Method | Training Approach | Primary Bias Effect | Computational Cost | Best-Suited Applications |
|---|---|---|---|---|
| Bagging | Parallel, homogeneous | Reduces variance | Moderate | High-dimensional data, overfitting prevention |
| Boosting | Sequential, homogeneous | Reduces bias & variance | High | Imbalanced data, complex relationships |
| Stacking | Parallel, heterogeneous | Optimizes bias-variance tradeoff | High | Heterogeneous data, maximizing accuracy |
Stacked Generalization: Theory and Implementation
Conceptual Framework and Mathematical Foundation
Stacked generalization, also known as stacking or Super Learning, represents an advanced ensemble technique that combines multiple predictive models through a meta-learning framework [62]. The core concept involves training diverse "level-zero" algorithms on the original data, then using their cross-validated predictions as input features for a "level-one" meta-learner that produces final predictions [62].
The mathematical foundation rests on V-fold cross-validation to generate the level-one data. For a library of K candidate algorithms, the process generates cross-validated predictions Ŷₖ⁻⁽ⁱ⁾ for each observation i, where the superscript -(i) indicates the model was trained without the fold containing observation i [62]. These predictions form the design matrix for the meta-learner:
Z = [Ŷ₁⁻⁽ⁱ⁾, Ŷ₂⁻⁽ⁱ⁾, ..., Ŷₖ⁻⁽ⁱ⁾]
The meta-learner then learns the optimal combination of these predictions, typically under constraints such as non-negative coefficients summing to 1 (convex combination) [62]. Theoretical results guarantee that in large samples, the stacked ensemble will perform at least as well as the best individual algorithm in the library [62].
Implementation Protocol for Computational Screening
Implementing stacked generalization for high-throughput DFT screening follows a structured protocol:
Step 1: Library Definition
- Select a diverse collection of level-zero algorithms including both parametric and non-parametric methods
- Ensure algorithmic diversity (regression models, tree-based methods, neural networks, etc.)
- Include algorithms with different inductive biases and regularization approaches
Step 2: V-Fold Cross-Validation
- Split data into V mutually exclusive and exhaustive folds (typically V=5 or V=10)
- For each fold v:
- Define fold v as validation set, remaining folds as training set
- Train each level-zero algorithm on the training set
- Generate predictions for the validation set
- Compile cross-validated predictions for entire dataset
Step 3: Meta-Learner Training
- Use cross-validated predictions as features for meta-learner
- Employ non-negative least squares with sum-to-one constraints
- Alternatively, use more flexible meta-learners with regularization to prevent overfitting
Step 4: Final Ensemble Construction
- Retrain all level-zero algorithms on full dataset
- Combine predictions using weights from meta-learner
- Validate ensemble performance on held-out test set
This protocol systematically reduces reliance on any single modeling assumption, effectively mitigating biases inherent to individual algorithms [62].
Application to High-Throughput DFT Screening
Workflow Integration
Integrating stacked generalization into high-throughput DFT workflows requires careful architectural planning. The complete pipeline encompasses data generation, feature engineering, model training, and validation, with ensemble methods acting as the predictive core.
Diagram 1: Stacked Generalization Workflow for DFT Screening. This diagram illustrates the integration of stacked generalization into high-throughput DFT screening pipelines, showing the flow from initial calculations through ensemble construction to final predictions.
Case Study: Perovskite Stability Prediction
A prominent application of ensemble methods in computational materials science involves screening ABO₃ perovskites for thermodynamic stability. In a comprehensive study screening 5,329 perovskite compositions, researchers employed high-throughput DFT to calculate formation energies, stability metrics, and oxygen vacancy formation energies [11]. The dataset included both cubic and distorted structures, with stability assessed against all known competing phases in the Open Quantum Materials Database [11].
When applying ensemble methods to this challenge, the stacked generalization approach would typically incorporate diverse algorithms including:
- Kernel ridge regression for capturing nonlinear relationships
- Random forests for handling complex feature interactions
- Gradient boosted trees for sequential refinement
- Neural networks for high-dimensional pattern recognition
In practice, similar ensemble approaches have identified 395 thermodynamically stable perovskites, many previously unreported experimentally [11]. The ensemble methodology proved particularly valuable for handling the diverse chemical space spanned by 73 different elements occupying A and B sites [11].
Case Study: MOF-74 Screening for Gas Separation
Another compelling application involves screening metal-organic frameworks (MOFs) for gas separation and storage applications. In a high-throughput DFT screening of single-metal and high-entropy MOF-74 variants, researchers identified Mn-MOF-74 as exhibiting superior CO₂/N₂ separation capability (ΔECO₂ - ΔEN₂ = -0.19 eV) [24]. Notably, the high-entropy MnFeCoNiMgZn-MOF-74 demonstrated enhanced H₂ storage capability (ΔEH₂ = -0.16 eV) due to synergistic effects between metal centers [24].
Ensemble methods would be particularly valuable in this context for several reasons:
- Predicting binding energies across diverse metal combinations
- Quantifying uncertainty in DFT-calculated adsorption properties
- Identifying promising high-entropy combinations beyond intuitive chemical rules
- Mitigating functional-driven biases in guest-host interaction energies
The demonstrated success of individual DFT screenings highlights the potential for ensemble approaches to further enhance predictive accuracy and reliability in porous materials discovery.
Experimental Protocols
Protocol 1: Stacked Generalization for Formation Energy Prediction
Purpose: Accurately predict DFT-calculated formation energies while mitigating functional-driven biases.
Materials and Computational Environment:
- DFT-calculated formation energy dataset (e.g., OQMD [11])
- Computing cluster with minimum 32 CPU cores, 128GB RAM
- Python/R programming environment with scikit-learn, MLens, or SuperLearner libraries
Procedure:
- Data Preparation:
- Compile formation energies and feature set (compositional, structural, electronic descriptors)
- Perform train-test split (80-20 ratio) with stratification by chemical system
- Standardize features to zero mean and unit variance
Library Definition:
- Implement 8-10 diverse algorithms including:
- Linear models with regularization (Ridge, Lasso)
- Kernel methods (Support Vector Regression)
- Tree-based ensembles (Random Forest, Gradient Boosting)
- Neural networks (Multilayer Perceptron)
- Implement 8-10 diverse algorithms including:
Cross-Validation Setup:
- Configure 5-fold cross-validation
- For each fold, train base learners on 80% of training data
- Generate predictions on remaining 20% of training data
- Repeat until all training instances have out-of-fold predictions
Meta-Learner Training:
- Compile cross-validated predictions as level-one data
- Train meta-learner (typically linear model with non-negative constraints)
- Validate on held-out training fold to optimize meta-learner hyperparameters
Evaluation:
- Retrain entire system on full training set using optimized weights
- Assess performance on held-out test set using MAE, RMSE, and R² metrics
- Compare against individual model performance to quantify improvement
Validation: Perform learning curve analysis to ensure adequate training data, and conduct residual analysis to identify systematic error patterns.
Protocol 2: Bias-Aware Ensemble for Stability Classification
Purpose: Classify materials as stable/unstable while addressing data representation biases.
Materials and Computational Environment:
- Labeled dataset of stable/unstable materials with balanced representation
- Feature set including ionic radii, electronegativity, coordination environments
- Computing environment with GPU acceleration for deep learning components
Procedure:
- Bias Assessment:
- Analyze training data for representation gaps across chemical spaces
- Identify underrepresented element combinations and structural types
- Quantify potential measurement biases in reference DFT calculations
Bias-Aware Library Construction:
- Include algorithms with varying sensitivity to data imbalances
- Implement weighted loss functions for underrepresented classes
- Incorporate synthetic data generation for sparse regions of feature space
Stratified Cross-Validation:
- Implement stratified sampling to maintain class balance across folds
- Ensure proportional representation of chemical systems in each fold
- Monitor performance consistency across data subgroups
Ensemble Optimization:
- Optimize for balanced accuracy rather than overall accuracy
- Incorporate fairness constraints into meta-learner objective function
- Validate on external datasets with different composition distributions
Uncertainty Quantification:
- Estimate prediction uncertainties through bootstrap aggregating
- Flag high-uncertainty predictions for potential experimental prioritization
- Calibrate confidence scores using Platt scaling or isotonic regression
Validation: Conduct subgroup analysis to verify consistent performance across chemical systems, and compare against experimental observations when available.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Ensemble Learning in Materials Screening
| Tool/Category | Specific Examples | Function | Implementation Considerations |
|---|---|---|---|
| DFT Codes | VASP [11], Quantum ESPRESSO | Generate training data: formation energies, electronic structures | Accuracy-efficiency tradeoffs, functional selection |
| Feature Generation | matminer, pymatgen | Compute compositional, structural, electronic descriptors | Feature relevance, dimensionality reduction |
| Ensemble Libraries | scikit-learn, MLens, H2O.ai | Implement bagging, boosting, stacking algorithms | Computational scalability, parallelization |
| Meta-Learners | Non-negative least squares, Regularized linear models | Optimally combine base model predictions | Constraint implementation, regularization strength |
| Validation Frameworks | Cross-validation, Bootstrap | Estimate performance, quantify uncertainty | Data splitting strategy, performance metrics |
| High-Throughput Infrastructure | AiiDA, FireWorks | Automate workflow management | Scalability, fault tolerance, data provenance |
Performance Benchmarking and Validation
Quantitative Performance Metrics
Rigorous validation is essential for evaluating ensemble methods in computational materials science. Key metrics include:
Predictive accuracy: Mean absolute error (MAE) and root mean squared error (RMSE) for continuous properties; accuracy, F1-score, and AUC for classification tasks
Uncertainty quantification: Calibration curves, confidence interval coverage, and proper scoring rules
Computational efficiency: Training and inference time, memory footprint, and scaling behavior
Bias mitigation effectiveness: Performance consistency across data subgroups, fairness metrics
In practical applications, stacked generalization has demonstrated significant performance improvements. For instance, in psychosocial maladjustment prediction, a stacked ensemble achieved accuracy of 0.834 and AUC of 0.909, outperforming individual base learners [63]. Similar improvements have been observed in materials contexts, though specific quantitative benchmarks for DFT applications remain limited in the literature.
Comparative Analysis of Ensemble Strategies
Different ensemble strategies offer distinct tradeoffs for high-throughput screening applications:
Bagging vs. Boosting: Experimental comparisons show that boosting typically achieves higher accuracy but with substantially greater computational cost (approximately 14× longer training time with 200 base learners) [64]. Bagging provides more consistent improvement across datasets and is less prone to overfitting [64].
Stacking vs. Simpler Ensembles: Stacked generalization generally outperforms single-model approaches and simpler averaging ensembles, particularly for heterogeneous datasets where different algorithms excel in different regions of feature space [62]. The performance advantage comes with increased complexity in implementation and tuning.
Diagram 2: Stacking Architecture for Bias Reduction. This diagram illustrates how stacked generalization combines predictions from multiple base models through a meta-learner to produce more accurate and robust predictions than any single model.
Ensemble machine learning methods, particularly stacked generalization, offer powerful approaches for addressing systematic biases in high-throughput DFT screening. By combining diverse models through sophisticated aggregation strategies, these techniques mitigate functional-driven, configuration-based, and data-representation biases that plague individual predictors. The integration of these methods into computational materials science and drug discovery pipelines enhances prediction reliability, reduces false leads, and accelerates the discovery process.
Future research directions include developing domain-adapted ensemble methods specifically designed for materials science challenges, integrating active learning with ensemble approaches to strategically guide data acquisition, and creating more interpretable ensemble architectures that provide physical insights alongside predictions. As high-throughput computational screening continues to expand across materials and pharmaceutical domains, ensemble methods will play an increasingly vital role in ensuring the reliability and actionable insights derived from these powerful approaches.
Mitigating Errors in Solvation and Non-Equilibrium Process Modeling
Accurate modeling of solvation effects and non-equilibrium processes is crucial for high-throughput density functional theory (DFT) screening, particularly in applications ranging from drug development to materials design. These simulations present unique challenges because they must capture the complex interplay between solute electronic structure and solvent environment, often under conditions where the solvent cannot reach instantaneous equilibrium with the changing solute charge distribution. The reliability of high-throughput predictions of thermodynamic stability, spectroscopic properties, and reactivity directly depends on effectively mitigating errors in these computationally demanding scenarios. This application note outlines theoretical frameworks, practical protocols, and error-mitigation strategies to enhance the reliability of solvation models in high-throughput DFT workflows, with particular emphasis on non-equilibrium processes relevant to spectroscopic properties and charge transfer events.
Theoretical Framework
Equilibrium versus Non-Equilibrium Solvation
In solvation modeling, a critical distinction exists between equilibrium and non-equilibrium regimes:
Equilibrium solvation applies to processes slow enough to allow complete relaxation of all solvent polarization mechanisms (electronic and nuclear) to the solute's charge distribution. This regime uses the static dielectric constant (ε₀) and is appropriate for geometry optimizations and property calculations of stable states [65].
Non-equilibrium solvation governs ultrafast processes like electronic excitations and charge transfer where only the solvent's electronic polarization can adjust during the event. The slower orientational and vibrational components remain frozen in a non-equilibrium state. This approach requires the optical dielectric constant (ε∞), typically set as the square of the solvent's refractive index (ε∞ = n_refr²) [66] [65].
State-Specific Solvation Models
Traditional linear response (LR)-PCM methods couple the solvent response to the transition density between ground and excited states, making them suitable only for optically allowed transitions. However, state-specific (SS) approaches offer a more generalized framework by directly modeling the solvent response to the entire charge density of a specific electronic state, making them essential for charge-transfer states, triplet excitations, and other optically forbidden transitions [65].
The novel non-equilibrium solvation theory based on constrained equilibrium thermodynamics provides a rigorous foundation for SS methods, properly accounting for solvent reorganization energy in ultrafast processes [66] [67]. This framework enables more accurate calculation of solvatochromic Stokes shifts and other spectroscopic properties by correctly capturing the non-equilibrium polarization response during electronic transitions.
Computational Protocols
Protocol for Spectroscopic Properties in Solution
This protocol details the calculation of absorption and emission energies in solution, including Stokes shifts, using state-specific TD-DFT with non-equilibrium solvation.
Table 1: Key Computational Parameters for Spectroscopic Calculations
| Parameter | Recommended Setting | Purpose |
|---|---|---|
| Solvation Method | SS-PCM with non-equilibrium correction | Captures solvent electronic polarization during fast excitation |
| Ground State Optimization | Equilibrium solvation (ε₀) | Relaxed initial state geometry |
| Vertical Excitation | Non-equilibrium solvation (ε∞) | Models instantaneous electronic transition |
| Excited State Optimization | Equilibrium solvation (ε₀) | Relaxed excited state geometry |
| Emission Energy | Non-equilibrium solvation (ε∞) | Models emission from relaxed excited state |
Workflow Implementation:
Ground State Geometry Optimization
- Employ DFT with a hybrid functional (e.g., PBE0)
- Use equilibrium solvation with the static dielectric constant
- Verify convergence and absence of imaginary frequencies
Vertical Excitation Energy Calculation
- Perform TD-DFT with state-specific non-equilibrium solvation
- Set OpticalDielectric to the square of solvent's refractive index
- Activate StateSpecific keyword in $pcm section [65]
Excited State Geometry Optimization
- Optimize geometry in the target excited state
- Use equilibrium solvation (static dielectric constant)
- Confirm convergence with excited-state frequency analysis
Emission Energy Calculation
- Compute vertical emission from optimized excited state geometry
- Apply non-equilibrium solvation with optical dielectric constant
Stokes Shift Calculation
- Calculate as the difference between absorption and emission maxima [66]
High-Throughput Screening Protocol for Thermodynamic Stability
This protocol enables high-throughput assessment of thermodynamic stability for materials discovery applications.
Table 2: DFT Parameters for High-Throughput Stability Screening
| Parameter | Recommended Setting | Rationale |
|---|---|---|
| Functional | PBE with DFT+U for transition metals | Balanced accuracy/efficiency for diverse compositions |
| Basis Set | Plane-wave or localized basis appropriate for system | Consistent treatment across chemical space |
| Solvation | Implicit solvation model (e.g., C-PCM) for solution properties | Efficiency for high-throughput screening |
| Convergence Criteria | Tightened for energy and forces (e.g., 10⁻⁶ eV) | Reliable formation energies |
Workflow Implementation:
Structure Generation
Formation Energy Calculation
- Compute Hf(ABO₃) = E(ABO₃) - μA - μB - 3μO [11]
- Use appropriate reference states with experimental corrections where needed
Stability Assessment
- Calculate convex hull distance: Hstab(ABO₃) = Hf(ABO₃) - H_hull [11]
- Identify stable compounds (H_stab < 0.025 eV/atom)
Property Prediction
- Compute target properties (band gap, oxygen vacancy formation, binding energies)
- For MOF gas adsorption: calculate ΔE_ads = E(MOF+gas) - E(MOF) - E(gas) [24]
Error Mitigation Strategies
Addressing Functional and Density-Driven Errors
DFT errors in solvation energy calculations can be decomposed into functional errors and density-driven errors, with the latter arising from inaccuracies in the electron density due to self-interaction error:
- Functional Error (ΔE_func): Inaccuracy due to the exchange-correlation functional approximation
- Density-Driven Error (ΔE_dens): Error resulting from inaccuracies in the self-consistent electron density [68]
Mitigation Approaches:
Hybrid Functional Selection: Use modern hybrid functionals (e.g., ωB97X-D, B3LYP-D3) with demonstrated performance for main-group chemistry [68]
HF-DFT Analysis: For systems prone to delocalization error, evaluate using HF densities in the DFT functional (HF-DFT) to identify density-sensitive cases [68]
Error Decomposition: Perform DFT error decomposition to identify whether functional or density-driven errors dominate for specific chemical systems
Reference Comparisons: Where feasible, compare with local coupled cluster [LNO-CCSD(T)] references to quantify and identify error patterns [68]
Uncertainty Quantification in High-Throughput Workflows
In high-throughput screening, incorporate uncertainty estimates through:
Phonon-Induced Fluctuations: Account for thermal fluctuations using ab initio molecular dynamics (AIMD), particularly for adsorption energies and reaction barriers where standard deviations can reach ~0.16 eV [39]
Functional Sensitivity Analysis: Compute properties with multiple functionals to assess prediction variability
Machine Learning Enhancement: Integrate graph neural networks with semi-supervised learning to reconcile computational and experimental data discrepancies [69]
Research Reagent Solutions
Table 3: Essential Computational Tools for Solvation Modeling
| Tool Category | Specific Implementation | Application |
|---|---|---|
| Quantum Chemistry Software | Q-Chem with SS-PCM/TD-DFT | State-specific solvation for excitation energies [66] [67] [65] |
| High-Throughput Frameworks | OQMD database with qmpy python package | Thermodynamic stability screening [11] |
| Wavefunction Methods | Local natural orbital CCSD(T) (LNO-CCSD(T)) | Gold-standard references for error analysis [68] |
| Cloud-Native DFT | Accelerated DFT (GPU-optimized) | Rapid screening of large materials spaces [70] |
| Solvation Databases | Semi-supervised graph neural networks | Predicting solvation free energies [69] |
Workflow Visualization
Application Examples
Solvatochromic Stokes Shift in Fluorescent Dyes
The SS-PCM/TD-DFT method with novel non-equilibrium solvation theory successfully predicted solvatochromic Stokes shifts for the BBTA fluorescent dye in DMSO solvent. The calculated absorption and emission energies as well as the Stokes shift showed excellent agreement with experimental values, with electrostatic and polarization components identified as crucial contributors to spectral shifts in polar solvents [66]. This demonstrates the protocol's utility for designing fluorescent probes with tailored photophysical properties.
High-Entropy MOF Screening for Gas Separation
High-throughput DFT screening of single-metal and high-entropy MOF-74 structures identified Mn-MOF-74 as having superior CO₂/N₂ separation capability (ΔECO₂ - ΔEN₂ = -0.19 eV) and MnFeCoNiMgZn-MOF-74 as exhibiting enhanced H₂ storage (ΔE_H₂ = -0.16 eV) due to synergistic effects between metal centers [24]. The workflow incorporated binding energy calculations, thermodynamic stability assessment, and AIMD simulations for uncertainty quantification.
Implementing robust error mitigation strategies in solvation and non-equilibrium process modeling is essential for reliable high-throughput DFT screening. The state-specific solvation approaches, rigorous protocols for spectroscopic properties, and comprehensive error analysis frameworks outlined in this application note provide researchers with practical tools to enhance prediction accuracy across diverse applications from drug development to materials design. As high-throughput computational screening continues to expand, integrating these methodologies will be crucial for bridging the gap between computational predictions and experimental realization.
Strategies for Improving Sample Efficiency and Model Generalization
In high-throughput density functional theory (DFT) calculations for thermodynamic stability screening, two intertwined challenges are paramount: achieving sample efficiency to make thousands of computations tractable, and ensuring model generalization so that predictions remain accurate for novel, unseen compositions. High-throughput DFT involves computations on hundreds, thousands, or even tens of thousands of compounds, a scale that demands new calculation and data management methodologies [21]. This Application Note details protocols that leverage machine learning (ML) and strategic descriptor selection to simultaneously enhance sample efficiency and model generalizability, thereby accelerating the reliable discovery of new materials.
Theoretical Background and Key Concepts
The Sample Efficiency and Generalization Problem in High-Throughput Screening
In the context of high-throughput DFT, sample efficiency refers to minimizing the number of computationally expensive, first-principles calculations required to explore a vast chemical space or to optimize simulation parameters. Model generalization is the ability of a model or descriptor trained on a finite set of calculations to make accurate predictions for materials outside its training set.
These concepts are deeply connected. An inefficient screening process that relies on brute-force calculation limits the diversity and quantity of training data, which can hamper the development of generalizable models. Conversely, poor generalization renders high-throughput efforts ineffective for genuine discovery. The core strategy for addressing this dual problem involves replacing expensive calculations with cheaper, physically-relevant proxies or optimization procedures where possible, without sacrificing predictive fidelity [71] [25].
Foundational Principles
The following principles underpin the protocols in this document:
- Physical Insight as a Regularizer: Incorporating known physical laws and principles into ML models intrinsically limits the search space to physically plausible solutions. This structured approach, as seen in custom thermodynamics platforms, improves interpretability and generalization compared to fully unstructured black-box models [72].
- Data-Efficient Optimization: Bayesian optimization (BO) is a powerful derivative-free method for finding the optimum of complex, expensive-to-evaluate functions with minimal data points. Its data-efficiency stems from building a surrogate probabilistic model of the objective function and using it to strategically select the most informative points to evaluate next [71].
- Descriptor Similarity for Property Transfer: Materials with similar electronic structures tend to exhibit similar properties. Using the similarity in electronic structure descriptors, such as the density of states (DOS), as a screening criterion allows for the identification of novel materials with catalytic or stability properties comparable to a known reference, effectively transferring knowledge from a few calculations to a broader chemical space [25].
Protocols for Enhanced Sample Efficiency and Generalization
Protocol 1: Bayesian Optimization of DFT Charge Mixing Parameters
- Aim: To significantly reduce the number of self-consistent field (SCF) iterations required for DFT convergence by optimizing charge mixing parameters, thereby lowering the computational cost of individual simulations [71].
- Background: The convergence of a DFT calculation depends sensitively on the parameters controlling the mixing of the charge density between SCF iterations. Default parameters are often suboptimal, leading to excessive iterations. Manually tuning them is tedious. Bayesian optimization provides a systematic and automated approach.
Diagram 1: Bayesian optimization workflow for DFT parameters.
Experimental Procedure:
- Define Parameter Space: Identify the charge mixing parameters to optimize (e.g.,
AMIX,BMIX,AMIX_MAG,BMIX_MAGin VASP) and define a plausible range for each. - Set Objective Function: The objective is to minimize the number of SCF iterations required to reach convergence. A single evaluation of the objective function involves running a VASP calculation with a proposed set of parameters and recording the final SCF iteration count.
- Initialize BO: Select an initial set of parameter combinations (e.g., via Latin Hypercube sampling) and run the corresponding DFT calculations to seed the BO surrogate model.
- BO Loop: For a fixed number of iterations or until convergence: a. The Gaussian process surrogate model is updated with all available (parameters, SCF iterations) data. b. The next parameter set to evaluate is chosen by maximizing an acquisition function (e.g., Expected Improvement). c. A new DFT calculation is run with the proposed parameters. d. The result is added to the dataset.
- Output: The parameter set yielding the lowest number of SCF iterations is identified as optimal.
- Define Parameter Space: Identify the charge mixing parameters to optimize (e.g.,
Validation: The optimized parameters should be tested on a held-out set of material systems (insulators, semiconductors, metals) to confirm they consistently outperform default settings without compromising physical accuracy [71].
Protocol 2: High-Throughput Screening via Electronic Structure Similarity
- Aim: To discover novel bimetallic catalysts or stable alloys with properties comparable to a precious-metal reference (e.g., Pd) by screening thousands of candidates based on electronic structure similarity, minimizing the need for exhaustive DFT-based property calculation [25].
- Background: This protocol leverages the principle that the full electronic density of states (DOS) pattern is a rich descriptor of a material's chemical properties. By quantifying similarity to a target DOS, one can rapidly prioritize candidates for full validation.
Diagram 2: High-throughput screening using DOS similarity.
Experimental Procedure:
- Candidate Generation & Stability Pre-screening:
- Generate a large set of candidate structures (e.g., 4350 ordered bimetallic alloys).
- Perform high-throughput DFT calculations to compute the formation energy (ΔEf) for each.
- Filter out thermodynamically unstable structures (e.g., ΔEf > 0.1 eV/atom) [25].
- Descriptor Calculation:
- For all thermodynamically stable candidates, perform a single, static DFT calculation to obtain the projected DOS on the close-packed surface atoms.
- Calculate the reference material's DOS (e.g., Pd(111)) using the same computational parameters.
- Similarity Quantification:
- Compute the similarity metric (ΔDOS) between each candidate and the reference. The metric should emphasize states near the Fermi level using a Gaussian weighting function [25]:
ΔDOS = { ∫ [DOS_candidate(E) - DOS_reference(E)]² · g(E; σ) dE }^(1/2)whereg(E; σ)is a Gaussian centered at the Fermi energy with a standard deviation of ~7 eV.
- Compute the similarity metric (ΔDOS) between each candidate and the reference. The metric should emphasize states near the Fermi level using a Gaussian weighting function [25]:
- Candidate Selection:
- Rank all candidates by their ΔDOS value (lowest to highest).
- Select the top candidates (e.g., 10-20) with the highest similarity for subsequent experimental validation and targeted, high-accuracy DFT analysis of the property of interest (e.g., catalytic activity, thermodynamic stability).
- Candidate Generation & Stability Pre-screening:
Validation: Experimental synthesis and testing of the top candidates is crucial. For example, this protocol successfully identified a novel Ni-Pt catalyst with performance comparable to Pd for H₂O₂ synthesis, validating the generalization power of the DOS similarity descriptor [25].
Data Analysis and Interpretation
Quantitative Comparison of Methods
Table 1: Comparison of Protocols for Improving Efficiency and Generalization.
| Protocol | Primary Efficiency Gain | Generalization Mechanism | Reported Efficacy |
|---|---|---|---|
| Bayesian Optimization of DFT Parameters [71] | Reduces SCF iterations per calculation (computational cost). | Produces robust parameters that work across diverse systems (metals, insulators, semiconductors). | "Significant time savings" achieved; algorithm is system-agnostic. |
| Screening via DOS Similarity [25] | Reduces the number of systems requiring full property calculation (throughput). | Electronic structure similarity as a physically-motivated descriptor for property transfer. | 4 out of 8 top candidates showed catalytic performance comparable to Pd; discovery of Ni61Pt39. |
| Thermodynamic Regularization [73] | Improves data efficiency by preventing overfitting to limited training data. | Physical constraints act as a regularizer, guiding model to generalizable solutions. | Mitigates "thermodynamic overfitting"; improves predictive performance on test data. |
Key Research Reagent Solutions
Table 2: Essential Computational Tools and Their Functions.
| Research Reagent / Tool | Function in Protocol |
|---|---|
| VASP (Vienna Ab initio Simulation Package) [71] | Performs the underlying DFT calculations for both electronic structure determination and energy evaluation. |
| Bayesian Optimization Library (e.g., scikit-optimize) | Implements the surrogate model and acquisition function to efficiently search parameter space. |
| Inorganic Crystal Structure Database (ICSD) [21] | Provides the initial crystal structures for high-throughput screening pipelines. |
| Stochastic OnsagerNet (S-OnsagerNet) [72] | A deep learning platform to discover interpretable, low-dimensional thermodynamic descriptions from high-dimensional data, enforcing physical laws. |
Implementation Guidelines
Integrating Protocols into a High-Throughput Workflow
A robust high-throughput infrastructure should manage data flow through several stages: Data Selection → Data Generation → Data Storage & Retrieval → Data Analysis [21]. The protocols described herein can be integrated as follows:
- Pre-Screening: Use Protocol 2 (DOS similarity) during the Data Selection phase to filter large candidate pools before committing to full property calculations.
- Efficient Calculation: Apply Protocol 1 (Bayesian optimization) within the Data Generation phase to ensure each individual DFT calculation runs at peak efficiency.
- Model Building: When building ML models on calculated data, incorporate thermodynamic regularizers [73] or physical architectures like S-OnsagerNet [72] during Data Analysis to ensure learned models are generalizable and physically consistent.
Troubleshooting and Mitigating Overfitting
A key risk in data-driven materials science is overfitting, where a model performs well on training data but fails to predict new materials accurately. In a thermodynamic context, this manifests as "thermodynamic overfitting," where an overly complex model dissipates more energy in testing than expected from training [73].
- Identification: A significant drop in performance (e.g., predicted stability or work output) between training data and a held-out test set of compositions indicates overfitting.
- Solution: Thermodynamic Regularization: Introduce a cost term during model training that penalizes excessive model complexity. This aligns with the principle of building models with "requisite complexity"—complex enough to capture the environment's structure but not excessively so [73]. Physically, this regularizer corresponds to the work dissipated by an overly complex predictive model.
Validating Predictions and Benchmarking Against Experimental Data
The integration of high-throughput Density Functional Theory (DFT) calculations with experimental validation has emerged as a transformative paradigm in materials science and drug development. This protocol details a robust framework for transitioning from computationally predicted stability to experimentally synthesized materials, a critical pathway within broader thesis research on high-throughput DFT and thermodynamic stability screening. The core challenge lies in effectively bridging the gap between theoretical predictions and synthetic reality, ensuring that computationally identified candidates are not only stable but also synthesizable and functionally valid. The following sections provide a comprehensive, step-by-step guide for researchers and drug development professionals, enabling the efficient experimental validation of materials discovered through computational screening.
Quantitative Screening Metrics and Data Triage
The initial phase of the validation pipeline involves the critical assessment of computational outputs to identify the most promising candidates for experimental synthesis. The key quantitative metrics used for this triage are summarized in Table 1.
Table 1: Key Quantitative Metrics for Experimental Triage from Computational Screening
| Metric | Target Value | Interpretation & Experimental Implication |
|---|---|---|
| DFT Formation Energy (ΔEƒ) | < 0 eV (Negative) | Thermodynamically favorable formation; primary indicator of stability. [74] |
| Formation Energy Margin | ΔEƒ < 0.1 eV | A considered margin for potentially metastable or nanosize-stabilized phases that may still be synthesizable. [74] |
| Energy Above Hull (Ehull) | ~ 0 meV/atom | Phase stability; lower values indicate the material is stable or nearly stable with respect to decomposition into other phases. [75] |
| DOS Similarity (ΔDOS) | → 0 | As the value approaches zero, the electronic structure is more similar to a reference catalyst (e.g., Pd), suggesting comparable catalytic properties. [74] |
| Synthesizability Score (from SynthNN) | High Probability | Classifier prediction of synthetic accessibility, informed by all known synthesized materials; significantly outperforms human experts and formation energy alone. [76] |
The decision to proceed with experimental validation should be based on a consensus across these metrics. For instance, a material with a negative formation energy and a high synthesizability score is a high-priority candidate. A material with a slightly positive formation energy (< 0.1 eV) but a high DOS similarity to a known active material might still be considered for synthesis, leveraging kinetic stabilization effects, particularly in nanoscale systems. [74]
Integrated Computational-Experimental Workflow
A successful validation campaign follows a tightly coupled workflow where computation and experiment inform each other iteratively. The overall process is depicted in the diagram below.
Diagram 1: Integrated Computational-Experimental Validation Workflow.
Detailed Experimental Protocols
Protocol 1: Synthesis of Bimetallic Alloy Catalysts
This protocol is adapted from a successful high-throughput discovery of Pd-substitute bimetallic catalysts. [74]
4.1.1 Principle To synthesize bimetallic alloy nanoparticles predicted via DFT to have electronic density of states (DOS) similar to a reference catalyst (e.g., Pd) and a negative formation energy.
4.1.2 Reagents and Materials
- Metal precursors: e.g., Chloroplatinic acid (H₂PtCl₆), Nickel chloride (NiCl₂), etc.
- Reducing agent: e.g., Sodium borohydride (NaBH₄), Ethylene glycol.
- Stabilizing agent: e.g., Polyvinylpyrrolidone (PVP).
- Support material (if applicable): e.g., Carbon black, Alumina.
- Solvents: e.g., Ethanol, Deionized water.
4.1.3 Procedure
- Solution Preparation: Dissolve the metal precursors in a suitable solvent (e.g., water/ethylene glycol mixture) at a stoichiometric ratio corresponding to the predicted stable composition (e.g., Ni₆₁Pt₃₉).
- Stabilization: Add a stabilizing agent (e.g., PVP) to the solution and stir vigorously to ensure uniform mixing and to prevent nanoparticle agglomeration.
- Reduction: Under an inert atmosphere (e.g., N₂), slowly add a cold, freshly prepared aqueous solution of reducing agent (e.g., NaBH₄) with continuous stirring. The rapid reduction promotes the formation of alloyed phases rather than phase-separated structures.
- Aging and Collection: Allow the reaction mixture to stir for several hours. Recover the nanoparticles by centrifugation, and wash thoroughly with ethanol and water to remove residual reagents.
- Post-processing: Dry the collected nanoparticles under vacuum. For catalytic applications, the material may be calcined and/or activated under a specific gas atmosphere (e.g., H₂) at a controlled temperature to remove surface organics and activate the surface.
4.1.4 Key Considerations
- The nonequilibrium alloyed phases, even with slightly positive formation energy (ΔEƒ < 0.1 eV), can be stabilized by the nanosize effect during this wet-chemical synthesis. [74]
- The composition and structure must be verified before functional testing.
Protocol 2: Structural and Chemical Characterization
4.2.1 Principle To confirm the successful synthesis of the target material, including its phase, composition, and morphology.
4.2.2 Techniques and Methodology
- X-ray Diffraction (XRD):
- Purpose: To identify the crystalline phase and confirm alloy formation (evidenced by a peak shift relative to pure metal peaks).
- Procedure: Prepare a thin film of the powder sample on a glass slide. Acquire a diffraction pattern using a Cu Kα source. Analyze the peak positions and widths using Rietveld refinement.
- Transmission Electron Microscopy (TEM/HRTEM):
- Purpose: To determine nanoparticle size, morphology, and elemental distribution.
- Procedure: Disperse the sample in ethanol and drop-cast onto a lacey carbon TEM grid. Acquire high-resolution images and perform Energy-Dispersive X-ray Spectroscopy (EDS) mapping to confirm the homogeneous distribution of both metals in the alloy.
- X-ray Photoelectron Spectroscopy (XPS):
- Purpose: To analyze the surface composition and electronic state of the elements.
- Procedure: Mount powder on a conductive tape. Acquire spectra under ultra-high vacuum. Use C 1s peak (284.8 eV) for charge correction. Deconvolute peaks for relevant elements to identify oxidation states.
Protocol 3: Functional Property Validation
4.3.1 Principle To test the synthesized material for its intended functional property, thereby validating the initial computational prediction.
4.3.2 Example: Catalytic Testing for H₂O₂ Synthesis [74]
- Reactor Setup: Use a fixed-bed or batch reactor system equipped with mass flow controllers for gases and safety measures for handling H₂ and O₂ mixtures.
- Reaction Conditions:
- Catalyst mass: 10-50 mg.
- Reactant gases: H₂ and O₂ in a non-explosive ratio (e.g., 4:96 or 5:95), diluted in an inert gas (e.g., N₂).
- Pressure: 1 - 10 bar.
- Temperature: 20 - 50 °C.
- Solvent: Often methanol or water.
- Product Quantification:
- Method: Liquid chromatography or colorimetric methods.
- Procedure: Periodically sample the reaction solution. Quench the reaction in the sample. For colorimetry, mix the sample with a titanium oxalate reagent, which forms a yellow complex with H₂O₂, and measure the absorbance at 407 nm. Compare against a standard calibration curve.
- Data Analysis: Calculate key performance indicators such as H₂O₂ productivity (mol·g⁻¹·h⁻¹) and selectivity. Compare the performance of the newly discovered catalyst (e.g., Ni₆₁Pt₃₉) against the reference catalyst (e.g., Pd).
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key materials and reagents essential for executing the described experimental protocols.
Table 2: Key Research Reagents and Materials for Experimental Validation
| Reagent/Material | Function/Application | Example/Notes |
|---|---|---|
| Metal Salts & Precursors | Source of metal elements for nanoparticle synthesis. | Chloroplatinic acid (H₂PtCl₆), Nickel Chloride (NiCl₂), Palladium Acetate (Pd(OAc)₂). [74] |
| Chemical Reducing Agents | To reduce metal ions to their zero-valent state in solution synthesis. | Sodium Borohydride (NaBH₄ - strong), Ethylene Glycol (mild, also serves as solvent). |
| Surfactants & Stabilizers | To control nanoparticle growth and prevent agglomeration. | Polyvinylpyrrolidone (PVP), Cetyltrimethylammonium bromide (CTAB). [74] |
| Porous Support Materials | To provide a high-surface-area matrix for dispersing catalyst nanoparticles. | Carbon black, Mesoporous silica (SBA-15), Alumina (Al₂O₃). |
| Reference Catalysts | Benchmark for performance comparison during functional testing. | Commercial Pd/C catalyst used as a reference in H₂O₂ synthesis studies. [74] |
| Characterization Standards | For calibration of instrumentation. | Silicon powder for XRD calibration; Gold nanoparticles for TEM resolution testing. |
| Thermodynamic Databases | Source of critically evaluated thermodynamic data for analysis. | NIST Web Thermo Tables (WTT). [77] |
The discovery of advanced catalytic materials is pivotal for the development of efficient chemical processes and sustainable energy technologies. Traditional experimental approaches to catalyst development are often time-consuming and resource-intensive, creating a bottleneck in materials innovation. High-throughput computational screening, particularly when integrated with experimental validation, has emerged as a powerful paradigm to accelerate this discovery process. This protocol details a integrated computational-experimental screening framework specifically designed for the discovery of bimetallic catalysts, using the replacement of palladium (Pd) in hydrogen peroxide (H₂O₂) direct synthesis as a demonstrative case study [25]. The methodology is grounded in the fundamental principle that materials with similar electronic structures tend to exhibit similar catalytic properties, enabling the efficient computational prioritization of candidate materials for experimental testing.
Computational Screening Protocol
Initial Candidate Generation and Thermodynamic Stability Screening
The first stage of the protocol involves generating a comprehensive set of candidate structures and evaluating their thermodynamic stability using first-principles calculations.
- Candidate Pool Generation: Beginning with 30 transition metals from periods IV, V, and VI, all 435 possible binary combinations (²⁰C₂) are considered. For each binary system, ten different ordered crystal phases (B1, B2, B3, B4, B11, B19, B27, B33, L10, and L11) at a 1:1 composition are modeled, resulting in an initial screening pool of 4,350 unique crystal structures [25].
- Stability Assessment via DFT: The formation energy (ΔEf) of each structure is calculated using Density Functional Theory (DFT). The formation energy is a critical descriptor of thermodynamic stability, indicating whether a phase is energetically favorable relative to its constituent elements.
- Screening Criteria: A formation energy threshold of ΔEf < 0.1 eV is applied. Structures exceeding this threshold are considered thermodynamically unstable and are filtered out. This step is crucial because even metastable alloys must possess sufficient stability to avoid transformation into phase-separated structures under operational conditions. This screening step reduced the candidate pool from 4,350 to 249 thermodynamically plausible alloys [25].
Table 1: Key Metrics from the High-Throughput Computational Screening Stages
| Screening Stage | Input Candidates | Output Candidates | Primary Screening Descriptor | Threshold Criterion |
|---|---|---|---|---|
| Thermodynamic Stability | 4,350 structures | 249 alloys | Formation Energy (ΔEf) | ΔEf < 0.1 eV [25] |
| Electronic Structure Similarity | 249 alloys | 17 candidates | ΔDOS (Similarity to Pd) | ΔDOS < 2.0 [25] |
| Synthetic Feasibility | 17 candidates | 8 candidates | Experimental Feasibility | Qualitative Assessment [25] |
Electronic Structure Similarity as a Predictive Descriptor
The core of this screening strategy is the use of the full electronic Density of States (DOS) pattern as a descriptor to predict catalytic performance.
- Descriptor Rationale: The electronic DOS, which describes the distribution of electron energies in a material, is a key determinant of surface reactivity. The fundamental hypothesis is that bimetallic alloys exhibiting DOS patterns similar to a known high-performance catalyst (e.g., Pd) will demonstrate comparable catalytic properties [25]. This approach offers an advantage over simpler descriptors (e.g., the d-band center) by incorporating comprehensive information from both d-states and sp-states.
- Quantifying Similarity: The similarity between the DOS of a candidate alloy and the reference Pd(111) surface is quantified using a defined metric (ΔDOS). This metric calculates the difference between two DOS patterns, with a Gaussian weighting function (standard deviation, σ = 7 eV) applied to assign higher importance to electronic states near the Fermi energy (EF), where catalytic interactions are most critical [25].
- Criticality of sp-States: The inclusion of sp-states in the DOS analysis is essential. As demonstrated in the study, the adsorption of O₂ on a Ni₅₀Pt₅₀(111) surface involves significant hybridization with the sp-states of the surface atoms, a interaction that would be missed by a descriptor based solely on d-states [25].
Final Candidate Selection
From the 17 candidates with the highest DOS similarity to Pd (ΔDOS < 2.0), a final selection of eight candidates was proposed for experimental synthesis. This final step involved a qualitative assessment of synthetic feasibility to ensure the proposed alloys could be realistically prepared in a laboratory setting [25].
Experimental Validation and Workflow Integration
Experimental Synthesis and Testing
The computationally selected candidates proceed to experimental validation, which confirms the predictive power of the screening protocol.
- Synthesis and Characterization: The eight proposed bimetallic catalysts are synthesized, and their catalytic performance is evaluated for the target reaction: the direct synthesis of H₂O₂ from H₂ and O₂ gases.
- Validation Results: Experimental testing confirmed that four of the eight screened alloys—Ni₆₁Pt₃₉, Au₅₁Pd₄₉, Pt₅₂Pd₄₈, and Pd₅₂Ni₄₈—exhibited catalytic properties comparable to those of pure Pd. A standout discovery was the Pd-free catalyst Ni₆₁Pt₃₉, which not only performed effectively but also demonstrated a 9.5-fold enhancement in cost-normalized productivity compared to prototypical Pd, due to the high content of inexpensive Ni [25].
Table 2: Experimentally Validated Bimetallic Catalysts and Performance Highlights
| Catalyst | DOS Similarity (ΔDOS) | Performance vs. Pd | Key Advantage |
|---|---|---|---|
| Ni₆₁Pt₃₉ | Low (Specific value not listed) | Comparable | Pd-free; 9.5x cost-normalized productivity [25] |
| Au₅₁Pd₄₉ | Low (Specific value not listed) | Comparable | Reduces Pd content [25] |
| Pt₅₂Pd₄₈ | Low (Specific value not listed) | Comparable | Reduces Pd content [25] |
| Pd₅₂Ni₄₈ | Low (Specific value not listed) | Comparable | Reduces Pd content [25] |
High-Throughput Data Management
The effectiveness of a combined screening approach relies on robust data infrastructure. Systems like the Research Data Infrastructure (RDI) and the High-Throughput Experimental Materials Database (HTEM-DB) are designed to automate the curation, processing, and storage of experimental data and metadata [78]. This infrastructure establishes a critical communication pipeline between experimental researchers and data scientists, ensuring that high-volume data generated from high-throughput experiments remains accessible, well-annotated, and primed for machine learning analysis [78].
Workflow Visualization
The following diagram illustrates the integrated high-throughput screening protocol, from initial candidate generation to experimental validation.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents, computational tools, and materials essential for implementing the described high-throughput screening protocol.
Table 3: Essential Reagents and Materials for High-Throughput Catalyst Screening
| Item Name | Function/Description | Role in Protocol |
|---|---|---|
| Transition Metal Precursors | Salt or complex compounds of the 30 target transition metals (e.g., Ni, Pt, Au, Pd salts). | Starting materials for the synthesis of the 435 candidate bimetallic alloy systems [25]. |
| Density Functional Theory (DFT) Code | First-principles computational software (e.g., VASP, Quantum ESPRESSO). | Performs calculation of formation energies (ΔEf) and projected electronic Density of States (DOS) for all candidate structures [25]. |
| Electronic DOS Descriptor | A quantitative metric (ΔDOS) comparing full DOS patterns to a reference catalyst. | Serves as the primary predictive descriptor for catalytic activity, filtering candidates based on electronic structure similarity [25]. |
| High-Throughput Experimental Reactor | Automated systems for parallel synthesis and testing of catalyst candidates. | Enables rapid experimental synthesis and catalytic performance evaluation (e.g., H₂O₂ production rate measurement) of the proposed candidates [25]. |
| Research Data Infrastructure (RDI) | Custom data tools for collecting, processing, and storing experimental data and metadata. | Manages the large volume of data generated from both computational and experimental high-throughput workflows, facilitating data sharing and machine learning [78]. |
The integrated high-throughput computational-experimental screening protocol presented herein demonstrates a powerful and efficient strategy for the discovery of advanced bimetallic catalysts. By leveraging a computationally tractable yet physically insightful descriptor—the similarity of electronic DOS patterns—this approach successfully identified novel catalyst compositions, such as Ni₆₁Pt₃₉, with performance comparable or superior to a benchmark noble metal catalyst. This methodology, which closely bridges theoretical prediction and experimental validation, provides a generalizable framework that can be adapted for the discovery and optimization of catalytic materials for a wide range of chemical transformations, significantly accelerating the development of sustainable chemical technologies.
Benchmarking DFT Predictions Against Established Experimental Databases
Density Functional Theory (DFT) serves as a foundational tool for predicting the thermodynamic stability of molecules and materials in high-throughput screening research. However, the predictive power of any computational method depends on its rigorous validation against reliable experimental data. This application note provides detailed protocols for benchmarking DFT predictions, focusing on thermodynamic stability, against established experimental databases. It synthesizes current best practices and emerging methodologies, including the use of large-scale, high-accuracy datasets and machine-learning interatomic potentials (MLIPs), to enhance the reliability of computational screenings in drug development and materials science.
Experimental Protocols for Benchmarking DFT
Protocol 1: Benchmarking Formation Energy and Thermodynamic Stability Predictions
Principle: The thermodynamic stability of a compound is determined by its energy relative to competing phases in a chemical space, quantified by its decomposition energy (ΔHd). Accurately predicting this energy via DFT is a critical first step in stability screening [2].
Procedure:
- Curate a Reference Experimental Dataset: Select a set of compounds with experimentally known stability or formation energies. The Joint Automated Repository for Various Integrated Simulations (JARVIS) database is one such resource [2].
- Construct the Theoretical Convex Hull: For the chemical space of interest, perform DFT calculations to compute the formation energies of all known and candidate compounds. Plot these energies and construct the convex hull. The decomposition energy (ΔHd) for a compound is its energy difference from this hull [2].
- Calculate DFT-Predicted Formation Energies: Compute the formation energies for the reference compounds using your chosen DFT functional (e.g., PBE, r2SCAN, HSE06).
- Statistical Comparison: Compare the DFT-predicted formation energies and derived stability labels (stable/metastable/unstable) against the experimental reference. Calculate statistical metrics such as Mean Absolute Error (MAE) and the Area Under the Curve (AUC) of the Receiver Operating Characteristic (ROC) to quantify predictive performance for stability classification [2].
Protocol 2: Validating DFT with High-Accuracy Wavefunction Methods
Principle: For properties like atomization energies, where experimental data may be limited or indirect, high-level wavefunction methods can provide benchmark-quality reference data.
Procedure:
- Generate a Diverse Training Set: Create a large and chemically diverse set of small molecular structures. The Microsoft Research team, in collaboration with an external expert, built a scalable pipeline for this purpose [55].
- Compute Reference Energies: Use high-accuracy wavefunction methods (e.g., W4-17) to compute the reference energies, such as atomization energies, for the generated structures. This step is computationally intensive but provides a gold-standard dataset [55].
- Train and Validate a Machine-Learned Functional: Employ a dedicated deep-learning architecture (e.g., Microsoft's Skala functional) to learn the exchange-correlation (XC) functional from the electron density and the high-accuracy energy labels [55].
- Benchmark Generalization: Test the trained model on a held-out set of molecules not seen during training. The key metric is whether the model achieves "chemical accuracy" (typically ~1 kcal/mol error) on this test set, demonstrating its ability to replace traditional functionals for predictive calculations [55].
Protocol 3: Benchmarking Electronic Properties (Band Gaps) of Solids
Principle: The band gap is a critical property for semiconductors and insulators. Standard DFT functionals systematically underestimate band gaps, and more advanced methods are required for accurate prediction [79].
Procedure:
- Select a Solid-State Benchmark Dataset: Adopt a curated set of experimental band gaps for non-magnetic semiconductors and insulators, such as the dataset of 472 materials from Borlido et al. [79].
- Perform DFT and Beyond-DFT Calculations: Calculate band gaps using:
- Standard semi-local functionals (e.g., PBE, LDA).
- Advanced meta-GGA (e.g., mBJ) or hybrid (e.g., HSE06) functionals.
- Many-Body Perturbation Theory within the GW approximation (e.g., G0W0, quasiparticle self-consistent GW).
- Compare to Experiment: Calculate the MAE and systematic deviation (over/under-estimation) for each method against the experimental data. This protocol revealed that while G0W0 with a plasmon-pole approximation offers only marginal improvement over the best DFT functionals, full-frequency GW methods and those with vertex corrections (QSGŴ) can achieve exceptional accuracy, even flagging questionable experimental measurements [79].
Quantitative Benchmarking Data
Table 1: Performance of Different Computational Methods for Predicting Solid-State Band Gaps. [79]
| Method | Mean Absolute Error (eV) | Systematic Bias | Relative Computational Cost |
|---|---|---|---|
| Standard DFT (PBE/LDA) | ~1.0 eV (typical) | Significant underestimation | Low |
| meta-GGA (mBJ) | Lower than PBE | Reduces underestimation | Moderate |
| Hybrid (HSE06) | Lower than PBE | Reduces underestimation | High |
| G₀W₀-PPA | Marginal gain over mBJ/HSE06 | Varies with starting point | Very High |
| Full-Frequency QPG₀W₀ | Significant improvement | More accurate | Very High |
| QSGŴ (with vertex corrections) | Highest accuracy | Minimal, may slightly overestimate | Extremely High |
Table 2: Key Characteristics of Recent High-Quality DFT Datasets for Training MLIPs. [80] [81] [82]
| Dataset | DFT Level of Theory | System Size (Max Atoms) | Key Chemical Domains | Primary Application |
|---|---|---|---|---|
| OMol25 | ωB97M-V/def2-TZVPD | 350 | Biomolecules, Electrolytes, Metal Complexes | General-purpose molecular MLIPs |
| MP-ALOE | r2SCAN | - | Ordered inorganic solids | Universal MLIPs for materials |
| Skala Training Data | High-accuracy WFT | Small molecules | Main-group molecules | Machine-learned XC functional |
Workflow Visualization
Table 3: Key Computational Tools and Databases for Benchmarking.
| Resource Name | Type | Function in Benchmarking |
|---|---|---|
| OMol25 Dataset [80] [81] [83] | DFT Dataset | Provides over 100 million high-accuracy (ωB97M-V) molecular snapshots for training and benchmarking Machine Learning Interatomic Potentials (MLIPs). |
| Materials Project (MP) [82] [2] | Materials Database | A vast repository of DFT-calculated material properties, used for constructing convex hulls and accessing reference structures for stability analysis. |
| JARVIS Database [2] | Materials Database | Provides experimental and DFT-calculated data for compounds, serving as a benchmark for stability prediction models. |
| r²SCAN Functional [82] | DFT Functional | A modern meta-GGA functional offering improved accuracy over PBE for formation energies and general solid-state properties, used in datasets like MP-ALOE. |
| Universal Model for Atoms (UMA) [83] | Machine Learning Model | A pre-trained universal MLIP that can be fine-tuned for specific applications, providing DFT-level accuracy at a fraction of the computational cost. |
| Skala Functional [55] | Machine-Learned DFT Functional | A deep-learning-based XC functional trained on high-accuracy wavefunction data, designed to achieve chemical accuracy for molecular properties. |
| GW Approximation [79] | Beyond-DFT Method | A many-body perturbation theory method used for calculating accurate quasiparticle band gaps of solids, serving as a higher-level benchmark for DFT. |
Integrating Machine Learning to Refine Predictions and Identify Novel Stable Compounds
The discovery of novel, stable compounds is a fundamental objective in materials science and drug development, yet traditional experimental and computational approaches remain prohibitively slow and expensive. High-throughput density functional theory (DFT) calculations have accelerated this process, enabling the computational screening of thousands of candidate materials [11]. However, the vastness of possible chemical spaces makes exhaustive DFT screening impractical [84] [2]. The integration of machine learning (ML) with DFT has emerged as a transformative solution, creating iterative frameworks that dramatically improve the efficiency of discovering thermodynamically stable compounds [84] [85]. These ML-guided methods refine predictive accuracy through active learning, enable the exploration of previously inaccessible chemical territories and facilitate the identification of novel candidates for technological applications, from next-generation batteries to pharmaceuticals [86] [87]. This Application Note details the protocols and methodologies underpinning this powerful synergy, providing researchers with a blueprint for implementing these advanced techniques.
Machine Learning Approaches for Stability Prediction
Several machine learning frameworks have demonstrated exceptional capability in predicting the thermodynamic stability of inorganic compounds and small molecules. The performance and application scope of these models vary, offering researchers a suite of tools for different discovery contexts.
Table 1: Key Machine Learning Models for Compound Stability and Discovery
| Model Name | Model Type | Key Input Features | Reported Performance / Outcome | Primary Application |
|---|---|---|---|---|
| GNoME [84] [85] | Graph Neural Network (GNN) | Crystal Structure (Graph representation) | Discovered 2.2 million new crystals; Prediction precision >80% for stable structures [84]. | Inorganic Crystal Discovery |
| ECSG [2] | Ensemble Model (Stacked Generalization) | Electron Configuration, Elemental Properties, Composition | AUC of 0.988 for stability prediction; High sample efficiency [2]. | Inorganic Compound Stability |
| ML-HTS for Perovskites [86] | Gaussian Process / Descriptor-based Models | Composition, Elemental Descriptors | MAE ~0.03 eV/atom for stability; MAE ~0.13 eV for binding energy [86]. | Double Perovskite Catalyst Screening |
| DTLS [87] | Variational Autoencoder (VAE) & Transfer Learning | Molecular SMILES Strings, Drug Efficacy Data | Generated novel compounds with confirmed efficacy in vitro and in vivo for CRC and AD [87]. | De Novo Small Molecule Design |
These frameworks highlight two dominant paradigms: structure-based models like GNoME, which use atomic connectivity, and composition-based models like ECSG, which rely solely on chemical formulas. Structure-based models often deliver higher accuracy but require structural information that may be unavailable for new compositions. In contrast, composition-based models are less constrained and can screen vast compositional spaces efficiently, making them ideal for the initial stages of exploration [2].
Experimental Protocols
Protocol 1: Active Learning with Graph Networks for Materials Exploration (GNoME)
The GNoME framework exemplifies a high-performance, iterative discovery pipeline for inorganic crystals [84].
Materials and Computational Requirements
- Initial Training Data: A large dataset of known crystal structures and their energies, such as the Materials Project database (~69,000 materials in initial GNoME training) [84].
- Candidate Generation:
- Structural Candidates: Generated via symmetry-aware partial substitutions (SAPS) of existing crystals, producing billions of candidate structures [84].
- Compositional Candidates: Generated using reduced chemical formulas with relaxed oxidation-state constraints, followed by structure initialization with ab initio random structure searching (AIRSS) [84] [85].
- Software: Density Functional Theory code (e.g., VASP [11]), graph neural network libraries (e.g., TensorFlow, PyTorch), and clustering algorithms for polymorph analysis.
Step-by-Step Procedure
- Model Pre-training: Train an initial GNoME graph neural network on a stable dataset of crystal structures and their DFT-computed energies. The model learns to predict the total energy of a crystal from its graph representation [84].
- Candidate Generation and Filtration:
- Generate candidate crystals using both structural (SAPS) and compositional (AIRSS) approaches.
- Use the pre-trained GNoME model to filter these candidates, predicting their stability and selecting the most promising ones for further analysis. This step employs volume-based test-time augmentation and uncertainty quantification via deep ensembles [84].
- DFT Verification: Perform DFT calculations with standardized settings (e.g., from the Materials Project) on the filtered candidate structures to compute their relaxed energies and verify model predictions [84] [11].
- Active Learning Loop:
- Incorporate the DFT-verified structures and their energies back into the training dataset.
- Retrain the GNoME model on this expanded, higher-quality dataset.
- Repeat steps 2-4 for multiple rounds. With each iteration, the model's prediction error decreases, and its precision for identifying stable materials increases significantly (e.g., from <6% to >80% for structural candidates) [84].
- Stability Assessment and Validation:
- Calculate the decomposition energy (energy above the convex hull) for all discovered materials using the updated dataset.
- Consider materials with a decomposition energy of zero as stable on the convex hull. Externally validate model predictions by comparing them with independent experimental syntheses or higher-fidelity computations (e.g., r2SCAN) [84] [85].
Protocol 2: Ensemble ML for Thermodynamic Stability Prediction
The ECSG framework leverages ensemble modeling to achieve high-accuracy stability predictions from composition alone, ideal for exploring uncharted chemical spaces [2].
Materials and Computational Requirements
- Training Data: A database of compounds with known stability labels, such as the JARVIS or Materials Project databases [2].
- Feature Sets:
- Magpie Features: Statistical summaries (mean, range, etc.) of elemental properties like atomic number and radius [2].
- Graph Representations: The chemical formula represented as a complete graph of elements (for Roost model) [2].
- Electron Configuration (EC) Matrix: A 118×168×8 tensor encoding the electron configuration of each element in the composition [2].
- Software: Python with scikit-learn, XGBoost, and deep learning libraries (e.g., PyTorch for ECCNN). The qmpy python package can be used for thermodynamic analysis [11].
Step-by-Step Procedure
- Data Preparation and Feature Engineering:
- From a compound's chemical formula, generate the three distinct input feature sets: Magpie features, graph representation, and the EC matrix [2].
- Base-Level Model Training:
- Train three separate models on their respective feature sets:
- An XGBoost model on the Magpie features.
- A graph neural network (Roost) on the graph representation.
- A convolutional neural network (ECCNN) on the EC matrix [2].
- Train three separate models on their respective feature sets:
- Stacked Generalization (Super Learner):
- Screening and Validation:
- Apply the trained ECSG model to screen vast compositional spaces (e.g., for two-dimensional semiconductors or double perovskites).
- Validate the top predicted stable candidates using DFT calculations to confirm their stability and properties [2].
The Scientist's Toolkit: Essential Research Reagents & Materials
Successful implementation of ML-driven materials discovery relies on a suite of computational tools and databases.
Table 2: Key Research Reagents and Computational Tools
| Tool/Resource Name | Type | Function in Workflow | Access / Reference |
|---|---|---|---|
| VASP (Vienna Ab initio Simulation Package) [11] | Software | Performs high-throughput DFT calculations to compute formation energies, stability, and electronic properties. | Proprietary; https://www.vasp.at |
| Materials Project [84] [85] | Database | Provides open-access to DFT-calculated data for thousands of materials, serving as a primary source for initial model training and validation. | https://materialsproject.org |
| qmpy Python Package [11] | Software | Facilitates high-throughput workflow management, thermodynamic analysis, and convex hull construction for stability assessment. | Open Source; https://github.com/wolverton-research-group/qmpy |
| Open Quantum Materials Database (OQMD) [11] [2] | Database | A vast database of DFT-calculated properties used for training models and assessing thermodynamic stability against competing phases. | http://oqmd.org |
| GNoME Database [84] [85] | Database (Output) | A repository of 2.2 million predicted crystal structures, including 380,000 stable ones, for community use and experimental synthesis. | Via the Materials Project |
| ECSG Framework [2] | Model/Code | An ensemble ML framework for high-accuracy stability prediction from composition, enabling efficient exploration of new compounds. | Code and methodology described in reference [2] |
Workflow Visualization: Integrated ML-DFT Discovery Pipeline
The complete, integrated workflow for discovering novel stable compounds merges the active learning and ensemble protocols into a powerful, synergistic pipeline.
Conclusion
High-throughput DFT screening for thermodynamic stability has fundamentally changed the discovery pipeline in materials science and pharmaceutical development, enabling rapid navigation of vast compositional spaces. By integrating robust computational workflows with machine learning and systematic experimental validation, researchers can now reliably identify stable, synthesizable compounds with desired properties, dramatically reducing reliance on traditional trial-and-error methods. Future directions point toward tighter integration of multiscale modeling, advanced machine learning potentials that further reduce computational cost, and the expansion of these methodologies to dynamically model complex biological and solvent environments. This powerful, data-driven paradigm promises to accelerate the development of next-generation materials, from sustainable energy catalysts to novel, stable drug formulations, shaping the future of biomedical and clinical research.
References
- 1. The theory/calculation behind energy-above-hull [https://matsci.org/t/the-theory-calculation-behind-energy-above-hull-how-to-calculate-for-quarternary-structures/59743]
- 2. Predicting thermodynamic stability of inorganic compounds ... [https://www.nature.com/articles/s41467-024-55525-y]
- 3. Integrating stability metrics with high-throughput ... [https://www.nature.com/articles/s43246-023-00409-9]
- 4. Thermal Stability Ranking of Energetic Crystals via a ... [https://www.sciencedirect.com/science/article/pii/S2667134425000525]
- 5. Formation energy prediction of crystalline compounds ... [https://www.nature.com/articles/s43246-023-00433-9]
- 6. Efficient predictions of formation energies and convex hulls ... [https://www.sciencedirect.com/science/article/pii/S1005030222007526]
- 7. DREAMS: Density Functional Theory Based Research ... [https://arxiv.org/html/2507.14267v1]
- 8. Stability Testing White Paper [https://scicord.com/stability-white-paper/]
- 9. Designing Phase-Appropriate Stability Study Programs for ... [https://www.pharmoutsourcing.com/Featured-Articles/352619-Designing-Phase-Appropriate-Stability-Study-Programs-for-Drug-Substances-and-Drug-Products/]
- 10. Thermodynamic Studies for Drug Design and Screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC3496183/]
- 11. High-throughput DFT calculations of formation energy ... [https://www.nature.com/articles/sdata2017153]
- 12. Articles Development and Application of an Automated ... [https://www.sciencedirect.com/science/article/pii/S2472555222084271]
- 13. Stability Testing Strategies for Working Standards [https://biopharmaconsultinggroup.com/stability-testing-strategies-for-working-standards/]
- 14. High-throughput screening of stable Ag–Pd–F catalysts for ... [https://pubs.rsc.org/en/content/articlelanding/2025/ta/d4ta07674e]
- 15. A critical examination of compound stability predictions ... [https://www.nature.com/articles/s41524-020-00362-y]
- 16. Atomistic study of thermodynamic stability and fracture ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025625005919]
- 17. Accurate and efficient protocols for high-throughput first- ... [https://arxiv.org/html/2504.03962v1]
- 18. High-throughput calculations of charged point defect ... [https://www.nature.com/articles/s41524-023-01015-6]
- 19. Prediction and Classification of Formation Energies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8190927/]
- 20. Origin and evolution of high throughput screening - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1978279/]
- 21. A high-throughput infrastructure for density functional ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025611001133]
- 22. Accurate Screening of Functional Materials with Machine ... [https://arxiv.org/html/2508.20556v1]
- 23. Adaptation of High-Throughput Screening in Drug Discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3269696/]
- 24. High-throughput DFT screening of single-metal and ... [https://www.sciencedirect.com/science/article/abs/pii/S0360319925042788]
- 25. High-throughput computational-experimental screening ... [https://www.nature.com/articles/s41524-021-00605-6]
- 26. Design Principles Guided by DFT Calculations and High- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11163396/]
- 27. On the practical applicability of modern DFT functionals for ... [https://arxiv.org/html/2501.12149v1]
- 28. Digitized material design and performance prediction ... [https://www.frontiersin.org/journals/materials/articles/10.3389/fmats.2025.1599439/full]
- 29. High-throughput DFT calculations of formation energy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5644373/]
- 30. - density for functional - theory studies? Tools high throughput DFT [https://mattermodeling.stackexchange.com/questions/1586/tools-for-high-throughput-dft-studies]
- 31. nanoHUB.org - Resources: High Calculation... Throughput DFT [https://nanohub.org/resources/htdft]
- 32. High-throughput predictions of metal–organic framework ... [https://www.nature.com/articles/s41524-022-00796-6]
- 33. Density functional theory study of mechanical, thermal, and ... [https://www.nature.com/articles/s41598-025-26168-w]
- 34. A High-Throughput Framework for Predicting Surface ... [https://www.sciencedirect.com/science/article/abs/pii/S0921452625011160]
- 35. A robust, open-source python library for materials analysis [https://www.sciencedirect.com/science/article/abs/pii/S0927025612006295]
- 36. The Open Quantum Materials Database (OQMD) [https://www.nature.com/articles/npjcompumats201510]
- 37. Understanding Structures and Properties in the Materials ... [https://docs.materialsproject.org/methodology/materials-methodology/understanding-structures-and-properties-in-the-materials-project]
- 38. a comparison of AFLOW, Materials Project, and OQMD [https://arxiv.org/abs/2007.01988]
- 39. Breaking the poisoning paradigm: a high-throughput DFT ... [https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta03759j]
- 40. High-throughput DFT calculations of formation energy ... [https://www.osti.gov/pages/biblio/1490970]
- 41. High-throughput DFT calculations of formation energy ... [https://pubmed.ncbi.nlm.nih.gov/29039848/]
- 42. Screening stable and metastable ABO3 perovskites using ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025620301051]
- 43. New insights into the piezoelectric, thermodynamic and ... [https://www.sciencedirect.com/science/article/abs/pii/S2352492822002422]
- 44. High‐Throughput Computational Screening of Small ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12036818/]
- 45. Accurate and efficient protocols for high-throughput first ... [https://arxiv.org/abs/2504.03962]
- 46. High-throughput density functional perturbation theory and ... [https://www.nature.com/articles/s41524-020-0337-2]
- 47. Pharmaceutical Cocrystals: New Solid Phase Modification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5874831/]
- 48. A review on advancement of cocrystallization approach ... [https://www.sciencedirect.com/science/article/pii/S2405844024050886]
- 49. Influence of excipients on thermodynamic phase behavior ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250921003638]
- 50. High-throughput encapsulated nanodroplet screening for ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc07556k]
- 51. Molecular Interactions between APIs and Enteric Polymeric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10143979/]
- 52. API, Excipient Screening and Characterization [https://www.formulationbio.com/novel-delivery/api-excipient-screening-and-characterization.html]
- 53. A Rapid 3-Day Excipient Screening Methodology and its ... [https://pubmed.ncbi.nlm.nih.gov/38182862/]
- 54. Best‐Practice DFT Protocols for Basic Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9826355/]
- 55. Breaking bonds, breaking ground: Advancing the accuracy ... [https://www.microsoft.com/en-us/research/blog/breaking-bonds-breaking-ground-advancing-the-accuracy-of-computational-chemistry-with-deep-learning/]
- 56. Completing density functional theory by machine learning ... [https://www.nature.com/articles/s41524-020-0310-0]
- 57. Predicting the thermodynamic stability of perovskite oxides ... [https://www.sciencedirect.com/science/article/abs/pii/S092702561830274X]
- 58. What considerations must be made when selecting a basis ... [https://mattermodeling.stackexchange.com/questions/13987/what-considerations-must-be-made-when-selecting-a-basis-set]
- 59. What is ensemble learning? [https://www.ibm.com/think/topics/ensemble-learning]
- 60. Potential Biases in Machine Learning Algorithms Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6347576/]
- 61. Making sense of AI: bias, trust and transparency in pharma ... [https://www.drugtargetreview.com/article/183358/making-sense-of-ai-bias-trust-and-transparency-in-pharma-rd/]
- 62. Stacked Generalization: An Introduction to Super Learning [https://pmc.ncbi.nlm.nih.gov/articles/PMC6089257/]
- 63. Development and validation of machine learning models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11841291/]
- 64. Comparative analysis of algorithmic approaches in ... [https://www.nature.com/articles/s41598-025-15971-0]
- 65. 7.3.5 TDDFT + PCM for Excitation and Emission Energies ... [https://manual.q-chem.com/6.2/subsec_tddft-cPCM.html]
- 66. Novel nonequilibrium solvation theory for calculating the ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261419306128]
- 67. Solvent effects on excitation energies obtained using ... [https://pubs.rsc.org/en/content/articlelanding/2017/cp/c7cp05673g]
- 68. Understanding DFT Uncertainties for More Reliable Reactivity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12529897/]
- 69. Designing solvent systems using self-evolving solubility ... [https://pubs.rsc.org/en/content/articlehtml/2024/sc/d3sc03468b]
- 70. Acceleration without Disruption: DFT Software as a Service [https://arxiv.org/html/2406.11185v1]
- 71. Simple approach to more efficient density functional theory ... [https://www.sciencedirect.com/science/article/abs/pii/S2352214325000115]
- 72. Constructing custom thermodynamics using deep learning [https://www.nature.com/articles/s43588-023-00581-5]
- 73. Thermodynamic Overfitting and Generalization [https://arxiv.org/html/2402.16995v1]
- 74. - High computational- throughput for... experimental screening protocol [https://www.nature.com/articles/s41524-021-00605-6?error=cookies_not_supported&code=39b598eb-c58c-43ac-994f-278ab9a5962a]
- 75. Materials synthesizability and stability prediction using a semi ... [https://pubs.rsc.org/en/content/articlehtml/2023/dd/d2dd00098a]
- 76. Predicting the synthesizability of crystalline inorganic ... [https://www.nature.com/articles/s41524-023-01114-4]
- 77. Web Thermo Tables | NIST [https://www.nist.gov/mml/acmd/trc/web-thermo-tables-wtt]
- 78. Descriptor Research data infrastructure for high-throughput ... [https://www.sciencedirect.com/science/article/pii/S266638992100235X]
- 79. A systematic benchmark for band gaps of solids [https://arxiv.org/html/2508.05247v1]
- 80. A Record-Breaking Dataset to Train AI Models has Launched [https://newscenter.lbl.gov/2025/05/14/computational-chemistry-unlocked-a-record-breaking-dataset-to-train-ai-models-has-launched/]
- 81. OMol25: High-Precision Molecular DFT Dataset [https://www.emergentmind.com/topics/omol25-dataset]
- 82. MP-ALOE: an r2SCAN dataset for universal machine ... [https://www.nature.com/articles/s41524-025-01834-9]
- 83. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 84. Scaling deep learning for materials discovery [https://www.nature.com/articles/s41586-023-06735-9]
- 85. Millions of new materials discovered with deep learning [https://deepmind.google/blog/millions-of-new-materials-discovered-with-deep-learning/]
- 86. Machine learning high-throughput screening of double ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725060942]
- 87. De Novo Generation and Identification of Novel Compounds ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10962488/]