Harnessing Thermodynamic Driving Force to Master Solid-State Reactions: A Guide for Researchers
This article provides a comprehensive exploration of how increasing the thermodynamic driving force dictates outcomes in solid-state reactions, a cornerstone of inorganic materials synthesis.
Harnessing Thermodynamic Driving Force to Master Solid-State Reactions: A Guide for Researchers
Abstract
This article provides a comprehensive exploration of how increasing the thermodynamic driving force dictates outcomes in solid-state reactions, a cornerstone of inorganic materials synthesis. We delve into the foundational principles distinguishing thermodynamic and kinetic control, drawing on recent in situ characterization studies that validate the max-ΔG theory. The content outlines practical methodologies for calculating and applying driving force to predict reaction pathways, addresses common troubleshooting and optimization challenges, and presents a framework for validating predictions against experimental data. Tailored for researchers, scientists, and drug development professionals, this guide synthesizes cutting-edge research to empower the rational design and synthesis of advanced materials, from pharmaceuticals to energy storage compounds.
The Principles of Thermodynamic Control in Solid-State Chemistry
Defining Thermodynamic Driving Force (ΔG) in Solid-State Systems
Fundamental FAQs: Understanding ΔG
What is the Thermodynamic Driving Force (ΔG) in solid-state systems?
The thermodynamic driving force, represented by the Gibbs free energy change (ΔG), determines the spontaneity and direction of solid-state reactions. It is defined by the equation ΔG = ΔH - TΔS, where ΔH is the enthalpy change, T is the absolute temperature, and ΔS is the entropy change of the system [1] [2]. In solid-state synthesis, this driving force dictates which intermediate phases form first, ultimately determining the success of synthesizing target materials [3] [4].
How does ΔG predict reaction spontaneity in solid-state synthesis?
The sign and magnitude of ΔG provide crucial information about reaction feasibility [1] [5]:
- ΔG < 0: The reaction proceeds spontaneously in the forward direction
- ΔG > 0: The reaction proceeds spontaneously in the reverse direction
- ΔG = 0: The reaction is at equilibrium with no net change
In solid-state systems, research has demonstrated that when the driving force to form one product exceeds that of all competing phases by ≥60 meV/atom, thermodynamic control dominates, and this product forms predictably as the initial phase [3].
What is the "max-ΔG theory" in solid-state reactions?
The max-ΔG theory states that when two solid phases react, the initial product formed will be the one that leads to the largest decrease in Gibbs energy (most negative ΔG), regardless of reactant stoichiometry [3]. This approach calculates ΔG for each possible reaction in a compositionally unconstrained manner, normalized per atom of material formed, based on the observation that solid products form locally at particle interfaces without knowledge of the sample's overall composition [3].
How do temperature and entropy affect ΔG in solid-state systems?
Temperature influences ΔG primarily through the entropy term (TΔS) in the Gibbs free energy equation [5] [2]. The relationship between ΔH, ΔS, and temperature creates four possible scenarios for solid-state reactions:
Table: Temperature Dependence of Solid-State Reactions
| ΔH | ΔS | Temperature Effect | Example Reaction Type |
|---|---|---|---|
| < 0 | > 0 | Spontaneous at all temperatures | Graphite + O₂ → CO₂ |
| < 0 | < 0 | Spontaneous only at low temperatures | Freezing, condensation |
| > 0 | > 0 | Spontaneous only at high temperatures | Melting, decomposition |
| > 0 | < 0 | Non-spontaneous at all temperatures | - |
Troubleshooting Guides: Common Experimental Issues
Problem: Unpredictable intermediate phase formation
Issue: Competing phases appear instead of the desired initial intermediate, despite calculations suggesting favorable thermodynamics.
Solution:
- Calculate driving force differences: Determine if the ΔG difference between competing phases exceeds the 60 meV/atom threshold for thermodynamic control [3].
- Analyze the interface reaction hull: Construct a convex hull in free energy and composition space using normalized compositions and energies to identify all possible competing phases [4].
- Apply selectivity metrics: Use primary and secondary competition metrics to assess the favorability of target versus impurity phase formation:
- Primary competition: Measures competition to form the initial product
- Secondary competition: Measures competition after the initial product forms [4]
Problem: Persistent impurity phases despite favorable target ΔG
Issue: Impurity phases remain even when thermodynamic calculations predict a selective reaction to the desired target material.
Solution:
- Evaluate secondary interfaces: When the target phase forms, it creates new interfaces (α|γ and γ|β) that can lead to impurity phase formation through secondary reactions [4].
- Check precursor selection: Non-standard precursors containing additional elements beyond those in the target composition can sometimes provide better selectivity by altering the reaction pathway [4].
- Assess interfacial energy effects: The nucleation rate depends on both bulk reaction energy (ΔG) and interfacial energy (γ), according to the equation:
( Q = A \exp\left(\frac{-16\pi\gamma^3}{3n^2k_BT\Delta G^2}\right) ) [3]
Even with favorable ΔG, high interfacial energy can significantly reduce nucleation rates.
Problem: Inconsistent results between different laboratory conditions
Issue: Solid-state reactions yield different products when scale, mixing method, or heating rates are modified.
Solution:
- Distinguish thermodynamic vs. kinetic control:
- Thermodynamic control: Operates when one product's driving force significantly exceeds competitors (≥60 meV/atom)
- Kinetic control: Dominates when multiple competing phases have similar driving forces [3]
Standardize powder processing:
- Maintain consistent grinding/milling protocols to control particle interfaces
- Ensure homogeneous mixing to avoid local composition variations
- Control powder morphology to minimize variable interfacial contacts [4]
Optimize thermal profile:
Quantitative Data Tables for Experimental Planning
Table: Thermodynamic Control Thresholds in Solid-State Systems
| Parameter | Threshold Value | Experimental Significance | Validation Method |
|---|---|---|---|
| ΔG Difference for Thermodynamic Control | ≥60 meV/atom | Predictable initial product formation | In situ XRD on 37 reactant pairs [3] |
| Percentage of Reactions in Thermodynamic Control | 15% of possible reactions | Scope of predictive synthesis applicability | Materials Project data analysis [3] |
| Primary Competition Metric | Lower values indicate higher selectivity | Predicts success in forming target versus impurities | Applied to 3520 literature reactions [4] |
Table: Thermodynamic Parameter Calculations for Solid-State Reactions
| Parameter | Calculation Method | Experimental Consideration | Data Source |
|---|---|---|---|
| Compositionally Unconstrained ΔG | Normalized per atom of material formed | Determines initial phase regardless of stoichiometry | Materials Project [3] [4] |
| Interface Reaction Hull | Convex hull in free energy-composition space | Identifies all possible intermediate phases | First-principles thermodynamic data [4] |
| Reaction Selectivity | Primary and secondary competition metrics | Ranks synthesis approaches by success likelihood | High-throughput reaction enumeration [4] |
Experimental Protocols & Methodologies
Protocol: Determining Thermodynamic Control in Solid-State Reactions
Objective: Experimentally validate whether a solid-state reaction falls under thermodynamic control where the max-ΔG theory applies.
Materials & Equipment:
- High-purity precursor powders
- In situ X-ray diffraction (XRD) capability
- Controlled atmosphere furnace
- Mortar and pestle or ball mill for mixing
- Synchrotron radiation source (for high-resolution studies)
Procedure:
- Sample Preparation:
- Select precursor pairs based on computational screening of reaction energies
- Mix powders in stoichiometric ratios using consistent grinding protocol
- Prepare multiple samples for statistical reliability
In Situ Characterization:
- Heat samples at controlled rate (e.g., 10°C/min) to target temperature (e.g., 700°C)
- Perform XRD measurements at frequent intervals (e.g., two scans per minute)
- Monitor phase formation sequence using weight fraction calculations from XRD intensities [3]
Data Analysis:
- Identify the first crystalline phase detected
- Compare with computational predictions of phase formation based on ΔG
- Calculate driving force differences between competing phases
- Classify as thermodynamic control if the observed initial phase matches the max-ΔG prediction and its driving force exceeds competitors by ≥60 meV/atom [3]
Protocol: Constructing Interface Reaction Hulls for Synthesis Planning
Objective: Create interface reaction hulls to predict intermediate phase formation and assess synthesis selectivity.
Computational Methods:
- Data Acquisition:
- Obtain formation enthalpies (ΔHf) from Materials Project database
- Extend to Gibbs free energies (ΔGf(T)) using machine learning models and experimental thermochemistry data [4]
Hull Construction:
- For binary systems: Use binary compositional phase diagram
- For ternary+ systems: Compute slices of full compositional phase diagram along tie-line connecting precursors
- Use combinatorial reaction enumeration to identify balanced reactions [4]
Selectivity Analysis:
- Calculate primary competition metric measuring favorability of initial target formation
- Calculate secondary competition metric measuring phase competition after initial product forms
- Rank potential synthesis reactions by selectivity metrics [4]
The Scientist's Toolkit: Essential Research Reagents & Materials
Table: Key Materials for Solid-State Thermodynamics Research
| Material/Reagent | Function in Experimental Research | Application Example | Considerations |
|---|---|---|---|
| LiOH vs. Li₂CO₃ | Lithium sources with different reactivity | Li-Nb-O system studies | LiOH provides larger ΔG differences than Li₂CO₃ [3] |
| Nb₂O₅ | Niobium source for ternary oxide formation | Test system for thermodynamic control | Forms LiNb₃O₈, LiNbO₃, Li₃NbO₄ with Li sources [3] |
| BaS / BaCl₂ | Non-standard precursors for titanate synthesis | BaTiO₃ formation | Unconventional precursors can provide faster, purer synthesis [4] |
| Na₂TiO₃ | Titanium source with additional elements | Alternative BaTiO₃ synthesis | Enables reactions not possible with conventional precursors [4] |
Advanced Concepts: Thermodynamic vs. Kinetic Control
Understanding the Regimes of Control
Solid-state reactions operate in two distinct regimes with different predictive capabilities:
Thermodynamic Control Regime:
- Occurs when the driving force (ΔG) to form one product exceeds all competitors by ≥60 meV/atom
- The initial product formed can be predicted using the max-ΔG theory
- Represents approximately 15% of possible reactions [3]
- Enables predictive synthesis from first principles
Kinetic Control Regime:
- Occurs when multiple competing products have comparable driving forces
- Initial product formation depends on kinetic factors like diffusion barriers and nucleation kinetics
- Outcomes cannot be reliably predicted using thermodynamics alone [3]
- Requires explicit modeling of diffusion and nucleation processes
Practical Implications for Synthesis Design
The distinction between these regimes has significant practical implications:
- Reaction Selection: Focus on reactions falling within the thermodynamic control regime for predictable outcomes
- Precursor Choice: Consider unconventional precursors that create larger ΔG differences to push reactions into thermodynamic control
- Temperature Optimization: Use temperature to manipulate the balance between ΔH and TΔS terms to enhance selectivity
- Interface Engineering: Control powder interfaces to minimize secondary reactions that lead to impurity phases
By applying these concepts and methodologies, researchers can systematically approach solid-state synthesis with greater predictive power, reducing the traditional trial-and-error approach and accelerating materials discovery.
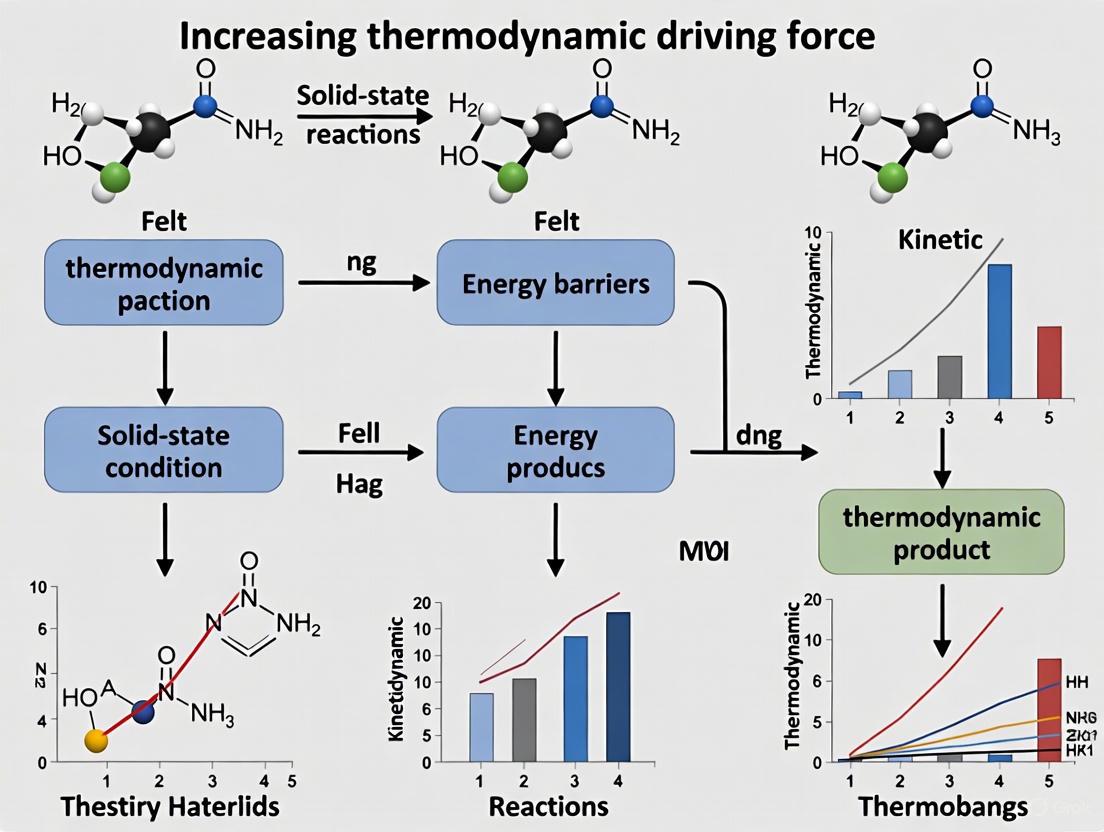
The Max-ΔG Theory provides a predictive framework for determining the initial product formed in solid-state reactions. This principle states that when two solid phases react, the first product to form is the one that leads to the largest decrease in the Gibbs free energy (ΔG), regardless of the stoichiometric amounts of reactants present. This approach calculates the thermodynamic driving force in a compositionally unconstrained manner, normalizing the energy per atom of material formed. The theory's significance lies in its ability to predict reaction pathways, which is crucial for rational synthesis planning in materials science and solid-state chemistry. When the thermodynamic driving force to form one product sufficiently exceeds that of all competing phases, thermodynamics primarily dictates the initial reaction outcome, bypassing kinetic limitations that often complicate solid-state reactions.
Theoretical Foundation: Gibbs Free Energy and Reaction Spontaneity
Fundamentals of Gibbs Free Energy
The Gibbs free energy (G) is a thermodynamic potential defined by the equation: [G = H - TS] where H is enthalpy, T is absolute temperature, and S is entropy. The change in Gibbs free energy, ΔG, for a process is given by: [\Delta G = \Delta H - T \Delta S] A negative ΔG value indicates a spontaneous process, while a positive value denotes non-spontaneity. In the context of the Max-ΔG theory, the relevant quantity is the Gibbs free energy change normalized per atom of material formed, which represents the thermodynamic driving force for product formation.
Maximum Work Interpretation
The Gibbs free energy change has a fundamental physical interpretation: it represents the maximum amount of non-pressure-volume (non-pV) work that can be extracted from a process at constant temperature and pressure. For a reversible process, the relationship is: [\Delta G = w{max}] where (w{max}) refers to all types of work except expansion (pressure-volume) work. This principle underscores why the phase with the most negative ΔG (largest decrease in free energy) is thermodynamically favored to form first, as it releases the maximum available energy.
Thermodynamic vs. Kinetic Control
The Max-ΔG theory operates most effectively in the regime of thermodynamic control, where the driving force to form one product greatly exceeds that of all competing products. According to classical nucleation theory, the nucleation rate (Q) for a product is estimated by: [Q = A \exp\left(\frac{-16\pi\gamma^3}{3n^2k_BT\Delta G^2}\right)] where:
- A is a prefactor related to thermal fluctuations and diffusion rates
- γ is the interfacial energy
- n is the atomic density
- k_B is Boltzmann's constant
- T is temperature
- ΔG is the bulk reaction energy
The exponential term typically dominates the nucleation rate, making ΔG a decisive factor in determining which product forms first when interfacial energies and prefactors are comparable between competing phases.
Experimental Validation and Quantitative Threshold
Research Methodology
Recent research has systematically validated the Max-ΔG theory through in situ X-ray diffraction (XRD) measurements on 37 pairs of reactants across multiple chemical spaces, including detailed studies of Li-Mn-O and Li-Nb-O systems. The experimental protocol involved:
- Sample Preparation: Reactant pairs were mixed in specific ratios (e.g., 1:1 Li:Nb ratio for Li-Nb-O systems)
- Heating Profile: Samples were heated to 700°C at 10°C/min, held at 700°C for 3 hours, then cooled to room temperature
- In Situ Characterization: XRD measurements were performed at a rate of two scans per minute using synchrotron radiation at Beamline 12.2.2 of the Advanced Light Source
- Data Analysis: Weight fractions of detected phases were quantified as a function of temperature to identify the first-formed product
Threshold for Thermodynamic Control
Experimental results revealed a precise threshold for thermodynamic control in solid-state reactions:
Table 1: Quantitative Threshold for Thermodynamic Control
| Parameter | Value | Interpretation |
|---|---|---|
| Energy Difference Threshold | ≥60 meV/atom | Minimum ΔG difference required for thermodynamic control |
| Percentage of Reactions Affected | 15% | Proportion of reactions falling in thermodynamic control regime |
| Total Reactions Analyzed | 105,652 | Number of reactions assessed in Materials Project database |
When the driving force to form one product exceeds that of all competing phases by ≥60 meV/atom, the initial product formation can be reliably predicted using the Max-ΔG theory. Below this threshold, kinetic factors often dominate, and predictions based solely on thermodynamics become less reliable.
Experimental Protocols for Verification
In Situ XRD Monitoring of Solid-State Reactions
Materials and Equipment:
- High-purity reactant powders (e.g., LiOH, Li₂CO₃, Nb₂O₅ for Li-Nb-O system)
- In situ XRD stage with heating capability
- Synchrotron radiation source or laboratory XRD with heating attachment
- Temperature controller with programmable heating rates
Step-by-Step Procedure:
- Sample Preparation: Mechanically mix reactant powders in desired molar ratios using mortar and pestle or ball milling
- Loading: Place homogeneous mixture in in situ XRD heating stage
- Temperature Program: Apply controlled heating ramp (typically 10°C/min) to target temperature (e.g., 700°C)
- Data Collection: Continuously collect XRD patterns throughout heating process (recommended: 2 scans/minute)
- Phase Identification: Analyze sequential XRD patterns to identify the first crystalline phase that appears
- Validation: Compare experimentally observed first product with theoretical prediction based on Max-ΔG calculations
Computational Prediction Method
Procedure for Calculating Compositionally Unconstrained ΔG:
- Identify Possible Products: Compile list of all thermodynamically stable phases in the chemical space of interest using databases like the Materials Project
- Calculate Reaction Energies: For each possible product, compute the Gibbs free energy change using the formula: [\Delta G{rxn} = G{product} - \sum G_{reactants}]
- Normalize Per Atom: Divide ΔG_{rxn} by the number of atoms in the product formula unit to obtain ΔG per atom
- Compare Driving Forces: Identify the product with the most negative ΔG (largest driving force)
- Apply Threshold: Verify that the ΔG difference between the top candidate and competing phases meets or exceeds 60 meV/atom for reliable prediction
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: Why does my experiment sometimes not follow the Max-ΔG prediction, even when one product has a significantly more negative ΔG? A1: Several kinetic factors can interfere with thermodynamic predictions:
- Diffusion Limitations: If ion mobility is severely restricted, the product requiring the least atomic rearrangement may form first
- Structural Templating: Phases with high structural similarity to precursors may have reduced nucleation barriers
- Interfacial Energy Variations: Differences in interfacial energy (γ) between competing products can significantly affect nucleation rates despite similar bulk ΔG values
- Precursor Decomposition: If reactants decompose before reacting, the decomposition products may become the relevant reactants
Q2: How can I increase the likelihood that my reaction will follow the Max-ΔG prediction? A2: To favor thermodynamic control:
- Increase Temperature: Higher temperatures enhance diffusion and reduce kinetic barriers
- Extended Annealing: Longer reaction times allow systems to overcome kinetic limitations
- Reactant Preparation: Use finely ground, well-mixed powders to maximize contact and minimize diffusion paths
- Choose Appropriate Precursors: Select reactants with high reactivity and low decomposition temperatures
Q3: Can the Max-ΔG theory predict metastable phase formation? A3: The standard Max-ΔG approach only considers thermodynamically stable phases (those on the convex hull). Metastable phases, which by definition have less negative ΔG values than stable phases at the same composition, are not typically predicted. However, in cases where metastable phases have significantly lower interfacial energies, they may form due to kinetic preferences.
Q4: How does reactant stoichiometry affect Max-ΔG predictions? A4: The Max-ΔG theory specifically neglects overall reactant stoichiometry in its predictions, as it assumes products form locally at particle interfaces. This approach is justified by experimental observations that solid products form at interfacial contacts without global knowledge of the sample's overall composition.
Common Experimental Issues and Solutions
Table 2: Troubleshooting Guide for Max-ΔG Experiments
| Problem | Possible Causes | Solutions |
|---|---|---|
| Unexpected initial product | Insufficient ΔG difference (<60 meV/atom), kinetic dominance | Calculate ΔG difference; increase temperature; extend reaction time |
| No reaction observed | Diffusion limitations, low reactivity | Improve powder mixing; decrease particle size; increase temperature |
| Multiple simultaneous products | Comparable ΔG values for competing phases | Modify precursors to increase ΔG difference for target product |
| Inconsistent results between batches | Particle size variations, mixing inhomogeneity | Standardize powder processing; use controlled milling protocols |
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Materials for Max-ΔG Studies
| Material/Reagent | Function/Application | Specific Examples |
|---|---|---|
| In Situ XRD Capability | Real-time phase identification during heating | Synchrotron beamlines; laboratory XRD with heating stage |
| High-Purity Precursors | Ensure reproducible reaction thermodynamics | LiOH, Li₂CO₃, Nb₂O₅, MnO₂ for lithium metal oxide systems |
| Computational Databases | Source of thermodynamic data for ΔG calculations | Materials Project, OQMD, AFLOW |
| Temperature-Controlled Stages | Precise thermal profiles for reproducible kinetics | Programmable furnaces with controlled atmosphere capability |
Theoretical Framework and Practical Implications
The quantitative framework established for the Max-ΔG theory enables researchers to classify reactions into distinct control regimes:
Thermodynamic Control Regime (ΔG difference ≥60 meV/atom):
- Initial product formation can be reliably predicted from thermodynamics alone
- Reactions are selective and typically yield high amounts of the predicted product
- Comprises approximately 15% of possible solid-state reactions
Kinetic Control Regime (ΔG difference <60 meV/atom):
- Multiple competing products have comparable driving forces for formation
- Kinetic factors (diffusion, nucleation, interfacial energies) dominate product selection
- Explicit modeling of kinetic factors is necessary for prediction
This classification provides practical guidance for synthesis planning. When designing reactions to produce specific target materials, researchers should first calculate the ΔG landscape to determine if the system falls within the thermodynamic control regime. If it does, synthesis outcomes can be predicted with high confidence. If not, kinetic manipulations (e.g., careful control of particle size, temperature profiles, or precursor selection) become essential for directing the reaction pathway.
Troubleshooting Guides
FAQ 1: How can I determine if my solid-state reaction is under kinetic or thermodynamic control?
Answer: You can determine the control mechanism by observing how the first-formed product changes with reaction conditions and by calculating the thermodynamic driving forces of competing reactions [6] [3].
- Observe Product Formation Over Time: Monitor the reaction with in situ techniques like high-temperature XRD. If the initial product is different from the final, stable product, the initial formation is under kinetic control, while the final state represents thermodynamic control [6] [3].
- Check for Temperature Dependence: Perform the reaction at different temperatures. A product that forms faster at lower temperatures is likely the kinetic product. A product that becomes dominant at higher temperatures or after prolonged heating is the thermodynamic product [6] [7].
- Calculate Driving Forces: Compute the compositionally unconstrained Gibbs free energy change (∆G) per atom for all possible intermediate phases. If the driving force to form one product exceeds that of all others by ≥ 60 meV/atom, thermodynamics will likely control the initial product formation. If multiple phases have similar driving forces, kinetics will likely determine the outcome [3].
FAQ 2: The initial product of my synthesis is a metastable phase, not the target material. How can I steer the reaction toward the thermodynamic product?
Answer: To favor the thermodynamic product, you need to provide conditions that allow the system to reach equilibrium.
- Increase Reaction Temperature: Higher temperatures provide the thermal energy needed for reactants and metastable products to overcome reverse activation barriers and reorganize into the most stable configuration [6] [7].
- Prolong Reaction Time: Thermodynamic control requires sufficient time for equilibration. Extending the reaction duration allows for the dissolution and reformation of phases [6].
- Ensure Reversibility: The reaction pathway must allow for the interconversion between products. If the kinetic product is trapped (e.g., by forming an insoluble or stable phase), it cannot convert to the thermodynamic product. Adjusting precursors or conditions to ensure all steps are reversible can help [6].
FAQ 3: My reaction yields a mixture of products. How can I increase selectivity for a single phase?
Answer: Selectivity depends on creating a large enough energy difference in the controlling regime.
- For Thermodynamic Selectivity: Design your reactant pairs so that the driving force (∆G) to form your desired product is maximized and exceeds that of the nearest competing phase by the 60 meV/atom threshold. This large energy difference makes the desired product the dominant initial phase, bypassing kinetic intermediates [3].
- For Kinetic Selectivity: If you need to isolate a metastable kinetic product, use low temperatures and short reaction times. This provides insufficient energy for the system to equilibrate, trapping the product that forms fastest. Using sterically hindered reactants or bases can also favor one transition state over another [6] [3].
The following table summarizes key quantitative relationships and thresholds for thermodynamic and kinetic control.
Table 1: Quantitative Thresholds and Relationships for Reaction Control
| Parameter | Description | Value or Relationship | Application |
|---|---|---|---|
| Driving Force Threshold [3] | The minimum difference in ∆G (per atom) required for one product to be predictably favored under thermodynamic control. | ≥ 60 meV/atom | Predicts initial product formation in solid-state reactions when one phase's driving force is significantly larger. |
| Product Ratio (Kinetic Control) [6] | The ratio of products under kinetic control is a function of the difference in their activation energies. | (\ln\left(\frac{[A]t}{[B]t}\right) = -\frac{\Delta E_a}{RT}) | Determines the product mixture when the reaction is irreversible. |
| Product Ratio (Thermodynamic Control) [6] | The product ratio at equilibrium is a function of the difference in their standard Gibbs free energies. | (\ln\left(\frac{[A]\infty}{[B]\infty}\right) = -\frac{\Delta G^\circ}{RT}) | Determines the product mixture when the reaction is reversible and has reached equilibrium. |
Experimental Protocols
Protocol 1: Differentiating Kinetic and Thermodynamic Products in a Solid-State Reaction
This protocol uses in situ X-ray Diffraction (XRD) to identify the reaction regime, as validated in recent literature [3].
1. Objective: To determine whether a solid-state reaction between two precursors is under kinetic or thermodynamic control by identifying the first-formed product and its evolution.
2. Materials:
- Precursor powders (e.g., LiOH and Nb₂O₅, or other relevant reactants).
- Mortar and pestle or ball mill for mixing.
- Inert atmosphere furnace compatible with in situ XRD.
- Synchrotron or laboratory in situ XRD setup with a heating stage.
3. Methodology:
- Sample Preparation: Mix the precursor powders homogeneously in the desired stoichiometry.
- Data Collection: Load the mixed powder into the in situ XRD stage.
- Ramp Experiment: Heat the sample from room temperature to a target temperature (e.g., 700°C) at a controlled rate (e.g., 10°C/min) while collecting XRD patterns at frequent intervals (e.g., every 30 seconds or 2 minutes) [3].
- Isothermal Experiment: Rapidly heat the sample to a specific temperature and hold it for an extended period (e.g., 3-12 hours), collecting XRD patterns periodically [3].
- Data Analysis:
- Identify the temperature at which the first crystalline product appears.
- Track the phase fractions (weight fractions from Rietveld refinement) of all phases as a function of time and temperature.
- If the first product is a metastable phase that later disappears in favor of a different, stable phase, the initial formation is kinetically controlled.
- If the first product to form is the final stable phase and persists, the reaction is likely under thermodynamic control from the outset.
Protocol 2: Applying the max-∆G Theory to Predict Initial Products
This computational protocol helps plan syntheses by predicting which phase will form first.
1. Objective: To computationally predict the initial product of a solid-state reaction between two precursors using the max-∆G theory.
2. Materials:
- Computational resources and access to a thermodynamic database (e.g., the Materials Project).
- Software for calculating phase diagrams (e.g., pymatgen).
3. Methodology:
- Identify Competing Phases: List all known stable and metastable ternary (or higher) compounds in the chemical space of your two precursors.
- Calculate Reaction Energies: For each possible product phase, calculate the compositionally unconstrained reaction energy. This is the Gibbs free energy change, ∆G, for the reaction forming one mole of the product from the precursors, normalized per atom of the product formed. Use the formula: ∆G(per atom) = [G(product) - ΣG(reactants)] / number of atoms in the product [3].
- Apply the Threshold: Compare the calculated ∆G values. The phase with the most negative ∆G (largest driving force) is predicted to form first if its driving force exceeds that of the next most favorable phase by ≥ 60 meV/atom [3].
Signaling Pathways and Workflows
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Investigating Reaction Control in Solid-State Synthesis
| Item | Function | Brief Explanation |
|---|---|---|
| Precursor Oxides/Carbonates (e.g., Nb₂O₅, Li₂CO₃) | Reactants for synthesis. | The choice of precursor (e.g., LiOH vs. Li₂CO₃) can significantly alter the reaction's driving force (∆G), influencing whether the reaction falls under kinetic or thermodynamic control [3]. |
| In Situ XRD Setup | For real-time phase analysis. | Allows for the direct observation of the first crystalline phase to form and its subsequent transformation, which is critical for distinguishing kinetic and thermodynamic products [3]. |
| Thermodynamic Database (e.g., Materials Project) | Source of computed Gibbs free energies. | Provides the necessary data (G) to calculate the driving force (∆G) for potential products, enabling the application of the max-∆G prediction theory [3]. |
| Controlled Atmosphere Furnace | For precise thermal treatment. | Enables the application of specific temperature-time profiles (e.g., rapid heating vs. long isothermal holds) to manipulate the reaction regime [6] [3]. |
The Critical 60 meV/atom Threshold for Predictive Synthesis
Frequently Asked Questions (FAQs)
1. What is the 60 meV/atom threshold in solid-state synthesis? The 60 meV/atom threshold is a quantitative boundary for thermodynamic control in solid-state reactions. Experimental validation on 37 pairs of reactants demonstrated that the initial product formation can be reliably predicted when its thermodynamic driving force (ΔG) exceeds that of all other competing phases by at least 60 milli–electron volt per atom [3]. When multiple phases have formation energies within this threshold, kinetic factors often determine the reaction outcome [3] [8].
2. How does this threshold help in predicting synthesis outcomes? This threshold helps identify which reactions will proceed under thermodynamic control, meaning the product with the largest negative formation energy forms first. Analysis of the Materials Project database indicates that approximately 15% of possible reactions (105,652 reactions) fall within this regime of thermodynamic control, where the initial product can be predicted from computed reaction energies alone [3].
3. What is the "max-ΔG theory" and how is it related? The "max-ΔG theory" states that when two solid phases react, they initially form the product with the largest compositionally unconstrained thermodynamic driving force (ΔG) per atom of material formed, irrespective of the overall reactant stoichiometry [3]. The 60 meV/atom threshold validates and refines this principle, defining the conditions where it reliably applies [3].
4. What happens when competing phases have driving forces closer than 60 meV/atom? When multiple competing phases have formation driving forces within 60 meV/atom of each other, the reaction enters a kinetic control regime [3] [8]. In this case, factors such as ionic diffusion rates, structural templating, and interfacial energies become decisive in determining the initial product, making outcomes harder to predict with thermodynamics alone [3] [8].
5. Can this framework help in synthesizing metastable polymorphs? Yes. The reaction energy (ΔGrxn) is a key handle for polymorph selection. A large thermodynamic driving force can lower the critical radius required for nucleation, favoring metastable polymorphs with lower surface energy, even when their bulk energy is higher than the stable polymorph [9]. This principle was demonstrated with LiTiOPO4, where precursor selection controlled the reaction energy and determined which polymorph was obtained [9].
Troubleshooting Guide for Solid-State Experiments
Problem: Unpredicted Phase Forms Despite Favorable Thermodynamics
| Potential Cause | Diagnostic Steps | Solution |
|---|---|---|
| Kinetic Control Regime | Calculate the energy difference between the predicted phase and its closest competitor. | If ΔΔG < 60 meV/atom, modify synthesis to influence kinetics: increase temperature to enhance diffusion or select precursors that lower nucleation barriers [3] [8]. |
| Precursor Decomposition | Use TGA/DSC to analyze the thermal behavior of precursors. | Choose precursors that decompose cleanly to the desired reactive species before the target reaction occurs. For example, BaCO3 decomposes to BaO before ternary reaction in Ba-Ti-O systems [8]. |
| Diffusion-Limited Selectivity | Check if the product layer has low ionic conductivity. | Model ionic fluxes using Onsager analyses. Consider a two-step synthesis or a different precursor that forms a more permeable intermediate phase [8]. |
Problem: Inconsistent Results or Poor Yield of Target Phase
| Potential Cause | Diagnostic Steps | Solution |
|---|---|---|
| Uncontrolled Nucleation of Metastable Polymorph | Use in situ XRD to identify the first phase that crystallizes. | If a metastable polymorph forms first, reduce the reaction driving force by using less reactive precursors. This increases the critical nucleus size, favoring the stable polymorph [9]. |
| Insufficient Driving Force | Calculate the reaction energy (ΔGrxn) for your target from the chosen precursors. | Select more reactive precursors that provide a larger thermodynamic driving force (more negative ΔGrxn) to overcome nucleation barriers for the desired phase [9]. |
| Inhomogeneous Reactant Mixing | Characterize precursor particle size and mixing uniformity. | Improve solid-solid contact by using finer powders, ball milling reactants, or employing sol-gel methods to achieve atomic-scale mixing [3]. |
Problem: Difficulty in Reproducing Literature Syntheses
| Potential Cause | Diagnostic Steps | Solution |
|---|---|---|
| Unreported Kinetic Factors | The synthesis may be sensitive to hidden variables (e.g., moisture, heating rate). | Perform a sensitivity analysis on heating rates and atmospheric conditions. Use in situ characterization (XRD, DSC) to monitor the reaction pathway in real-time [3] [10]. |
| Unidentified Polymorphic Transformation | Use in situ XRD to check for phase transitions during heating/cooling. | The first-formed phase may not be the final one. Identify the temperature of transformation and adjust annealing conditions to stabilize the desired polymorph [9]. |
Experimental Protocols & Methodologies
Protocol 1: Validating the 60 meV/atom Threshold via In Situ XRD
This protocol is adapted from the experimental work that established the 60 meV/atom threshold [3].
1. Research Reagent Solutions
| Item | Function/Benefit |
|---|---|
| High-Purity Oxide/Carbonate Precursors | Ensures well-defined starting chemistry and accurate thermodynamic calculations. |
| Synchrotron Radiation Source (e.g., ALS beamline 12.2.2) | Enables high-resolution, fast-scanning XRD for real-time monitoring of phase evolution. |
| Programmable Tube Furnace with XRD Holder | Allows for controlled heating profiles (e.g., 10°C/min) during data collection. |
| Rietveld Refinement Software | Quantifies the weight fraction of each crystalline phase present throughout the reaction. |
2. Procedure
- Sample Preparation: Mix reactant powders (e.g., LiOH and Nb₂O₅) in a 1:1 cation ratio using a mortar and pestle or ball mill.
- In Situ Data Collection: Load the mixture into a capillary or a flat-plate holder in the synchrotron XRD setup. Heat the sample to a target temperature (e.g., 700°C) at a controlled rate (e.g., 10°C/min), holding at the peak temperature for several hours. Collect XRD patterns at frequent intervals (e.g., every 30 seconds).
- Data Analysis:
- Identify the first crystalline phase(s) to appear from the XRD patterns.
- Using calculated formation energies from databases like the Materials Project, determine the thermodynamic driving force (ΔG) for all possible ternary products from the reactants.
- Correlate the first-formed phase with the phase that has the largest ΔG. The 60 meV/atom threshold represents the point where this correlation becomes robust.
Protocol 2: Targeting a Metastable Polymorph by Controlling Reaction Energy
This protocol is based on research demonstrating selective formation of LiTiOPO₄ polymorphs [9].
1. Research Reagent Solutions
| Item | Function/Benefit |
|---|---|
| Alternative Precursor Sets (e.g., Li₃PO₄ + TiO₂ vs. Li₂CO₃ + TiO₂ + (NH₄)₂HPO₄) | Different precursors create different reaction pathways and intermediates, thereby tuning the overall ΔGrxn for the final product. |
| Density Functional Theory (DFT) Calculations | Used to compute the surface energies (γ) of competing polymorphs and the reaction energies for different precursor combinations. |
| Classical Nucleation Theory Model | Applies Equation (1) to predict conditions (ΔGrxn, T) under which the metastable polymorph will nucleate faster. |
2. Procedure
- Precursor Selection: Based on DFT calculations, select a precursor combination that provides a large, negative reaction energy (ΔGrxn) to form the final compound. A large driving force favors the nucleation of metastable polymorphs with low surface energy [9].
- Synthesis and Characterization: Heat the precursor mixture while monitoring with in situ XRD.
- Analysis: Confirm that the metastable polymorph nucleates first, as predicted by the nucleation theory model. The use of highly reactive precursors keeps the critical nucleus size small, amplifying the advantage of the polymorph with the lowest surface energy [9].
Thermodynamic and Kinetic Parameters in Solid-State Synthesis
The following table consolidates key quantitative findings from recent research, providing a reference for experimental planning [3] [9] [8].
| Parameter | Typical/Threshold Value | Significance & Context |
|---|---|---|
| Threshold for Thermodynamic Control | 60 meV/atom | The difference in driving force (ΔΔG) required to reliably predict the first-formed product [3]. |
| Reactions under Thermodynamic Control | ~15% | The proportion of possible reactions in the Materials Project database that meet the 60 meV/atom threshold [3]. |
| Polymorph Bulk Energy Difference (ΔGi→j) | 10 - 100 meV/atom | The energy range between a stable and a metastable polymorph. Smaller differences make metastable phases more accessible [9]. |
| Reaction Energy to Access Metastable Polymorph (ΔGrxn) | -20 to < -200 meV/atom | The required driving force to nucleate a metastable polymorph, depending on its energy and surface energy advantage [9]. |
| Surface Energy Difference (γi - γj) | Up to ~150 meV/Ų | The surface energy advantage a metastable polymorph may have over the stable one (e.g., in ZrO₂ and HfO₂) [9]. |
| Formation Energy Difference in Ba-Ti-O | ≈51 meV/atom (Ba₂TiO₄ vs BaTiO₃) | An example of a system below the 60 meV/atom threshold, where kinetics and diffusion dominate product formation [8]. |
Conceptual Diagrams
Diagram 1: Decision Framework for Reaction Control
Diagram 2: Nucleation Control for Metastable Polymorphs
The Role of Intermediate Phases in Consuming Free Energy
Frequently Asked Questions
What is the fundamental role of an intermediate phase in a solid-state reaction? An intermediate phase is a state of matter that forms between the initial reactants and the final stable product during a solid-state reaction. Its primary role in the context of free energy is to provide a lower-energy pathway for the reaction to proceed. The formation of the first intermediate phase is often crucial, as it determines the remaining driving force available to form the desired final material by consuming a portion of the total free energy change [11].
Why does my reaction sometimes get "stuck" at an intermediate phase instead of progressing to the final product? A reaction may stall at an intermediate phase if the thermodynamic driving force to form the subsequent phase is insufficient. Recent research has quantified a threshold for thermodynamic control: the initial product formation is predictable when its driving force exceeds that of all other competing phases by at least 60 milli-electron volt per atom (meV/atom) [11]. If multiple phases have comparable driving forces (below this threshold), kinetic factors, rather than thermodynamics, tend to dominate, potentially halting the reaction at the intermediate stage [11].
How does the composition of an intermediate phase evolve during a reaction, and why is this important? During reactions, particularly in complex multicomponent systems, metal cations can counter-diffuse between different phases at the interface. This leads to composition evolution of the intermediate phases. This evolution is critical because it enhances the overall thermodynamic stability of the system, allowing the multicomponent phases to remain stable under more extreme conditions, such as higher oxygen partial pressures during ablation [12]. This process improves the material's final performance.
What is the relationship between nucleation, growth, and intermediate phases? The formation of a system of intermediate-phase crystals involves simultaneous kinetic processes. Nucleation is the initial formation of stable crystals of the new solid phase. Growth is the subsequent increase in size of these nuclei. During the intermediate stage of a phase transition, growth and ongoing nucleation occur concurrently while the metastability of the parent phase decreases. The rates of these processes directly influence the size distribution and volume fraction of the intermediate phases that consume the system's free energy [13].
Troubleshooting Guides
Problem: Unpredictable or Irreproducible Formation of Initial Intermediate Phases
Issue: The first intermediate phase that forms varies between experiments, leading to inconsistent results.
Possible Causes and Solutions:
- Cause 1: Insufficient Driving Force Differentiation. The driving forces to form multiple competing intermediate phases are too similar, placing the reaction outside the regime of thermodynamic control.
- Solution: Use thermodynamic databases to calculate the free energy of formation (ΔG) for all plausible intermediate phases. Adjust the reactant composition or use precursor materials that create a larger ΔG (≥ 60 meV/atom) for your desired intermediate phase to bring the reaction into the thermodynamic control regime [11].
- Cause 2: Inconsistent Reactant Morphology.
- Solution: Standardize the chemical and morphological properties of your solid reagents. Use reagents with consistently high surface area and reactivity to ensure reproducible interfacial contact and diffusion distances [14].
Problem: Reaction Kinetics are Too Slow
Issue: The solid-state reaction does not proceed at a practical rate or appears to halt.
Possible Causes and Solutions:
- Cause 1: Low Temperature.
- Solution: Increase the reaction temperature. High temperatures are typically employed to overcome kinetic barriers by providing the thermal energy required for significant atomic or ionic diffusion [14].
- Cause 2: Large Diffusion Distances.
- Solution: Reduce the particle size of the solid reactants. Finer powders decrease the diffusion distance that atoms or ions must travel, significantly accelerating the reaction rate [14]. Techniques like high-energy ball milling can be used to mix and reduce particle size simultaneously.
Problem: Formation of Metastable or Undesired Intermediate Phases
Issue: The reaction pathway leads to a metastable intermediate that hinders the formation of the target material.
Possible Causes and Solutions:
- Cause: Kinetic Control Over Thermodynamics. The reaction conditions favor a kinetically accessible product rather than the thermodynamically most stable one.
- Solution: Modify the reaction pathway by using a reactive atmosphere [14] or by designing a highly exothermic reaction, such as in Self-propagating High-temperature Synthesis (SHS), where the large heat release can help overcome energy barriers to the stable phases [14]. Slowing the heating rate can also allow the system more time to reach thermodynamic equilibrium.
Experimental Protocols & Data
Protocol: Constructing a Phase Diagram via Thermal Analysis
This methodology allows you to map the stability regions of intermediate phases.
- Sample Preparation: Prepare a series of mixtures covering the entire composition range of the two-component (binary) system.
- Heating/Cooling Cycles: For each composition, subject the sample to a controlled heating or cooling cycle while carefully monitoring temperature.
- Data Collection: Plot temperature vs. time curves (cooling curves) for each composition. Identify arrest points on these curves where the temperature remains constant despite heat being removed; this indicates a phase transformation (e.g., liquid to solid) due to the evolution of latent heat, which consumes free energy [15].
- Diagram Construction: Plot the temperatures of these arrest points against the composition to construct the liquidus and solidus lines of the phase diagram. The regions between these lines are where intermediate phases or two-phase mixtures coexist [15].
Protocol: Investigating Solid-State Reactions in Multicomponent Ceramics
This protocol is based on research into multiphase multicomponent ultra-high temperature ceramics [12].
- Synthesis: Fabricate a ceramic containing multiple distinct phases (e.g., Hf-rich carbide, Nb-rich carbide, Zr-rich silicide).
- Ablation/Treatment: Subject the material to high-temperature treatment (e.g., ablation in a high-speed flame or high-temperature annealing).
- Interface Analysis: Use electron microscopy (SEM/TEM) and spectroscopy (EDS) to analyze the matrix/oxide scale interface region.
- Measurement: Track the counter-diffusion of metal cations (e.g., Hf, Nb, Zr) between the different multicomponent phases and the resulting composition evolution of each phase.
- Correlation: Correlate the degree of composition evolution with measurements of the improved thermodynamic stability and ablation performance of the material.
Quantitative Threshold for Thermodynamic Control
The following table summarizes key quantitative findings on predicting intermediate phase formation.
- Table 1: Threshold for Thermodynamic Control in Solid-State Reactions
| Parameter | Value | Significance |
|---|---|---|
| Threshold Driving Force | ≥ 60 meV/atom | When the driving force to form one phase exceeds all others by this value, the initial product is predictable by thermodynamics [11]. |
| Reactions Under Thermodynamic Control | ~15% | The estimated proportion of possible reactions that fall within this predictable regime based on Materials Project data [11]. |
The Scientist's Toolkit: Essential Research Reagents & Materials
- Table 2: Key Materials and Their Functions in Solid-State Reaction Research
| Item | Function in Experiment |
|---|---|
| High-Purity Solid Precursors | Ensure reproducible reactions free from contamination that could alter reaction pathways or stabilize unwanted intermediates [14]. |
| Surfactants (e.g., Tween series) | Used in precursor preparation to control particle size and inhibit particle growth during thermal treatment; longer chains better prevent growth [14]. |
| Inert/Reactive Atmosphere Gases | Control the reaction environment; can suppress sublimation, influence reaction reversibility, or react with powders to form new phases (e.g., nitrides) [14]. |
| MnO₂, Mn₂O₃, or MnCO₃ Microspheres | Act as morphological templates for creating hollow/porous oxide structures via impregnation and solid-state reaction, facilitating shorter diffusion paths [14]. |
Thermodynamic and Kinetic Workflows
Diagram: Free Energy Consumption Pathway
Free Energy Pathway
This diagram illustrates the pathway of free energy consumption during a solid-state reaction. The formation of an intermediate phase provides a kinetic pathway with a lower activation barrier, consuming a portion of the total free energy change (ΔG_total) in stages (ΔG₁ and ΔG₂) [15] [11].
Diagram: Solid-State Reaction Experimental Workflow
Experimental Workflow
This workflow outlines the key stages in a solid-state reaction experiment, highlighting steps critical to controlling intermediate phases. Precursor preparation and mixing determine initial diffusion distances [14]. The high-temperature treatment enables diffusion and nucleation, which can be monitored to identify the formation of intermediate phases that consume the system's free energy [12] [14].
Calculating and Applying Driving Force for Predictive Synthesis
Leveraging Ab Initio Computations and Materials Project Data
Frequently Asked Questions (FAQs)
FAQ 1: How can I identify which failed synthesis experiments still provide valuable data for optimizing precursor selection?
Answer: Failed experiments are highly valuable when they reveal the formation of stable intermediate phases that consume the thermodynamic driving force. The ARROWS3 algorithm is specifically designed to learn from such outcomes [16].
- Procedure for Learning from Failed Experiments:
- After a reaction fails to produce the target, use X-ray diffraction (XRD) to identify all crystalline phases present in the product.
- Input the identified intermediate phases into a synthesis optimization algorithm.
- The algorithm will determine the pairwise reactions that led to these intermediates.
- Subsequent precursor selections are then optimized to avoid these specific energy-draining pathways, thereby preserving a larger driving force (ΔG′) for the target material formation [16].
FAQ 2: My target material is metastable. How can I use computational data to find a viable synthesis pathway?
Answer: Synthesizing metastable materials requires careful kinetic control to avoid the formation of more stable, competing phases. The key is to identify precursors and a reaction pathway that bypasses these stable intermediates.
- Protocol for Metastable Target Synthesis:
- Calculate Stability: Use DFT data from sources like the Materials Project to confirm your target is metastable (positive energy above the convex hull).
- Screen for Low-Temperature Pathways: Use an algorithm like ARROWS3 to rank precursor sets based on their ability to form the target while avoiding stable intermediates, even if the absolute driving force is smaller.
- Validate Experimentally: Test the top-ranked precursor sets at low temperatures (e.g., 300-400 °C) to kinetically trap the metastable phase. Successful synthesis of Na₂Te₃Mo₃O₁₆ and LiTiOPO4 using this method demonstrates its efficacy [16].
FAQ 3: What is the relationship between the thermodynamic driving force of a reaction and its kinetics in solid-state synthesis?
Answer: While a more negative ΔG (larger driving force) often correlates with a faster reaction rate, the relationship is not always linear. A derived global model shows that the activation energy (ΔE‡) is a non-linear function of the reaction energy (ΔEᵣ) [17].
- Key Parameters: The relationship is governed by three physical parameters:
- Eₘᵢₙ: A minimum preorganisational barrier.
- Eₑq: A reaction symmetry offset.
- θ: A kinetic curvature factor [17].
- Practical Implication: In highly exergonic regimes (very negative ΔG), further increasing the driving force may offer little rate improvement. In these cases, kinetic control shifts to structural factors beyond simple thermodynamics [17].
FAQ 4: How can I model a full solid-state reaction pathway before running an experiment?
Answer: The ReactCA simulation framework allows for the prediction of time-dependent phase evolution.
- ReactCA Workflow:
- Input Recipe: Specify your precursors, their ratios, and a heating profile.
- Automate Data Collection: The framework pulls thermodynamic data (e.g., from the Materials Project) and estimates reaction rates.
- Run Simulation: The cellular automaton models reactions at pairwise interfaces between particles, predicting the emergence and consumption of intermediates and products over time [18].
- Outcome: This simulation provides a quantitative prediction of reaction outcomes, which can be used as a digital twin to pre-screen recipes in silico [18].
Troubleshooting Guides
Problem 1: Persistent formation of inert byproducts blocking target formation.
| Step | Action | Expected Outcome |
|---|---|---|
| 1. Diagnosis | Use XRD to identify the specific inert byproduct(s) formed. | Confirmation of the stable intermediate phase(s) consuming reactants. |
| 2. Analysis | Input the byproducts into a precursor optimization algorithm (e.g., ARROWS3). | The algorithm identifies and flags precursor combinations that lead to these byproducts. |
| 3. Intervention | Switch to a new precursor set proposed by the algorithm, which is predicted to avoid the identified byproducts. | A reaction pathway with a retained driving force (ΔG′) at the target-forming step. |
| 4. Validation | Execute the new synthesis recipe and analyze the product with XRD. | Increased yield of the target material and suppression of the previous byproducts [16]. |
Problem 2: Low yield of the target material despite a large calculated thermodynamic driving force.
| Potential Cause | Solution | Principle |
|---|---|---|
| Rapid formation of kinetic intermediates | Lower the reaction temperature and use a longer annealing time. | Favors kinetic products by reducing diffusion rates and bypassing high-temperature stable intermediates [16]. |
| Poor diffusion between precursor particles | Improve mixing through thorough grinding or use precursor solutions. | Increases interfacial contact area between reactants, promoting complete reaction [18]. |
| Inaccurate driving force estimation | Use computational tools that consider temperature-dependent vibrational entropy. | Gibbs free energy (ΔG = ΔH - TΔS) is temperature-dependent. Machine-learned entropy estimates provide more accurate ΔG at synthesis temperatures [18]. |
Experimental Protocols & Data
Protocol: Optimizing Precursors using the ARROWS3 Algorithm
Objective: To automatically select precursor sets that maximize the thermodynamic driving force for a target material by learning from experimental intermediates [16].
Workflow:
Materials and Data Requirements:
- Target Material: Composition and structure.
- Precursor Pool: A list of potential solid precursor powders.
- Thermochemical Data: DFT-calculated formation energies (e.g., from the Materials Project).
- Characterization: X-ray Diffraction (XRD) for phase identification.
Quantitative Data from Synthesis Validation Studies
The following table summarizes experimental data from the validation of the ARROWS3 algorithm, demonstrating its performance across different target materials [16].
Table 1: ARROWS3 Algorithm Experimental Validation Data
| Target Material | Number of Precursor Sets Tested | Synthesis Temperatures (°C) | Total Number of Experiments | Key Finding |
|---|---|---|---|---|
| YBa₂Cu₃O₆.₅ (YBCO) | 47 | 600, 700, 800, 900 | 188 | Identified all effective synthesis routes with fewer iterations than black-box methods. |
| Na₂Te₃Mo₃O₁₆ (NTMO) | 23 | 300, 400 | 46 | Successfully guided synthesis of a metastable phase. |
| LiTiOPO₄ (t-LTOPO) | 30 | 400, 500, 600, 700 | 120 | Achieved high-purity synthesis of a polymorph prone to phase transition. |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational and Experimental Resources
| Item | Function in Research | Application Example |
|---|---|---|
| Materials Project Database | Provides access to pre-calculated DFT data for a vast range of inorganic materials, including formation energies and phase stability. | Used for the initial ranking of precursor sets based on the thermodynamic driving force (ΔG) to form the target [16]. |
| ARROWS3 Algorithm | An active learning algorithm that optimizes precursor selection by leveraging experimental data to avoid energy-draining intermediate phases. | Guided the synthesis of YBa₂Cu₃O₆.₅ and metastable Na₂Te₃Mo₃O₁₆ by iteratively learning from failed experiments [16]. |
| ReactCA Simulation Framework | A cellular automaton model that predicts phase evolution in solid-state reactions, incorporating precursor choice, atmosphere, and heating profile. | Allows for in silico testing of synthesis recipes and identification of potential intermediates before lab work [18]. |
| X-ray Diffraction (XRD) | The primary experimental technique for identifying crystalline phases present in a reaction product, including the target, intermediates, and byproducts. | Critical for providing feedback to optimization algorithms like ARROWS3 on the outcome of each synthesis experiment [16]. |
| Ab Initio Code (e.g., CRYSTAL) | Software for performing quantum-mechanical calculations to predict material properties, such as total energy, electronic structure, and vibrational frequencies. | Used for calculating fundamental thermodynamic properties of materials from first principles [19]. |
The Compositionally Unconstrained Approach to ΔG Calculation
Troubleshooting Guides
FAQ 1: How is the Gibbs Free Energy (ΔG) defined and calculated for predicting reaction spontaneity?
Answer: The Gibbs Free Energy (G) is a thermodynamic potential that combines enthalpy and entropy to predict the direction of chemical reactions at constant temperature and pressure. It is defined by the equation [1] [20]:
[ G = H - TS ]
where:
- ( G ) is Gibbs free energy
- ( H ) is enthalpy
- ( T ) is absolute temperature
- ( S ) is entropy
The change in Gibbs Free Energy, ΔG, for a process is calculated as [1]:
[ \Delta G = \Delta H - T \Delta S ]
A negative ΔG value indicates a spontaneous reaction, while a positive ΔG signifies a non-spontaneous reaction that requires energy input to occur [1].
Experimental Protocol for Determining ΔG:
- Calorimetry Measurement: Use isothermal calorimetry to directly measure the enthalpy change (ΔH) of the solid-state reaction [21].
- Entropy Estimation: Determine the entropy change (ΔS) through heat capacity measurements or computational methods.
- Calculation: Apply the measured ΔH and ΔS values to the Gibbs free energy equation at your experimental temperature.
FAQ 2: What experimental setup is recommended for direct thermodynamic characterization of solid-state reactions?
Answer: Direct thermodynamic characterization of solid-state reactions requires specialized approaches to overcome the challenges of limited diffusion and slow reaction kinetics in solid systems.
Experimental Protocol for Solid-State Thermodynamic Characterization [21]:
- Reagent Preparation:
- Select and dry solid reactants thoroughly
- Use fine-grained materials to increase surface area
- Weigh reactants in required stoichiometric amounts
Mixing Procedure:
- For small quantities (<20g): Use agate mortar and pestle
- Add volatile organic liquid (acetone or alcohol) to aid homogenization
- Mix thoroughly until organic solvent evaporates completely (10-15 minutes)
- For larger quantities: Use mechanical ball milling
Reaction Container Selection:
- High temperatures: Use noble metals (platinum, gold)
- Lower temperatures (<600-700°C): Nickel containers may be suitable
Heat Treatment:
- Pellet samples to increase contact area between grains
- Heat to appropriate temperature (typically 1000-1500°C)
- Control atmosphere as needed for specific reactions
Analysis:
- Characterize products using X-ray diffraction (XRD)
- Use scanning electron microscopy (SEM) for morphological analysis
FAQ 3: What are the common challenges in measuring ΔG for solid-state reactions and how can they be resolved?
Answer: Solid-state reactions present unique challenges for thermodynamic characterization due to their slow kinetics and complex reaction mechanisms.
Troubleshooting Table for Solid-State ΔG Measurements:
| Challenge | Cause | Solution |
|---|---|---|
| Incomplete Reaction | Limited diffusion between solid reactants | Increase surface area through fine grinding; pelletize samples to enhance contact [22] |
| Inconsistent Results | Poor mixing homogeneity | Use volatile organic liquids during mixing; employ mechanical ball milling for larger quantities [22] |
| Container Reactivity | Chemical interaction at high temperatures | Select appropriate container material (Pt, Au for high T; Ni for lower T) [22] |
| Slow Reaction Kinetics | Low atomic mobility in solid state | Increase temperature (typically 1000-1500°C); extend reaction time [22] |
| Heat Measurement Challenges | Small thermal effects in solid-state | Use sensitive isothermal calorimetry; verify via solution calorimetry with Hess's law [21] |
The Scientist's Toolkit: Research Reagent Solutions
Essential Materials for Solid-State Reaction Research:
| Item | Function | Application Notes |
|---|---|---|
| Agate Mortar and Pestle | Homogeneous mixing of solid reactants | Ideal for small quantities (<20g); chemically inert [22] |
| Volatile Organic Solvents | Aiding homogenization during mixing | Acetone or alcohol preferred; evaporates completely after mixing [22] |
| Ball Mill | Mechanical mixing of larger quantities | Essential for quantities >20g; ensures uniform mixing [22] |
| High-Temperature Furnace | Controlled heat treatment | Capable of reaching 1000-1500°C; atmosphere control capability [22] |
| Isothermal Calorimeter | Direct measurement of reaction enthalpy | Enables direct thermodynamic characterization of solid-state reactions [21] |
| Platinum/Gold Containers | Chemically inert reaction vessels | Essential for high-temperature reactions; prevent container contamination [22] |
Workflow Visualization
Solid-State ΔG Calculation Workflow
Thermodynamic Relationships Diagram
Thermodynamic Decision Pathway
Frequently Asked Questions (FAQs)
Q1: What is the primary challenge when selecting a lithium source for the solid-state synthesis of Li-Nb-O phases? The primary challenge is managing lithium loss, especially during high-temperature calcination steps. Lithium is a volatile element, and its loss during synthesis can shift the final product's stoichiometry away from the desired LiNbO3 phase, leading instead to the formation of Li-deficient phases like LiNb3O8 or unreacted Nb2O5. This loss directly reduces the thermodynamic driving force for forming the target ternary oxide, as the reaction cannot proceed to completion without sufficient lithium. Precise control of the initial Li/Nb ratio and the use of a finely mixed precursor are critical to compensate for and minimize this loss [23] [24].
Q2: How does the initial Li/Nb ratio influence the final phase in a simple solid-state reaction? The initial Li/Nb ratio is a critical parameter that determines the crystallographic phase of the final product. Research has demonstrated a clear trend:
- Low Li/Nb ratio: Favors the formation of the LiNb3O8 phase.
- High Li/Nb ratio: Favors the formation of the LiNbO3 phase.
- Intermediate ratios: Produce a mixture of both phases, with the proportion of LiNbO3 increasing as the lithium content increases. This occurs because a higher lithium concentration increases the thermodynamic driving force for the formation of the lithium-richer LiNbO3 phase [23].
Q3: Why is the thermodynamic driving force important in the synthesis of ternary oxides like LiNbO3? Recent research has established that the success of solid-state synthesis often depends on the first intermediate phase that forms. When the reaction energy (driving force) to form a particular phase is significantly larger than that of all competing phases—specifically, exceeding them by ≥60 milli-electron volt per atom—thermodynamics primarily dictates the initial product. This "regime of thermodynamic control" means the outcome becomes predictable. For Li-Nb-O synthesis, ensuring a high driving force for LiNbO3, for instance by using a lithium-rich precursor, can help steer the reaction toward the desired product [11].
Q4: What are the advantages of using lithium nitrate (LiNO3) over other lithium sources? Lithium nitrate is often preferred in solid-state synthesis for several reasons:
- Reactivity: It is a highly reactive compound that can be easily dissolved in water to create a homogeneous slurry with Nb2O5 powder, ensuring excellent mixing at the molecular level.
- Decomposition: Upon heating, LiNO3 decomposes, providing a highly reactive lithium species that can readily diffuse and react with Nb2O5 to form the desired lithium niobate phases.
- Simplicity: Using LiNO3 with Nb2O5 in a simple slurry mixing, drying, and calcination procedure has been successfully demonstrated to produce both LiNbO3 and LiNb3O8 phases, offering a straightforward and direct synthesis route [23].
Troubleshooting Guides
Problem 1: Formation of Unwanted LiNb3O8 Impurity Phase
| Symptom | Cause | Solution |
|---|---|---|
| XRD analysis shows a mixture of LiNbO3 and LiNb3O8, or pure LiNb3O8, when phase-pure LiNbO3 was the target. | Insufficient Lithium: The initial Li/Nb ratio in the precursor is too low, favoring the Li-deficient LiNb3O8 phase.Lithium Loss: Significant lithium volatilization occurred during calcination, effectively reducing the Li/Nb ratio.Inhomogeneous Mixing: The lithium source and Nb2O5 are not intimately mixed, leading to local variations in stoichiometry. | - Increase the initial Li/Nb ratio to provide a lithium excess and compensate for anticipated losses [23].- Ensure a finely powdered and homogeneous precursor mixture. The slurry method using dissolved LiNO3 is recommended for improved mixing [23].- Verify the calcination temperature and duration are not excessively high or long, which can exacerbate lithium loss. |
Problem 2: Incomplete Reaction and Presence of Unreacted Nb2O5
| Symptom | Cause | Solution |
|---|---|---|
| Unreacted Nb2O5 is detected in the final product via XRD. | Insufficient Thermodynamic Driving Force: The reaction energy may be too low, potentially due to a low Li/Nb ratio or kinetic barriers.Low Reactivity of Precursors: The physical mixture of solids has poor interfacial contact, limiting diffusion and reaction.Insufficient Calcination Temperature/Time: The energy provided was not enough to overcome the kinetic barriers to reaction. | - Optimize the Li/Nb ratio to increase the driving force for the ternary oxide formation [23] [11].- Switch to a wet-chemical or slurry mixing method to maximize reactant contact [23].- Conduct a TGA/DTA analysis to identify the appropriate reaction temperature and ensure the calcination is performed at a sufficient temperature (e.g., 800°C) [23]. |
Problem 3: Lithium Loss and Stoichiometry Control in Thin-Film Synthesis
| Symptom | Cause | Solution |
|---|---|---|
| Thin films are Li-deficient, leading to poor ionic conductivity or incorrect phase formation. | Volatilization: Lithium evaporates during high-temperature post-deposition annealing, a common issue in solution-based methods [24].Energetic Processes: In physical vapor deposition, preferential re-sputtering of the light lithium atoms can occur. | - Sputtering Solution: Use reactive magnetron co-sputtering with separate Li and Nb targets. This allows independent control of the lithium flux and enables the creation of Li-rich films to counteract losses [24].- Low-Temperature Processing: Develop synthesis protocols that minimize high-temperature steps. For example, co-sputtering can produce amorphous Li-Nb-O films with good ionic conductivity at room temperature, bypassing the need for high-temperature crystallization [24]. |
Experimental Protocol: Solid-State Synthesis via Slurry Mixing
This protocol is adapted from the simple method described in the search results to prepare LiNbO3 and LiNb3O8 phases [23].
Objective: To synthesize crystalline LiNbO3 and/or LiNb3O8 powders via a simple solid-state reaction using a slurry mixing technique.
Materials and Equipment:
- Niobium Oxide (Nb2O5·xH2O), fine powder (100–150 mesh)
- Lithium Nitrate (LiNO3), P.A. grade
- Distilled water
- Analytical balance
- Magnetic stirrer and stir bar
- Drying oven
- High-temperature furnace (capable of 800°C)
- Alumina crucibles
Procedure:
- Solution Preparation: Dissolve a predetermined mass of LiNO3 in distilled water. The mass should be calculated based on the desired Li/Nb mass ratio, which is typically varied between 1% and 10% [23].
- Slurry Formation: Add 2 grams of hydrated Nb2O5 powder to the LiNO3 solution. Stir the resulting slurry vigorously for 4 hours at room temperature to ensure homogeneous mixing.
- Drying: Transfer the slurry to a drying container and evaporate the water in an oven to obtain a dry, mixed precursor powder.
- Calcination: Place the dried precursor powder into an alumina crucible and calcine it in a furnace at 800°C for a defined period (e.g., 4 hours) to crystallize the lithium niobate phases.
- Characterization: The final product can be characterized by X-ray diffraction (XRD) to determine the present phases (LiNbO3 or LiNb3O8), and by scanning electron microscopy (SEM) for morphological analysis [23].
Workflow Diagram: Li-Nb-O Synthesis and Phase Control
The diagram below illustrates the synthesis pathway and the key factors influencing the final Li-Nb-O phase.
| Initial Li/Nb Ratio | Dominant Crystalline Phase | Notes |
|---|---|---|
| Low | LiNb3O8 | Favored under lithium-deficient conditions. |
| Intermediate | Mixture of LiNbO3 and LiNb3O8 | The proportion of LiNbO3 increases with increasing Li content. |
| High | LiNbO3 | Favored under lithium-rich conditions. |
| Film Type | Crystallinity | Li-to-Nb Atomic Ratio | Room-Temperature Ionic Conductivity (S cm⁻¹) |
|---|---|---|---|
| Li-Rich Film | Amorphous | 2.31 | Data not explicitly stated, but enhanced diffusion reported. |
| Nb-Rich Film | Amorphous | 1.36 | 1.33 × 10⁻⁷ |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Li-Nb-O Synthesis Experiments
| Material / Reagent | Function in Synthesis | Key Considerations |
|---|---|---|
| Lithium Nitrate (LiNO3) | Lithium source in solid-state and slurry reactions. | Highly reactive and soluble, allowing for homogeneous precursor preparation. Helps increase thermodynamic driving force for Li-rich phases [23]. |
| Niobium Oxide (Nb2O5) | Niobium source in solid-state and slurry reactions. | Should be a fine powder (e.g., 100-150 mesh) to maximize surface area for reaction. Hydrated form (Nb2O5·xH2O) can be used [23]. |
| Metallic Lithium (Li) Target | Lithium source in physical vapor deposition (co-sputtering). | Allows independent control of Li flux. Highly reactive; requires careful handling and storage under inert conditions or in paraffin oil [24]. |
| Metallic Niobium (Nb) Target | Niobium source in physical vapor deposition (co-sputtering). | DC power applied to the Nb target is a key parameter to control the Nb/O atomic ratio in the deposited films [24]. |
| Argon and Oxygen Gases | Sputtering atmosphere for thin-film deposition. | Argon is the primary sputtering gas; oxygen is introduced reactively to form the oxide film. The flow ratio is critical [24]. |
Using Convex Hull Diagrams to Visualize Reaction Energetics
Frequently Asked Questions
FAQ 1: What is the "energy above hull" (E*hull) and what does it tell me about my material's stability?
The energy above hull (Ehull) is the vertical energy distance from a material's formation energy to the convex hull in energy-composition space. A material with an Ehull of 0 meV/atom is thermodynamically stable and lies on the convex hull. A material with an E*hull greater than 0 meV/atom is metastable and has a driving force to decompose into a mixture of the stable phases located at the points on the hull directly beneath it [25]. The magnitude of E_hull indicates the degree of instability; a higher value means the material is more likely to decompose.
FAQ 2: How is the energy above hull actually calculated for a multi-element system?
The convex hull is a geometric construction that identifies the minimum energy "envelope" in energy-composition space, regardless of whether the system has 2, 3, or 4 elemental species [25]. The Ehull for a specific compound is its energy difference relative to this envelope. For a stable phase on the hull, Ehull is zero. For a phase above the hull, E*hull is the energy difference per atom between the phase and its decomposition products. The decomposition pathway is the one that minimizes the energy, which can involve a combination of several stable phases [25]. For example, the oxynitride BaTaNO₂ is calculated to be 32 meV/atom above the hull, decomposing into a mixture of 2/3 Ba₄Ta₂O₉ + 7/45 Ba(TaN₂)₂ + 8/45 Ta₃N₅ [25].
FAQ 3: I calculated a negative decomposition energy (E*d) for my synthesis reaction. Does this mean my target material is synthesizable?
Not necessarily. A negative Ed for a specific synthesis reaction only indicates that the reaction is *spontaneous as written [25]. However, it does not guarantee that your target material will form, as it might be thermodynamically favored to decompose into other, more stable phases not considered in your reaction equation. The definitive metric for thermodynamic synthesizability is the Ehull. A phase with Ehull > 0 is metastable and might still be synthesizable under kinetic control, but it is not the equilibrium product [25].
FAQ 4: What is the relationship between the convex hull and predicting solid-state reaction outcomes?
Recent research has validated the "max-ΔG theory," which states that when two solid reactants are combined, the initial product formed is the one that leads to the largest decrease in Gibbs energy (ΔG) per atom, regardless of the overall reactant stoichiometry [3]. This product is the one on the convex hull with the largest formation energy from the precursors. This thermodynamic control is proven to be predictive when the driving force to form one product exceeds that of all other competing phases by ≥60 meV/atom [3]. Below this threshold, kinetic factors often determine the initial product.
FAQ 5: Why does my convex hull calculation require so many reference structures, and how can I ensure its quality?
The convex hull is built from all possible phases in a chemical system. An incomplete set of reference energies will result in an inaccurate hull, potentially misidentifying stable materials and yielding incorrect E*hull values. To ensure quality [25]:
- Comprehensive Data: Use a large database like the Materials Project or calculate all known stable and metastable phases in your system.
- Consistent Settings: All energy calculations must use identical computational parameters (e.g., DFT functional, convergence criteria).
- Normalization: All energies must be normalized per atom for a valid comparison.
Key Quantitative Data for Experimental Design
Table 1: Quantitative Stability and Reactivity Thresholds
| Concept | Key Threshold | Interpretation | Source |
|---|---|---|---|
| Thermodynamic Control Threshold | ≥60 meV/atom | The driving force difference needed for the thermodynamically favored phase to be the initial reaction product predictably. | [3] |
| Metastability Boundary | >0 meV/atom | Any material with an energy above hull greater than zero is metastable with respect to decomposition. | [25] |
Table 2: Example Decomposition and Energy Above Hull
| Material | Energy Above Hull | Decomposition Products (Stoichiometry) | Source |
|---|---|---|---|
| BaTaNO₂ | 32 meV/atom | 2/3 Ba₄Ta₂O₉ + 7/45 Ba(TaN₂)₂ + 8/45 Ta₃N₅ | [25] |
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Computational Resources for Solid-State Reaction Research
| Item Name | Function/Explanation |
|---|---|
| LiOH & Li₂CO₃ | Common lithium sources in solid-state synthesis. The choice of precursor can significantly impact the reaction pathway and driving force due to different decomposition energies [3]. |
| Nb₂O₅ | A common Nb₂O₅ (Niobium pentoxide) precursor used in the synthesis of ternary oxides like those in the Li-Nb-O system [3]. |
| NH₃ | Used as a nitriding agent in the synthesis of oxynitrides, where it provides a source of nitrogen [25]. |
| DFT+VASP | First-principles calculation suite for computing the formation energies of crystals, which are the foundational data for constructing convex hulls [25]. |
| pymatgen | Python library for materials analysis. It contains robust tools for generating phase diagrams and calculating the energy above hull from computed data [25]. |
| Materials Project Database | A vast repository of computed crystal structures and their properties, providing the reference data needed to build convex hulls for most inorganic systems [25]. |
Visualizing the Convex Hull and Reaction Energetics
The following diagram illustrates the relationship between formation energy, the convex hull, and the energy above hull in a hypothetical ternary system.
Integrating In Situ Characterization (XRD) for Pathway Validation
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of using in situ XRD over ex situ methods for studying solid-state reactions? In situ X-ray diffraction (XRD) allows researchers to detect and identify intermediate crystalline phases as they form during a reaction in real-time. This is crucial because these transient phases often consume the driving force of the reaction and dictate the subsequent synthesis pathway. Ex situ methods, which analyze samples after the reaction is complete, can miss these critical intermediates, leading to an incomplete understanding of the reaction mechanism [3].
Q2: Under what conditions can the thermodynamic driving force reliably predict the initial product of a solid-state reaction? Recent research has validated a quantitative threshold for thermodynamic control. The initial reaction product can be predicted with high confidence when its thermodynamic driving force (ΔG) exceeds that of all other competing phases by at least 60 meV/atom. When multiple phases have comparable driving forces (a difference of less than 60 meV/atom), kinetic factors like diffusion and structural templating dominate, and the outcome is less predictable by thermodynamics alone [3].
Q3: How does the choice of precursor materials influence the reaction pathway? The precursor can significantly alter the reaction energy landscape. For example, in the Li-Nb-O system, using LiOH as a Li source creates a large thermodynamic driving force to form Li3NbO4, favoring thermodynamic control. In contrast, using Li2CO3 results in much smaller differences between the driving forces to form competing phases like LiNb3O8, LiNbO3, and Li3NbO4, pushing the reaction into a kinetically-controlled regime where the initial product is less predictable [3].
Q4: What does the "max-ΔG theory" state? The "max-ΔG theory" posits that when two solid phases react, the initial product formed will be the one that leads to the largest decrease in Gibbs free energy (ΔG) per atom, irrespective of the overall stoichiometry of the reactant mixture. This is because solid products often form locally at particle interfaces without knowledge of the bulk composition [3].
Troubleshooting Guides
Issue 1: Unpredictable or Irreproducible Initial Reaction Products
Problem: The first crystalline phase that appears in your in situ XRD data does not match thermodynamic predictions, or varies between experiments.
Possible Causes and Solutions:
- Cause: The reaction is operating in a kinetically-controlled regime. The driving forces to form multiple competing phases are too similar (difference < 60 meV/atom), so kinetic factors dictate the outcome [3].
- Solution: Consult computational databases (e.g., the Materials Project) to calculate the reaction energies (ΔG) for all possible products. If the differences are small, thermodynamic prediction is not reliable for your system.
- Solution: Modify the reaction conditions to favor thermodynamics. This could involve increasing the reaction temperature to overcome kinetic barriers or switching to precursor materials that provide a larger thermodynamic driving force for your desired product [3].
- Cause: Structural templating is reducing the nucleation barrier for a specific phase. A product with a similar crystal structure to the precursor may form first, even if it is not the most thermodynamically stable phase [3].
- Solution: Analyze the structural relationships between your precursors and the observed intermediate phases. In situ XRD can identify this templating effect.
Issue 2: Difficulty in InterpretingIn SituXRD Data
Problem: The diffraction patterns are complex, with overlapping peaks from multiple phases, making it hard to identify the sequence of phase formation.
Possible Causes and Solutions:
- Cause: The time or temperature resolution of the XRD measurements is too coarse, missing brief intermediate phases.
- Solution: If using synchrotron radiation, increase the scan frequency. In lab-based instruments, slow the heating rate or use longer counting times to improve signal-to-noise, balancing this against the need for temporal resolution [3].
- Cause: The formation of non-crystalline or poorly crystalline intermediates that do not produce sharp diffraction peaks.
- Solution: Employ complementary in situ techniques. Pair XRD with X-ray absorption fine structure (XAFS) spectroscopy, which is sensitive to short-range order and local coordination, even in amorphous materials [26].
Issue 3: Failure to Form the Target Equilibrium Phase
Problem: The reaction pathway gets "stuck" at an intermediate phase, and the final target material does not form, even with prolonged heating.
Possible Causes and Solutions:
- Cause: The initial intermediate phase consumes too much of the driving force, leaving insufficient energy to propel the reaction to the final equilibrium product [3].
- Solution: Use in situ XRD to identify the first-forming intermediate. Design a modified synthesis route that avoids this phase, for example, by using a different set of precursors that provide a more direct and energetically favorable pathway to the target [3].
- Solution: Apply an altered thermal profile, such as a two-step heating process, to bypass the kinetic trap of a specific intermediate.
Quantitative Data on Thermodynamic and Kinetic Control
The following table summarizes key quantitative findings from recent research on predicting solid-state reaction pathways.
Table 1: Quantitative Threshold for Thermodynamic Control in Solid-State Reactions
| Parameter | Value | Significance and Implication |
|---|---|---|
| Threshold for Thermodynamic Control | ≥ 60 meV/atom | When the driving force (ΔG) for the most stable phase exceeds all others by this amount, the initial product is highly predictable. Below this, kinetic control dominates [3]. |
| Reactions in Thermodynamic Regime | 15% | Proportion of possible reactions analyzed in the Materials Project database that fall within the predictable, thermodynamic control regime [3]. |
| Key Influencing Factor | Precursor Reactivity | The chemical nature of the precursors (e.g., LiOH vs. Li2CO3) directly impacts the magnitude of ΔG differences between products, shifting the reaction between thermodynamic and kinetic control [3]. |
Experimental Protocols for Key Experiments
Protocol: Validating the Reaction Pathway withIn SituXRD
Objective: To identify the sequence of crystalline phase formations during a solid-state synthesis and determine if the initial product aligns with thermodynamic predictions.
Materials:
- High-purity precursor powders (e.g., LiOH·H2O and Nb2O5, or Li2CO3 and Nb2O5).
- Mortar and pestle or ball mill for mixing.
- Synchrotron beamline equipped with a high-temperature stage and fast XRD detector, or a laboratory XRD with a non-ambient reaction chamber.
Methodology:
- Sample Preparation: Mix the reactant powders thoroughly in the desired stoichiometric ratio. For hygroscopic materials, perform mixing in an inert atmosphere.
- Loading: Load a small amount of the mixed powder into a capillary or onto a sample holder compatible with the in situ heating stage.
- Data Collection Setup: Program the heating stage to mimic the planned synthesis conditions (e.g., heat to 700°C at 10°C/min, followed by an isothermal hold). Configure the XRD to collect patterns at a high frequency (e.g., every 30 seconds or 2 scans per minute) to capture rapid phase transformations [3].
- Data Acquisition: Initiate the heating program and begin collecting sequential XRD patterns throughout the entire thermal cycle.
- Data Analysis:
- Use reference patterns (e.g., ICDD database) to identify the crystalline phases present in each collected pattern.
- Track the appearance and disappearance of diffraction peaks to construct a timeline of phase formation.
- The first new set of crystalline diffraction peaks to appear corresponds to the initial reaction product.
- Validation: Compare the identified initial product with the one predicted to have the largest compositionally unconstrained ΔG (calculated from a database like the Materials Project). Agreement suggests thermodynamic control, especially if the ΔG difference exceeds 60 meV/atom [3].
Research Reagent Solutions
Table 2: Essential Materials for In Situ XRD Studies of Solid-State Reactions
| Item / Reagent | Function / Explanation |
|---|---|
| High-Purity Precursors | Ensures that unintended side reactions with impurities do not obscure the true reaction pathway. Critical for reproducible and interpretable results. |
| Synchrotron Radiation Source | Provides a high-intensity, high-brightness X-ray beam. This enables the fast data collection and high resolution needed to capture rapid reaction kinetics and identify subtle intermediate phases [3] [26]. |
| Environmental Stage (Heating) | Allows for real-time simulation of synthesis conditions (high temperature) while the XRD measurement is ongoing. This is the core of the in situ experiment. |
| Computational Database (e.g., Materials Project) | Provides access to calculated thermodynamic data (formation energies, ΔG) for reactants and potential products. This is essential for making initial predictions about reaction pathways and driving forces [3]. |
Signaling Pathways and Workflow Diagrams
Diagram 1: Framework for predicting and validating solid-state reaction pathways based on thermodynamic driving force. The 60 meV/atom threshold determines the regime of control [3].
Diagram 2: The core experimental workflow for using in situ XRD to validate a solid-state synthesis pathway in real-time [3].
Overcoming Kinetic Barriers and Optimizing Reaction Selectivity
Identifying and Managing Kinetic Competition in Low-ΔG Scenarios
Troubleshooting Guides
Common Problems and Solutions
Problem 1: Persistent Impurity Phases
- Issue: Unwanted by-products, such as Bi₂Fe₄O₉ and Bi₂₅FeO₃₉ when synthesizing BiFeO₃, persist despite being within the target's thermodynamic stability region [4].
- Diagnosis: This indicates strong kinetic competition. The driving force to form the impurity is similar to or greater than that of your target phase [27].
- Solution: Recalculate the Interface Reaction Hull for your precursor system. Identify the most thermodynamically competitive impurity and select new precursors that minimize its driving force or maximize the driving force to your target (the MTC principle) [4] [27].
Problem 2: Reaction Stalling Before Completion
- Issue: The reaction starts but does not go to completion, resulting in a mixture of precursors, intermediates, and the target phase [4].
- Diagnosis: The initial product layer creates a diffusion barrier, and the local thermodynamic driving force at the new interfaces is insufficient to continue the reaction [4].
- Solution: Consider using a precursor that forms a high-energy amorphous intermediate, which can provide a greater overall driving force to the final crystalline target [28]. Alternatively, use finer precursor powders or intermittent grinding to refresh interfaces.
Problem 3: Failure to Form a Metastable Target
- Issue: The reaction consistently forms the thermodynamically stable phase instead of the desired metastable phase [29] [16].
- Diagnosis: Conventional precursors and high-temperature conditions favor the equilibrium phase.
- Solution: Employ kinetically controlled synthesis routes, such as Solid-State Metathesis Reactions, which can provide a large initial driving force under mild conditions to bypass the stable phase [29]. Use algorithms like ARROWS³ to identify precursors that avoid forming stable intermediates [16].
Frequently Asked Questions (FAQs)
FAQ 1: What is kinetic competition in solid-state synthesis? Kinetic competition occurs when multiple solid phases can nucleate and grow from the same precursors. Even if a target phase is thermodynamically stable, a competing phase with a similar or larger thermodynamic driving force (more negative ΔG) may form faster, leading to persistent impurities [4] [27].
FAQ 2: My target phase is on the convex hull. Why do I get impurities? Being on the convex hull means your phase is thermodynamically stable, but it does not guarantee a large kinetic driving force for its formation. Impurities often appear as kinetic by-products because they have a lower nucleation barrier or a more favorable initial driving force under your specific synthesis conditions [4] [27].
FAQ 3: How can I computationally predict if my reaction will have kinetic competition? You can assess this using thermodynamic selectivity metrics:
- Construct an Interface Reaction Hull: This is a slice of the phase diagram between your two precursors. The non-precursor vertices represent possible intermediate or product phases [4].
- Calculate the Minimum Thermodynamic Competition (MTC): For a given set of conditions, this is the free energy difference between your target phase and the most stable competing phase. A smaller (more negative) value indicates weaker competition [27].
FAQ 4: Are there automated tools to help with precursor selection? Yes, computational workflows are being developed for this purpose. For example, the ARROWS³ algorithm combines thermodynamic data from sources like the Materials Project with experimental outcomes to actively learn which precursors avoid forming stable intermediates, thereby retaining a large driving force to form the target [16].
Quantitative Data Tables
Table 1: Thermodynamic Selectivity Metrics for Synthesis Planning
| Metric Name | Formula | Interpretation | Application Example |
|---|---|---|---|
| Primary Competition [4] | Derived from interface reaction hull | Ranks the favorability of initial phase formation from precursors. | Used to analyze 3,520 solid-state reactions, identifying optimal synthesis routes for compounds like BaTiO₃ [4]. |
| Secondary Competition [4] | Derived from interface reaction hull | Assesses the favorability of subsequent phase formation after an initial product layer exists. | Explains why reactions stall and how impurity phases like δ in Figure 1b can block full conversion [4]. |
| Minimum Thermodynamic Competition (MTC) [27] | (\Delta \varPhi (Y) = \varPhi{k}(Y) - \min\limits{i\in I{c}}\varPhi{i}(Y)) | Identifies synthesis conditions (Y) that maximize the free energy difference between the target phase (k) and its most competitive neighboring phase. | Validated by text-mining 331 aqueous recipes; phase-purity for LiIn(IO₃)₄ and LiFePO₄ was achieved only near MTC-predicted conditions [27]. |
Table 2: Experimentally Validated Alternative Precursors for Common Targets
| Target Material | Conventional Precursor(s) | Alternative Precursor(s) | Performance Outcome |
|---|---|---|---|
| Barium Titanate (BaTiO₃) | BaO + TiO₂ | BaS/BaCl₂ + Na₂TiO₃ | Faster reaction kinetics and fewer impurities compared to conventional methods [4]. |
| YBa₂Cu₃O₆₅ (YBCO) | Various in Y-Ba-Cu-O system | Optimized sets identified by ARROWS³ algorithm | Algorithm identified all effective synthesis routes from a pool of 47 precursor combinations with fewer experiments [16]. |
| Metastable CuSe₂ | Standard solid-state | Kinetically-controlled metathesis | Isolated a metastable superconducting phase under mild conditions, inaccessible via standard synthesis [29]. |
Experimental Protocols
Protocol 1: Constructing and Using an Interface Reaction Hull for Precursor Selection
This methodology helps predict and visualize potential kinetic competitors [4].
- Gather Thermodynamic Data: Acquire formation enthalpies (ΔHf) for all relevant phases in your chemical system from a database like the Materials Project. Convert these to Gibbs free energies of formation (ΔGf(T)) using machine learning models or experimental thermochemistry data [4].
- Define the Precursor Tie-Line: For your chosen precursor pair, define the tie-line in composition space that connects them.
- Enumerate Balanced Reactions: Using combinatorial reaction enumeration, list all stoichiometrically balanced reactions between the precursors that yield possible product phases or phase mixtures. The maximum number of products for a vertex is one less than the number of elements in the system [4].
- Calculate Reaction Free Energies: Compute the Gibbs free energy of reaction (ΔGrxn) for each enumerated reaction.
- Plot the Hull: Create a plot with the atomic mixing ratio of precursors (x) on the x-axis and ΔGrxn on the y-axis. The vertices of the hull represent the most stable reaction products at given compositions [4].
- Analyze for Competition: The non-precursor vertices on this hull are potential kinetic competitors. The reaction with the most negative ΔGrxn is often predicted to form first. Select precursors where your target is the vertex with the largest driving force [4].
Protocol 2: Implementing the Minimum Thermodynamic Competition (MTC) Principle
This framework identifies synthesis conditions that minimize the formation of kinetic by-products [27].
- Define the System: Identify your target phase and the intensive variables of your synthesis (e.g., pH, redox potential E, and aqueous metal ion concentrations for solution synthesis) [27].
- Compute Free Energy Landscapes: Calculate the free-energy surfaces for the target and all competing phases. For aqueous systems, this involves calculating the Pourbaix potential (Ψ) [27].
- Calculate Thermodynamic Competition: At a given set of conditions Y, the thermodynamic competition experienced by the target phase k is calculated as (\Delta \varPhi (Y) = \varPhi{k}(Y) - \min\limits{i\in I{c}}\varPhi{i}(Y)), where the second term is the minimum free energy of all competing phases [27].
- Optimize Conditions: Use a computational algorithm to find the conditions Y* that minimize ΔΦ(Y). This is the point of Minimum Thermodynamic Competition, where the energy difference between the target and its closest competitor is maximized [27].
- Experimental Validation: Perform synthesis at the predicted MTC point and at other control points. Characterization (e.g., XRD) should show highest phase purity at the MTC point [27].
Workflow Visualization
Diagram 1: Autonomous Precursor Optimization Workflow
Diagram 2: Kinetic Competition Leading to Reaction Stalling
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Kinetic Control
| Item | Function in Managing Kinetic Competition |
|---|---|
| Modulated Elemental Reactant (MER) Method | A thin-film deposition technique that allows atomic-level control over precursor layer sequence and local composition, enabling the isolation and study of fundamental reaction mechanisms and access to novel phases [28]. |
| Solid-State Metathesis Reactions | A class of reactions that are often highly exothermic, providing a large kinetic driving force to form metastable products under relatively mild conditions, thus bypassing more stable phases [29]. |
| ARROWS³ Algorithm | An active learning algorithm that uses thermodynamic data and experimental feedback to autonomously select optimal precursors which avoid the formation of energy-draining intermediate phases [16]. |
| Interface Reaction Hull Model | A computational model that visualizes the free energies of all possible products between two precursors, serving as a predictive tool for identifying kinetic competitors during the initial reaction design phase [4]. |
| Materials Project Database | A core repository of computed materials data (e.g., formation energies, phase diagrams) used to calculate thermodynamic driving forces and construct interface reaction hulls for synthesis planning [4] [16] [27]. |
The Impact of Diffusion Limitations and Structural Templating
Frequently Asked Questions (FAQs) and Troubleshooting Guide
Fundamental Concepts
Q1: What are the primary diffusion mechanisms in solids, and how do they impact solid-state reaction rates?
Solid-state diffusion occurs via several distinct mechanisms, each with different kinetics that critically influence reaction rates [30].
- Substitutional (Vacancy) Diffusion: Atoms exchange with vacancies in the crystal lattice. This mechanism has a high activation energy and is relatively slow, as it depends on both vacancy formation and migration energies [30].
- Interstitial Diffusion: Smaller atoms (e.g., C, N, H) move through spaces between host atoms. This is faster than substitutional diffusion and does not require vacancies [30].
- Grain Boundary and Surface Diffusion: Atoms migrate along high-energy interfaces between crystals (grain boundaries) or on free surfaces. These are short-circuit pathways with lower activation energies, leading to significantly higher diffusion rates, especially at lower temperatures and in nanocrystalline materials [30].
Q2: What is structural templating, and when does it dominate reaction outcomes?
Structural templating is a kinetic phenomenon where a precursor material acts as a structural template, lowering the nucleation barrier for a product phase that has a similar crystal structure. This occurs when the interfacial energy (γ) between the reactant and product is reduced due to structural similarity [3]. Templating often dominates the reaction pathway when the thermodynamic driving forces (∆G) for multiple competing product phases are comparable, placing the reaction in a regime of kinetic control [3].
Troubleshooting Common Experimental Issues
Q3: My solid-state reaction is not proceeding to completion, even at high temperatures. What could be the cause?
This is a classic symptom of severe diffusion limitations.
- Potential Cause 1: Low Reactant Contact. In powder mixtures, the reaction relies on point contacts between particles. Limited contact area drastically reduces the reaction interface.
- Troubleshooting Solution:
- Improve Mixing: Use high-energy milling or ball milling to create finer, more intimately mixed powders. This reduces diffusion distances and can enhance reactivity [14].
- Increase Surface Area: Source reactants with smaller particle sizes (e.g., nanopowders) to increase the contact area and shorten diffusion paths.
- Potential Cause 2: Product Layer Formation. A dense product layer can form at the interface, physically separating the reactants and acting as a diffusion barrier.
- Troubleshooting Solution:
- Cyclic Grinding: Periodically interrupt the heating process to cool and re-grind the sample. This fractures the product layer and exposes fresh reactant surfaces [14].
- Use a Flux: Incorporate a low-melting-point flux that can dissolve the reactants and facilitate transport between them.
Q4: My reaction consistently forms an unexpected intermediate phase instead of the thermodynamically stable target. How can I steer the reaction toward the desired product?
This issue arises from kinetic competition and often involves structural templating.
- Potential Cause: Kinetic Control and Templating. The unexpected phase likely has a lower nucleation barrier due to structural similarity to a reactant, even if it is not the most thermodynamically stable phase [3].
- Troubleshooting Solution:
- Increase Thermodynamic Driving Force: The key is to push the reaction into the regime of thermodynamic control. According to recent research, this is achieved when the driving force (∆G) for your desired product exceeds that of all competing phases by a threshold of ≥60 meV/atom [3].
- Select Alternative Precursors: Choose reactant pairs that maximize the ∆G for your target material. Use computational databases like the Materials Project to screen for precursor combinations with a large energy difference to competing phases [3].
- Design a Multi-Step Pathway: If a direct reaction is not feasible, design a synthesis pathway where the initial kinetically favored intermediate subsequently reacts with high driving force to form the final target, preserving much of the initial free energy [3].
Q5: How can I control the morphology (e.g., hollow spheres) of my solid-state reaction product?
Morphology control can be achieved by leveraging diffusion-driven phenomena and templating.
- Strategy 1: Kirkendall Effect. This is a classic approach for creating hollow structures.
- Experimental Protocol: Select a reactant system where one species diffuses outwards much faster than the other diffuses inwards. The imbalance in diffusion fluxes leads to a net mass transport outward, creating vacancies that coalesce into a central pore. For example, hollow LiNi0.5Mn1.5O4 microspheres have been synthesized by impregnating MnO2 templates with Li and Ni precursors, where the fast outward diffusion of Mn and Ni atoms leads to hollowing [14].
- Strategy 2: Sacrificial Templating.
- Experimental Protocol: Use a pre-formed template with the desired morphology (e.g., spherical CaCO3 particles [31]) and coat it with your reactant material. Subsequently, remove the core template through calcination or selective dissolution, leaving behind a hollow replica of the original template.
Quantitative Data for Experimental Planning
Table 1: Comparative Diffusion Parameters in Metallic Systems [30]
| Diffusing Element | Host Metal | Mechanism | Activation Energy, Q (kJ/mol) | Pre-exponential Factor, D₀ (m²/s) |
|---|---|---|---|---|
| Carbon (C) | FCC-Fe | Interstitial | 155 | 2.0 × 10⁻⁵ |
| Nickel (Ni) | FCC-Fe | Substitutional | 279 | 1.3 × 10⁻¹ |
| Iron (Fe) | BCC-Fe | Substitutional | 240 | 2.0 × 10⁻¹ |
| Copper (Cu) | Aluminum (Al) | Substitutional | 136 | 7.8 × 10⁻⁵ |
Table 2: Thresholds for Solid-State Reaction between 4H-SiC and Metals [32]
| Metal | Temperature Threshold (°C) | Key Reaction Product |
|---|---|---|
| Cobalt (Co) | 550 (on C-face) | Co₂Si + C |
| Nickel (Ni) | 600 | Ni₃₁Si₁₂ + C |
| Iron (Fe) | 650 | Fe₃Si + C |
Essential Visual Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Solid-State Reaction Studies
| Reagent / Material | Function in Experiment | Key Considerations |
|---|---|---|
| LiOH / Li₂CO₃ | Lithium source for oxide synthesis (e.g., in Li-Mn-O or Li-Nb-O systems) [3] | LiOH is more reactive, provides a larger thermodynamic driving force than Li₂CO₃ [3]. |
| Nb₂O₅ | Niobium source for synthesizing ternary oxides (e.g., LiNbO₃) [3] | Particle size and morphology affect diffusion distance and reaction onset temperature. |
| Polycaprolactone (PCL) | A biodegradable polymer used in template-based fabrication of drug carriers and scaffolds [33] [34] | Provides a controllable matrix for encapsulation; properties can be tuned with molecular weight. |
| Calcium Carbonate (CaCO₃) | Versatile sacrificial template for creating micro/nano-spheres and capsules [31] | Can be synthesized in various morphologies (spherical, needle-like); easily removed with mild acid. |
| 4H-SiC Substrate | A wide-bandgap semiconductor used in diffusion couple studies with metals [32] | Anisotropic diffusion is observed; C-face and Si-face react at different temperatures with metals like Co [32]. |
| Fe, Ni, Co Metals | For studying solid-state diffusion reactions and silicide formation with SiC [32] | The metal type significantly influences the diffusion rate and temperature threshold for reaction. |
Strategies for Selecting Reactants and Precursors to Maximize ΔG
Frequently Asked Questions (FAQs)
FAQ 1: Why is maximizing the thermodynamic driving force (ΔG) important in solid-state synthesis? A more negative ΔG (a larger thermodynamic driving force) leads to faster reaction kinetics and a higher likelihood of successfully forming the target material. In solid-state reactions, a large driving force helps overcome kinetic barriers and reduces the chance of the reaction becoming trapped in a metastable state with impurity phases [35] [36].
FAQ 2: My reaction is thermodynamically feasible (ΔG < 0), but I still get impure products. Why? A negative ΔG for the overall reaction is necessary but not always sufficient. The issue often lies in the reaction pathway. If low-energy intermediate phases form first, they can consume the available driving force, leaving insufficient energy to complete the transformation to the desired pure final product. The key is to select precursors that not only provide a large overall ΔG but also avoid these deep kinetic traps [35] [4].
FAQ 3: What is the difference between "reaction energy" and "inverse hull energy"?
- Reaction Energy (ΔE): The total energy released for the direct reaction from your chosen precursors to the final target material.
- Inverse Hull Energy (ΔE_inv): The energy difference between your target material and its nearest competing stable phases on the convex hull. A large, negative inverse hull energy means the target is the most stable phase in its local compositional space, making it more selective against impurities [35].
FAQ 4: Can I use computational methods to predict the best precursors? Yes. Using thermodynamic databases (e.g., the Materials Project), you can construct interface reaction hulls for potential precursor pairs. This allows you to calculate and compare the reaction energy and selectivity metrics for different synthesis routes before any lab work [4].
Troubleshooting Guides
Problem: Persistent Impurity Phases
Possible Causes and Solutions:
- Cause 1: Formation of low-energy intermediates.
- Solution: Redesign your precursor strategy. Instead of starting from simple binaries or stable compounds, consider synthesizing a higher-energy, metastable precursor. This precursor should be chosen so that its reaction path to the final product has a large driving force and avoids stable intermediate compounds on the phase diagram [35].
- Cause 2: The reaction pathway intersects multiple competing phases.
- Solution: Use computational selectivity metrics to navigate the phase diagram. Choose a precursor pair whose compositional tie-line (the straight line connecting the two precursors in composition space) goes through as few competing phases as possible. The ideal path has the target phase as the lowest energy point [35] [4].
Problem: Slow or Incomplete Reaction Kinetics
Possible Causes and Solutions:
- Cause: Insufficient thermodynamic driving force.
- Solution: Select higher-energy (less stable) precursors. The larger the energy difference between the precursors and the products, the greater the driving force for the reaction, leading to faster transformation kinetics. Avoid using precursors that are already very stable on the convex hull [35].
Quantitative Data and Principles
The following table summarizes the key principles for precursor selection, prioritized from most to least critical, based on large-scale experimental validation [35].
Table 1: Principles for Selecting Precursors to Maximize Driving Force and Selectivity
| Principle | Description | Rationale |
|---|---|---|
| 1. Target at the Deepest Point | The target material should be the lowest energy point on the convex hull along the reaction path between the two precursors. | Ensures the thermodynamic driving force for nucleating the target is greater than for any competing phase. |
| 2. Maximize Inverse Hull Energy | Prioritize reactions where the target has a large, negative inverse hull energy. | A large inverse hull energy makes the target phase highly selective against impurities, even if they momentarily form. |
| 3. Initiate with Two Precursors | Whenever possible, design the reaction to begin between only two precursors. | Minimizes simultaneous pairwise reactions that can lead to multiple, difficult-to-remove impurity phases. |
| 4. Use High-Energy Precursors | Choose metastable or less stable compounds as precursors. | Maximizes the overall reaction energy (ΔE), providing a larger driving force for fast kinetics. |
| 5. Minimize Competing Phases | Select a precursor pair whose compositional tie-line intersects as few other stable phases as possible. | Reduces the opportunity for the reaction to be sidetracked by forming undesired by-products. |
Experimental Protocols
Protocol: Robotic High-Throughput Validation of Precursor Principles
This protocol is adapted from a large-scale study that validated thermodynamic precursor selection principles for 35 quaternary oxides using an automated synthesis lab [35].
1. Objective: To experimentally test and compare the phase purity of target materials synthesized from traditional precursors versus precursors selected via thermodynamic design principles.
2. Research Reagent Solutions
Table 2: Essential Materials for Robotic Synthesis Validation
| Item | Function in the Experiment |
|---|---|
| Robotic Inorganic Materials Synthesis Laboratory | Automates powder handling, ball milling, oven firing, and X-ray characterization for high-throughput, reproducible experiments. |
| Precursor Library | A collection of 28 unique precursor powders spanning 27 elements, including binary oxides, carbonates, and pre-synthesized metastable compounds. |
| X-ray Diffractometer (XRD) | Integrated into the robotic workflow for high-throughput phase identification and quantification of reaction products. |
| Computational Thermodynamics Database | (e.g., Materials Project): Source of formation enthalpies to construct convex hulls and calculate reaction energies for precursor selection. |
3. Methodology:
Step 1: Computational Precursor Selection
- For a given target material (e.g., a quaternary oxide), enumerate all possible precursor combinations.
- Using formation energies from a thermodynamic database, construct the relevant pseudo-ternary or higher-dimensional phase diagram (convex hull).
- For each potential precursor pair, calculate the reaction energy (ΔE) and the inverse hull energy (ΔE_inv) of the target.
- Rank the precursor pairs based on the principles in Table 1, prioritizing those where the target is the deepest point on the hull and has the largest inverse hull energy.
Step 2: Robotic Synthesis
- The robotic system takes the selected precursor powders (e.g., both traditionally selected and thermodynamically designed ones).
- It automatically performs weighing, transfers powders to reaction vessels, and adds grinding media.
- The precursors are ball-milled to ensure intimate mixing and then fired in a furnace according to a pre-programmed temperature profile.
Step 3: Automated Characterization and Analysis
- The robotic system transfers the reacted powder to an X-ray diffractometer for analysis.
- The resulting XRD patterns are analyzed for phase purity. The success of a synthesis is determined by the strength and purity of the diffraction signals corresponding to the target phase.
4. Workflow Visualization
Protocol: Computational Assessment of Reaction Selectivity
This protocol describes how to use the "interface reaction hull" to calculate selectivity metrics for a proposed solid-state reaction [4].
1. Objective: To compute the primary and secondary competition metrics for a reaction between two proposed precursors, providing a thermodynamic measure of its selectivity.
2. Methodology:
Step 1: Gather Thermodynamic Data
- Acquire the Gibbs free energies of formation, ΔG_f(T), for all known phases in the chemical system containing your precursors and target. This can be done using first-principles data (e.g., from the Materials Project) corrected for temperature with machine learning models or experimental data [4].
Step 2: Construct the Interface Reaction Hull
- For a pair of solid precursors, plot a "quasibinary" phase diagram. The x-axis is the atomic mixing ratio between the two precursors, and the y-axis is the Gibbs free energy of the system.
- The vertices of this hull represent the most stable phases (or phase mixtures) at any given composition.
Step 3: Identify the Target and Competing Phases
- Locate your target phase on this hull. If it is a vertex, it is thermodynamically stable.
- Identify all other stable vertices (competing phases) on the hull.
Step 4: Calculate Selectivity Metrics
- Primary Competition: Analyze the driving force to form the target phase versus the driving force to form the first competing impurity phase that would appear along the reaction path.
- Secondary Competition: Model the interfaces that form after the target phase nucleates (e.g., between the target and an unreacted precursor) to see if additional impurity phases are thermodynamically favored at those secondary interfaces.
3. Workflow Visualization
The Interplay of Interfacial Energy and Nucleation Rates
Frequently Asked Questions (FAQs)
FAQ 1: What determines whether a solid-state reaction will be under thermodynamic or kinetic control? The outcome is determined by the difference in thermodynamic driving force (∆G) between competing potential products. When the driving force to form one product exceeds that of all other competing phases by ≥60 meV/atom, the reaction falls under thermodynamic control, and the initial product formed can be reliably predicted. When multiple phases have comparable driving forces (below this threshold), the reaction enters a kinetic control regime, where factors like interfacial energy and structural templating dominate the outcome [3].
FAQ 2: How does interfacial energy specifically affect my nucleation rates?
Interfacial energy (γ) has an exponential impact on nucleation rates, as shown in the classical nucleation theory equation [3]:
Q = A * exp(-16πγ³ / (3n²kₜT∆G²))
Where Q is the nucleation rate. Because γ is raised to the third power, even small reductions in interfacial energy can lead to dramatic increases in nucleation rates. For example, pre-adsorbing human serum albumin on hydroxyapatite surfaces reduced the induction period for calcium oxalate monohydrate nucleation from 230 minutes to 65 minutes—a ~72% reduction—by modifying the interfacial properties [37].
FAQ 3: Can I predict which phase will form first in my solid-state synthesis? Yes, but only for a specific subset of reactions. Recent high-throughput studies indicate that approximately 15% of possible solid-state reactions (representing 105,652 reactions analyzed) fall within the regime of thermodynamic control where initial product formation can be predicted using the max-ΔG theory. For these reactions, the first product formed will be the one with the largest compositionally unconstrained thermodynamic driving force, regardless of reactant stoichiometry [3].
FAQ 4: What practical methods can I use to reduce interfacial energy in my experiments?
- Surface pre-treatment: Utilize surfaces with high Lewis base parameters (γ⁻) and low interfacial tension with water (γ_SL), which have been shown to be more effective for nucleation [37]
- Heterogeneous nucleation: Design reactions to occur on surfaces or impurities rather than homogeneously, as the nucleation barrier for heterogeneous nucleation is significantly lower [38]
- Structural templating: Leverage crystalline precursors with structural similarity to your desired phase to reduce nucleation barriers [3]
FAQ 5: Why does increasing thermodynamic driving force not always increase my reaction rate? The relationship between thermodynamic driving force (∆G) and reaction rate is non-linear. Recent research has revealed that in highly exergonic regimes, further increases in driving force offer diminishing returns for rate improvement, and control shifts to structural factors. The global kinetic-thermodynamic relationship is characterized by three physically meaningful parameters: a minimum preorganisational barrier (Emin), a reaction symmetry offset (Eeq), and a kinetic curvature factor (θ) [17].
Troubleshooting Guides
Problem: Unexpected Intermediate Phases Forming
Symptoms:
- Formation of metastable intermediates not predicted by equilibrium phase diagrams
- Failure to form desired target phase despite sufficient driving force
- Inconsistent results between different precursor batches
Diagnosis and Solution:
| Step | Procedure | Expected Outcome |
|---|---|---|
| 1 | Calculate driving force difference between competing phases using computed data (e.g., Materials Project) | Identify if your reaction falls in kinetic (∆G difference <60 meV/atom) or thermodynamic control regime (∆G difference ≥60 meV/atom) [3] |
| 2 | For kinetic control regime, analyze structural similarities between precursors and potential products | Identify which phases likely benefit from reduced nucleation barriers via structural templating [3] |
| 3 | Modify synthesis parameters to increase selectivity: increase temperature, alter precursor morphology, or introduce dopants | Shift reaction toward thermodynamic control or selectively promote desired phase through kinetic pathways |
Preventive Measures:
- Screen potential precursor pairs using computational databases before experimental work
- When possible, select precursor systems where your target phase has a significantly higher driving force than competitors
- Use in situ characterization (XRD, spectroscopy) to monitor early-stage phase formation
Problem: Inconsistent Nucleation Rates Between Experiments
Symptoms:
- Variable induction periods for phase formation
- Poor reproducibility between apparently identical experiments
- Sensitivity to minor changes in precursor preparation
Diagnosis and Solution:
| Factor | Investigation Method | Corrective Action |
|---|---|---|
| Interfacial Energy Variations | Characterize surface properties of precursors (contact angle, surface energy) | Standardize precursor synthesis and storage conditions; implement surface modification if necessary [37] |
| Heterogeneous Nucleation Sites | Analyze impurity content and surface morphology of precursors | Use purified precursors or intentionally introduce controlled nucleation sites [38] |
| Thermal History | Review heating rates and temperature profiles | Implement standardized thermal protocols with controlled heating rates [3] |
Experimental Verification:
- Perform control experiments with systematically varied interfacial properties
- Use constant composition kinetics techniques to quantitatively compare nucleation rates [37]
- Characterize solid phases during reaction using XRD, SEM, and diffuse reflectance FTIR [37]
Quantitative Data Tables
Table 1: Thermodynamic Control Threshold in Solid-State Reactions
| Parameter | Value | Significance | Experimental Basis |
|---|---|---|---|
| Minimum ΔG difference for thermodynamic control | ≥60 meV/atom | Predictable initial product formation | In situ XRD of 37 reactant pairs [3] |
| Percentage of predictable reactions | 15% (105,652 reactions) | Scope of max-ΔG theory applicability | Analysis of Materials Project data [3] |
| Temperature dependence of nucleation rate | ∝ T/η (near T_m) | Strong temperature sensitivity near melting point | Classical nucleation theory [38] |
Table 2: Interfacial Energy Effects on Experimental Nucleation Parameters
| System | Interfacial Modification | Induction Period Change | Growth Rate Change |
|---|---|---|---|
| Calcium oxalate on hydroxyapatite | None (pure HAP) | 230 minutes (reference) | 0.56 × 10⁻⁷ mol/(min·m²) [37] |
| Calcium oxalate on albumin-treated HAP | Albumin immobilization | 65 minutes (~72% reduction) | 2.14 × 10⁻⁷ mol/(min·m²) [37] |
Experimental Protocols
Protocol 1: Determining Thermodynamic vs. Kinetic Control in Solid-State Reactions
Objective: Establish whether a given precursor system will react under thermodynamic or kinetic control.
Materials:
- High-purity precursor powders
- Computational access to Materials Project database
- In situ XRD capability (synchrotron or laboratory source)
Methodology:
- Calculate Driving Forces: For your precursor pair, compute the compositionally unconstrained ΔG for all possible ternary reaction products using DFT-computed formation energies [3]
- Identify Competing Phases: Rank potential products by driving force and calculate differences between competing phases
- Experimental Validation:
- Mix precursors in appropriate stoichiometry (typically 1:1 cation ratio)
- Heat to target temperature (e.g., 700°C) at controlled rate (10°C/min)
- Perform in situ XRD measurements every 30 seconds during heating
- Identify first crystalline phase formed
- Correlation Analysis: Compare experimentally observed first phase with predicted phase from max-ΔG theory
Interpretation:
- If ΔG difference ≥60 meV/atom and observed phase matches prediction → Thermodynamic control
- If ΔG difference <60 meV/atom and observed phase doesn't match prediction → Kinetic control
Protocol 2: Quantifying Nucleation Rate Modifications from Interfacial Energy Changes
Objective: Systematically measure how surface modifications affect nucleation kinetics.
Materials:
- Constant composition kinetics apparatus
- Contact angle measurement capability
- XRD, SEM, and diffuse reflectance FTIR characterization
Methodology:
- Surface Characterization:
- Measure contact angles using sessile drop method
- Calculate Lewis base parameters (γ⁻) and interfacial tension with water (γ_SL) [37]
- Controlled Nucleation Experiments:
- Prepare solutions with fixed supersaturation (e.g., σ = 1.44 for COM) [37]
- Introduce substrates with systematically varied interfacial properties
- Monitor induction periods until detectable nucleation occurs
- Measure initial growth rates following nucleation
- Phase Characterization:
- Analyze solid phases by XRD to confirm identity
- Examine morphology by SEM
- Verify composition by diffuse reflectance FTIR
Data Analysis:
- Plot induction period versus interfacial parameters
- Correlate growth rates with measured surface properties
- Fit data to classical nucleation theory equations to extract interfacial energy changes
Research Reagent Solutions
Table 3: Essential Materials for Interfacial Energy and Nucleation Studies
| Reagent/Material | Function | Application Notes |
|---|---|---|
| LiOH & Li₂CO₃ | Lithium sources for solid-state reactions | Demonstrate different thermodynamic behavior despite similar chemistry; LiOH provides larger driving forces [3] |
| Nb₂O₅ | Niobium source for model reactions | Forms three stable ternaries (LiNb₃O₈, LiNbO₃, Li₃NbO₄) with distinct nucleation behaviors [3] |
| Human Serum Albumin | Surface modification agent | Lowers interfacial energy when immobilized on surfaces; significantly reduces nucleation induction periods [37] |
| Hydroxyapatite (HAP) | Model substrate for heterogeneous nucleation | Well-characterized surface for studying interfacial energy effects on mineralization [37] |
Visualization Diagrams
Temperature and Processing Conditions to Favor Thermodynamic Products
In solid-state synthesis, the final product of a reaction is often determined by the delicate balance between kinetic and thermodynamic control. A thermodynamic product is the most stable phase under the reaction conditions, possessing the lowest Gibbs free energy. In contrast, a kinetic product forms faster because it has a lower activation energy barrier, even if it is less stable. The pathway taken depends heavily on the processing conditions, especially temperature. This guide provides troubleshooting and best practices for researchers aiming to steer solid-state reactions toward the desired thermodynamic product, thereby increasing the yield and purity of target materials in drug development and materials science.
Understanding Kinetic vs. Thermodynamic Control
FAQ: What is the fundamental difference between a kinetic and a thermodynamic product?
- Kinetic Product: This is the product that forms the fastest. Its formation is favored under conditions that do not allow for reversibility, typically at lower temperatures. The reaction pathway with the lowest activation energy is favored, leading to the product that is easiest to form, though not necessarily the most stable [39].
- Thermodynamic Product: This is the most stable product, with the lowest overall energy. Its formation is favored when the reaction is reversible and given enough time (or thermal energy) to reach equilibrium. This typically occurs at higher temperatures [39].
A useful analogy is purchasing a can opener. Buying a cheap, flimsy one is the "kinetic" decision—it requires less initial investment (energy). Buying an expensive, durable one is the "thermodynamic" decision—it costs more upfront but is more stable and reliable in the long run [39].
Visualizing the Reaction Pathways
The diagram below illustrates the energy landscape for a system that can form both kinetic and thermodynamic products.
Reaction Coordinate Diagram for Product Formation. This diagram shows that the kinetic product forms via a pathway with a lower activation energy (Eₐ), while the thermodynamic product is more stable but requires overcoming a higher energy barrier [40] [39].
The Role of Temperature and Processing Conditions
FAQ: How does temperature influence which product I obtain?
Temperature is the primary lever for controlling the reaction outcome.
- Low Temperature (Kinetic Control): At lower temperatures, molecules lack the thermal energy to overcome the higher activation barrier leading to the thermodynamic product. More importantly, they cannot re-cross the barrier once a product is formed. This makes the reaction irreversible, freezing the system in the kinetically favored state [39].
- High Temperature (Thermodynamic Control): At higher temperatures, molecules have sufficient energy to surmount all energy barriers. The reaction becomes reversible, allowing the system to sample all possible states. Over time, the reaction will equilibrate toward the most stable (thermodynamic) product [39].
The table below summarizes key temperature-related parameters to favor thermodynamic products.
| Parameter | Condition to Favor Thermodynamic Product | Rationale |
|---|---|---|
| Reaction Temperature | High temperature | Provides energy to overcome higher activation barriers and enables reversibility [39]. |
| Annealing Time | Long duration (hours to days) | Allows sufficient time for the system to reach thermodynamic equilibrium [16]. |
| Cooling Rate | Slow cooling (annealing) | Prevents the system from being quenched into a metastable kinetic state. |
Troubleshooting Common Experimental Issues
Problem: Reaction Yields a Metastable Impurity Instead of the Target Phase
- Potential Cause: The reaction is under kinetic control due to insufficient temperature or time.
- Solutions:
- Increase Synthesis Temperature: Raise the furnace temperature in a step-wise manner to provide the necessary thermal energy for the thermodynamic phase to nucleate and grow.
- Lengthen Annealing Time: Hold the sample at the target temperature for a longer duration (e.g., 12-48 hours) to allow atomic rearrangements.
- Use a "Chemical Oven": In Self-propagating High-temperature Synthesis (SHS), a highly exothermic mixture can be added to provide intense, localized heat, pushing the system toward the thermodynamic product [41].
Problem: The Desired Thermodynamic Product Forms but is Impure
- Potential Cause: Competing side reactions consume precursors, forming stable, inert intermediates that block the path to the final target [16].
- Solutions:
- Optimize Precursor Selection: Choose precursors that minimize the formation of stable, competing byproducts. Algorithms like ARROWS3 are designed specifically for this, identifying precursors that retain a large thermodynamic driving force to form the target [16].
- Use a Flux or Mineralizer: A small amount of a flux material can be added to enhance solid-state diffusion and promote the growth of the stable crystalline phase.
Problem: Inconsistent Results Between Batches
- Potential Cause: Inhomogeneous precursor mixtures or slight variations in temperature profiles.
- Solutions:
- Mechanical Activation: Use high-energy ball milling to create nanocomposite precursor particles. This ensures intimate mixing and can lower the effective activation energy for the desired reaction [41].
- Strict Temperature Profiling: Calibrate furnaces and use controlled heating rates with precise temperature holds.
Methodologies for Favouring Thermodynamic Products
Protocol: High-Temperature Solid-State Synthesis
Objective: To synthesize a phase-pure thermodynamic product.
- Precursor Preparation: Weigh out high-purity, finely powdered reactants. For enhanced reactivity, use a planetary mill for high-energy ball milling to create a homogenous mixture [41].
- Pelletization: Press the powder mixture into a pellet using a hydraulic press. This increases inter-particle contact, improving solid-state diffusion.
- Reaction Setup: Place the pellet in a crucible (e.g., alumina, platinum) suitable for the required temperature and atmosphere.
- Thermal Treatment:
- Heating Rate: Use a moderate heating rate (e.g., 5-10°C/min) to the target temperature.
- Reaction Temperature: Heat to a high temperature, often 60-80% of the melting point of the target product (following Tamman's rule).
- Annealing Time: Hold at the target temperature for an extended period (e.g., 24 hours).
- Cooling Rate: Cool the sample slowly (e.g., 1-2°C/min) to room temperature to prevent thermal stress and formation of metastable phases.
- Product Characterization: Analyze the final product using X-ray Diffraction (XRD) to confirm phase purity.
Protocol: Leveraging Thermodynamic Driving Force with Precursor Selection
Objective: To select precursors that avoid the formation of stable intermediates and maximize the driving force to form the target.
This methodology is based on the ARROWS3 algorithm [16], which can be guided by the following steps:
Workflow for Optimizing Precursor Selection. This iterative process uses experimental failure data to learn which precursor combinations avoid the formation of inert intermediates, thereby preserving the thermodynamic driving force (ΔG) for the target material [16].
The Scientist's Toolkit: Essential Research Reagent Solutions
The table below lists key materials and their functions in solid-state synthesis aimed at thermodynamic products.
| Reagent/Material | Function in Synthesis |
|---|---|
| High-Purity Oxide/Carbonate Precursors | Provides the primary cation sources for ceramic materials. High purity minimizes interference from impurities. |
| Inert Salt (e.g., NaCl) | Acts as a high-temperature flux or a diluent to moderate the reaction temperature by absorbing heat upon melting [41]. |
| High-Energy Ball Mill | Used for mechanical activation of precursors, creating nanocomposites that react more readily and completely [41]. |
| Planetary Centrifuge | Enables the creation of an artificial high-gravity environment, which can be used to control the phase composition of SHS products [41]. |
| Chemical Oven Mixture | A highly exothermic mixture placed around the sample in SHS to provide additional, sustained heat for the reaction [41]. |
Validating Predictions and Comparing Thermodynamic vs. Kinetic Outcomes
Experimental Validation via In Situ X-ray Diffraction (XRD)
Frequently Asked Questions (FAQs)
FAQ 1: What is the primary advantage of using in situ XRD over ex situ methods for studying solid-state reactions? In situ XRD allows for the direct observation of structural changes, phase transitions, and reaction intermediates as they occur under non-ambient conditions. This is crucial for capturing metastable intermediates and understanding the real-time reaction pathway, which can be missed when analysis is performed after the fact (ex situ) [42]. For research on increasing thermodynamic driving force, it enables direct validation of whether the desired high-driving-force phase forms first, as predicted by theory [3].
FAQ 2: My in situ XRD data shows unexpected peak shifts during a heating experiment. What is the most likely cause? This is most commonly caused by thermal height expansion of the sample holder. As the temperature increases, the sample holder expands, moving the sample out of the optimal focusing plane of the X-ray beam. This causes systematic peak shifts [42]. Most modern systems require a Z-stage to automatically correct for this expansion. It is recommended to calibrate the thermal height expansion of your specific sample holder under the same conditions (atmosphere, pressure) used for your measurements [42].
FAQ 3: Under what conditions can the thermodynamic driving force (max-ΔG) reliably predict the first product formed in a solid-state reaction? Recent in situ XRD studies have quantified a threshold for thermodynamic control. The initial product can be predicted when its driving force exceeds that of all other competing phases by ≥60 meV/atom [3]. When the driving forces of competing phases are closer than this, kinetic factors (e.g., diffusion barriers, structural templating) are more likely to determine the initial outcome [3].
FAQ 4: How do I choose between a direct heater and an environmental heater for my high-temperature in situ experiment? The choice involves a trade-off between maximum temperature and temperature homogeneity/accuracy.
| Heater Type | Best For | Advantages | Limitations |
|---|---|---|---|
| Direct Heater | Very high temperatures (up to 2300°C), fast heating rates [42] | Can achieve extreme temperatures; compatible with cryostats for cooling in some designs [42] | Less homogeneous temperature distribution; measured temperature may deviate from actual sample surface temperature [42] |
| Environmental Heater | Homogeneous temperature distribution, accurate temperature measurement, sample spinning [42] | Excellent temperature homogeneity; minimal temperature deviation between sensor and sample; allows for sample spinning to improve statistics [42] | Generally lower maximum temperature compared to direct heaters [42] |
FAQ 5: The temperature readout on my instrument does not match the true sample temperature. How can I correct for this? The difference between the displayed temperature (measured at the sample holder) and the true sample surface temperature is a known challenge. This requires a process called temperature validation [42]. Since certified standard materials for XRD are not available, researchers use commonly accepted reference materials with well-known thermal properties (e.g., phase transition points) to create a validation curve for their specific instrument and setup [42].
Troubleshooting Guides
Issue 1: Poor Powder Statistics or Preferred Orientation in Patterns
Problem: XRD patterns show deviations from expected relative peak intensities, making phase identification and quantification difficult.
Possible Causes and Solutions:
- Cause: Preferred orientation in the powder sample, where crystallites align non-randomly.
- Solution: Use an environmental heater with a sample spinner if possible. This rotates the sample during measurement, averaging out orientation effects and producing more representative intensities [42].
- Solution: For direct heaters, ensure the powder is finely ground and packed as randomly as possible.
- Cause: The sample is not remaining in the ideal focusing plane during the experiment (e.g., due to uneven thermal expansion or sample warping).
- Solution: Ensure the thermal height expansion of the sample holder has been properly calibrated and that a Z-stage is actively correcting the sample position [42].
Issue 2: Failure to Observe an Expected Phase Transition
Problem: A known phase transition is not detected at its expected temperature in an in situ experiment.
Possible Causes and Solutions:
- Cause: The reaction or transition is kinetically hindered (too slow) at the selected heating rate.
- Solution: Incorporate isothermal holds at key temperatures into your heating protocol to allow sufficient time for the transformation to occur and be detected.
- Cause: The temperature validation is inaccurate for your current setup, meaning the true sample temperature is different from what you think.
- Solution: Perform temperature validation using a standard material under conditions as close as possible to your actual experiment (same heating rate, atmosphere, sample geometry) [42].
- Cause: The detection limit of the diffractometer is insufficient for a small amount of the new phase.
- Solution: Use longer counting times or a higher X-ray flux (if available) to improve signal-to-noise and detect minor phases.
Issue 3: Unanticipated Phases Forming as First Intermediates
Problem: The initial phase that forms does not align with the thermodynamic prediction (max-ΔG), even when the driving force seems sufficient.
Possible Causes and Solutions:
- Cause: The reaction is operating in the kinetic control regime, where multiple phases have a comparable driving force (e.g., within ~60 meV/atom of each other) [3].
- Solution: Analyze the driving forces for all competing phases. If they are similar, kinetic factors like structural similarity (templating) or low diffusion pathways will dominate. The experimental outcome in this regime may not be predictable by thermodynamics alone [3].
- Cause: The interfacial energy (γ) for the nucleation of a different phase is significantly lower, for instance, due to structural templating from a reactant.
- Solution: Consider the structural relationships between the reactants and potential products. Phases that share a similar structure to a reactant may nucleate more easily, even if they are not the thermodynamic ground state [3].
Experimental Protocols
Protocol 1: Temperature Validation for a Non-Ambient XRD Attachment
Objective: To establish a correlation between the instrument's temperature readout and the actual temperature at the sample surface.
Materials:
- Non-ambient XRD attachment (e.g., furnace or cryostat)
- Reference materials with well-characterized, sharp phase transitions (e.g., certain pure metals or salts with known melting points or solid-state transitions)
- Standard sample holder
Methodology:
- Preparation: Lightly load a thin, even layer of the reference material onto the sample holder to minimize temperature gradients.
- Initial Alignment: At room temperature, align the height of the sample holder to the goniometer center using the primary beam method (finding the position where the beam intensity is halved) [42].
- Data Collection: Program a temperature ramp that passes through the known transition point of the reference material. Collect XRD patterns continuously at a rapid rate (e.g., every few degrees or seconds).
- Analysis: Identify the temperature on the instrument readout at which the diffraction pattern of the reference material changes (e.g., peaks disappear upon melting or new peaks appear in a solid-state transition).
- Validation Curve: Plot the known transition temperature against the instrument readout temperature. This curve is used to correct future experimental temperatures.
Protocol 2: In Situ Monitoring of a Solid-State Reaction with High Thermodynamic Driving Force
Objective: To validate whether the initial product formed in a solid-state reaction is the one with the largest thermodynamic driving force (max-ΔG).
Materials:
- Precursor powders (e.g., LiOH and Nb₂O₅ for Li-Nb-O systems) [3]
- Non-ambient XRD attachment with temperature control and a validated temperature profile
- Z-stage for automatic height correction
Methodology:
- Sample Preparation: Mix precursor powders thoroughly in the desired stoichiometry. For interfacial reactions, this can include gentle mixing to create particle-particle contacts without fully homogenizing.
- Experimental Setup: Load the mixed powder into the non-ambient sample holder. Ensure the Z-stage is enabled and the correct thermal expansion calibration is selected [42].
- In Situ Reaction: Program a temperature ramp (e.g., 10°C/min) to the desired reaction temperature. Continuously collect XRD patterns throughout the heating and any subsequent isothermal hold.
- Data Analysis:
- Identify the first new crystalline phase to appear in the XRD patterns.
- Compare this phase against a list of all possible products, ranked by their computed compositionally unconstrained reaction energy (ΔG, in meV/atom) [3].
- Determine if the observed first product is the one with the max-ΔG and if its driving force is ≥60 meV/atom greater than its nearest competitor, confirming thermodynamic control [3].
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function / Relevance |
|---|---|
| Environmental Heater | Provides a homogeneous temperature environment around the sample, enabling accurate temperature measurement and minimizing gradients that can complicate kinetic and thermodynamic analysis [42]. |
| Z-Stage | An automatic height correction system that is essential for non-ambient experiments. It counteracts thermal expansion of the sample holder, keeping the sample in focus and preventing artifactual peak shifts [42]. |
| Reference Materials for Temperature Validation | Materials with known, sharp phase transitions (e.g., melting points) are used to correlate the instrument's temperature readout with the true sample temperature, a critical step for quantitative work [42]. |
| Strip Heater (Direct Heater) | Enables experiments at very high temperatures (up to 2300°C) and with fast heating rates, useful for studying high-temperature synthesis or materials with very high melting points [42]. |
| Gas/Atmosphere Control System | Allows the study of reactions under specific atmospheres (e.g., inert, oxidizing, reducing) or controlled relative humidity, which is critical for simulating real-world conditions or investigating hydration reactions [42]. |
Workflow and Conceptual Diagrams
In Situ XRD Experimental Workflow
Thermodynamic vs Kinetic Control Regime
Threshold for Thermodynamic Control in Solid-State Reactions
The following data is derived from a 2024 study that used in situ characterization on 37 pairs of reactants to validate the principles of thermodynamic control [3].
| Parameter | Value | Significance |
|---|---|---|
| Threshold for Thermodynamic Control | ≥60 meV/atom | The initial product formation can be predicted when its driving force exceeds that of all other competing phases by this amount [3]. |
| Reactions in Thermodynamic Control | 15% | The percentage of possible reactions analyzed from the Materials Project database that fall within this predictable regime [3]. |
| Key Technique for Validation | In situ XRD | Enabled the identification of the first crystalline phase to form during heating, allowing for direct comparison with computed reaction energies [3]. |
Comparison of Heat Transfer Mechanisms in Non-Ambient XRD
Understanding heat transfer is critical for interpreting temperature-dependent data [42].
| Mechanism | Process | Key Dependence in NA-XRD |
|---|---|---|
| Conduction | Heat transfer through atomic motion within a solid [42] | Thermal conductivity of the sample and sample holder; critical for temperature accuracy in direct heaters [42]. |
| Convection | Heat transfer via movement of a fluid (gas/liquid) [42] | Pressure and type of gas in the sample chamber; low molecular weight gases have higher conductivity [42]. |
| Radiation | Heat transfer via electromagnetic waves [42] | Strongly dependent on temperature (T⁴); becomes dominant at very high temperatures [42]. |
Within the context of increasing thermodynamic driving force in solid-state reactions, the selection of lithium precursors plays a critical role in determining reaction kinetics, product purity, and overall energy efficiency. This technical support center document provides a comparative analysis of lithium hydroxide (LiOH) and lithium carbonate (Li2CO3) as reactants in materials synthesis, with particular emphasis on solid-state battery electrolyte fabrication and lithium extraction processes. The thermodynamic and kinetic behaviors of these precursors differ substantially, influencing nucleation barriers, reaction pathways, and ultimate material performance. Understanding these differences is essential for researchers aiming to optimize solid-state reactions for enhanced ionic conductivity, phase purity, and processing efficiency in advanced energy storage systems. The following sections provide detailed experimental protocols, troubleshooting guidance, and quantitative comparisons to assist researchers in selecting and optimizing lithium precursor usage in their specific experimental contexts.
Quantitative Data Comparison
Table 1: Comparative Properties of Lithium Reactants in Solid-State Systems
| Property | Li₂CO₃ | LiOH | Measurement Conditions | Impact on Solid-State Energetics |
|---|---|---|---|---|
| Solubility Trend | Decreases with temperature [43] | Increases with temperature (typical behavior) | Aqueous systems, 20-90°C | Affects precursor mobility and reactive interfaces |
| Conductivity Achievement | 1.28 × 10⁻⁵ S·cm⁻¹ [44] | Typically higher in final products | LLZAO pellets, 1175°C, 2h sintering [44] | Indirect impact through final product density and purity |
| Extraction Efficiency | 90.4% from spodumene [45] | Data limited as product | With Na₂CO₃ + Al₂O₃ additive [45] | Thermodynamic driving force with additives |
| Reactive Byproduct Risk | Li₂SiO₃ (traps Li⁺) [45] | Not typically reported | α-spodumene direct processing [45] | Competes with desired reaction pathway |
| Role in Sintering | Intrinsic sintering aid [44] | Not typically used | SSRS method, 1100-1200°C [44] | Enhances densification and grain growth |
Table 2: Thermodynamic Driving Force Optimization Strategies
| Challenge | Li₂CO₃-Based Solution | LiOH-Based Solution | Thermodynamic Principle |
|---|---|---|---|
| Limited Lithium Yield | Add Al₂O₃ to trap silicate anions [45] | Not specifically addressed in search results | Shifts equilibrium by removing competitive phase |
| Low Final Density | One-step SSRS with 10wt% excess Li₂CO₃ [44] | Not specifically addressed in search results | Creates liquid phase enhancing diffusion |
| Phase Impurity | Sinter at 1175°C for 2 hours [44] | Not specifically addressed in search results | Optimizes kinetics for pure cubic phase formation |
| Common Ion Suppression | Avoid high LiCl concentrations [43] | Not specifically addressed in search results | Le Chatelier's principle governing solubility |
Experimental Protocols & Methodologies
Solid-State Reactive Sintering with Li₂CO₃
Objective: Fabricate Al-doped Li₇La₃Zr₂O₁₂ (LLZAO) solid electrolytes using Li₂CO₃ as both lithium source and intrinsic sintering aid [44].
Materials:
- Li₂CO₃ powder (additional 10 wt% beyond stoichiometry)
- La₂O₃ (pre-dried to remove moisture)
- ZrO₂
- Al₂O₃ (30 mol% dopant source)
Procedure:
- Stoichiometric Calculation: Calculate required masses based on desired product composition of Al-doped LLZO.
- Powder Mixing: Combine all powders including excess Li₂CO₃ in appropriate solvent (e.g., ethanol or acetone) and mill for 12-24 hours using zirconia balls to ensure homogeneous mixing.
- Binder Removal: Dry the mixed slurry and remove organic binders at 500-600°C for 1-2 hours if used.
- Pellet Formation: Uniaxially press the mixed powder at 100-300 MPa in a die of desired diameter.
- Reactive Sintering: Heat pellets in alumina crucibles to 1175°C for 2 hours in air atmosphere with heating and cooling rates of 3-5°C/min [44].
- Characterization: Analyze phase purity by XRD, microstructure by SEM, and ionic conductivity by electrochemical impedance spectroscopy.
Critical Parameters:
- Maintain precise temperature control (±5°C) at 1175°C for optimal results
- Use excess Li₂CO₃ (10 wt%) to compensate for lithium volatilization
- Ensure homogeneous mixing to avoid secondary phase formation
Direct Lithium Extraction from α-Spodumene Using Solid-State Reactions
Objective: Extract lithium directly from α-spodumene ore as Li₂CO₃ through solid-state reactions without acid treatment or high-temperature phase conversion [45].
Materials:
- α-spodumene ore (finely ground <100μm)
- Na₂CO₃ (anhydrous)
- Al₂O₃ (additive to prevent Li trapping)
- Distilled water (for washing)
Procedure:
- Feedstock Preparation: Grind α-spodumene to particle size <100μm to enhance reaction kinetics.
- Reactant Mixing: Combine α-spodumene with Na₂CO₃ and 10-20 wt% Al₂O₃ additive based on total mass. Mix thoroughly by dry milling or in minimal ethanol.
- Solid-State Reaction: Heat mixture to 850-900°C for 1-2 hours in air atmosphere in a muffle furnace.
- Product Isolation: Cool the reaction product to room temperature and wash with distilled water to dissolve soluble Li₂CO₃.
- Recovery: Filter the solution to remove insoluble silicates and other residues, then concentrate and recover Li₂CO₃ by crystallization [45].
Critical Parameters:
- Al₂O₃ addition is essential to prevent Li₂SiO₃ formation and increase extraction yield to >90%
- Optimize Na₂CO₃ stoichiometry (typically 20-30% excess) to ensure complete reaction
- Control washing parameters (liquid-to-solid ratio, temperature) to maximize Li₂CO₃ recovery
Troubleshooting Guides & FAQs
Common Experimental Challenges & Solutions
Table 3: Troubles Guide for Lithium Reactant-Based Solid-State Synthesis
| Problem | Possible Causes | Solutions | Applicable to |
|---|---|---|---|
| Low Lithium Extraction Yield | Li₂SiO₃ byproduct formation trapping Li⁺ | Add Al₂O₃ (10-20 wt%) to trap silicate anions [45] | Li₂CO₃ production |
| Low Ionic Conductivity in Final Pellet | Low relative density, secondary phases | Optimize sintering profile (1175°C, 2h), use excess Li₂CO₃ (10wt%) as sintering aid [44] | Li₂CO₃ in LLZO |
| Poor Reactivity in Solid-State Synthesis | Large particle size, insufficient mixing | Reduce precursor particle size, extend milling time, ensure homogeneous mixing | Both |
| Lithium Loss During Processing | Volatilization at high temperature | Use 10-15% excess lithium precursor, create sacrificial lithium atmosphere [44] | Both |
| Unwanted Byproduct Phases | Incorrect temperature profile, stoichiometry | Optimize heating rate (3-5°C/min), verify precursor stoichiometry | Both |
Frequently Asked Questions
Q1: Why does Li₂CO₃ solubility decrease with increasing temperature, and how does this affect solid-state reactions?
A: The unusual retrograde solubility of Li₂CO₃ (decreasing with temperature increase) is attributed to its strong temperature-dependent hydration behavior and ion-pair formation [43]. In solid-state reactions, this property can be advantageous during cooling and washing stages, where higher yields can be obtained at lower temperatures, but may limit precursor mobility during high-temperature processing.
Q2: What is the mechanism by which Al₂O₃ increases lithium extraction yield from spodumene?
A: Al₂O₃ acts as a thermodynamic driver by reacting with silicate anions to form stable aluminosilicates, preventing the formation of Li₂SiO₃ which traps lithium in an inactive phase. This shifts the reaction equilibrium toward Li₂CO₃ formation, increasing extraction yield from 76% to over 90% [45].
Q3: How does Li₂CO₃ function as an intrinsic sintering aid in LLZO synthesis?
A: Excess Li₂CO₃ creates a liquid phase at sintering temperatures (above its melting point) which enhances mass transport through solution-precipitation mechanisms, leading to higher density and larger grain size in the final pellet. This enables relative densities up to 90% without advanced sintering techniques [44].
Q4: What are the key advantages of the one-step solid-state reactive sintering (SSRS) method?
A: SSRS eliminates the separate powder synthesis step, reduces processing time and energy consumption, utilizes Li₂CO₃ as both reactant and sintering aid, and produces high-purity (96.3%) cubic LLZO with simplified processing [44].
Q5: Why is controlling the precipitation temperature important for Li₂CO₃ recovery from solutions?
A: Due to its retrograde solubility, Li₂CO₃ recovery is more efficient at higher temperatures where solubility is lower. Operating at 90°C instead of 20°C can significantly increase precipitation yield without additional chemicals [43].
Signaling Pathways & Workflow Diagrams
Research Reagent Solutions
Table 4: Essential Materials for Lithium Reactant Energetics Research
| Reagent/Material | Function in Research | Key Considerations | Application Examples |
|---|---|---|---|
| Li₂CO₃ (Lithium Carbonate) | Primary lithium source, sintering aid | Use 10wt% excess to compensate for Li loss; retrograde solubility [43] | LLZO solid electrolyte synthesis [44] |
| LiOH (Lithium Hydroxide) | Alternative lithium source | Different decomposition behavior; hygroscopic | Battery cathode material synthesis |
| Al₂O₃ (Aluminum Oxide) | Thermodynamic driving force enhancer | Traps silicate anions; prevents Li trapping [45] | Direct Li extraction from spodumene [45] |
| Na₂CO₃ (Sodium Carbonate) | Alkaline reactant for Li exchange | Enables direct extraction without acid [45] | Solid-state reactions with spodumene |
| α-Spodumene (LiAlSi₂O₆) | Natural lithium source | Requires additives to prevent Li₂SiO₃ formation [45] | Direct lithium extraction studies |
| La₂O₃/ZrO₂ | Oxide precursors for LLZO | Must be pre-dried; stoichiometric precision critical [44] | Garnet-type solid electrolyte synthesis |
Quantifying Predictive Success Across 37 Reactant Pairs
Frequently Asked Questions & Troubleshooting
Q1: My solid-state reaction formed an unexpected initial product, even though the computed driving force seemed favorable. What could have gone wrong? Your reaction likely fell outside the regime of thermodynamic control. The max-ΔG theory only applies when the driving force for one product exceeds all others by a threshold of ≥60 meV/atom [3]. If multiple competing phases have a comparable driving force (within this 60 meV/atom window), the outcome is determined by kinetic factors, and the initial product cannot be reliably predicted by thermodynamics alone [3]. Please verify the energy differences between all potential products in your chemical space.
Q2: What are the primary kinetic factors that can override thermodynamic predictions? When thermodynamic selectivity is low, the initial phase formed is often the one that is kinetically most accessible [3]. This can be influenced by:
- Minimal Diffusion Pathways: The product requiring the least atomic diffusion forms first [3].
- Structural Templating: Phases with structural similarity to the precursors have reduced nucleation barriers, encouraging their formation [3].
Q3: According to the research, for what percentage of possible reactions can the initial product be thermodynamically predicted? Analysis of the Materials Project data indicates that approximately 15% of all possible reactions fall within the regime of thermodynamic control where the initial product can be predicted using the max-ΔG theory [3]. This highlights a significant opportunity for predictive synthesis, but also shows that most reactions require consideration of kinetic factors.
Q4: How can I determine if my reaction is under thermodynamic or kinetic control? You must compute the compositionally unconstrained reaction energy (ΔG) for every potential product phase. The reaction is under thermodynamic control only if one product has a driving force that is ≥60 meV/atom greater than all others [3]. The table below summarizes the key differentiators between these regimes.
| Feature | Regime of Thermodynamic Control | Regime of Kinetic Control |
|---|---|---|
| Driving Force Difference | ≥ 60 meV/atom [3] | < 60 meV/atom [3] |
| Predictive Method | max-ΔG theory | Requires explicit modeling of diffusion/nucleation |
| Initial Product | The phase with the largest ΔG [3] | The kinetically most accessible phase |
| Experimental Outcome | Highly selective | Can form multiple competing intermediates |
Experimental Protocols & Methodologies
This section details the key experimental methods from the foundational study, which validated the thermodynamic threshold across 37 reactant pairs [3].
Protocol 1: In Situ Synchrotron XRD for the Li-Nb-O System
- Objective: To determine the first intermediate phase formed and compare outcomes between different Li-source reactants.
- Reactants: Two pairs were investigated:
- LiOH + Nb₂O₅
- Li₂CO₃ + Nb₂O₅ [3]
- Sample Preparation: Reactants were mixed in a 1:1 Li:Nb molar ratio [3].
- Heating Profile: Heated to 700°C at a rate of 10°C/min, held at 700°C for 3 hours, then naturally cooled to room temperature [3].
- Data Collection: XRD measurements were performed at beamline 12.2.2 at the Advanced Light Source (ALS) at a rate of two scans per minute [3].
- Data Analysis: The weight fraction of each detected phase was plotted as a function of temperature to identify the initial crystallization event.
Protocol 2: High-Throughput In Situ XRD Guided by Machine Learning
- Objective: To expand validation across a broader range of chemical spaces efficiently.
- Scale: 26 additional pairs of reactants from 12 different chemical spaces [3].
- Characterization: In situ XRD measurements.
- Automation: Machine learning (ML) algorithms were used to steer the diffractometer toward features in each pattern that facilitated the automated identification of reaction intermediates [3].
Summary of Quantitative Findings from 37 Reactant Pairs The following table consolidates the key quantitative results from the cited study.
| Experimental Metric | Value | Significance |
|---|---|---|
| Total Reactant Pairs Studied | 37 pairs | Provides a substantial experimental basis for validating the threshold [3]. |
| Chemical Spaces Probed | 12+ spaces (e.g., Li-Mn-O, Li-Nb-O) | Demonstrates the principle across multiple systems [3]. |
| Threshold for Thermodynamic Control | ≥60 meV/atom | A quantitative criterion for predicting the initial product [3]. |
| Reactions under Thermodynamic Control | 105,652 (15% of analyzed reactions) | Assessed from a large-scale analysis of the Materials Project database [3]. |
The Scientist's Toolkit: Research Reagent Solutions
The following table details key materials and their functions in conducting these solid-state synthesis experiments.
| Item | Function in the Experiment |
|---|---|
| LiOH / Li₂CO₃ | Common Li-source precursors; the choice influences the reaction's total driving force and pathway [3]. |
| Nb₂O₅ | A representative Nb-source reactant used in the Li-Nb-O test case [3]. |
| Synchrotron X-ray Source | Provides high-intensity, high-resolution X-rays for rapid in situ diffraction measurements, enabling the observation of transient intermediates [3]. |
Workflow and Conceptual Diagrams
The following diagrams illustrate the core concepts and experimental workflows using the specified color palette.
Decision Flow: Thermodynamic vs. Kinetic Control
Workflow: In Situ Reaction Analysis
Welcome to the Thermodynamic Control Technical Support Center
This support center is designed for researchers working to predict and control solid-state reaction pathways. A pivotal 2024 study analyzing 37 reactant pairs established a quantitative framework for this, identifying that approximately 15% of possible solid-state reactions fall within a predictable regime of thermodynamic control [3]. Our troubleshooting guides and FAQs below will help you diagnose and resolve common experimental challenges related to this concept.
Frequently Asked Questions (FAQs) & Troubleshooting
FAQ 1: What does it mean that only 15% of reactions are in the "thermodynamic regime"?
- Answer: This statistic, derived from an analysis of the Materials Project database, indicates that only a minority of possible solid-state synthesis reactions have a thermodynamic driving force (∆G) large enough to reliably overcome kinetic factors and dictate the first intermediate phase that forms [3]. For these select reactions, you can predict the initial product using computational thermodynamics alone, significantly streamlining synthesis planning.
FAQ 2: How can I predict if my reaction will be thermodynamically controlled?
Answer: Calculate and compare the driving force. Your reaction is likely under thermodynamic control if the driving force (∆G) to form one product exceeds that of all other competing phases by ≥60 meV/atom [3]. This 60 meV/atom threshold was validated experimentally and serves as the key criterion.
Table 1: Threshold for Thermodynamic Control in Solid-State Reactions
Parameter Value Significance Threshold Driving Force ≥ 60 meV/atom Minimum difference in ∆G required for thermodynamic control [3] Reactions in Thermodynamic Regime ~15% Percentage of predictable reactions via thermodynamics alone [3]
FAQ 3: My experiment did not form the predicted phase, despite a high driving force. What went wrong?
- Troubleshooting Guide:
- Verify the Driving Force Calculation: Re-check your computed ∆G values. Ensure you are using compositionally unconstrained, normalized (per atom) reaction energies, as stoichiometry is often irrelevant for the initial product forming at particle interfaces [3].
- Check the ∆G Difference: Confirm that the difference between your predicted phase and its closest competitor is at least 60 meV/atom. If multiple phases have a comparable driving force (difference < 60 meV/atom), you are in the kinetic control regime, and the max-∆G theory does not apply [3].
- Investigate Kinetic Factors:
- Diffusion: The predicted phase might require more long-range atomic diffusion than a kinetically favored alternative [3].
- Structural Templating (Nucleation): A phase with high structural similarity to your precursors may have a lower nucleation barrier, forming first even if it is not the thermodynamic favorite [3].
FAQ 4: What is the fundamental kinetic-thermodynamic relationship I should understand?
- Answer: The relationship is intrinsically non-linear. While classical models like the Bell-Evans-Polanyi principle or Leffler equation assume a linear relationship, a 2025 study derived a more universal, non-linear equation from microscopic reversibility [17]. This model explains that the sensitivity of the reaction rate to the thermodynamic driving force (the Brønsted slope) changes across the exergonic and endergonic spectrum. In highly exergonic regimes, further increasing the driving force yields diminishing returns on the reaction rate, and control shifts to structural factors [17].
Experimental Protocol: Determining the First Intermediate Phase
This protocol is adapted from the in-situ XRD methodology used to validate the 60 meV/atom threshold [3].
Objective: To experimentally identify the first crystalline intermediate phase formed during a solid-state reaction.
Materials and Equipment:
- High-purity precursor powders
- Mortar and pestle or ball mill for mixing
- In-situ X-ray Diffraction (XRD) setup with a heating stage (e.g., synchrotron beamline for high resolution and speed)
- or Ex-situ XRD setup and a programmable furnace with quench capability
Procedure:
- Sample Preparation: Mix reactant powders thoroughly. For in-situ XRD, load the mixture into a capillary or a stage-compatible holder. For ex-situ studies, divide the mixture into multiple identical aliquots.
- Thermal Treatment:
- In-situ Method: Place the sample in the in-situ XRD stage. Heat the sample at a controlled rate (e.g., 10°C/min) to a target temperature while collecting XRD patterns at frequent intervals (e.g., every 30 seconds or two scans per minute) [3].
- Ex-situ Method: Place each aliquot in a preheated furnace for a specific, short duration (e.g., 1, 2, 5, 10 minutes) at a target temperature. Immediately quench each sample after its allotted time to halt the reaction.
- Data Collection: Collect XRD patterns for all samples (in-situ time/temperature points or ex-situ quenched samples).
- Data Analysis: Analyze the sequence of XRD patterns. The first intermediate phase is identified as the first new set of crystalline diffraction peaks that appears, after the dehydration or decomposition of precursors, but before any other new phase [3].
The following workflow diagrams the experimental and decision process for applying these concepts.
Diagram 1: Workflow for reaction planning and validation.
Diagram 2: Relationship between driving force and reaction control.
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Materials for Solid-State Synthesis and In-Situ Analysis
| Item | Function / Relevance |
|---|---|
| High-Purity Precursor Oxides/Carbonates (e.g., Nb₂O₅, Li₂CO₃, LiOH) | Starting materials for model solid-state reactions; purity is critical to avoid spurious phase formation [3]. |
| In-Situ XRD Capillary/Heating Stage | Specialized sample holder that allows for simultaneous heating and X-ray diffraction analysis to track phase formation in real-time [3]. |
| Synchrotron Radiation Source | Provides high-intensity, high-resolution X-rays for in-situ XRD, enabling fast data collection with clear resolution of intermediate phases [3]. |
| Computational Database Access (e.g., Materials Project) | Source for ab initio computed thermodynamic data (formation energies) required to calculate the driving force (∆G) for competing reactions [3]. |
Benchmarking Computed Predictions Against Real-World Synthesis Outcomes
Frequently Asked Questions (FAQs)
General Principles
What is the "thermodynamic driving force" in solid-state synthesis? The thermodynamic driving force, often represented as ΔG (change in Gibbs free energy), is the decrease in energy when reactants transform into a product. A larger (more negative) ΔG means a reaction is more thermodynamically favorable. Recent research has established that when the driving force to form one product exceeds that of all other competing phases by at least 60 meV/atom, the reaction enters a "regime of thermodynamic control," making the initial product predictable. When multiple phases have comparable driving forces, kinetic factors often determine the outcome. [3]
When can I trust computational predictions to guide my synthesis? Computational predictions from databases like the Materials Project are most reliable when used to identify reactions within the thermodynamic control regime. Analysis shows that approximately 15% of possible reactions fall into this predictable category. For the remaining reactions, kinetic factors like diffusion barriers and structural templating can lead to outcomes that differ from computational predictions, so experimental validation is crucial. [3]
Troubleshooting Failed Syntheses
My synthesis did not yield the computationally predicted product. What went wrong? This common issue can arise from several factors. The table below summarizes potential causes and solutions.
Table: Troubleshooting Failed Syntheses
| Problem Category | Specific Issue | Recommended Solution |
|---|---|---|
| Thermodynamics | Competing phases have similar driving forces (ΔG < 60 meV/atom difference). | Use algorithmic precursor selection (e.g., ARROWS3) to find a route with a larger driving force. [46] |
| Kinetics | Low ion mobility or high nucleation barrier favors a metastable intermediate. | Increase reaction temperature or extend heating time to overcome kinetic barriers. |
| Precursor Selection | Initial intermediates consume too much driving force, leaving none for the target. | Change precursors to avoid forming highly stable, inert intermediate phases. [46] |
| Data Discrepancy | DFT-computed formation energies differ from experimental conditions. | Leverage AI models trained on both DFT and experimental data for more accurate property predictions. [47] [48] |
How can I select better precursors to avoid unwanted intermediates? The ARROWS3 algorithm provides a structured methodology for this. It first ranks potential precursor sets by their calculated ΔG to form the target. If experiments fail because of intermediate formation, the algorithm learns from these outcomes and re-ranks precursors based on the predicted driving force remaining after accounting for intermediate formation (ΔG'). This prioritizes precursor sets that avoid energy-consuming side reactions. [46]
Table: Key Questions for Precursor Selection
| Question | Consideration |
|---|---|
| Does the precursor combination have a large ΔG to form the target? | Check computational databases (e.g., Materials Project). |
| Does the reaction pathway involve highly stable intermediates? | Perform in-situ XRD on initial experiments to identify early-formed phases. [3] |
| Can a different precursor avoid a key intermediate? | Use an active learning algorithm to suggest alternatives. [46] |
Data and Methodology
What is the typical discrepancy between DFT-computed and experimental formation energies? The error between DFT computations (at 0K) and experimental measurements (at room temperature) is well-documented. The table below summarizes mean absolute errors (MAE) for major databases against experimental data.
Table: Discrepancy of DFT-Computed Formation Energies vs. Experiments [47] [48]
| Computational Database | Mean Absolute Error (eV/atom) |
|---|---|
| Open Quantum Materials Database (OQMD) | 0.083 - 0.136 eV/atom |
| Materials Project (MP) | 0.078 - 0.172 eV/atom |
| JARVIS | 0.095 eV/atom |
How can I reduce the impact of DFT-computation discrepancies in my research? Leverage Artificial Intelligence (AI) models that use deep transfer learning. These models are first trained on large DFT-computed datasets to learn the underlying patterns, then fine-tuned on smaller, more accurate experimental datasets. This approach has been shown to predict formation energy from a material's structure and composition with an MAE of 0.064 eV/atom on experimental test sets, outperforming standard DFT computations. [47]
Experimental Protocols & Workflows
Workflow 1: Determining the Regime of Thermodynamic Control
This protocol is used to validate whether a reaction is under thermodynamic control, based on in-situ characterization. [3]
Step-by-Step Guide:
- Sample Preparation: Select pairs of solid reactant powders.
- In-Situ Characterization: Load the sample into a synchrotron or laboratory X-ray diffractometer equipped with a heating stage.
- Data Collection: Heat the sample to a target temperature (e.g., 700°C) at a controlled rate (e.g., 10°C/min) while performing XRD scans frequently (e.g., every 30 seconds).
- Phase Identification: Analyze the XRD patterns to determine the first crystalline product that forms.
- Energy Calculation: Compute the compositionally unconstrained ΔG for all possible products from the initial reactants using a database like the Materials Project.
- Validation: Compare the experimental first product with the predicted product. Thermodynamic control is confirmed when the observed product is the one with the largest ΔG, and its driving force exceeds that of the next most stable competing phase by ≥60 meV/atom.
Determining the Regime of Thermodynamic Control
Workflow 2: Autonomous Precursor Selection with ARROWS3
This protocol uses the ARROWS3 algorithm to autonomously select precursors that avoid unfavorable intermediates. [46]
Step-by-Step Guide:
- Define Target: Input the composition and structure of the target material.
- Initial Ranking: The algorithm generates a list of stoichiometric precursor sets and ranks them by their computed ΔG to form the target directly.
- Experimental Testing: Perform synthesis with the top-ranked precursor sets across a range of temperatures.
- Pathway Analysis: Use in-situ or ex-situ XRD with machine-learned analysis to identify all intermediate phases that form.
- Algorithm Learning: ARROWS3 updates its model to predict which pairwise reactions lead to these intermediates.
- Re-ranking: The algorithm re-ranks all precursor sets based on the predicted driving force remaining for the target after accounting for the formation of intermediates (ΔG').
- Iteration: Steps 3-6 are repeated, with the algorithm proposing new precursor sets predicted to avoid energy-draining intermediates until the target is synthesized with high purity or all options are exhausted.
Autonomous Precursor Selection Workflow
The Scientist's Toolkit: Key Research Reagents & Materials
Table: Essential Computational and Experimental Resources
| Item Name | Function / Purpose | Key Details |
|---|---|---|
| Materials Project Database | Provides computed thermodynamic data for inorganic compounds. | Used to calculate reaction energies (ΔG) and identify stable phases. [3] [46] |
| In-Situ XRD Setup | Enables real-time monitoring of phase formation during heating. | Critical for identifying the first reaction intermediate; often uses synchrotron radiation for high resolution. [3] |
| AI Property Predictors | Predicts materials properties (e.g., formation energy) more accurately than DFT alone. | Uses deep transfer learning on combined DFT and experimental data; can achieve MAE < 0.07 eV/atom. [47] [48] |
| ARROWS3 Algorithm | Autonomously selects optimal precursors by learning from failed experiments. | Actively avoids precursors that form stable intermediates, maximizing driving force for the target. [46] |
Conclusion
Mastering the thermodynamic driving force is paramount for transitioning from empirical trial-and-error to predictive solid-state synthesis. The establishment of a quantitative 60 meV/atom threshold provides a clear criterion for identifying reactions under thermodynamic control, where the max-ΔG theory reliably predicts the initial product. For researchers in biomedicine and drug development, these principles offer a powerful framework for the rational design of crystalline APIs, multicomponent pharmaceutical materials, and excipients with targeted properties. Future directions will involve expanding these predictive models to more complex multi-component systems, integrating machine learning for high-throughput pathway discovery, and explicitly accounting for metastable phase formation. This paradigm shift towards thermodynamics-driven synthesis holds immense potential for accelerating the development of next-generation materials with tailored functionalities.
References
- 1. Gibbs (Free) Energy [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Thermodynamics/Energies_and_Potentials/Free_Energy/Gibbs_(Free)_Energy]
- 2. Gibbs Free Energy [https://www.chem1.com/acad/webtext/thermeq/TE4.html]
- 3. Quantifying the regime of thermodynamic control for solid- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11221506/]
- 4. Assessing Thermodynamic Selectivity of Solid-State ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10604012/]
- 5. 19.6: Gibbs Energy Change and Equilibrium [https://chem.libretexts.org/Bookshelves/General_Chemistry/Map%3A_General_Chemistry_(Petrucci_et_al.)/19%3A_Spontaneous_Change%3A_Entropy_and_Gibbs_Energy/19.6%3A_Gibbs_Energy_Change_and_Equilibrium]
- 6. Thermodynamic and kinetic reaction control [https://en.wikipedia.org/wiki/Thermodynamic_and_kinetic_reaction_control]
- 7. 14.3: Kinetic vs. Thermodynamic Control of Reactions [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/14%3A_Conjugated_Compounds_and_Ultraviolet_Spectroscopy/14.03%3A_Kinetic_vs._Thermodynamic_Control_of_Reactions]
- 8. Ion correlations explain kinetic selectivity in diffusion ... [https://arxiv.org/html/2501.08560v2]
- 9. Selective formation of metastable polymorphs in solid-state ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10793945/]
- 10. Can we predict materials that can be synthesised? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8178993/]
- 11. Quantifying the regime of thermodynamic control for solid ... [https://pubmed.ncbi.nlm.nih.gov/38959320/]
- 12. Revealing the solid-state reaction process among ... [https://www.sciencedirect.com/science/article/pii/S2772834X24000204]
- 13. On the theory of transient nucleation at the intermediate ... [https://www.sciencedirect.com/science/article/pii/S0375960114003284]
- 14. Solid State Reaction - an overview [https://www.sciencedirect.com/topics/chemistry/solid-state-reaction]
- 15. Phase Diagrams and Solidification (all content) [https://www.doitpoms.ac.uk/tlplib/phase-diagrams/printall.php]
- 16. Autonomous and dynamic precursor selection for solid- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10618174/]
- 17. The global kinetic–thermodynamic relationship derived from ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04829j]
- 18. ReactCA: A Cellular Automaton for Predicting Phase ... [https://arxiv.org/html/2407.19124v1]
- 19. Ab initio Modelling in Solid State Chemistry [https://www.bsc.es/news/events/ab-initio-modelling-solid-state-chemistry]
- 20. Gibbs free energy [https://en.wikipedia.org/wiki/Gibbs_free_energy]
- 21. Direct thermodynamic characterization of solid-state ... [https://pubs.rsc.org/en/content/articlelanding/2024/cp/d3cp03933a]
- 22. Solid-state reaction route [https://en.wikipedia.org/wiki/Solid-state_reaction_route]
- 23. A simple method to prepare lithium niobates (LiNbO 3 and ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884225039033]
- 24. Reactive co-sputtered Li–Nb–O thin films with tunable ionic ... [https://www.nature.com/articles/s41598-025-21732-w]
- 25. The theory/calculation behind energy-above-hull [https://matsci.org/t/the-theory-calculation-behind-energy-above-hull-how-to-calculate-for-quarternary-structures/59743]
- 26. Recent advances in battery characterization using in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10867397/]
- 27. Optimal thermodynamic conditions to minimize kinetic by- ... [https://www.nature.com/articles/s44160-023-00479-0]
- 28. Investigating Solid-State Reactions via Kinetic Control of ... [https://scholarsbank.uoregon.edu/items/3b5d9e41-9b8b-4488-9441-1184421c34bb]
- 29. Toward a paradigm of materials design: kinetic control in solid ... [https://www.chem.colostate.edu/seminars/tba-jamie-neilson-ph-d/]
- 30. Mechanisms and modelling of diffusion in solids [https://link.springer.com/article/10.1007/s43621-025-01746-0]
- 31. Calcium carbonate particles: Template-driven structural ... [https://www.sciencedirect.com/science/article/abs/pii/S030881462501698X]
- 32. Mechanism of solid-state diffusion reaction in vacuum ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884224008022]
- 33. Template-Free Manufacturing of Defined Structure and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10674349/]
- 34. Scaffolds as Structural Tools for Bone-Targeted Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6161191/]
- 35. Navigating phase diagram complexity to guide robotic ... [https://www.nature.com/articles/s44160-024-00502-y]
- 36. Thermodynamic Driving Forces and Chemical Reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7036932/]
- 37. Nucleation at surfaces: the importance of interfacial energy [https://pubmed.ncbi.nlm.nih.gov/10541263/]
- 38. Classical nucleation theory [https://en.wikipedia.org/wiki/Classical_nucleation_theory]
- 39. Thermodynamic and Kinetic Products [https://www.masterorganicchemistry.com/2012/02/09/kinetic-thermodynamic-products-can-openers/]
- 40. 6.3: A Quick Review of Thermodynamics and Kinetics [https://chem.libretexts.org/Courses/Westminster_College/CHE_261_-_Organic_Chemistry_I/06%3A_Introduction_to_Reaction_Mechanisms/6.3%3A_A_Quick_Review_of_Thermodynamics_and_Kinetics]
- 41. Self-propagating high-temperature synthesis [https://en.wikipedia.org/wiki/Self-propagating_high-temperature_synthesis]
- 42. Non-Ambient X-ray Diffraction [https://wiki.anton-paar.com/us-en/non-ambient-x-ray-diffraction/]
- 43. Solubility of Li2CO3 in Na–K–Li–Cl brines from 20 to 90 °C [https://www.sciencedirect.com/science/article/abs/pii/S0021961413002723]
- 44. One-step solid-state reactive sintering with Li2CO3 as an ... [https://www.sciencedirect.com/science/article/pii/S2949822825009335]
- 45. Energy-Saving, Acid-Free, Hard-Rock Lithium Extraction [https://als.lbl.gov/energy-saving-acid-free-hard-rock-lithium-extraction/]
- 46. Autonomous and dynamic precursor selection for solid- ... [https://www.nature.com/articles/s41467-023-42329-9]
- 47. Moving closer to experimental level materials property ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9279333/]
- 48. Enhancing materials property prediction by leveraging ... [https://www.nature.com/articles/s41467-019-13297-w]