Beyond Simple Rules: Energy Above Hull vs. Charge Balancing for Predicting Material Synthesizability in Drug Development
This article provides a comprehensive analysis of two pivotal approaches for predicting material synthesizability: the thermodynamic metric of energy above hull and the heuristic rule of charge balancing.
Beyond Simple Rules: Energy Above Hull vs. Charge Balancing for Predicting Material Synthesizability in Drug Development
Abstract
This article provides a comprehensive analysis of two pivotal approaches for predicting material synthesizability: the thermodynamic metric of energy above hull and the heuristic rule of charge balancing. Aimed at researchers and drug development professionals, we explore the foundational principles of each method, their computational and experimental applications, and their respective limitations. By comparing their performance through benchmarking studies and real-world case studies, we offer a clear framework for selecting the appropriate synthesizability assessment tool. The article concludes with an outlook on how integrated, machine-learning-enhanced models are shaping the future of reliable and efficient material discovery, directly impacting the development of novel pharmaceuticals and functional materials.
Demystifying Synthesizability: From Charge Neutrality to Thermodynamic Stability
Defining Synthesizability in Materials Science and Drug Discovery
The discovery of new functional materials and active pharmaceutical ingredients is fundamentally limited by a critical challenge: synthesizability. This concept refers to whether a proposed chemical compound can be successfully synthesized and isolated in practical, experimentally-realizable conditions. In computational materials science and drug discovery, researchers increasingly rely on predictive models to identify promising candidates from vast chemical spaces, making accurate synthesizability assessment crucial for reducing costly experimental failures. The core problem revolves around bridging the gap between theoretical predictions, which often rely on thermodynamic stability metrics like energy above hull, and practical synthetic feasibility, for which charge-balancing in inorganic crystals serves as a simple historical proxy [1] [2].
This guide examines the defining frameworks, computational methodologies, and experimental validation protocols for synthesizability prediction, with a specific focus on the comparative analysis between energy above hull and charge-balancing approaches. As materials and drug discovery increasingly leverage high-throughput computational screening, the development of robust, data-driven synthesizability models represents a pivotal step toward realizing autonomous discovery pipelines [1] [3].
Core Concepts and Definitions
Fundamental Principles of Synthesizability
Synthesizability encompasses multiple dimensions that determine whether a theoretical material or drug candidate can be translated into an experimentally accessible compound. Key aspects include:
- Thermodynamic Stability: Often assessed through energy above hull calculations, which measure a compound's stability relative to its competing phases [1].
- Kinetic Accessibility: Concerns the energy barriers and pathways for compound formation, which may enable synthesis of metastable phases [3].
- Synthetic Feasibility: Considers practical constraints including precursor availability, reaction conditions, and the existence of viable synthetic routes [3] [2].
In drug discovery, synthesizability additionally encompasses synthetic complexity, step count, yield, and the commercial availability of required building blocks and reagents.
Charge-Balancing as a Synthesizability Proxy
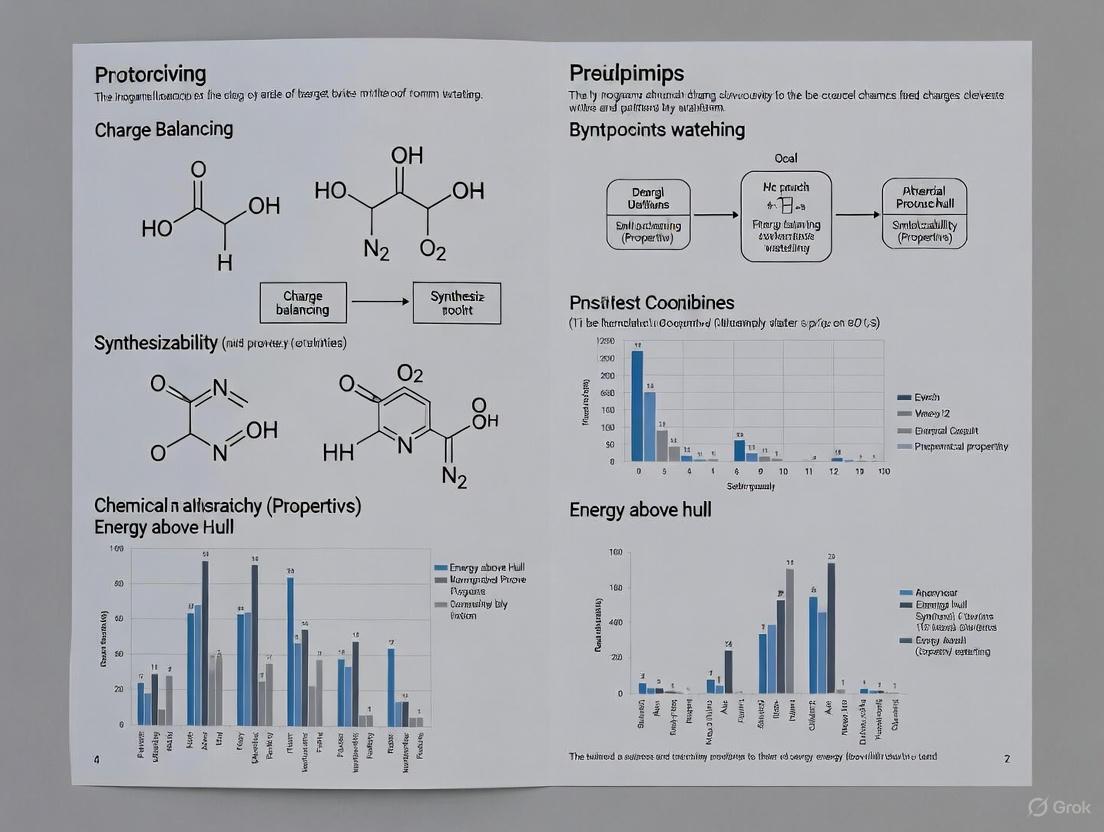
Charge-balancing represents a chemically intuitive approach to predicting synthesizability, particularly for inorganic crystalline materials. This method applies the principle of charge neutrality, assuming that synthesizable ionic compounds must have a net neutral charge when elements are assigned their common oxidation states [1].
However, this approach demonstrates significant limitations. Analysis of known inorganic materials reveals that only approximately 37% of synthesized compounds are charge-balanced according to common oxidation states. Even among typically ionic binary cesium compounds, only 23% adhere to charge-balancing principles [1]. This poor performance stems from the method's inability to account for diverse bonding environments in metallic alloys, covalent materials, and complex ionic solids where non-stoichiometry and mixed bonding character prevail.
Energy Above Hull as a Stability Metric
The energy above hull (Eₕᵤₗₗ) represents a computational approach to synthesizability assessment based on thermodynamic stability. Calculated using density functional theory (DFT), Eₕᵤₗₗ represents the energy difference between a compound and its most stable decomposition products at zero temperature [1] [3].
Materials with Eₕᵤₗₗ = 0 eV/atom are considered thermodynamically stable, while those with Eₕᵤₗₗ > 0 are metastable or unstable. However, this approach presents significant limitations. Many technologically crucial materials (including virtually all key magnet technologies like Nd₂Fe₁₄B, SrFe₁₂O₁₉, and SmCo₅) are metastable at 0 K but are successfully synthesized at elevated temperatures where kinetic factors dominate [2]. Studies indicate that Eₕᵤₗₗ-based screening captures only approximately 50% of known synthesizable inorganic crystalline materials [1].
Table 1: Comparative Analysis of Synthesizability Prediction Methods
| Method | Theoretical Basis | Accuracy | Limitations | Applicability |
|---|---|---|---|---|
| Charge-Balancing | Charge neutrality principle | ~37% with known materials [1] | Inflexible to different bonding environments; cannot account for metallic/covalent systems | Primarily ionic crystalline materials |
| Energy Above Hull | Thermodynamic stability via DFT | ~50% with known materials [1] | Fails for kinetically stabilized phases; computationally expensive | Crystalline materials with known decomposition pathways |
| SynthNN | Deep learning on composition data | 1.5× higher precision than human experts [1] | Requires large training datasets; limited to composition-based predictions | Inorganic crystalline materials |
| CSLLM | Large language models on crystal structures | 98.6% accuracy [3] | Requires structure information; complex training process | 3D crystal structures with defined atomic positions |
Computational Methodologies
Machine Learning Approaches
SynthNN: Deep Learning for Synthesizability Classification
The SynthNN model represents a significant advancement in synthesizability prediction through deep learning applied to chemical compositions without requiring structural information. The model employs the atom2vec framework, which learns optimal chemical representations directly from the distribution of synthesized materials [1].
Experimental Protocol:
- Data Collection: Training data is extracted from the Inorganic Crystal Structure Database (ICSD), representing synthesized crystalline inorganic materials [1].
- Data Augmentation: Artificially generated unsynthesized materials are created to form a balanced dataset [1].
- Model Architecture: Utilizes atom embedding matrices optimized alongside neural network parameters [1].
- Training Approach: Implements positive-unlabeled (PU) learning to handle incomplete labeling of unsynthesized examples [1].
- Validation: Benchmarking against random guessing and charge-balancing baselines using standard classification metrics [1].
In comparative evaluations, SynthNN demonstrated 1.5× higher precision than the best human experts and completed synthesizability assessment tasks five orders of magnitude faster [1]. Remarkably, without explicit programming of chemical rules, the model autonomously learned fundamental principles including charge-balancing, chemical family relationships, and ionicity [1].
SynthNN Model Architecture
CSLLM: Large Language Models for Crystal Synthesis
The Crystal Synthesis Large Language Model (CSLLM) framework represents a transformative approach to synthesizability prediction, achieving state-of-the-art 98.6% accuracy by leveraging specialized large language models fine-tuned on crystal structure data [3].
Experimental Protocol:
- Data Curation:
- Text Representation: Development of "material string" representation integrating space group, lattice parameters, atomic species, Wyckoff positions, and fractional coordinates [3].
- Model Architecture: Three specialized LLMs for synthesizability prediction, synthetic method classification, and precursor identification [3].
- Training Procedure: Fine-tuning on the comprehensive dataset using the material string representation [3].
- Validation: Comparative benchmarking against thermodynamic (74.1% accuracy) and kinetic (82.2% accuracy) stability methods [3].
The CSLLM framework demonstrates exceptional generalization capability, achieving 97.9% accuracy on complex structures with large unit cells considerably exceeding training data complexity [3].
Table 2: CSLLM Framework Performance Metrics
| Model Component | Task | Accuracy | Dataset Size | Comparative Performance |
|---|---|---|---|---|
| Synthesizability LLM | Binary classification (synthesizable/non-synthesizable) | 98.6% [3] | 150,120 structures | Outperforms energy above hull (74.1%) and phonon stability (82.2%) |
| Method LLM | Synthetic method classification (solid-state/solution) | 91.0% [3] | Not specified | N/A |
| Precursor LLM | Precursor identification for binary/ternary compounds | 80.2% success rate [3] | Not specified | N/A |
Positive-Unlabeled Learning for Solid-State Synthesis
Positive-unlabeled (PU) learning has emerged as a particularly effective framework for synthesizability prediction, addressing the fundamental challenge that while positive examples (synthesized materials) are well-documented, negative examples (unsynthesizable materials) are rarely reported in the literature.
Experimental Protocol for Solid-State Synthesizability Predictions:
- Data Extraction: Synthesis information for 4,103 ternary oxides was manually extracted from literature sources, including synthesis success/failure and reaction conditions [4].
- Data Quality Validation: The human-curated dataset identified 156 outliers in a text-mined dataset of 4,800 entries, with only 15% of these outliers correctly extracted by automated methods [4].
- Model Training: Implementation of PU learning to handle the inherent data imbalance and uncertainty in negative examples [4].
- Prediction Application: The trained model identified 134 out of 4,312 hypothetical compositions as likely synthesizable [4].
This approach demonstrates the critical importance of data quality in synthesizability prediction, with human-curated data significantly outperforming automated text-mining approaches for training reliable models [4].
Experimental Validation and Synthesis Planning
Synthesis Route Prediction
Beyond binary synthesizability classification, comprehensive synthesis planning requires predicting specific synthetic routes and precursors. The CSLLM framework addresses this through specialized models for method classification and precursor identification [3].
Method LLM Experimental Protocol:
- Task: Classify appropriate synthetic methods (solid-state vs. solution) for given crystal structures.
- Performance: Achieves 91.0% accuracy in synthetic method classification [3].
- Implementation: Fine-tuned LLM using material string representations of crystal structures paired with synthetic method data.
Precursor LLM Experimental Protocol:
- Task: Identify suitable solid-state synthesis precursors for binary and ternary compounds.
- Performance: Achieves 80.2% accuracy in precursor prediction [3].
- Implementation: Combinatorial analysis of potential precursor combinations coupled with reaction energy calculations.
High-Throughput Experimental Validation
The ultimate validation of synthesizability predictions requires experimental verification. Automated laboratories enable high-throughput synthesis and characterization, dramatically accelerating the feedback loop between prediction and validation [2].
Experimental Workflow:
- Candidate Selection: AI-driven selection of novel materials from millions of possibilities using machine-learning interatomic potentials and DFT [2].
- Recipe Generation: In-house reaction modeling generates plausible synthesis recipes [2].
- Automated Synthesis: Robotic systems perform hundreds of experiments per week [2].
- Characterization and Feedback: Automated analysis characterizes results and feeds back to recipe prediction models [2].
This approach reduces materials discovery timelines from decades to approximately two years while cutting costs by approximately 90% [2].
High-Throughput Validation Workflow
The Scientist's Toolkit: Research Reagents and Materials
Table 3: Essential Research Materials for Synthesizability Investigations
| Reagent/Material | Function | Application Context |
|---|---|---|
| Ternary Oxide Precursors | Metal oxide powders serving as reactants for solid-state synthesis | Experimental validation of ternary oxide synthesizability predictions [4] |
| ICSD Database | Comprehensive repository of experimentally characterized inorganic crystal structures | Training and benchmarking data for synthesizability models [1] [3] |
| MatSyn25 Dataset | Large-scale dataset of 2D material synthesis processes extracted from 85,160 research articles | Training specialized AI models for 2D material synthesis prediction [5] |
| CLscore Model | Pre-trained PU learning model for identifying non-synthesizable structures | Generating negative examples for balanced training datasets [3] |
| Material String Representation | Text-based encoding of crystal structure information | Fine-tuning LLMs for synthesizability and synthesis route prediction [3] |
The evolving landscape of synthesizability prediction demonstrates a clear trajectory from simple heuristic approaches like charge-balancing to sophisticated data-driven models leveraging deep learning and large language models. While energy above hull calculations provide valuable thermodynamic insights, their limitations in predicting kinetically stabilized phases have motivated the development of complementary approaches that directly learn synthesizability patterns from experimental data.
The integration of computational predictions with high-throughput experimental validation represents the most promising path forward, creating closed-loop discovery systems that continuously refine synthesizability models. As these technologies mature, they will dramatically accelerate the translation of theoretical materials and drug candidates into experimentally accessible compounds, ultimately transforming the pace of innovation across materials science and pharmaceutical development.
The critical importance of data quality cannot be overstated—whether for traditional machine learning models or modern LLMs. Human-curated datasets [4] and comprehensive repositories like MatSyn25 for 2D materials [5] provide the essential foundation for developing reliable synthesizability predictors that can genuinely transform materials discovery workflows.
Charge balancing serves as a foundational heuristic in materials science for initially assessing the synthesizability of inorganic crystalline compounds. This whitepaper examines the principle that materials with a net neutral ionic charge—based on common oxidation states—are more likely to be synthetically accessible. While computationally inexpensive and chemically intuitive, this method possesses significant limitations in predictive accuracy. Quantitative analysis reveals that only 37% of known synthesized inorganic materials in the Inorganic Crystal Structure Database (ICSD) are charge-balanced according to common oxidation states, dropping to just 23% for binary cesium compounds [1]. Contemporary research increasingly integrates charge-balancing with advanced computational models, such as deep learning synthesizability classifiers and density functional theory (DFT), to create more reliable frameworks for predicting viable materials. This evolution reflects a broader paradigm shift in synthesizability research from simple heuristic filters toward multi-faceted, data-driven approaches that better capture the complex thermodynamic and kinetic factors governing material synthesis.
The accelerating demand for novel materials to enable sustainable technologies has placed unprecedented focus on computational discovery methods. With computational databases now containing millions of predicted crystalline structures—far exceeding the number of experimentally synthesized compounds—the critical challenge lies in identifying which candidates are truly synthesizable [6] [1]. In this screening process, simple heuristics like charge balancing provide initial triage mechanisms for navigating vast chemical spaces.
Charge balancing operates on the chemically intuitive principle that inorganic compounds tend toward charge neutrality, where the total positive charge from cations balances the total negative charge from anions according to their expected oxidation states. This approach requires minimal computational resources compared to first-principles calculations, making it attractive for initial filtering. However, its reliability as a standalone synthesizability criterion remains questionable, as it fails to account for the complex bonding environments and kinetic factors that ultimately determine synthetic accessibility [1].
Within the broader context of energy above hull versus charge balancing synthesizability research, this whitepaper examines the technical foundations, quantitative performance, and fundamental limitations of the charge-balancing heuristic. By comparing its performance against stability-based metrics and contemporary machine learning approaches, we aim to provide researchers with a comprehensive framework for selecting appropriate synthesizability assessment methods based on their specific discovery objectives and computational resources.
Theoretical Foundations of Charge Balancing
Chemical Principles Underlying Charge Neutrality
The charge balancing heuristic originates from fundamental chemical principles of ionic bonding, where electrons are transferred from electropositive elements to electronegative elements, resulting in stable electron configurations. The approach assumes that elements exhibit predictable oxidation states based on their position in the periodic table and that the sum of oxidation states across all atoms in a compound should equal zero for a stable crystal to form [1].
This formalism applies most directly to strongly ionic compounds where chemical bonding can be accurately described through complete electron transfer. For such materials, charge balancing provides a reasonable first approximation of stability, as large charge imbalances would create unfavorable electrostatic potentials. The heuristic is implemented computationally by assigning common oxidation states to each element (e.g., +1 for alkali metals, +2 for alkaline earth metals, -2 for oxygen) and verifying that the sum of oxidation states multiplied by their stoichiometric coefficients equals zero [1].
Computational Implementation
The computational implementation of charge balancing is exceptionally lightweight compared to first-principles quantum mechanical calculations. The algorithm requires only:
- Elemental Composition Input: The chemical formula of the compound
- Oxidation State Assignment: A predefined database of common oxidation states for each element
- Stoichiometric Calculation: Multiplication of oxidation states by stoichiometric coefficients and summation
This process involves simple arithmetic operations without the need for structural information or iterative calculations, making it scalable to billions of candidate materials with minimal computational resources. However, this simplicity comes at the cost of chemical accuracy, particularly for materials with significant covalent character, metallic bonding, or uncommon oxidation states [1].
Table 1: Key Components of Charge Balancing Implementation
| Component | Description | Example Values |
|---|---|---|
| Oxidation State Database | Common oxidation states for elements | Na: +1, Mg: +2, Al: +3, O: -2, F: -1 |
| Calculation Method | Sum of (oxidation state × stoichiometric coefficient) | Na₂O: 2×(+1) + 1×(-2) = 0 |
| Decision Rule | Compound is plausible if sum equals zero | Zero = plausible; Non-zero = implausible |
Quantitative Performance Assessment
Limitations in Predicting Known Synthesized Materials
The most significant limitation of charge balancing emerges when evaluating its performance against databases of experimentally synthesized materials. Comprehensive analysis of the Inorganic Crystal Structure Database (ICSD) reveals that only approximately 37% of all known synthesized inorganic crystalline materials are charge-balanced according to common oxidation states [1]. This surprisingly low success rate indicates that the majority of experimentally accessible materials violate simple charge-balancing rules.
The performance further deteriorates when examining specific material classes. For binary cesium compounds—typically considered highly ionic—only 23% of known synthesized compounds are charge-balanced [1]. This demonstrates that even for material classes where ionic bonding dominates, charge balancing fails to accurately predict synthesizability, suggesting that factors beyond simple electron transfer govern synthetic accessibility.
Comparison with Alternative Screening Methods
When evaluated against other material screening approaches, charge balancing demonstrates distinct performance characteristics across precision and recall metrics:
Table 2: Performance Comparison of Synthesizability Prediction Methods
| Method | Precision for Synthesized Materials | Computational Cost | Key Limitations |
|---|---|---|---|
| Charge Balancing | Very Low (37% coverage of ICSD) [1] | Negligible | Overlooks covalent/metallic bonding, kinetic effects |
| Formation Energy (DFT) | Moderate (~50% coverage of ICSD) [1] | Very High | Requires crystal structure; misses kinetically stabilized phases |
| SynthNN (ML Model) | 7× higher precision than charge balancing [1] | Low | Requires training data; composition-only limitation |
The precision advantage of machine learning approaches like SynthNN becomes particularly evident in head-to-head comparisons with human experts. In controlled material discovery evaluations, SynthNN achieved 1.5× higher precision than the best human expert while completing the assessment five orders of magnitude faster [1].
Diagram 1: Method performance versus cost comparison. Charge balancing offers low computational cost but poor coverage of known synthesized materials, while ML approaches like SynthNN provide better coverage with moderate computational requirements.
Fundamental Limitations and Chemical Exceptions
Bonding Environments Beyond Ionic Approximations
The charge balancing heuristic fails most prominently for materials with substantial covalent character, where electron sharing rather than complete transfer dominates bonding. In such cases, formal oxidation states become poorly defined, and the assignment of integer charges to atoms provides an inaccurate representation of the electronic structure. Metallic alloys and intermetallic compounds represent another significant challenge, as their bonding involves delocalized electrons that cannot be adequately described using localized oxidation state models [1].
Compounds with uncommon oxidation states further complicate the charge-balancing approach. For instance, the heuristic would incorrectly reject prussian blue analogues containing Fe(II)/Fe(III) mixtures or rare-earth compounds with mixed valency, despite these materials being synthetically accessible. The rigid assignment of common oxidation states cannot accommodate the nuanced electron configurations that occur in complex solid-state materials [1].
Kinetic and Thermodynamic Considerations
Charge balancing operates as a purely geometric constraint without consideration of the energetic landscape governing material formation. It ignores:
- Thermodynamic Stability: A charge-balanced compound may still be thermodynamically unstable with respect to decomposition into other phases.
- Kinetic Accessibility: Synthesizability depends on viable formation pathways and energy barriers, not just thermodynamic stability.
- Finite-Temperature Effects: Entropic contributions and temperature-dependent phase stability are not captured.
The limitations of stability-based screening alone are evidenced by the existence of numerous metastable phases that persist due to kinetic barriers rather than thermodynamic stability. These materials would be incorrectly excluded by both charge-balancing and formation-energy filters despite being experimentally synthesizable [6].
Experimental versus Computational Charge Assessment
Recent advances in experimental charge determination highlight the complexity of real-world charge distributions. The innovative iSFAC (ionic scattering factors) modeling method using electron diffraction has enabled direct experimental measurement of partial atomic charges in crystalline materials [7]. This technique has revealed counterintuitive charge distributions, such as negative partial charges on carbon atoms within carboxylate groups due to electron delocalization—contradicting simple oxidation state predictions [7].
These experimental findings demonstrate that real charge distributions in materials are more nuanced than integer oxidation states suggest. The iSFAC method has successfully quantified partial charges in diverse systems including antibiotic molecules (ciprofloxacin), amino acids (tyrosine, histidine), and inorganic frameworks (ZSM-5 zeolite), providing experimental validation of complex charge distribution phenomena that transcend simple charge-balancing heuristics [7].
Integrated Workflows: Beyond Simple Heuristics
Modern Synthesizability Prediction Frameworks
Contemporary materials discovery pipelines have evolved beyond standalone heuristics to integrated frameworks that combine multiple assessment methods. The synthesizability-guided pipeline demonstrates this approach by employing a unified model that integrates complementary signals from both composition and crystal structure [6]. This method uses:
- Compositional Model: A fine-tuned compositional MTEncoder transformer that processes stoichiometric information
- Structural Model: A graph neural network fine-tuned from the JMP model that analyzes crystal structure graphs
- Rank-Average Ensemble: Aggregates predictions from both models to generate enhanced synthesizability rankings [6]
This integrated approach successfully identified several hundred highly synthesizable candidates from over 4.4 million computational structures, with experimental validation confirming 7 of 16 attempted syntheses—demonstrating substantially improved performance over single-modality assessments [6].
Diagram 2: Modern synthesizability assessment workflow integrating multiple computational models with experimental validation, demonstrating higher success rates than single-heuristic approaches.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Tools for Advanced Synthesizability Assessment
| Tool/Category | Function | Application in Synthesizability Research |
|---|---|---|
| iSFAC Modeling | Experimental partial charge determination via electron diffraction | Quantifies real charge distributions; validates computational predictions [7] |
| DFT Calculations | First-principles electronic structure analysis | Provides formation energies, density of states, and thermodynamic stability [8] |
| Graph Neural Networks | Structure-aware machine learning models | Encodes crystal structure information for synthesizability classification [6] |
| Compositional Transformers | Stoichiometry-based deep learning | Processes chemical formulas without structural information [6] [1] |
| High-Throughput Automation | Parallel synthesis and characterization | Rapid experimental validation of computational predictions [8] |
Charge balancing remains a computationally efficient heuristic for initial material screening but possesses fundamental limitations that restrict its utility as a standalone synthesizability criterion. Quantitative analysis reveals its poor performance in predicting known synthesized materials, with only 37% of ICSD compounds satisfying charge-balance criteria [1]. Its inability to account for diverse bonding environments, kinetic factors, and complex charge distributions underscores the need for more sophisticated assessment methods.
The evolving paradigm in synthesizability research integrates multiple complementary approaches—combining composition-based and structure-aware machine learning models with experimental validation [6]. These integrated frameworks demonstrate substantially improved performance over single-heuristic methods, successfully guiding experimental synthesis of novel materials. As materials discovery accelerates to meet global technological challenges, the role of simple heuristics like charge balancing will increasingly shift from primary screening criteria to complementary components within more comprehensive, multi-faceted discovery workflows.
In the computational design of new materials, from inorganic crystals to pharmaceutical compounds, predicting thermodynamic stability is a fundamental challenge. Energy above hull (Ehull) has emerged as the gold-standard metric for this purpose, providing a rigorous measure of a material's stability against decomposition into competing phases [9]. Defined as the energy difference between a material's formation energy and the minimum formation energy possible for its composition within a chemical system, Ehull serves as a critical filter for prioritizing candidate materials for synthesis [10].
This metric is derived from a mathematical construction known as the convex hull, which represents the minimum energy "envelope" in energy-composition space [11]. A material with an Ehull of zero meV/atom lies precisely on this hull and is considered thermodynamically stable. Conversely, a positive Ehull indicates the material is metastable or unstable, with the magnitude quantifying the energy cost of its decomposition [11] [10]. Values exceeding 200 meV/atom are generally considered large and suggest a material may be unsynthesizable, though this threshold varies across chemical systems [10].
Within the broader context of synthesizability research, Ehull provides a foundational thermodynamic perspective that complements other critical considerations, such as kinetic barriers and synthetic accessibility. While charge balancing and other chemical intuition-based approaches offer valuable insights, Ehull delivers a quantitative, first-principles stability assessment that is essential for rational materials design.
Theoretical Foundation and Calculation Methodology
Fundamental Concepts
The calculation of energy above hull rests upon several key thermodynamic concepts:
Formation Energy (Ef): The energy required to form a compound from its elemental constituents at standard conditions, typically expressed in eV/atom [11]. For a compound AₓBᵧCz, it is calculated as ΔHf = Etotal - xμA^0 - yμB^0 - zμC^0, where Etotal is the DFT total energy and μ_i^0 are the reference chemical potentials of the elements [12].
Decomposition Energy (Ed): The energy released or required for a phase to decompose into other more stable compounds in the system [11]. This represents the actual energy landscape for phase decomposition, making it a more direct measure of stability than formation energy alone.
Convex Hull: A geometric construction representing the set of phases with the lowest possible formation energies across all compositions in a chemical system [11]. The hull is built in normalized energy versus composition space, with every composition represented as totaling one atom [11].
Computational Framework
The standard methodology for Ehull determination involves several systematic steps:
Table 1: Key Computational Components for Ehull Calculation
| Component | Description | Common Tools/Methods |
|---|---|---|
| Energy Calculations | Density Functional Theory (DFT) calculations at 0 K | VASP, Quantum ESPRESSO |
| Reference States | Elemental chemical potentials under standard conditions | Materials Project database |
| Correction Schemes | Addressing systematic DFT errors for specific elements | Materials Project GGA/GGA+U mixing scheme [10] |
| Hull Construction | Geometric determination of the stable phase envelope | Pymatgen PhaseDiagram class |
| Stability Assessment | Vertical distance measurement to hull surface | Automated builders in Materials Project infrastructure [10] |
Workflow for Energy Above Hull Determination
The following diagram illustrates the complete computational workflow for determining a material's energy above hull:
Advanced Calculation Considerations
For multi-component systems (ternary, quaternary), the convex hull construction becomes increasingly complex. The hull evolves from points (binary), to lines (ternary), to triangles and tetrahedra (quaternary), where multiple phases coexist in thermodynamic equilibrium [11]. The decomposition pathway for a metastable phase involves determining the precise mixture of stable phases that minimizes the total energy at that composition.
The calculation must preserve normalization per atom throughout. For example, considering BaTaNO₂ with decomposition products ⅔ Ba₄Ta₂O₉ + 7⁄₄₅ Ba(TaN₂)₂ + 8⁄₄₅ Ta₃N₅, the stoichiometric coefficients ensure conservation of elemental concentrations when using normalized (eV/atom) energies [11].
Experimental Protocols and Validation
Standardized DFT Calculation Parameters
To ensure consistency with major materials databases, the following protocol should be implemented:
Structural Relaxation: Full optimization of lattice parameters and atomic positions using plane-wave DFT with PAW pseudopotentials [9].
Electronic Structure Settings: Energy cutoff of 520 eV, k-point density of at least 64 k-points per Å⁻³, Gaussian smearing of 0.05 eV [12].
Exchange-Correlation Functional: Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA), with +U corrections applied to elements with localized d-or f-electrons [10].
Energy Convergence: Self-consistent field tolerance of 10⁻⁶ eV/atom, force convergence threshold of 0.01 eV/Å [12].
Reference Energies: Elemental chemical potentials derived from standard reference states in the Materials Project database [10].
Validation Against Experimental Data
Validating computational Ehull predictions requires correlation with experimental synthesizability data:
Table 2: Ehull Thresholds for Experimental Synthesizability
| Ehull Range (meV/atom) | Stability Classification | Experimental Synthesizability | Remarks |
|---|---|---|---|
| 0 | Thermodynamically stable | High | On the convex hull [10] |
| 0-80 | Metastable | Moderate to high | Common threshold for synthesizability filter [9] |
| 80-200 | Metastable to unstable | Low | May require kinetic stabilization |
| >200 | Unstable | Very low | Considered very large, unlikely to be synthesizable [10] |
Materials with Ehull values below 80 meV/atom have demonstrated successful experimental realization, with the majority of known stable compounds falling within this range [9]. However, exceptions exist, as some metastable phases (Ehull > 0) can be synthesized through kinetic control or non-equilibrium methods [11].
Ehull in the Context of Synthesizability Research
Comparison with Alternative Stability Metrics
While Ehull represents the thermodynamic gold standard, synthesizability research incorporates multiple complementary metrics:
Formation Energy (Ef): Provides an initial stability screening but fails to account for competing phases [9].
Charge Balancing: An empirical approach based on chemical intuition, particularly relevant for ionic compounds, but lacking quantitative predictive power for complex multi-component systems.
Energy Above Hull (Ehull): Comprehensive thermodynamic stability metric considering all decomposition pathways [11] [10].
Decomposition Energy (Ed): Directly quantifies the energy change for specific decomposition reactions [11].
The relationship between these metrics and their predictive capabilities can be visualized as follows:
Integration with Machine Learning Approaches
Recent advances have incorporated Ehull into machine learning pipelines for accelerated materials discovery:
Stability Prediction: Graph neural networks trained on DFT-calculated Ehull values can rapidly screen thousands of candidate structures [12].
Feature Representation: Fourier-transformed crystal properties (FTCP) incorporate both real-space and reciprocal-space information to predict synthesizability with over 82% accuracy [9].
High-Throughput Screening: ML models using Ehull as a target property enable efficient identification of promising materials from vast chemical spaces [9] [12].
Research Reagent Solutions for Computational Studies
Table 3: Essential Computational Tools for Ehull Research
| Tool/Resource | Type | Function in Ehull Research | Access Method |
|---|---|---|---|
| VASP | Software | First-principles DFT calculations for energy determination | Commercial license |
| pymatgen | Python library | Phase diagram analysis and convex hull construction | Open source |
| Materials Project API | Database | Access to calculated reference energies | Free web API |
| atomate2 | Workflow manager | Automated DFT calculation workflows | Open source |
| CGCNN/GNN Models | ML Architecture | Fast energy prediction for initial screening | Open source |
Energy above hull represents the most rigorous computational metric for assessing thermodynamic stability, serving as an indispensable tool in modern materials design and drug development. Its derivation from first principles and comprehensive consideration of all possible decomposition pathways provides a fundamental advantage over empirical approaches like charge balancing. As machine learning methodologies continue to evolve, Ehull remains the foundational stability metric upon which predictive synthesizability models are built, enabling the efficient exploration of vast chemical spaces for promising new materials with tailored properties. For researchers embarking on stability studies, implementing the standardized protocols outlined in this guide will ensure consistent, reliable Ehull determinations that align with established computational materials science practices.
The discovery of new functional materials is a cornerstone of technological advancement, spanning sectors from drug development to renewable energy. Within this pursuit, two distinct philosophies have emerged: the intuitive chemistry approach, rooted in empirical rules and human domain knowledge, and the computational physics paradigm, grounded in quantum mechanics and high-throughput simulation. This whitepaper provides an in-depth technical analysis of these methodologies, framing their core differences within a critical research axis: the use of charge balancing versus energy above hull as primary metrics for predicting material synthesizability. We dissect the fundamental principles, experimental protocols, and practical applications of each approach for an audience of researchers, scientists, and drug development professionals.
Core Philosophical and Methodological Divergences
At its heart, the distinction between intuitive chemistry and computational physics is a dichotomy between chemistry's pragmatic, rule-based worldview and physics' first-principles, axiomatic approach.
Intuitive Chemistry is characterized by its loyalty to predictive accuracy and practicality over any single philosophical school. As noted in analyses of quantum chemistry, the field operates with a "shut up and calculate" pragmatism, where the wavefunction is treated not as a literal description of reality but as a powerful computational device for predicting measurable outcomes [13]. This perspective aligns more naturally with the Copenhagen interpretation of quantum mechanics, where the "collapse of the wavefunction" upon measurement provides a clear operational link between mathematical formalism and experimental observables like energy, dipole moment, or excitation spectra [13]. The focus is on solving the time-independent Schrödinger equation for stationary states and interpreting the square modulus of wavefunctions as probability densities that can be compared with experimental data.
Computational Physics, particularly in materials science, embraces a more fundamentalist approach. It seeks to understand and predict material behavior from first principles, primarily through density functional theory (DFT) and related quantum mechanical methods. This paradigm treats the wavefunction as a physical entity and utilizes the full predictive power of quantum mechanics to compute properties from the ground up, often with minimal empirical input. The methodology is inherently computational and scales with increasing processor power, allowing for high-throughput screening of thousands of candidate materials based on fundamental physics.
Table 1: Fundamental Divergences Between the Two Paradigms
| Aspect | Intuitive Chemistry | Computational Physics |
|---|---|---|
| Primary Foundation | Chemical rules of thumb, empirical knowledge, and domain expertise [14] | First-principles quantum mechanics (e.g., Density Functional Theory) [6] |
| Philosophical Alignment | Copenhagen interpretation (pragmatic, measurement-focused) [13] | More aligned with fundamental, reality-describing interpretations [13] |
| Central Synthesizability Metric | Charge Balancing and Electronegativity Balance [14] | Energy Above Hull (thermodynamic stability) [6] |
| Typical Workflow | Application of sequential "filters" encoding human knowledge [14] | High-throughput DFT calculation and screening [6] |
| View of Wavefunction | Computational tool for prediction [13] | Literal description of physical reality [13] |
| Automation Potential | Challenging to fully codify human intuition | Highly amenable to automation and scaling via HPC/AI |
The Synthesizability Challenge: Charge Balancing vs. Energy Above Hull
A critical battlefield for these competing paradigms is the prediction of whether a hypothetical material can be successfully synthesized. This directly impacts the efficiency of discovery pipelines in drug development and materials science.
The Intuitive Chemistry Approach: Charge Balancing
The intuitive chemistry approach relies on heuristic "filters" derived from a chemist's knowledge. The most foundational of these is the charge neutrality or charge balancing principle.
- Theoretical Basis: This filter assesses whether a compound's elemental composition can yield a net neutral ionic charge using the common oxidation states of its constituents. It is predicated on the classical chemical concept that stable inorganic compounds tend to be electrically neutral overall [14].
- Experimental Protocol: The methodology for applying charge balance filters is straightforward and does not require intensive computation:
- Input: A candidate chemical composition (e.g., A(x)B(y)C(_z)).
- Oxidation State Assignment: Assign probable oxidation states to each element based on known chemical rules (e.g., O is typically -2, Alkali metals +1).
- Neutrality Check: Calculate the total charge from the cation and anion components. If the sum is zero, the composition is "Allowed"; otherwise, it is "Forbidden" [14].
- Electronegativity Check: A complementary filter often used verifies that the most electronegative ion in the compound also carries the most negative charge, ensuring local charge stability [14].
This approach is computationally inexpensive and can rapidly screen billions of compositions. However, its inflexibility is a significant limitation, as it fails to account for materials where bonding is not purely ionic, such as metallic alloys or covalent solids [1]. Studies have shown that only about 37% of known synthesized inorganic materials are charge-balanced according to common oxidation states, highlighting a high false-positive rate for this rule alone [1].
The Computational Physics Approach: Energy Above Hull
The computational physics paradigm frames synthesizability primarily in terms of thermodynamic stability, with the key metric being the energy above hull.
- Theoretical Basis: A material's stability is determined by its decomposition energy into other, more stable phases in the same chemical space. The energy above hull (E({\text{hull}})) is the energy difference per atom between the material's calculated formation energy and the tie-line (convex hull) constructed from the most stable phases at that composition. A low or zero E({\text{hull}}) indicates thermodynamic stability, suggesting the material is less likely to decompose [6] [1].
- Experimental Protocol: This method is computationally intensive and forms the backbone of modern high-throughput materials screening.
- Structure Generation: Propose a candidate crystal structure.
- DFT Relaxation: Perform a full DFT geometry optimization to find the structure's ground-state energy. This is often the most time-consuming step, though machine learning surrogate models are increasingly used to accelerate it [6] [14].
- Hull Construction: Calculate the formation energies of all known and competing phases in the relevant chemical system from databases like the Materials Project.
- E(_{\text{hull}}) Calculation: Determine the candidate's energy relative to the convex hull. Structures within a small threshold (e.g., 0-50 me/atom) are typically considered potentially stable [6].
While powerful, this method has a significant blind spot: it overlooks kinetic barriers and non-equilibrium synthesis pathways. A material can be metastable (positive E({\text{hull}})) yet still be synthesizable if kinetic barriers prevent its decomposition [6] [1]. It is estimated that E({\text{hull}}) calculations alone capture only about 50% of synthesized inorganic crystalline materials [1].
Diagram 1: Two pathways for predicting synthesizability. The computational physics path (red) relies on quantum mechanical calculations, while the intuitive chemistry path (blue) applies sequential heuristic filters.
Quantitative Comparison of Predictive Performance
The performance gap between these approaches is stark. The following table synthesizes quantitative data from recent benchmarking studies.
Table 2: Performance Metrics for Synthesizability Prediction Methods
| Methodology | Primary Metric | Approximate Precision | Key Strength | Key Weakness |
|---|---|---|---|---|
| Charge Balancing [1] | Charge Neutrality | Very Low (Baseline) | Computationally trivial, highly interpretable | Only 37% of known materials are charge-balanced |
| Energy Above Hull [1] | Thermodynamic Stability | ~50% Recall | Strong physical basis, identifies stable ground states | Fails for kinetically stabilized phases |
| Human Experts [1] | Domain Experience | Benchmark (1.0x precision) | Leverages deep, contextual knowledge | Slow, not scalable, expert-dependent |
| SynthNN (ML on Compositions) [1] | Data-Driven Likelihood | 7x higher precision than DFT E-hull; 1.5x higher than human experts | Learns implicit chemical rules from all known materials; highly scalable | "Black box" model; requires large, clean training data |
Emerging Hybrids and AI-Enhanced Workflows
The stark contrast between these paradigms is giving way to powerful hybrid approaches that leverage the strengths of both.
Embedded Human-Knowledge Pipelines
One modern strategy involves "stitching together" chemical rules and human intuition into a structured screening pipeline. A representative workflow, designed to discover "perovskite-inspired" materials, applies these filters in sequence [14]:
- Charge Neutrality Filter
- Electronegativity Balance Filter
- Unique Oxidation State Filter (excludes compounds with multiple oxidation states per element)
- Oxidation State Frequency Filter (excludes uncommon oxidation states)
- Intra-Phase Diagram Stoichiometry Filter (compares to known compounds in the same chemical diagram)
- Cross-Phase Diagram Stoichiometry Filter (assesses common stoichiometries across related diagrams)
This approach can start with over 100,000 hypothetical compounds and refine them down to a few dozen high-priority candidates, dramatically increasing the likelihood of successful synthesis [14].
AI as a Unifying Bridge
Artificial Intelligence is now serving as a powerful bridge between these worlds. New methods are emerging that learn the implicit "rules" of chemistry directly from data, bypassing the need for humans to explicitly codify them.
- Learning Chemical Intuition: Models like SynthNN demonstrate that deep learning can predict synthesizability from composition alone, achieving a precision 1.5 times higher than the best human experts and completing the task five orders of magnitude faster [1]. Remarkably, without being explicitly programmed with chemical knowledge, these models learn the principles of charge-balancing, chemical family relationships, and ionicity directly from the data of known materials [1].
- Encoding Physical Laws: Conversely, AI is also being used to make computational physics more robust and intuitive. For instance, the CEONet model was specifically designed with "physics-awareness" hardwired into its architecture, allowing it to adhere to core quantum rules like orbital parity that standard AI tools struggle with [15]. This enables superhuman intuition about molecular orbitals, predicting complex properties with chemical accuracy at a glance.
Diagram 2: A modern hybrid pipeline. AI performs initial high-throughput screening, which is then refined by human-knowledge filters to identify the most promising synthesizable candidates [6] [14].
The Scientist's Toolkit: Essential Research Reagents
The following table details key computational and data "reagents" essential for working in this field.
Table 3: Essential Research Tools and Resources
| Tool / Resource | Type | Primary Function | Relevance |
|---|---|---|---|
| Materials Project [6] [14] | Database | Repository of computed DFT properties for hundreds of thousands of known and hypothetical materials. | Provides data for hull construction, training ML models, and benchmarking. |
| ICSD [1] | Database | The Inorganic Crystal Structure Database, a comprehensive collection of experimentally characterized crystal structures. | Source of ground-truth data for "synthesized" materials; essential for training and validation. |
| pymatgen [14] | Software Library | A robust Python library for materials analysis. | Used for structure manipulation, analysis, and integrating with high-throughput workflows. |
| DFT (e.g., VASP) | Computational Method | The workhorse for first-principles energy calculations. | Used to compute the formation energy and electronic structure of candidate materials. |
| Charge Neutrality Filter [14] | Algorithm | A rule-based filter to check for net neutral charge in a composition. | A fast, initial screen to reduce the candidate pool before more expensive calculations. |
| SynthNN / ML Models [1] | AI Model | Deep learning models trained to predict synthesizability from composition or structure. | Provides a rapid, data-driven assessment of synthesizability that captures complex, learned chemical rules. |
The dichotomy between intuitive chemistry and computational physics is not merely academic but has profound practical implications for the pace and success of materials and drug discovery. The charge-balancing approach of intuitive chemistry offers speed and interpretability but suffers from low accuracy as a standalone metric. The energy above hull paradigm of computational physics provides a rigorous thermodynamic foundation but fails to account for kinetic synthesizability and is computationally costly. The future of the field lies not in choosing one over the other, but in strategically integrating them. The most powerful modern pipelines leverage scalable AI models that learn the implicit rules of chemistry, followed by targeted application of human-domain-knowledge filters and first-principles validation. This hybrid strategy maximizes the respective strengths of each philosophy, creating a more efficient and reliable path to discovering the next generation of functional materials.
Why Synthesizability Prediction is a Critical Bottleneck in Discovery Pipelines
Synthesizability prediction represents a critical bottleneck in modern discovery pipelines, standing between computational design and experimental realization. In both drug discovery and materials science, the ability to generate candidate molecules or materials computationally has dramatically outpaced our capacity to synthesize them in the laboratory. This disconnect creates a fundamental inefficiency where significant resources are wasted on characterizing hypothetically promising candidates that prove to be synthetically inaccessible. The core challenge lies in developing accurate predictive models that can distinguish between merely stable structures and those that can be practically synthesized. Traditional approaches have relied heavily on two main proxies: energy above hull (thermodynamic stability metrics) and charge balancing (heuristic chemical rules). However, as this technical guide will demonstrate, both approaches exhibit significant limitations, necessitating advanced machine learning solutions that can integrate multiple data sources and physical constraints to provide reliable synthesizability assessments.
Traditional Approaches and Their Limitations
Energy Above Hull as a Synthesizability Proxy
The energy above hull (Eₕᵤₗₗ) metric, derived from density functional theory (DFT) calculations, has served as a primary filter for synthesizability prediction in materials discovery. This approach calculates the energy difference between a material's formation enthalpy and the sum of formation enthalpies of its most stable decomposition products. The underlying assumption is that thermodynamically stable materials (those with low Eₕᵤₗₗ) are more likely to be synthesizable.
Key Limitations:
- Zero-Kelvin Approximation: Eₕᵤₗₗ is typically calculated at 0 K and 0 Pa, ignoring finite-temperature effects that govern actual synthetic accessibility [6] [16].
- Neglect of Kinetic Factors: The metric fails to account for kinetic barriers that may prevent an otherwise energetically favorable reaction from occurring [16] [17].
- Entropic Considerations: Eₕᵤₗₗ overlooks entropic contributions to materials stability that become significant under experimental conditions [16].
- High False Positive Rate: Studies demonstrate that numerous hypothetical materials with low Eₕᵤₗₗ have never been synthesized, while many metastable materials with higher Eₕᵤₗₗ exist experimentally [1] [17].
Charge Balancing as a Synthesizability Proxy
Charge balancing applies simple chemical heuristics, predicting synthesizability based on whether a material has a net neutral ionic charge according to common oxidation states. This approach mirrors traditional chemical intuition but proves insufficient for comprehensive synthesizability prediction.
Key Limitations:
- Limited Applicability: Only 37% of synthesized inorganic materials in the Inorganic Crystal Structure Database (ICSD) are charge-balanced according to common oxidation states [1].
- Domain-Specific Failure: Even among typically ionic compounds like binary cesium compounds, only 23% of known compounds are charge-balanced [1].
- Bonding Environment Insensitivity: The approach cannot account for different bonding environments in metallic alloys, covalent materials, or ionic solids [1].
- Over-simplification: Fails to capture the complex array of factors beyond charge neutrality that influence synthesizability.
Table 1: Performance Comparison of Traditional Synthesizability Prediction Methods
| Method | Underlying Principle | Key Limitations | Reported Precision |
|---|---|---|---|
| Energy Above Hull | Thermodynamic stability from DFT calculations | Ignores kinetic factors and temperature effects; computational expensive | Captures only ~50% of synthesized materials [1] |
| Charge Balancing | Net neutral ionic charge based on oxidation states | Inflexible; fails for many material classes; limited predictive value | 37% of known synthesized materials meet criteria [1] |
| SynthNN | Deep learning on known material compositions | Requires quality training data; limited by known chemical space | 7× higher precision than DFT-based methods [1] |
Advanced Machine Learning Approaches
Positive-Unlabeled Learning Frameworks
The scarcity of confirmed negative examples (verified unsynthesizable materials) has led to the adoption of Positive-Unlabeled (PU) learning frameworks. These approaches treat the synthesizability prediction as a semi-supervised problem, using known synthesized materials as positive examples and treating the rest of chemical space as unlabeled.
SynCoTrain Framework: This innovative approach employs a dual-classifier co-training framework using two complementary graph convolutional neural networks: SchNet and ALIGNN [17]. SchNet uses continuous convolution filters suitable for encoding atomic structures (a "physicist's perspective"), while ALIGNN directly encodes atomic bonds and bond angles (a "chemist's perspective"). The model iteratively exchanges predictions between classifiers to mitigate individual model bias and enhance generalizability [17].
Implementation Methodology:
- Data Collection: Curate known synthesized materials from databases like ICSD and Materials Project
- Feature Encoding: Represent crystal structures as graphs with nodes (atoms) and edges (bonds)
- Co-training Loop:
- Each classifier predicts on unlabeled data
- High-confidence predictions are exchanged between classifiers
- Models are retrained on expanded labeled sets
- Prediction Aggregation: Final synthesizability scores are determined by averaging classifier predictions
Performance: SynCoTrain demonstrates robust performance on oxide crystals, achieving high recall on both internal and leave-out test sets, establishing it as a reliable tool for synthesizability prediction while balancing dataset variability and computational efficiency [17].
Composition-Based and Structure-Based Models
Machine learning approaches for synthesizability prediction can be broadly categorized into composition-based and structure-based methods:
Composition-Based Models (e.g., SynthNN):
- Operate solely on chemical formulas without structural information
- Use learned atom embeddings (atom2vec) to represent chemical formulas
- Train on databases of synthesized materials augmented with artificially generated unsynthesized materials
- Can screen billions of candidate materials efficiently [1]
Structure-Based Models:
- Utilize crystal structure graphs as input
- Incorporate both compositional and structural features
- Generally more accurate but require structural information that may be unknown for novel materials [6]
Table 2: Machine Learning Approaches for Synthesizability Prediction
| Model | Input Type | Methodology | Applications | Key Advantages |
|---|---|---|---|---|
| SynthNN [1] | Composition | Deep learning with atom embeddings | General inorganic crystalline materials | No structural information required; high throughput |
| SynCoTrain [17] | Crystal structure | Dual-classifier PU learning with co-training | Oxide crystals | Reduced model bias; improved generalizability |
| Unified Composition-Structure [6] | Both composition and structure | Ensemble of composition and structure encoders | General materials discovery | Enhanced ranking accuracy; state-of-the-art performance |
Experimental Protocols and Validation
Validating Synthesizability Predictions
Robust experimental validation is crucial for assessing synthesizability prediction models. Recent research has established rigorous protocols for model evaluation and experimental verification.
Experimental Validation Workflow:
- Candidate Screening: Apply synthesizability models to large computational databases (e.g., Materials Project, GNoME)
- Precursor Selection: Use precursor-suggestion models (e.g., Retro-Rank-In) to identify viable solid-state precursors [6]
- Synthesis Planning: Predict synthesis parameters (temperature, atmosphere) using models trained on literature-mined corpora [6]
- High-Throughput Synthesis: Execute syntheses in automated laboratory platforms
- Characterization: Verify products using X-ray diffraction and other analytical techniques
Recent Experimental Results: In one landmark study, researchers applied a unified synthesizability model to screen 4.4 million computational structures, identifying 24 highly synthesizable candidates [6]. Subsequent synthesis experiments characterized 16 targets, successfully synthesizing 7 matched structures, including one completely novel and one previously unreported structure [6]. This demonstrates the practical utility of modern synthesizability prediction pipelines.
Drug Discovery Applications
In pharmaceutical research, synthesizability prediction has been integrated into generative AI workflows for molecular design. The Variational Autoencoder with Active Learning (VAE-AL) framework incorporates synthesizability assessment through nested active learning cycles [18]:
Workflow Implementation:
- Initial Generation: VAE generates novel molecular structures
- Inner AL Cycle: Generated molecules evaluated for drug-likeness and synthetic accessibility using chemoinformatic predictors
- Outer AL Cycle: Promising molecules undergo docking simulations as affinity oracles
- Iterative Refinement: Molecules meeting thresholds fine-tune the VAE in subsequent cycles
Experimental Success: This approach generated novel scaffolds for CDK2 and KRAS targets. For CDK2, 9 molecules were synthesized yielding 8 with in vitro activity, including one with nanomolar potency [18]. This demonstrates the tangible impact of integrating synthesizability prediction directly into molecular design pipelines.
Table 3: Key Research Reagent Solutions for Synthesizability Research
| Resource | Type | Function | Application Context |
|---|---|---|---|
| Materials Project Database [16] [1] | Computational materials database | Provides crystal structures and calculated properties for known and predicted materials | Training data for machine learning models; benchmark comparisons |
| ICSD [16] [1] | Experimental crystal structure database | Comprehensive collection of experimentally determined inorganic crystal structures | Ground truth data for synthesizable materials; model training and validation |
| Retrosynthesis Platforms (SYNTHIA, AiZynthFinder) [19] | Retrosynthesis prediction tools | Propose viable synthetic routes for target molecules | Synthesizability assessment for organic molecules and drug candidates |
| Ollisim | Metric | Synthetic accessibility score based on molecular complexity | Rapid screening of generated molecules in drug discovery |
| GNoME Database [6] [20] | Computational materials database | Contains millions of predicted crystal structures | Source of candidate materials for synthesizability prediction |
| ALIGNN & SchNet [17] | Graph neural network architectures | Encode crystal structures for machine learning predictions | Base models for structure-based synthesizability classification |
Visualizing Synthesizability Prediction Workflows
Integrated Drug Discovery Pipeline
Generative AI Drug Discovery with Active Learning
Materials Synthesizability Prediction
Materials Synthesizability Prediction Pipeline
Synthesizability prediction remains a critical bottleneck in discovery pipelines, but significant progress has been made in developing sophisticated computational approaches that move beyond traditional proxies like energy above hull and charge balancing. Modern machine learning frameworks, particularly those utilizing positive-unlabeled learning and integrating both compositional and structural information, demonstrate substantially improved performance in identifying synthetically accessible candidates. The successful experimental validation of these approaches across both materials science and drug discovery domains highlights their growing practical utility. As these methods continue to mature and integrate more deeply with generative design workflows, they promise to significantly accelerate the discovery and development of novel functional materials and therapeutic agents by focusing experimental resources on targets with the highest probability of synthetic success.
A Practical Guide to Implementing Synthesizability Assessment
The accurate prediction of a material's thermodynamic stability is a cornerstone of computational materials science. For decades, the energy above hull, derived from the construction of a convex hull in energy-composition space, has served as a primary metric for assessing this stability. Concurrently, empirical rules like charge balancing have provided a chemist's intuition for synthesizability. This technical guide details the methodology for convex hull construction in multi-component systems, framing it within a broader research thesis that compares the efficacy of energy above hull against charge balancing for predicting synthesizability. While the convex hull provides a rigorous thermodynamic foundation, recent machine learning approaches demonstrate that synthesizability encompasses kinetic and experimental factors beyond pure ground-state stability [6] [1].
Theoretical Foundation of the Convex Hull
Definition and Thermodynamic Significance
The convex hull of a chemical system is the lower envelope of formation energies for all known compounds within that system [21]. A phase diagram is constructed by calculating the thermodynamic phase equilibria of multicomponent systems, and the convex hull represents the minimum energy "envelope" in energy-composition space [11] [21].
The formation energy, (ΔEf), is the energy change upon forming a phase of interest from its constituent elements. For a phase composed of (N) components, it is calculated as: [ΔEf = E - \sumi^N{niμi}] where (E) is the total energy of the phase, (ni) is the total number of moles of component (i), and (μ_i) is the total energy (chemical potential) of component (i) [21]. This energy is typically normalized per atom for comparative analysis.
The energy above hull ((E{hull})) is the vertical distance in energy from a phase's formation energy to the convex hull surface at the same composition [11]. A phase with (E{hull} = 0) is thermodynamically stable, meaning it has no stable decomposition products. A positive (E{hull}) value represents the decomposition energy, (ΔEd), which is the energy released (per atom) when the phase decomposes to the most stable phases on the hull [11] [21].
Geometric Interpretation in Multi-Dimensional Space
In a multi-component system, the convex hull is constructed in (M-1)-dimensional composition space for an M-component system. The hull comprises stable phases (vertices) and the facets connecting them. An unstable phase residing inside the hull will decompose into a combination of the stable phases located at the vertices of the facet directly beneath it.
- Binary Systems (1D composition space): The hull is a line segment connecting stable phases. The energy above hull is the vertical distance to this line.
- Ternary Systems (2D composition space): The hull is a surface of triangles. The decomposition path of a metastable phase is determined by the three stable phases at the vertices of the triangle below it.
- Quaternary Systems (3D composition space): The hull is a volume of tetrahedra. The energy above hull is the distance to this complex surface [11].
The following diagram illustrates the logical relationship between a compound's energy, the convex hull construction, and its derived stability property.
Computational Methodology
Workflow for Convex Hull Construction
Constructing a reliable convex hull requires a systematic workflow, from data acquisition to stability analysis. The process must ensure consistency in the computational data used for all entries within a chemical system.
Data Acquisition and Energy Calculation Protocols
The accuracy of the hull is contingent on the quality and consistency of the input formation energies.
- Density Functional Theory (DFT): The most common method, but computationally intensive. Standard protocol involves using a consistent set of pseudopotentials, exchange-correlation functionals (e.g., PBE), and calculation parameters (k-point mesh, energy cutoffs) for all structures in the system [21].
- Density Functional Tight Binding (DFTB): Can be an order of magnitude faster than DFT for calculating formation energies and convex hulls, making it suitable for pre-screening in complex systems like SiC and ZnO [22].
- Machine Learning Force Fields (ML-FF): Emerging tools like CHGNET, trained on Materials Project data, can approximate DFT-level energies for rapid screening [11].
Key Consideration: For databases like the Materials Project, which employs a mixed GGA/GGA+U approach, it is critical to use the consistently corrected ComputedStructureEntry objects from the API when building a phase diagram to ensure energies are comparable [21].
Convex Hull Calculation with Pymatgen
The Python Materials Genomics (pymatgen) library provides a robust implementation for convex hull construction. The code snippet below demonstrates the standard procedure.
Calculating Energy Above Hull and Decomposition
The energy above hull is the primary stability metric, but understanding the decomposition pathway is equally important.
The stoichiometric coefficients ensure mass and charge balance between the target phase and its decomposition products [11].
Energy Above Hull vs. Charge Balancing for Synthesizability
While energy above hull is a powerful thermodynamic tool, its limitation lies in equating thermodynamic stability with synthesizability. Charge balancing, a foundational chemical rule, offers a complementary perspective. The table below summarizes the distinct advantages and limitations of these two approaches, highlighting why neither is sufficient alone for accurate synthesizability prediction.
Table 1: Comparison of Energy Above Hull and Charge Balancing as Synthesizability Metrics
| Feature | Energy Above Hull | Charge Balancing |
|---|---|---|
| Basis | Quantum-mechanical total energies [21] | Empirical chemical rules (oxidation states) [14] |
| Primary Output | Decomposition energy (eV/atom) [11] | "Allowed" or "Forbidden" classification [14] |
| Strengths | Quantitative; accounts for complex competing phases; physically rigorous [21] | Computationally cheap; intuitive; applicable without structure [1] [14] |
| Limitations | Ignores kinetics and synthesis conditions; fails for metastable phases [6] [1] | Inflexible; poor performance (only 37% of known materials are charge-balanced) [1] |
| Synthesizability Link | Necessary but not sufficient for ground-state stability [21] | Neither necessary nor sufficient for synthesizability [1] |
The inadequacy of both methods as standalone synthesizability proxies has driven the development of advanced machine learning models. These models learn complex patterns from vast databases of synthesized materials, capturing factors beyond thermodynamics and simple chemical rules [1] [23]. For instance, the Crystal Synthesis Large Language Model (CSLLM) framework achieves 98.6% accuracy in predicting synthesizability, significantly outperforming thermodynamic stability (74.1% based on Ehull ≥0.1 eV/atom) and kinetic stability (82.2%) [23]. This underscores the complex nature of synthesizability.
Experimental Protocols and Reagent Solutions
Validating Synthesizability Predictions
The ultimate test of any prediction is experimental validation. A common protocol involves solid-state synthesis guided by computational predictions.
- Candidate Selection: Identify target compounds with low energy above hull (e.g., < 50 meV/atom) and/or high scores from ML synthesizability models [6] [23].
- Precursor Selection: Use a precursor-suggestion model (e.g., Retro-Rank-In) or literature mining to identify viable solid-state precursors [6].
- Reaction Planning: Balance the chemical reaction and compute precursor quantities. Predict calcination temperature using models like SyntMTE [6].
- Synthesis & Characterization: Execute the synthesis in a high-throughput laboratory platform. Characterize the products using X-ray Diffraction (XRD) to verify the formation of the target phase [6].
Research Reagent Solutions
Table 2: Essential Materials and Tools for Computational-Experimental Synthesis Validation
| Item | Function | Example Tools / Materials |
|---|---|---|
| DFT Software | Calculate formation energies for hull construction. | VASP, Quantum ESPRESSO [21] |
| Material Databases | Source of crystal structures and energies for hull input. | Materials Project, ICSD, OQMD [23] [21] |
| Analysis Library | Construct phase diagrams and calculate Ehull. | Pymatgen [21] |
| Synthesizability Model | Predict likelihood of successful synthesis. | CSLLM [23], SynthNN [1], SynCoTrain [24] |
| Precursor Suggestion | Recommend viable solid-state precursors. | Retro-Rank-In [6] |
| High-Throughput Lab | Execute and scale synthesis experiments rapidly. | Automated solid-state synthesis platforms [6] |
| Characterization Tool | Verify crystal structure of synthesized product. | X-ray Diffraction (XRD) [6] |
Calculating the energy above hull via convex hull construction remains an indispensable tool for assessing the thermodynamic stability of materials in multi-component systems. Its rigorous physical foundation provides critical insights into decomposition energies and phase equilibria. However, within the broader context of synthesizability research, it is clear that thermodynamic stability alone is an incomplete picture. The empirical rules of charge balancing, while chemically intuitive, are also insufficient. The future of accurate synthesizability prediction lies in integrated approaches that combine the physical rigor of the convex hull with data-driven models that capture the kinetic, experimental, and technological factors governing successful synthesis. Integrating Ehull as a feature within advanced ML frameworks like CSLLM represents a promising path forward for bridging the gap between computational prediction and experimental realization of novel materials.
The prediction of synthesizable inorganic crystalline materials represents a significant challenge in materials science and drug development. Traditional computational discovery methods, such as calculating formation energies via density-functional theory (DFT), have identified millions of candidate materials with promising properties. However, these methods often fail to accurately predict which materials are synthetically accessible, creating a critical bottleneck in transforming theoretical innovations into real-world applications. Within this context, charge-balancing—the principle that stable inorganic compounds typically exhibit a net neutral ionic charge based on common oxidation states—has served as a foundational, chemically-motivated heuristic for predicting synthesizability.
Charge-balancing provides a computationally inexpensive filter for identifying potentially stable materials by ensuring electrical neutrality in ionic compounds. This approach operates on the fundamental chemical principle that the sum of oxidation states in a neutral compound must equal zero, and in an ion must equal the charge on that ion. Despite its chemical intuition, recent research demonstrates that charge-balancing alone cannot accurately predict synthesizable inorganic materials. Remarkably, among all inorganic materials that have already been synthesized, only 37% can be charge-balanced according to common oxidation states, highlighting the limitations of this approach and necessitating a deeper understanding of both the rules and their exceptions [1].
Theoretical Foundation of Oxidation States
Defining Oxidation States
Oxidation states (oxidation numbers) represent the hypothetical charge an atom would have if all bonds to atoms of different elements were completely ionic. They simplify the process of determining what is being oxidized and reduced in redox reactions and provide a method for tracking electron transfer in chemical processes. The oxidation state of an atom equals the total number of electrons that have been removed from (producing a positive oxidation state) or added to (producing a negative oxidation state) an element to reach its present state [25].
The concept can be illustrated through vanadium chemistry. Starting from elemental vanadium (oxidation state 0), removal of two electrons produces the V²⁺ ion with an oxidation state of +2. Subsequent removal of another electron produces V³⁺ with an oxidation state of +3. Further oxidation can form VO²⁺, where the vanadium maintains an oxidation state of +4, demonstrating that the oxidation state doesn't always equal the simple ionic charge [25] [26].
Fundamental Rules for Determining Oxidation States
The systematic determination of oxidation states follows these established rules [25] [26]:
- Elemental State: The oxidation state of an uncombined element is zero (e.g., Xe, Cl₂, S₈, C, Si).
- Neutral Compounds: The sum of oxidation states of all atoms in a neutral compound is zero.
- Ions: The sum of oxidation states of all atoms in an ion equals the charge on the ion.
- Electronegativity: The more electronegative element in a substance is assigned a negative oxidation state, while the less electronegative element is assigned a positive state.
Table 1: Common Oxidation States with Standard Exceptions
| Element | Usual Oxidation State | Exceptions |
|---|---|---|
| Group 1 metals | Always +1 | Handful of obscure compounds (e.g., Na⁻) |
| Group 2 metals | Always +2 | |
| Oxygen | Usually -2 | Peroxides (e.g., H₂O₂, -1), F₂O (+2) |
| Hydrogen | Usually +1 | Metal hydrides (e.g., NaH, -1) |
| Fluorine | Always -1 | |
| Chlorine | Usually -1 | Compounds with O or F (variable) |
Practical Application of Charge-Balancing Rules
Worked Examples of Oxidation State Calculation
Example 1: Chromium in Dichromate Ion (Cr₂O₇²⁻) The oxidation state of oxygen is -2. Let the oxidation state of Cr be ( n ). Sum of oxidation states = charge on ion: ( 2n + 7(-2) = -2 ) ( 2n - 14 = -2 ) ( 2n = 12 ) ( n = +6 ) Thus, chromium has an oxidation state of +6 in the dichromate ion [26].
Example 2: Sulfur in Sulfate (SO₄²⁻) and Sulfite (SO₃²⁻) Ions For sulfate (SO₄²⁻): Let sulfur be ( n ). ( n + 4(-2) = -2 ) → ( n - 8 = -2 ) → ( n = +6 ) For sulfite (SO₃²⁻): Let sulfur be ( n ). ( n + 3(-2) = -2 ) → ( n - 6 = -2 ) → ( n = +4 ) This explains the modern nomenclature: sulfate(VI) and sulfate(IV) [26].
Charge-Balancing in Multi-Element Compounds
For compounds containing multiple elements with variable oxidation states, additional chemical knowledge may be required. In CuSO₄, recognizing the compound as ionic containing copper ions and sulfate ions (SO₄²⁻) indicates the copper must be present as Cu²⁺ to achieve neutrality, giving copper an oxidation state of +2 [26].
When dealing with ions of charge greater than one, charge balances must account for stoichiometric coefficients. For calcium chloride (CaCl₂) in water, the charge balance is: ( 2[\text{Ca}^{2+}] + [\text{H}_3\text{O}^+] = [\text{Cl}^-] + [\text{OH}^-] ) The calcium ion concentration is multiplied by two because each ion carries two positive charges [27].
Limitations of Charge-Balancing in Synthesizability Prediction
Quantitative Assessment of Charge-Balancing Efficacy
While charge-balancing provides an intuitive filter for material stability, its performance as a standalone synthesizability predictor is limited. Research evaluating charge-balancing against databases of known materials reveals significant shortcomings:
Table 2: Efficacy of Charge-Balancing for Synthesizability Prediction
| Material Category | Percentage Charge-Balanced | Key Insight |
|---|---|---|
| All synthesized inorganic materials | 37% | Majority of known materials violate simple charge-balancing |
| Ionic binary cesium compounds | 23% | Even highly ionic systems frequently violate the rule |
| Theoretical screening precision | Lower than SynthNN | Machine learning significantly outperforms the method |
The inflexibility of the charge neutrality constraint cannot account for diverse bonding environments present across material classes, including metallic alloys, covalent materials, or ionic solids with complex coordination environments [1].
Comparison with Thermodynamic and Machine Learning Approaches
Alternative approaches to synthesizability prediction include DFT-calculated formation energies and emerging machine learning models:
- Thermodynamic Stability: Assesses formation energy relative to decomposition products, assuming synthesizable materials lack thermodynamically stable decomposition products. This approach captures only approximately 50% of synthesized inorganic crystalline materials due to failure to account for kinetic stabilization [1].
- Machine Learning Models: Methods like SynthNN (a deep learning synthesizability model) leverage the entire space of synthesized inorganic chemical compositions without requiring structural information. SynthNN identifies synthesizable materials with 7× higher precision than charge-balancing and completes screening tasks five orders of magnitude faster than human experts [1].
- Large Language Models: The Crystal Synthesis LLM (CSLLM) framework achieves 98.6% accuracy in predicting synthesizability, significantly outperforming both charge-balancing and traditional stability screening methods [23].
Synthesizability Prediction Methods Comparison
Advanced Methodologies: Beyond Simple Charge-Balancing
Machine Learning Approaches to Synthesizability
Modern synthesizability prediction has evolved beyond simple rule-based approaches to data-driven methods that learn complex patterns from known materials:
- Positive-Unlabeled Learning: Addresses the challenge that unsuccessful syntheses are rarely reported by treating artificially generated materials as unlabeled data, probabilistically reweighting them according to synthesizability likelihood [1] [28].
- Atom2Vec Representation: Represents chemical formulas through learned atom embedding matrices optimized alongside neural network parameters, learning optimal representations directly from the distribution of synthesized materials without predefined assumptions [1].
- Semi-Supervised Learning: Achieves true positive rates of 83.4% with estimated precision of 83.6% for predicting synthesizable stoichiometries, enabling construction of continuous synthesizability phase maps [28].
Experimental Validation Protocols
Validating synthesizability predictions requires systematic experimental protocols:
- Candidate Selection: Prioritize candidates from computational screening based on synthesizability scores (e.g., CLscore > 0.5 from PU learning models) [23].
- Precursor Identification: For solid-state synthesis, identify appropriate precursor combinations with reaction energy calculations and combinatorial analysis [23].
- Synthesis Methods: Employ either solid-state (high-temperature ceramic processing) or solution-based methods depending on material system [23].
- Characterization: Validate successful synthesis and phase purity through X-ray diffraction, elemental analysis, and property measurement [28].
Table 3: Research Reagent Solutions for Synthesis Validation
| Reagent/Material | Function in Research Context |
|---|---|
| Inorganic Crystal Structure Database (ICSD) | Source of experimentally validated synthesizable materials for training and benchmarking |
| CLscore Threshold (0.5) | PU learning metric for distinguishing synthesizable from non-synthesizable structures |
| Material String Representation | Text-based crystal structure representation for LLM processing |
| Solid-State Precursors | Binary and ternary compounds used as reactants in ceramic synthesis |
| Positive-Unlabeled Learning Algorithm | Handles incomplete labeling of artificially generated unsynthesized examples |
While charge-balancing rules founded on oxidation state principles provide essential chemical intuition for materials stability, they serve as an incomplete proxy for synthesizability prediction. The poor performance of charge-balancing alone (capturing only 37% of known materials) underscores the complexity of synthetic accessibility, which depends on kinetic factors, precursor selection, and specific reaction conditions beyond simple electrostatic considerations.
However, rather than abandoning charge-balancing principles entirely, the most promising approaches integrate these fundamental chemical concepts with data-driven methodologies. Machine learning models like SynthNN and CSLLM internalize charge-balancing relationships alongside other chemical patterns, effectively learning the principles of charge-balancing, chemical family relationships, and ionicity directly from the data of known materials [1] [23]. This integration enables more reliable computational materials screening, guiding experimental efforts toward compositions with higher synthetic accessibility and accelerating the discovery of novel functional materials for energy applications, catalysis, and pharmaceutical development.
Integrating Synthesizability Predictions into High-Throughput Computational Screens
The central challenge in modern computational materials discovery is no longer generating candidate structures but identifying which are synthetically accessible. High-throughput computational screening (HTCS) has successfully identified millions of hypothetical materials with promising properties, yet a significant gap remains between theoretical prediction and experimental realization. Traditional synthesizability assessments have primarily relied on two competing approaches: calculating the energy above hull (a measure of thermodynamic stability) or applying charge balancing principles (a chemical intuition-based rule). This technical guide examines advanced strategies for integrating synthesizability predictions into HTCS workflows, moving beyond these traditional metrics to combine data-driven machine learning, human knowledge embedding, and synthesis pathway prediction.
Traditional vs. Advanced Synthesizability Metrics
Limitations of Conventional Approaches
Traditional screening methods have significant limitations in predicting synthesizability. The energy above hull metric, derived from density functional theory (DFT) calculations, assesses thermodynamic stability but fails to account for kinetic and synthetic accessibility. Studies show this approach captures only approximately 50% of synthesized inorganic crystalline materials [1]. Materials with favorable formation energies often remain unsynthesized, while various metastable structures with less favorable formation energies are successfully synthesized [23].
Charge balancing, while chemically intuitive, performs even worse as a synthesizability predictor. Analysis reveals that only 37% of known synthesized compounds are charge-balanced according to common oxidation states, with this percentage dropping to just 23% for binary cesium compounds [1]. This poor performance stems from the inflexibility of charge neutrality constraints in accounting for diverse bonding environments in metallic alloys, covalent materials, and ionic solids.
Quantitative Comparison of Synthesizability Assessment Methods
Table 1: Performance comparison of synthesizability assessment methodologies
| Method | Key Principle | Accuracy | Advantages | Limitations |
|---|---|---|---|---|
| Energy Above Hull | Thermodynamic stability relative to convex hull | ~74% (0.1 eV/atom threshold) [23] | Strong theoretical foundation; Quantitative | Misses kinetically stabilized phases; Computationally expensive |
| Charge Balancing | Net neutral ionic charge based on oxidation states | Covers only 37% of known materials [1] | Computationally inexpensive; Chemically intuitive | Poor accuracy; Inflexible for different bonding types |
| Machine Learning (SynthNN) | Pattern recognition from known material compositions | 7× higher precision than DFT [1] | High throughput; Learns complex patterns | Black box; Dependent on training data quality |
| CSLLM Framework | LLM fine-tuned on crystal structure representations | 98.6% accuracy [23] | Highest accuracy; Predicts methods/precursors | Requires structure input; Computational intensive |
| Human Knowledge Filters | Domain expertise encoded as rules | Varies by filter combination [14] | Interpretable; Incorporates chemical intuition | May miss novel chemistry; Requires expert curation |
Machine Learning Approaches for Synthesizability Prediction
Composition-Based Models
Composition-based machine learning models predict synthesizability directly from chemical formulas without requiring structural information. The SynthNN model exemplifies this approach, leveraging the entire space of synthesized inorganic chemical compositions from databases like the Inorganic Crystal Structure Database (ICSD) [1]. The model employs a semi-supervised positive-unlabeled (PU) learning approach that treats unsynthesized materials as unlabeled data and probabilistically reweights them according to their likelihood of being synthesizable.
Experimental Protocol for Composition-Based Screening:
- Data Curation: Extract known synthesizable compositions from ICSD
- Negative Sample Generation: Create artificially generated unsynthesized materials
- Model Training: Implement atom2vec framework to learn optimal composition representations
- Validation: Benchmark against charge-balancing and random guessing baselines
- Screening: Apply trained model to rank candidate compositions by synthesizability probability
In comparative evaluations, SynthNN demonstrated 1.5× higher precision than the best human experts and completed screening tasks five orders of magnitude faster, highlighting the transformative potential of ML approaches [1].
Structure-Aware Models
Structure-aware models incorporate crystallographic information to enhance synthesizability predictions. The Crystal Synthesis Large Language Models (CSLLM) framework represents the state-of-the-art, achieving 98.6% accuracy in predicting synthesizability of 3D crystal structures [23]. This framework utilizes three specialized LLMs to predict synthesizability, suggest synthetic methods (>90% accuracy), and identify suitable precursors (80.2% success rate).
Experimental Protocol for Structure-Aware Screening:
- Dataset Construction: Curate balanced dataset with 70,120 synthesizable structures from ICSD and 80,000 non-synthesizable structures
- Structure Representation: Convert crystals to "material string" text representation incorporating lattice parameters, composition, atomic coordinates, and symmetry
- Model Fine-tuning: Domain-adapt general LLMs on crystal structure data
- Validation: Test model performance against thermodynamic and kinetic stability metrics
- Application: Screen theoretical structures and predict synthesis pathways
The exceptional performance of CSLLM stems from its comprehensive structure representation and domain-focused fine-tuning, which aligns the broad linguistic capabilities of LLMs with material-specific features critical to synthesizability [23].
Integration Frameworks and Workflows
Unified Screening Pipeline Architecture
Effective integration of synthesizability predictions requires systematic pipelines that combine multiple complementary approaches. Recent research demonstrates that unified frameworks integrating compositional and structural signals outperform single-method assessments [6].
Synthesizability Guided Screening Workflow
Human Knowledge Embedding as Filters
An alternative approach embeds chemical domain knowledge directly into screening pipelines through systematically designed filters. This methodology encodes expert intuition as computable rules for downselecting candidate materials [14].
Six-Filter Pipeline for Perovskite-Inspired Materials:
- Charge Neutrality Filter: Ensures net neutral ionic charge
- Electronegativity Balance Filter: Verifies most electronegative ion has most negative charge
- Unique Oxidation State Filter: Eliminates compounds with multiple oxidation states per element
- Oxidation State Frequency Filter: Removes compounds with uncommon oxidation states
- Intra-Phase Diagram Stoichiometry: Compares to existing compounds within same ternary diagram
- Cross-Phase Diagram Stoichiometry: Assesses common stoichiometries across related diagrams
Application of this filter pipeline to >100,000 novel compounds in 60 "perovskite-inspired" ternary phase diagrams successfully reduced the candidate pool to just 27 high-confidence candidates meeting all criteria [14].
Experimental Validation and Case Studies
MOF Stability Integration Case Study
A comprehensive study integrating four stability metrics with HTCS of metal-organic frameworks (MOFs) for CO₂ capture demonstrates the practical utility of multi-metric synthesizability assessment [29]. The research evaluated 15,219 hypothetical MOFs using:
Stability Assessment Protocol:
- Thermodynamic Stability: Evaluated through free energy calculations using molecular dynamics
- Mechanical Stability: Calculated from elastic constants (bulk, shear, Young's moduli)
- Thermal Stability: Predicted using machine learning models
- Activation Stability: Assessed via synthesis feasibility metrics
The screening identified that while Zn₄O metal nodes represented 33.5% of the original database, they comprised only 0.7% of top-performing CO₂ capture candidates after stability screening. Conversely, V₃O₃ metal nodes increased from 12.4% in the original set to 46.6% in the final stable, high-performing candidates [29].
Synthesis Pipeline Validation
Recent research validates the complete synthesizability-guided pipeline from prediction to experimental realization. In one study, researchers applied a combined compositional and structural synthesizability score to evaluate structures from major materials databases, identifying several hundred highly synthesizable candidates [6].
Experimental Validation Protocol:
- Candidate Identification: Screen 4.4 million computational structures
- Synthesizability Scoring: Apply rank-average ensemble of composition and structure models
- Precursor Selection: Use Retro-Rank-In model for precursor suggestion
- Condition Optimization: Apply SyntMTE for calcination temperature prediction
- High-Throughput Synthesis: Execute synthesis in automated laboratory platform
- Characterization: Validate products via X-ray diffraction
This pipeline successfully synthesized 7 of 16 target compounds within just three days, demonstrating the practical efficiency of integrated synthesizability prediction [6].
Research Reagent Solutions
Table 2: Essential research reagents and computational tools for synthesizability screening
| Resource | Type | Function | Example Sources |
|---|---|---|---|
| Materials Databases | Data | Source of known and hypothetical structures | Materials Project, ICSD, COD, GNoME, Alexandria [6] [23] |
| Retrosynthesis Software | Software | Predict synthetic pathways and precursors | SYNTHIA (12M+ building blocks) [30] [31] |
| High-Throughput Screening | Experimental | Rapid experimental validation | PubChem BioAssay, UNCLE stability analyzer [32] [33] |
| Machine Learning Models | Computational | Predict synthesizability from composition/structure | SynthNN, CSLLM, MTEncoder, Graph Neural Networks [1] [23] |
| Stability Metrics | Analytical | Assess thermodynamic and mechanical stability | Free energy calculations, elastic constants [29] |
| Human Knowledge Filters | Algorithmic | Encode chemical intuition as rules | Charge neutrality, electronegativity balance [14] |
Integrating synthesizability predictions into high-throughput computational screens represents a critical advancement toward bridging the gap between theoretical materials discovery and experimental realization. The emerging paradigm moves beyond the traditional dichotomy of energy above hull versus charge balancing toward integrated frameworks that combine data-driven machine learning, human knowledge embedding, and synthesis pathway prediction. The most successful implementations leverage multiple complementary approaches—compositional and structural models, thermodynamic and kinetic metrics, computational and experimental validation—to achieve unprecedented accuracy in identifying synthetically accessible materials. As these methodologies continue to mature, they promise to significantly accelerate the discovery and deployment of novel materials for energy, electronics, and pharmaceutical applications.
The discovery of novel crystalline materials represents a cornerstone of advancements in various technological fields, from photonics to energy storage. Computational models, particularly density functional theory (DFT), have successfully predicted millions of hypothetical compounds with promising properties. However, a significant bottleneck persists: the synthesizability of these predicted structures. The central challenge lies in distinguishing materials that are merely thermodynamically stable on a computer from those that can be experimentally realized in a laboratory. This case study explores this critical disconnect, framing the discussion within the ongoing research tension between traditional stability metrics, such as the energy above hull, and chemistry-inspired approaches, like charge balancing.
Current materials databases, such as the Materials Project, GNoME, and Alexandria, now contain over 4.4 million computationally proposed crystal structures, vastly outnumbering the catalog of experimentally synthesized compounds [6]. The primary method for prioritizing these candidates has historically been the formation energy or energy above hull calculated using DFT. While this is a useful first filter, it operates on a significant limitation: it models stability at zero Kelvin, effectively ignoring finite-temperature effects, entropic factors, and kinetic barriers that govern real-world synthetic accessibility [6]. Consequently, this approach often fails to identify which theoretically stable compounds are experimentally accessible. For instance, despite 21 SiO₂ structures being listed within 0.01 eV of the convex hull in the Materials Project, the common cristobalite phase is not among them, highlighting the practical shortcomings of relying solely on thermodynamic stability [6].
In response to this challenge, new paradigms for predicting synthesizability have emerged. These can be broadly categorized into two families: data-driven machine learning models that learn from the entire corpus of known materials, and human-knowledge-driven "filters" that encode chemical intuition. This case study will examine a successful implementation of a synthesizability-guided pipeline that led to the experimental synthesis of novel materials, providing a practical comparison of these methodologies and their performance against traditional stability metrics.
Methodology: A Synthesizability-Guided Discovery Pipeline
The following section details the integrated computational and experimental methodology used in a recent successful discovery campaign.
Integrated Computational-Experimental Workflow
The discovery process followed a structured pipeline that moved from initial candidate screening to experimental synthesis and characterization, with synthesizability prediction as the core decision-making component. The overall workflow is depicted in the diagram below.
The Synthesizability Model Architecture
The core innovation of the pipeline was a unified synthesizability model that integrated complementary signals from a material's composition and its crystal structure. This approach recognized that synthesizability is influenced by both elemental chemistry (precursor availability, redox constraints) and structural motifs (local coordination, packing stability) [6].
- Problem Formulation: Each candidate material was represented by its composition ( xc ) and its relaxed crystal structure ( xs ). The goal was to learn a score ( s(x) \in [0, 1] ) that estimates the probability that the compound can be experimentally synthesized [6].
- Data Curation: Training data was sourced from the Materials Project. A composition was labeled as synthesizable (( y=1 )) if any of its polymorphs had a counterpart in the Inorganic Crystal Structure Database (ICSD). Compositions where all polymorphs were flagged as "theoretical" were labeled as unsynthesizable (( y=0 )). The final dataset contained 49,318 synthesizable and 129,306 unsynthesizable compositions [6].
- Model Implementation: The model used two specialized encoders:
- A compositional transformer (MTEncoder) fine-tuned to output a synthesizability score from the chemical formula alone.
- A structural graph neural network (based on the JMP model) fine-tuned to output a synthesizability score from the crystal structure graph.
- Rank-Average Ensemble: During screening, the probabilities from both models were aggregated using a rank-average ensemble (Borda fusion). This method converted probabilities to ranks across the candidate pool and computed an average rank, providing a robust mechanism for prioritizing candidates [6].
Synthesis Planning and Experimental Execution
For candidates passing the synthesizability filter, the next step was to predict viable synthesis routes.
- Precursor Selection: The Retro-Rank-In model was used to produce a ranked list of viable solid-state precursors for each target material. This model was trained on literature-mined corpora of solid-state synthesis recipes [6].
- Process Parameter Prediction: The SyntMTE model predicted the calcination temperature required to form the target phase. Reactions were balanced, and precursor quantities were computed accordingly [6].
- Experimental High-Throughput Synthesis: Selected candidates were synthesized in an automated solid-state laboratory platform. The resulting products were characterized automatically by X-ray diffraction (XRD) to verify the formation of the target crystal structure. The entire experimental process for 16 target candidates was completed in just three days, demonstrating the efficiency of the guided approach [6].
Comparative Analysis: Synthesizability Models vs. Traditional Metrics
The performance of modern synthesizability prediction models can be quantitatively compared against traditional stability metrics. The data below summarizes key performance indicators from recent state-of-the-art studies.
Table 1: Performance Comparison of Synthesizability Assessment Methods
| Method | Key Principle | Reported Accuracy/Precision | Primary Limitation |
|---|---|---|---|
| Energy Above Hull [6] [1] | Thermodynamic stability at 0 K | ~50-74% of synthesized materials captured | Neglects finite-temperature effects and kinetics |
| Charge Balancing [1] [14] | Net neutral ionic charge | 23-37% of known compounds are charge-balanced | Inflexible; fails for metallic/covalent materials |
| Compositional ML (SynthNN) [1] | Data-driven model from known compositions | 7x higher precision than DFT formation energy | Lacks structural information |
| Structure-Based PU Learning [23] | Machine learning on crystal structures | 87.9% - 92.9% accuracy | Requires known crystal structure |
| CSLLM Framework [23] | Large Language Model fine-tuned on material strings | 98.6% accuracy in synthesizability prediction | Requires extensive data curation and tuning |
| Unified Model (This Case Study) [6] | Combined composition & structure signals | 7 of 16 targets successfully synthesized | Integration complexity of multiple models |
The data reveals a clear progression. Traditional rules like charge balancing, while chemically intuitive, are poor predictors, correctly classifying only 23% of binary cesium compounds and 37% of all known inorganic materials [1]. DFT-based stability, though foundational, fails to account for the kinetic and entropic factors of real synthesis, explaining why many low-energy-above-hull structures remain unsynthesized.
Machine learning models mark a significant improvement. The SynthNN model, which uses only compositional data, demonstrated 1.5x higher precision than the best human experts and completed the screening task five orders of magnitude faster [1]. The most accurate results come from models that leverage structural information. The Crystal Synthesis Large Language Model (CSLLM) framework achieved a remarkable 98.6% accuracy in predicting the synthesizability of 3D crystal structures, significantly outperforming thermodynamic (74.1%) and kinetic (82.2%) stability methods [23].
Detailed Experimental Protocols
This section provides the technical methodologies underpinning the computational and experimental work cited in this case study.
Protocol: Training a Unified Synthesizability Model
This protocol is adapted from the pipeline that successfully identified novel synthesizable materials [6].
Data Collection and Labeling:
- Source raw data from the Materials Project API.
- Extract all entries and their associated polymorphs.
- Label a composition as synthesizable (
y=1) if any of its polymorphs is not flagged as "theoretical" (indicating an ICSD match). Label a composition as unsynthesizable (y=0) only if all its polymorphs are theoretical. - Stratify the data into training, validation, and test splits (e.g., 80/10/10).
Model Architecture and Training:
- Composition Encoder: Initialize a pre-trained composition transformer (e.g., MTEncoder). Attach a multi-layer perceptron (MLP) head with a sigmoid output activation.
- Structure Encoder: Initialize a pre-trained crystal graph neural network (e.g., JMP model). Attach a separate MLP head with a sigmoid output.
- Training Loop: Fine-tune both encoders and their heads end-to-end using a binary cross-entropy loss function. Utilize early stopping based on the validation Area Under the Precision-Recall Curve (AUPRC). Training requires substantial computational resources (e.g., an NVIDIA H200 cluster).
Inference and Candidate Screening:
- For each candidate in the screening pool, obtain the synthesizability probabilities from both the composition model, ( sc(i) ), and the structure model, ( ss(i) ).
- Convert these probabilities into ranks within the candidate pool. The rank for candidate
ifrom modelmis ( 1 + \sum{j=1}^N \mathbf{1}[sm(j) < s_m(i)] ). - Compute the final
RankAvg(i)score as the average of the two normalized ranks. - Screen candidates by setting a threshold on the
RankAvgscore (e.g., >0.95) rather than on raw probabilities.
Protocol: Solid-State Synthesis and XRD Characterization
This protocol details the experimental validation of computationally predicted materials, as described in the case study [6] and the synthesis of ErCo₂In [34].
Precursor Preparation:
- Select high-purity elemental powders or precursor compounds (typically ≥99.9% purity) based on the output of the synthesis planning model (e.g., Retro-Rank-In).
- Use an analytical balance to weigh precursors according to the stoichiometry of the target compound, accounting for any stoichiometric adjustments from reaction balancing.
Homogenization and Pelletization:
- Transfer the powder mixture to a mixing apparatus (e.g., a ball mill jar with grinding media).
- Mix thoroughly for a defined period (e.g., 30-60 minutes) to ensure homogeneity.
- Optionally, press the mixed powder into a pellet using a hydraulic press to increase inter-particle contact and reaction kinetics.
High-Temperature Reaction:
- Place the pellet or loose powder into a crucible suitable for high temperatures (e.g., alumina, quartz).
- Load the crucible into a tube or box furnace.
- Evacuate the furnace tube and backfill with an inert gas (e.g., argon) to prevent oxidation. Repeat this process several times.
- Heat the sample according to a temperature profile predicted by models like SyntMTE. A typical profile may include:
- Ramp to an intermediate temperature (e.g., 500°C) for decarbonation.
- Ramp to the final calcination temperature (e.g., 1000-1300°C).
- Hold at the target temperature for several hours to days (e.g., 12-48 hours) to facilitate complete reaction and crystallization.
- Cool the sample to room temperature, either by turning off the furnace (furnace cooling) or by removing it (quenching).
Product Characterization by X-ray Diffraction (XRD):
- Gently grind a portion of the synthesized product into a fine powder.
- Load the powder into a sample holder for a powder X-ray diffractometer.
- Collect diffraction data over a relevant 2θ range (e.g., 10° to 80°) using Cu Kα radiation.
- Compare the experimental diffraction pattern to the simulated pattern of the target crystal structure to confirm successful synthesis.
Table 2: Key Resources for Computational and Experimental Materials Discovery
| Resource Name | Type | Primary Function | Relevance to Discovery |
|---|---|---|---|
| Materials Project [6] | Database | Repository of computed material properties and structures. | Source of initial candidate structures and training data for ML models. |
| Inorganic Crystal Structure Database (ICSD) [1] [23] | Database | Repository of experimentally determined inorganic crystal structures. | Gold-standard source for "synthesizable" labels in model training. |
| Pymatgen [14] | Software | Python library for materials analysis. | Used for structure manipulation, analysis, and integrating with DFT codes. |
| SHELX [34] [35] | Software | Package for crystal structure determination and refinement. | Essential for solving and refining crystal structures from single-crystal XRD data. |
| VESTA [35] | Software | 3D visualization program for crystal structures and volumetric data. | Visualizing atomic models, electron densities, and crystal morphology. |
| High-Purity Elements | Reagent | Raw materials for solid-state synthesis. | Starting point for synthesizing target compounds; purity is critical. |
| Argon Gas | Reagent | Inert atmosphere gas. | Prevents oxidation of precursors and products during high-temperature synthesis. |
| Arc Melter | Equipment | Apparatus for melting high-temperature materials. | Used for initial sample preparation of intermetallic compounds (e.g., ErCo₂In) [34]. |
| Tube Furnace | Equipment | High-temperature oven with controlled atmosphere. | Standard equipment for solid-state reactions under inert gas or vacuum. |
| X-ray Diffractometer | Equipment | Instrument for structural characterization. | Primary tool for verifying the crystal structure of synthesized products. |
This case study demonstrates a paradigm shift in computational materials discovery, moving beyond the rigid constraints of thermodynamic stability and simple chemical rules. The successful synthesis of novel materials, including one completely novel and one previously unreported structure, validates the integrated approach of combining compositional and structural synthesizability models [6]. The quantitative data clearly shows that modern data-driven synthesizability models—such as the unified model featured here and the CSLLM framework—significantly outperform traditional metrics like energy above hull and charge balancing in predicting experimental outcomes [6] [23].
The implication for researchers is profound. While formation energy remains a valuable initial filter for stability, it is an insufficient predictor of synthetic accessibility. Integrating advanced synthesizability models into discovery pipelines dramatically increases the likelihood of experimental success, saving considerable time and resources. The future of materials discovery lies in hybrid strategies that leverage the physical insights of traditional metrics, the predictive power of machine learning trained on comprehensive experimental data, and the efficiency of high-throughput automated laboratories. This synergistic approach promises to accelerate the translation of promising computational predictions into tangible, novel materials that address pressing technological challenges.
The discovery of new functional materials, whether for renewable energy or modern medicine, is fundamentally constrained by a single, critical property: synthesizability. A material may exhibit exceptional theoretical properties on a computer, but its practical value is only realized upon successful synthesis in a laboratory. In computational materials science, the assessment of synthesizability has long been dominated by two competing paradigms: thermodynamic stability metrics, such as the energy above hull, and chemically intuitive rules, such as charge balancing [14]. The energy above hull, derived from Density Functional Theory (DFT) calculations, measures a compound's thermodynamic stability relative to competing phases on the convex hull [1]. In contrast, charge balancing leverages foundational chemical principles to assess whether a compound can achieve a net neutral ionic charge using common oxidation states of its constituent elements [14].
Recent research indicates that while energy above hull is a valuable filter, it alone is an insufficient predictor of synthetic accessibility. Studies reveal it captures only approximately 50% of synthesized inorganic crystalline materials, largely because it overlooks finite-temperature effects, kinetic factors, and non-equilibrium synthesis pathways [1]. Charge balancing, while chemically intuitive, performs even worse, correctly identifying only about 37% of known synthesized compounds [1]. This performance gap highlights a critical insight: synthesizability is a multi-faceted property influenced by factors beyond simple thermodynamics or ionic charge.
This whitepaper explores the translation of synthesizability assessment principles, developed for inorganic crystalline materials, to the domain of organic molecule and drug candidate discovery. We examine state-of-the-art computational models that integrate multiple data modalities, provide detailed experimental protocols for validation, and present a roadmap for leveraging these cross-disciplinary principles to accelerate the development of novel therapeutic agents.
Core Principles: Synthesizability Assessment in Inorganic Materials
The evolution of synthesizability prediction for inorganic crystals provides a foundational framework for cross-disciplinary translation. The limitations of single-principle approaches have spurred the development of sophisticated, data-driven models.
Table 1: Comparison of Traditional Synthesizability Assessment Methods for Inorganic Crystals
| Method | Fundamental Principle | Reported Precision/Accuracy | Key Limitations |
|---|---|---|---|
| Energy Above Hull | Thermodynamic stability relative to competing phases [1] | Identifies ~50% of synthesized materials [1] | Neglects kinetic factors and finite-temperature effects [6] |
| Charge Balancing | Net neutral ionic charge using common oxidation states [14] | Identifies 37% of known compounds [1] | Overly rigid; fails for metallic/covalent materials [1] |
| Human Expert Screening | Application of domain knowledge and intuition [14] | Outperformed by ML models (SynthNN) in precision [1] | Slow, subjective, and difficult to scale [1] |
The most significant advances have come from machine learning (ML) models that learn synthesizability directly from data of known materials, rather than relying on predefined physical proxies. For instance, the SynthNN model leverages a deep learning architecture to predict synthesizability from chemical composition alone, achieving a precision 7 times higher than using DFT-calculated formation energies and outperforming human experts in head-to-head comparisons [1]. Remarkably, without explicit programming of chemical rules, SynthNN was found to learn the principles of charge-balancing and ionicity from the data itself [1].
Further progress is demonstrated by models that integrate both compositional and structural data. A unified synthesizability score combining signals from a compositional transformer (MTEncoder) and a structural graph neural network (GNN) achieved state-of-the-art performance, successfully guiding the experimental synthesis of seven novel materials from a candidate pool of millions [6]. This multi-modal approach captures complementary information: composition governs precursor chemistry and elemental properties, while structure captures local coordination and motif stability [6].
Concurrently, large language models (LLMs) have been adapted for this task. The Crystal Synthesis LLM (CSLLM) framework utilizes a fine-tuned LLM to predict the synthesizability of arbitrary 3D crystal structures, achieving a remarkable 98.6% accuracy, significantly outperforming traditional stability-based screening [23]. This framework extends its capability to also predict viable synthetic methods and precursor sets, providing a more comprehensive guide for experimentalists [23].
Translation to Organic Molecules and Drug Discovery
The principles governing inorganic material synthesizability find powerful analogues in organic chemistry. The transition from static, rule-based filters to dynamic, multi-faceted, data-driven models is equally relevant for drug candidate design.
Translational Analogies
- From Composition to Molecular Scaffolds: In inorganic chemistry, composition-based models like SynthNN learn from the entire space of synthesized compositions [1]. The analogue for organic molecules is learning from the vast landscape of known molecular scaffolds and functional groups. Graph-based representations, which explicitly encode atoms as nodes and bonds as edges, have become the standard for capturing this structural information [36].
- From Crystal Structure to 3D Conformation: The 3D atomic coordinates of a crystal structure are critical for predicting its stability and properties [6] [23]. Similarly, the 3D conformation of a drug molecule determines its binding affinity to a biological target. Geometric deep learning models that incorporate molecular spatial structure are advancing the accuracy of property prediction [36].
- From Charge Balancing to Synthetic Accessibility Score (SAS): The charge balancing rule is a simple, hard filter [14]. In organic chemistry, rules-based filters for synthetic accessibility have also been developed, but modern approaches use ML models trained on vast reaction databases to score how readily a molecule can be synthesized, providing a more nuanced and accurate assessment.
Machine Learning and Representation Learning
The core of this translational effort lies in molecular representation learning. This field has catalyzed a paradigm shift from manual descriptor engineering to automated feature extraction using deep learning [36]. Key representation modalities include:
- String-Based Representations: SMILES and SELFIES provide compact, sequential encodings of molecular structure [36].
- Graph-Based Representations: Molecular graphs explicitly model atoms and bonds, forming the backbone of modern GNNs for property prediction [36].
- 3D Representations: These capture spatial geometry and electronic features, which are critical for modeling interactions, much like crystal structure is for inorganic materials [36].
Hybrid models that fuse multiple representation modalities—such as graphs, sequences, and quantum chemical descriptors—are emerging as the most powerful approach, mirroring the success of multi-modal models in inorganic crystallography [6] [36]. These models can be pre-trained on large, unlabeled molecular datasets via self-supervised learning (SSL) to learn rich, general-purpose representations before being fine-tuned for specific tasks like synthesizability prediction [36].
Experimental Protocols and Workflows
Translating computational predictions into tangible materials requires robust experimental workflows. The following section details a proven pipeline and the essential toolkit for experimental validation.
Detailed Experimental Protocol: A Synthesizability-Guided Pipeline
This protocol is adapted from high-throughput materials discovery campaigns [6].
1. Candidate Screening and Prioritization:
- Input Pool: Begin with a large database of candidate structures (e.g., 4.4 million for inorganic crystals [6] or a virtual chemical library for organic molecules).
- Synthesizability Scoring: Apply a trained ML model (e.g., a unified composition/structure model [6] or a specialized LLM like CSLLM [23]) to assign a synthesizability score to each candidate.
- Ranking and Filtering: Aggregate model predictions using a rank-average ensemble. Apply subsequent filters based on domain knowledge (e.g., removing compounds with toxic elements or unrealistic oxidation states) to arrive at a shortlist of high-priority targets (e.g., ~500 from an initial 4.4 million [6]).
2. Synthesis Planning:
- Precursor Identification: For a given target compound, apply a precursor-suggestion model (e.g., Retro-Rank-In [6]) to generate a ranked list of viable solid-state or solution precursors.
- Reaction Parameter Prediction: Use a reaction condition model (e.g., SyntMTE [6]) to predict key parameters such as calcination temperature for solid-state reactions or solvent, catalyst, and temperature for organic reactions. Balance the chemical reaction and compute precursor quantities.
3. High-Throughput Synthesis:
- Automated Laboratory Platform: Execute the synthesis recipes using an automated platform. For solid-state inorganic synthesis, this involves precise weighing, mixing, and heating in a furnace. For organic molecules, this may involve automated liquid-handling systems and parallel reactors.
- Process: The synthesis is conducted based on the predicted parameters from the previous step.
4. Product Characterization and Validation:
- Technique: Characterize the resulting products using X-ray Diffraction (XRD) for inorganic crystals or analytical techniques like Liquid Chromatography-Mass Spectrometry (LC-MS) and Nuclear Magnetic Resonance (NMR) for organic molecules.
- Analysis: Automate the analysis of the characterization data to verify if the synthesis produced the target compound. Successful matches confirm the prediction, while failures provide valuable data for model refinement.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents and Materials for Experimental Validation
| Reagent/Material | Function in Workflow | Specific Examples & Notes |
|---|---|---|
| Solid-State Precursors | Base reactants for inorganic solid-state synthesis; purity is critical for reproducibility. | Metal oxides, carbonates, etc. Platinoid group elements are often excluded for cost/availability [6]. |
| Organic Building Blocks | Functionalized molecules serving as precursors for organic synthesis or MOF construction. | MOF linkers, metallic centers; toxicity is a key screening parameter for biocompatible MOFs [37]. |
| Automated Synthesis Platform | Enables high-throughput, reproducible execution of synthesis recipes with minimal human error. | Robotic arms, automated furnaces, liquid-handling systems [6]. |
| Characterization Equipment | Verifies the success of synthesis by determining the structure and composition of the product. | X-ray Diffractometer (XRD) for crystals [6]; LC-MS, NMR for organic molecules. |
| Computational Resources | Runs large-scale ML models for screening and synthesis planning. | NVIDIA H200 cluster for model training/inference [6]. |
Data Presentation and Visualization
Quantitative benchmarking is essential for evaluating the performance of different synthesizability assessment methods.
Table 3: Quantitative Performance of Advanced Synthesizability Models
| Model Name | Input Data Type | Key Architectural Innovation | Reported Performance |
|---|---|---|---|
| SynthNN [1] | Composition only | Deep learning model (SynthNN) using atom2vec embeddings in a PU-learning framework. | 7x higher precision than DFT formation energy; outperformed all 20 human experts. |
| Unified Score Model [6] | Composition & Structure | Ensemble of compositional transformer (MTEncoder) and structural GNN (JMP-derived). | Successfully synthesized 7 of 16 predicted novel inorganic targets in 3-day experimental cycle. |
| CSLLM Framework [23] | Crystal Structure (Text) | Three specialized LLMs fine-tuned on a comprehensive dataset of material strings. | 98.6% accuracy in synthesizability prediction; >90% accuracy in method/precursor classification. |
| Filter Pipeline [14] | Composition (Human Rules) | Six sequential filters embedding chemical knowledge (e.g., charge neutrality, stoichiometry). | Downselected >100,000 novel compounds to 27 high-priority candidates. |
The logical relationship between different synthesizability concepts and the role of ML models in integrating them can be visualized as follows:
The transition from evaluating synthesizability based on single principles like energy above hull or charge balancing to integrated, multi-modal machine learning models represents a significant leap forward for computational materials discovery. The translation of these principles from inorganic crystals to organic molecules and drug candidates is not merely an analogy but a viable research program. By leveraging advanced molecular representations—particularly graph-based and 3D-aware models—and adopting hybrid ML architectures that fuse chemical, structural, and reaction data, the drug discovery pipeline can be substantially accelerated.
The future of this interdisciplinary field lies in the continued development of generative models for de novo molecular design constrained by synthesizability and property requirements, the creation of larger and more standardized datasets of successful and failed synthetic attempts, and the tighter integration of robotic experimentation for closed-loop discovery. As models like CSLLM demonstrate, the ultimate goal is a comprehensive system that not only identifies synthesizable candidates but also proposes viable synthesis routes and precursors, thereby bridging the gap between in-silico prediction and real-world laboratory synthesis for both advanced materials and life-saving therapeutics.
Overcoming Limitations and Enhancing Prediction Accuracy
The prediction of synthesizable materials is a cornerstone of computational materials discovery. While simple heuristics like charge balancing are historically used to assess stability, they frequently fail to identify known, experimentally realized compounds. This whitepaper examines the fundamental limitations of charge balancing as a standalone predictor and frames its shortcomings within the critical context of energy-above-hull (Ehull) and synthesizability research. We present quantitative evidence of these failures, detail advanced methodologies that overcome these limitations, and provide a practical toolkit for researchers navigating the complex landscape from theoretical prediction to experimental realization.
Charge balancing, rooted in the principle of achieving electroneutrality in ionic compounds, has long served as a first-pass filter for predicting stable inorganic materials. The underlying assumption is that compositions with a net charge of zero are more likely to form stable crystalline structures. However, the landscape of known materials is replete with examples that defy this simple rule, revealing its significant limitations.
The core issue is that charge balancing is a necessary but insufficient condition for stability. It is a static, compositional rule that ignores the dynamic, multi-dimensional nature of thermodynamic stability and kinetic synthesizability. As materials discovery increasingly relies on high-throughput computational screening, the failure of this simplistic metric creates a critical bottleneck, guiding researchers away from viable candidates and toward dead ends. This document explores the quantitative and theoretical underpinnings of these failures, providing a framework for more robust prediction methodologies essential for researchers and scientists in solid-state chemistry and related fields.
Quantitative Evidence: Documented Failures of Charge Balancing
Empirical data from large-scale materials databases and targeted experimental studies consistently demonstrate that charge balancing alone is a poor predictor of synthesizability. The following table summarizes key quantitative findings from recent research.
Table 1: Documented Evidence of Charge Balancing Limitations
| Evidence Type | Source/Study | Key Finding | Implication for Charge Balancing |
|---|---|---|---|
| Human-curated Ternary Oxides | Analysis of 4,103 ternary oxides [16] | Identified numerous synthesized compounds that would be deemed unstable by simple charge-balancing heuristics. | Charge balancing fails to account for complex bonding and kinetic stabilization. |
| Synthesizability-Guided Pipeline | Screening of 4.4M computational structures [6] | Successfully synthesized 7 of 16 predicted targets, many of which would not be prioritized by charge balancing alone. | Advanced, integrated models outperform single-metric rules. |
| Energy-Above-Hull Analysis | Community Discussion on Ehull [11] | A phase can be metastable (Ehull > 0) yet still be synthesizable (e.g., BaTaNO2 at 32 meV/atom above hull). | Thermodynamic metrics like Ehull provide a more nuanced view of stability than charge balancing. |
| "Balance" Challenges in SSEs | Review of Solid-State Electrolytes [38] | Highlights the need to balance multiple interdependent properties (e.g., cost vs. conductivity, mechanical property vs. conductivity), which single-factor analysis cannot address. | Real-world material viability depends on a "balance" of properties, not a single rule. |
The data clearly indicates that a paradigm shift is required, moving from isolated compositional checks to integrated models that account for thermodynamics, kinetics, and experimental feasibility.
Theoretical Foundations: Why Charge Balancing Fails
The failure of charge balancing can be attributed to its neglect of several fundamental principles of solid-state chemistry and synthesis.
Over-reliance on Thermodynamic Stability at 0K
Charge balancing is often used as a crude proxy for thermodynamic stability. However, true stability is more accurately assessed by a material's energy relative to all other competing phases in its chemical space—its energy above the convex hull (Ehull) [11]. Ehull is a rigorous metric that calculates the energy cost for a compound to decompose into a set of more stable phases on the convex hull. A compound with an Ehull of 0 meV/atom is thermodynamically stable, while one with a positive value is metastable. The crucial insight is that many metastable materials (Ehull > 0) are synthesizable because the kinetic barriers to decomposition are high [11]. Charge balancing cannot capture this nuance.
Neglect of Kinetic and Synthetic Factors
A material's existence is not solely determined by thermodynamics but also by the kinetic pathway of its synthesis. Key factors ignored by charge balancing include:
- Reaction Kinetics: The activation energy barriers of solid-state reactions can prevent the formation of the thermodynamic ground state and instead trap a metastable phase [16].
- Synthesis Conditions: Parameters such as temperature, pressure, and atmosphere dramatically influence which phase is formed. A compound may be unstable under ambient conditions but readily synthesized under high pressure or in a controlled atmosphere [38].
- Entropic Contributions: Stability at finite temperature is governed by Gibbs free energy, G = H - TS. Charge balancing, much like a 0K DFT calculation, ignores the entropic (TS) contributions that can stabilize a phase at higher temperatures [16].
The Complexity of Multi-element Systems
In ternary, quaternary, and higher-order systems, the concept of "charge balance" becomes increasingly ambiguous. The decomposition pathways are complex, and stable phases often exist in regions of the phase diagram where simple ionic charge counting does not apply. As explained in community discussions, the convex hull must be constructed in multi-dimensional composition space, and the decomposition products of a compound can be a mixture of several other phases with different stoichiometries [11].
Advanced Prediction Methodologies
To overcome the limitations of charge balancing, the field has moved toward integrated, data-driven models that directly predict synthesizability.
Integrated Synthesizability Models
A state-of-the-art approach involves building machine learning models that use both compositional and structural features. As demonstrated by Prein et al., a combined model can be represented as an ensemble of composition and structure encoders [6]:
𝐳c = fc(xc; θc), 𝐳s = fs(xs; θs)
where fc is a compositional transformer and fs is a crystal graph neural network. Their outputs are combined via a rank-average ensemble to prioritize candidates with the highest predicted synthesizability, a method proven to successfully guide experimental synthesis [6].
Positive-Unlabeled (PU) Learning from Literature Data
The scarcity of documented failed experiments presents a major challenge for model training. Positive-Unlabeled (PU) learning offers a solution by training on known synthesized materials (positives) and a large set of hypothetical materials (unlabeled, which may contain both positive and negative examples). This technique has been successfully applied to predict the solid-state synthesizability of ternary oxides, identifying 134 likely synthesizable compositions from a pool of over 4,000 hypotheticals [16].
Experimental Workflow for Validating Synthesizability
The following diagram illustrates a modern, closed-loop pipeline for materials discovery that integrates computational prediction with experimental validation.
Diagram 1: Synthesizability prediction and validation workflow. This integrated pipeline rapidly transitions from in-silico screening to experimental synthesis, successfully validating new materials in days [6].
For researchers embarking on synthesizability prediction and validation, the following tools and resources are essential.
Table 2: Key Research Reagent Solutions for Synthesizability Studies
| Item/Resource | Function/Brief Explanation | Relevance to Synthesizability |
|---|---|---|
| Computational Databases (MP, GNoME, Alexandria) | Provide pre-calculated formation energies, Ehull values, and crystal structures for hundreds of thousands of known and predicted materials [6] [11]. | Foundation for high-throughput screening and obtaining stability metrics like Ehull. |
| Pymatgen | A robust Python library for materials analysis. Essential for programmatically accessing database APIs and constructing phase diagrams to calculate Ehull [11]. | Critical for implementing the convex hull analyses and decomposition energy calculations that surpass charge balancing. |
| Solid-State Precursors (e.g., Carbonates, Oxides) | High-purity, fine-powder precursors are the starting materials for solid-state synthesis [16]. | The choice and quality of precursors directly impact the kinetics and success of a synthesis reaction. |
| Automated Synthesis Lab | High-throughput platform for executing solid-state reactions with precise control over temperature and atmosphere [6]. | Enables rapid experimental validation of computational predictions at scale, closing the discovery loop. |
| X-ray Diffractometer (XRD) | Instrument for determining the crystal structure of a synthesized powder and comparing it to the predicted target structure [6] [16]. | The definitive tool for verifying whether the synthesized product matches the predicted material. |
Charge balancing is an outdated and unreliable predictor for the synthesizability of known materials. Its failures are systematic and rooted in a fundamental oversimplification of the complex thermodynamic and kinetic realities of solid-state synthesis. The research community must embrace a new paradigm centered on rigorous metrics like the energy-above-hull and sophisticated, data-driven synthesizability models that integrate both compositional and structural information. The successful application of these advanced methods, leading to the rapid discovery of new materials in automated laboratories, marks the way forward. By moving beyond charge balancing, researchers can accelerate the identification of novel materials critical for technological advancement.
In the computational-driven paradigm of materials discovery, the energy above the convex hull (Ehull) has long served as a primary metric for assessing candidate material stability. This thermodynamic quantity measures a compound's energy distance from the lowest-energy phase equilibrium at zero temperature, with materials on the convex hull (Ehull = 0 eV/atom) considered thermodynamically stable and those with positive values deemed metastable or unstable [10] [11]. Conventional screening workflows, particularly those employed in high-throughput computational initiatives, have heavily relied on Ehull thresholds—often 0-200 meV/atom—as proxies for synthesizability, operating under the assumption that thermodynamic stability strongly correlates with experimental realizability [10] [39].
However, a significant and persistent gap exists between thermodynamic stability predictions and experimental synthesizability, creating a critical bottleneck in materials discovery pipelines. While materials with highly positive Ehull values are generally unsynthesizable, numerous metastable compounds (Ehull > 0) are routinely synthesized, and many computationally predicted "stable" materials (Ehull ≈ 0) remain elusive in the laboratory [23] [17] [39]. This discrepancy stems from the complex, multi-faceted nature of synthesis, which encompasses kinetic barriers, precursor availability, reaction pathways, and experimental conditions—factors largely unaccounted for in pure thermodynamic stability assessments [17] [6].
This whitepaper examines the fundamental limitations of Ehull as a standalone synthesizability metric and explores emerging computational frameworks that bridge this critical gap. By integrating insights from recent advances in machine learning and materials informatics, we present a more nuanced understanding of synthesizability that transcends traditional thermodynamic proxies, offering researchers in materials science and drug development more reliable guidance for prioritizing candidate materials.
The Theoretical Foundation: What Ehull Can and Cannot Predict
Computational Basis of Energy Above Hull
The energy above hull derives from the construction of convex hull phase diagrams in energy-composition space. For a given composition, the convex hull represents the set of lowest-energy phases and their mixtures, forming a multidimensional envelope [11]. The Ehull for any phase is calculated as the vertical energy distance to this lower envelope, representing the energy penalty if the phase were to decompose into the most stable competing phases at equilibrium [11]. In practice, for a compound ABO₂N with decomposition products 2/3 Ba₄Ta₂O₉ + 7/45 Ba(TaN₂)₂ + 8/45 Ta₃N₅, the Ehull is computed as:
Ehull = E(ABO₂N) - [⅔E(Ba₄Ta₂O₉) + 7/45E(Ba(TaN₂)₂) + 8/45E(Ta₃N₅)] [11]
where all energies are normalized per atom [11]. This calculation requires knowing both the target phase energy and the energies of all competing phases in the relevant chemical space, typically obtained through density functional theory (DFT) calculations [11] [40].
Inherent Limitations of the Ehull Metric
Despite its mathematical rigor, Ehull possesses several inherent limitations as a synthesizability predictor:
Zero-Kelvin Approximation: Standard Ehull calculations derive from DFT simulations at 0K, ignoring temperature-dependent entropic effects that significantly influence real-world phase stability [39]. Finite-temperature effects can dramatically alter relative phase stabilities, potentially stabilizing high-entropy phases at synthesis conditions [39].
Kinetic Blindness: The metric is purely thermodynamic and contains no information about kinetic barriers to phase formation or decomposition [17]. A material with favorable Ehull may have prohibitively high nucleation barriers, while metastable phases can persist indefinitely due to kinetic trapping [17] [39].
Synthesis Condition Insensitivity: Ehull calculations cannot account for the profound influence of synthesis environment (pressure, pH, precursor properties, external fields) on phase selection [17] [6]. Materials unstable under standard conditions may become accessible through specialized synthesis pathways.
Completeness Dependency: The accuracy of Ehull depends on complete knowledge of all competing phases in a chemical system [11]. Omitted phases—whether due to computational cost or undiscovered compounds—can lead to significant underestimation of the true decomposition energy [11].
Table 1: Quantitative Comparison of Synthesizability Prediction Approaches
| Method | Theoretical Basis | Key Metrics | Reported Accuracy | Primary Limitations |
|---|---|---|---|---|
| Ehull/DFT Stability [10] [11] | Thermodynamic equilibrium at 0K | Energy above convex hull (eV/atom) | 74.1% (as synthesizability proxy) [23] | Ignores kinetics, temperature effects, synthesis conditions |
| Phonon Stability [23] | Dynamic/kinetic stability | Presence of imaginary frequencies | 82.2% (as synthesizability proxy) [23] | Computationally expensive; synthesizable materials may have imaginary frequencies |
| PU Learning Models [17] [28] | Semi-supervised classification | CLscore, recall, precision | 83.4-87.9% recall [17] [28] | Dependent on quality of unlabeled data; model-specific biases |
| Dual Classifier (SynCoTrain) [17] | Co-training with GCNNs | Recall on test sets | High recall on oxides [17] | Architecture-dependent performance; requires careful hyperparameter tuning |
| CSLLM Framework [23] | Fine-tuned large language models | Classification accuracy | 98.6% synthesizability accuracy [23] | Requires extensive training data; computational resource demands |
Beyond Thermodynamics: The Multi-Faceted Nature of Synthesis
Kinetic and Technological Factors in Synthesis
The successful laboratory realization of a material depends on numerous factors beyond thermodynamic stability, creating the observed gap between Ehull predictions and experimental outcomes:
Kinetic Stabilization Mechanisms: Metastable phases (Ehull > 0) can be synthesized when kinetic barriers prevent transformation to more stable configurations. These barriers may arise from slow diffusion rates, high nucleation barriers, or intermediate phases that dominate the reaction pathway [17] [39]. For example, diamond remains metastable relative to graphite under ambient conditions but persists indefinitely due to immense transformation barriers [39].
Synthesis Method Dependence: Technological capabilities fundamentally constrain synthesizability. Materials requiring specific synthesis techniques (e.g., carbothermal shock, high-pressure synthesis, molecular beam epitaxy) may remain inaccessible until these methods are developed [17]. The recent synthesis of high-entropy alloys via carbothermal shock exemplifies how method innovation unlocks previously inaccessible materials [17].
Precursor and Pathway Considerations: Suitable precursor selection and reaction pathway design critically influence synthesis outcomes, independent of target phase thermodynamics [23] [6]. A compound with favorable Ehull may form only from specific precursors under narrow processing conditions, while computationally expensive decomposition energy calculations often fail to predict actual laboratory behavior [11].
The Data Challenge in Synthesizability Prediction
A fundamental obstacle in developing accurate synthesizability models is the absence of comprehensive negative data—reliable records of failed synthesis attempts [17]. This asymmetry arises from publication bias, where unsuccessful experiments are rarely documented in scientific literature or public databases [17]. Consequently, machine learning approaches must employ specialized techniques such as Positive-Unlabeled (PU) learning, which treats unreported materials as unlabeled rather than explicitly unsynthesizable [17] [28].
Emerging Computational Frameworks for Synthesizability Prediction
Machine Learning Approaches
Recent advances in machine learning have produced sophisticated models that directly address the limitations of pure thermodynamic assessments:
The CSLLM Framework: The Crystal Synthesis Large Language Model represents a breakthrough approach utilizing three specialized LLMs to predict synthesizability, synthetic methods, and suitable precursors respectively [23]. Trained on a balanced dataset of 70,120 synthesizable structures from the Inorganic Crystal Structure Database and 80,000 non-synthesizable structures identified through PU learning, the framework achieves 98.6% accuracy in synthesizability classification—significantly outperforming traditional Ehull (74.1%) and phonon stability (82.2%) metrics [23]. The model employs a novel "material string" representation that efficiently encodes essential crystal information for LLM processing [23].
SynCoTrain: This semi-supervised model employs a dual-classifier co-training framework with two graph convolutional neural networks (SchNet and ALIGNN) that iteratively exchange predictions to mitigate individual model biases [17]. By leveraging complementary architectural perspectives—ALIGNN encodes atomic bonds and angles while SchNet uses continuous convolution filters—the approach demonstrates robust performance on oxide crystals while balancing dataset variability and computational efficiency [17].
Composition-Structure Integrated Models: More recent frameworks integrate both compositional and structural descriptors through separate encoders (compositional transformers and graph neural networks) whose predictions are aggregated via rank-average ensemble methods [6]. This hybrid approach captures both elemental chemistry constraints and coordination-motif stability, enabling more holistic synthesizability assessments [6].
Table 2: Experimental Methodologies for Synthesizability Prediction
| Method/Protocol | Key Implementation Details | Data Requirements | Validation Approach | Domain Application |
|---|---|---|---|---|
| PU Learning with GCNNs [17] | Iterative labeling of unlabeled data using classifier confidence | Known synthesizable materials + large unlabeled set | Recall on internal/leave-out test sets | Oxide crystals; general inorganic crystals |
| CSLLM Fine-tuning [23] | Domain adaptation of LLMs using material string representation | 150,120 crystal structures with synthesizability labels | Hold-out test set accuracy (98.6%) | Arbitrary 3D crystal structures |
| Rank-Average Ensemble [6] | Borda fusion of composition and structure model predictions | 49,318 synthesizable + 129,306 unsynthesizable compositions | Prospective experimental validation (7/16 successes) | High-throughput screening of 4.4M structures |
| Retrosynthetic Planning [6] | Precursor suggestion + calcination temperature prediction | Literature-mined solid-state synthesis recipes | Experimental execution in automated laboratory | Oxide materials discovery |
Experimental Validation and Prospective Discovery
The ultimate validation of synthesizability models comes through prospective experimental testing—physically synthesizing predicted candidates in laboratory settings. In one notable demonstration, researchers applied a synthesizability-guided pipeline to screen over 4.4 million computational structures, identifying 24 high-priority candidates predicted to be highly synthesizable [6]. Through automated synthesis and characterization, they successfully realized 7 of 16 targeted compounds, including one completely novel and one previously unreported structure [6]. This successful translation from computational prediction to experimental realization highlights the practical utility of advanced synthesizability frameworks that transcend Ehull-based screening.
Table 3: Research Reagent Solutions for Synthesizability Assessment
| Resource/Tool | Function/Purpose | Application Context | Access/Implementation |
|---|---|---|---|
| CLscore [23] [17] | PU-learning based synthesizability score (0-1) | Initial screening of theoretical structures | Pre-trained models on materials databases |
| Material String Representation [23] | Compact text encoding of crystal structures | LLM-based synthesizability prediction | Custom conversion from CIF/POSCAR files |
| Retro-Rank-In [6] | Precursor suggestion model | Retrosynthetic planning for solid-state synthesis | Literature-mined precursor relationships |
| SyntMTE [6] | Calcination temperature prediction | Synthesis parameter optimization | Regression models trained on experimental data |
| Convex Hull Construction [11] | Phase stability assessment via Ehull | Thermodynamic stability screening | Pymatgen phase diagram module |
| Universal Interatomic Potentials [40] | Rapid energy and force estimation | High-throughput stability screening | MLIPs (CHGNET) trained on DFT data |
Integrated Workflows: Bridging the Gap Between Computation and Experiment
Effective synthesizability assessment requires integrating multiple computational and experimental approaches into a coherent workflow. The following diagrams illustrate both the conceptual framework and practical implementation of synthesizability-guided materials discovery:
Diagram 1: The Synthesizability Funnel - Progressive filtering from computational stability to experimental realization.
Diagram 2: CSLLM Synthesizability Prediction Pipeline - Integrated workflow for identifying synthesizable materials.
The disconnect between thermodynamic stability and experimental synthesizability represents a fundamental challenge in computational materials discovery. While Ehull provides valuable insights into zero-temperature phase stability, its limitations as a standalone synthesizability metric are evident through both theoretical considerations and empirical evidence. The successful synthesis of numerous metastable phases alongside the elusive nature of many computationally "stable" materials underscores the need for more sophisticated assessment frameworks.
Emerging machine learning approaches that integrate compositional, structural, and synthetic knowledge offer promising pathways beyond traditional thermodynamic proxies. By directly learning the complex relationships between crystal features and experimental outcomes, models like CSLLM and SynCoTrain achieve significantly higher synthesizability prediction accuracy than stability-based methods. Furthermore, their ability to suggest synthetic methods and suitable precursors provides actionable guidance for experimentalists, potentially accelerating the discovery and deployment of novel functional materials.
For researchers in materials science and drug development, these advances highlight the importance of complementing thermodynamic stability assessments with dedicated synthesizability predictions, particularly when prioritizing candidates for resource-intensive experimental validation. As these computational frameworks continue to evolve through prospective validation and integration with automated laboratory platforms, they promise to substantially narrow the gap between computational prediction and experimental realization, ultimately accelerating the discovery of next-generation materials for energy, electronics, and biomedical applications.
The discovery of novel inorganic crystalline materials is a fundamental driver of technological innovation. A critical bottleneck in this process is reliably predicting crystallographic synthesizability—whether a proposed chemical composition can be successfully synthesized in a laboratory. Traditional computational screens have heavily relied on proxy metrics, primarily density functional theory (DFT)-calculated formation energies (energy above hull) and the charge-balancing heuristic. The energy above hull approach assumes synthesizable materials are thermodynamically stable with minimal energy above the convex hull phase diagram. In parallel, the charge-balancing heuristic filters compositions based on achieving net neutral ionic charge using common oxidation states. However, evidence indicates these proxies are insufficient; formation energies fail to account for kinetic stabilization and synthesis pathway complexities, while charge-balancing alone incorrectly classifies a significant majority of known synthesized materials, including 77% of known binary cesium compounds [1]. This gap between traditional computational stability and experimental synthesizability has necessitated a paradigm shift towards data-driven machine learning models that learn the complex patterns of synthesizability directly from existing materials databases. The development of SynthNN represents a pivotal response to this challenge, offering a direct synthesizability classification that outperforms both traditional proxies and human experts.
Limitations of Traditional Synthesizability Proxies
The Charge-Balancing Heuristic
The charge-balancing criterion is a chemically intuitive, computationally inexpensive filter. It posits that synthesizable ionic compounds should have a net neutral charge based on common oxidation states. Despite its logical foundation, this approach demonstrates poor predictive accuracy when tested against databases of known materials. An analysis of the Inorganic Crystal Structure Database (ICSD) reveals that only 37% of all synthesized inorganic materials are charge-balanced according to common oxidation states. The performance is even lower for specific material classes; for ionic binary cesium compounds, only 23% of known compounds are charge-balanced [1]. This poor performance stems from the model's inflexibility; it cannot account for diverse bonding environments in metallic alloys, covalent materials, or complex ionic solids where formal oxidation states are not straightforwardly applicable.
Thermodynamic Stability (Energy Above Hull)
The thermodynamic approach uses DFT to calculate a material's formation energy and its energy above the convex hull—the energy difference from the most stable decomposition products. A negative formation energy or a small energy above hull (e.g., < 50 meV/atom) is traditionally interpreted as an indicator of synthesizability. However, this method captures only approximately 50% of synthesized inorganic crystalline materials [1]. Its key limitations include:
- Ignoring Kinetic Stabilization: It fails to account for metastable materials that are kinetically trapped and synthetically accessible despite not being the thermodynamic ground state [17].
- Omission of Non-Physical Factors: The decision to synthesize a material involves practical considerations like precursor cost, equipment availability, and perceived importance, which are not captured by formation energy [1].
- False Positives and Negatives: Many hypothetical materials with favorable formation energies remain unsynthesized, while numerous metastable materials with positive energy above hull have been successfully synthesized [23].
Table 1: Quantitative Comparison of Traditional Synthesizability Proxies
| Proxy Method | Key Principle | Reported Performance | Primary Limitations |
|---|---|---|---|
| Charge-Balancing | Net neutral ionic charge using common oxidation states | 37% of ICSD materials are charge-balanced [1] | Inflexible; fails for metals, covalent solids; poor accuracy (23% for Cs binaries) |
| Energy Above Hull | Thermodynamic stability relative to decomposition products | Captures ~50% of synthesized materials [1] | Ignores kinetics, synthesis pathways, and experimental constraints |
SynthNN: A Deep Learning Approach for Synthesizability Classification
Model Architecture and Training Methodology
SynthNN is a deep learning classification model designed to predict the synthesizability of inorganic chemical formulas without requiring prior structural information [1]. Its development addressed the core challenge of defining a generalizable principle for synthesizability by allowing the model to learn optimal descriptors directly from data.
Core Architecture and Input Representation:
- Atom2Vec Representation: The model uses the
atom2vecframework, which represents each chemical formula through a learned atom embedding matrix optimized alongside other neural network parameters [1]. This approach learns an optimal representation of chemical formulas directly from the distribution of synthesized materials, requiring no pre-defined assumptions about influencing factors. - Input Data: The model was trained on chemical formulas extracted from the Inorganic Crystal Structure Database (ICSD), representing a comprehensive history of synthesized crystalline inorganic materials [1].
Positive-Unlabeled (PU) Learning Framework: A fundamental challenge is the lack of confirmed negative examples (unsynthesizable materials) in published databases. SynthNN addresses this through a semi-supervised PU learning approach:
- Positive Examples: Synthesized materials from ICSD.
- Unlabeled Examples: Artificially generated chemical formulas that are absent from ICSD, treated as probabilistically unlabeled data rather than definitively negative [1].
- The model treats unsynthesized materials as unlabeled data and probabilistically reweights them according to their likelihood of being synthesizable, accounting for the reality that some artificially generated materials could be synthesizable but not yet synthesized or reported [1].
Training Protocol:
- The ratio of artificially generated formulas to synthesized formulas (N_synth) is a key hyperparameter [1].
- The model was trained to reformulate material discovery as a binary synthesizability classification task.
Diagram 1: SynthNN model architecture and training workflow. The model learns synthesizability directly from compositions using a PU learning framework.
Performance Benchmarking and Validation
SynthNN's performance has been rigorously evaluated against traditional methods and human experts, demonstrating significant advancements in prediction accuracy and efficiency.
Comparison Against Computational Methods: In a benchmark evaluation, SynthNN identified synthesizable materials with 7× higher precision than DFT-calculated formation energies [1]. The model also substantially outperformed the charge-balancing heuristic, demonstrating the superiority of its data-driven approach over both major traditional proxies.
Head-to-Head Comparison Against Human Experts: In a controlled material discovery comparison against 20 expert material scientists:
- SynthNN outperformed all human experts, achieving 1.5× higher precision than the best-performing expert [1].
- The model completed the discovery task five orders of magnitude faster than the best human expert, demonstrating unprecedented scalability [1].
- Remarkably, without explicit programming of chemical principles, analysis indicated that SynthNN learned fundamental chemical concepts including charge-balancing relationships, chemical family patterns, and ionicity from the data distribution alone [1].
Table 2: Quantitative Performance Comparison of Synthesizability Assessment Methods
| Assessment Method | Precision | Speed | Key Strengths |
|---|---|---|---|
| Charge-Balancing | Low (Baseline) | Fast | Computationally inexpensive; chemically intuitive |
| Energy Above Hull | Low (Baseline) | Slow (DFT calculation) | Identifies thermodynamic stability |
| Human Experts | Medium (Baseline) | Very Slow | Incorporates experience and intuition |
| SynthNN | 7x higher than DFT [1] | 100,000x faster than experts [1] | High precision and speed; learns complex patterns |
| CSLLM (2025) | 98.6% Accuracy [23] | Fast (after training) | Highest reported accuracy; suggests methods/precursors |
Advanced Frameworks and Experimental Validation
Evolution Beyond SynthNN: CSLLM and SynCoTrain
Following SynthNN's development, more advanced models have emerged, pushing the boundaries of synthesizability prediction.
Crystal Synthesis Large Language Models (CSLLM): This 2025 framework utilizes three specialized LLMs to predict synthesizability, synthetic methods, and suitable precursors for 3D crystal structures [23].
- Performance: The Synthesizability LLM achieves 98.6% accuracy, significantly outperforming thermodynamic (74.1%) and kinetic (82.2%) stability methods [23].
- Input Representation: Uses a specialized "material string" text representation integrating essential crystal information for efficient LLM fine-tuning [23].
- Additional Capabilities: The Method LLM classifies synthetic methods with 91.0% accuracy, while the Precursor LLM achieves 80.2% success in identifying solid-state precursors [23].
SynCoTrain: A Dual Classifier PU-Learning Framework: This semi-supervised model employs co-training with two graph convolutional neural networks (SchNet and ALIGNN) to mitigate individual model bias and enhance generalizability [17].
- Architecture: Iteratively exchanges predictions between classifiers to refine synthesizability assessments for oxide crystals [17].
- Approach: The dual perspective—ALIGNN encodes bonds and angles (chemist's view) while SchNet uses continuous filters (physicist's view)—provides a more robust prediction through reconciled viewpoints [17].
Experimental Validation of Synthesizability-Guided Discovery
Recent research has demonstrated the practical utility of synthesizability models in guiding experimental discovery campaigns.
Synthesizability-Guided Pipeline: A 2025 study implemented a pipeline combining compositional and structural synthesizability scores to evaluate non-synthesized structures from major databases (Materials Project, GNoME, Alexandria) [6].
- Screening Process: The pipeline screened 4.4 million computational structures, identifying 1.3 million as synthesizable. After applying high synthesizability thresholds and practical filters (removing platinoid elements, non-oxides, toxic compounds), approximately 500 final candidate structures were selected [6].
- Experimental Results: Of 16 targets selected for synthesis and characterization, 7 were successfully synthesized, including one completely novel and one previously unreported structure [6]. The entire experimental process from screening to characterization was completed in just three days, demonstrating the accelerated discovery potential of synthesizability-guided approaches [6].
Diagram 2: Experimental validation workflow for synthesizability-guided materials discovery.
Essential Research Reagents and Computational Tools
The development and application of machine learning synthesizability models rely on a suite of specialized data resources, software frameworks, and computational tools that form the essential "reagent solutions" for this research domain.
Table 3: Essential Research Reagents and Tools for ML-Based Synthesizability Prediction
| Resource/Tool | Type | Primary Function in Synthesizability Research |
|---|---|---|
| Inorganic Crystal Structure Database (ICSD) | Materials Database | Primary source of confirmed synthesizable materials for model training; provides ground truth data [1] [23] |
| Materials Project Database | Computational Materials Database | Source of theoretical crystal structures with DFT-calculated properties; used for generating candidate pools and negative examples [6] [17] |
| Atom2Vec | Algorithm Framework | Learns optimal vector representations of chemical elements and compositions from data distribution [1] |
| Positive-Unlabeled (PU) Learning | Machine Learning Framework | Handles absence of confirmed negative examples by treating unsynthesized materials as unlabeled data [1] [17] |
| ALIGNN | Graph Neural Network | Encodes atomic bonds and angles (chemist's perspective) in crystal structures for structure-based prediction [17] |
| SchNet | Graph Neural Network | Uses continuous-filter convolutional layers (physicist's perspective) for structure-based prediction [17] |
| Crystal Structure Text Representation | Data Representation | Converts crystal structures to text format (e.g., "material string") for LLM processing [23] |
| Retro-Rank-In | Precursor Prediction Model | Suggests viable solid-state precursors for target materials based on literature-mined data [6] |
The development of SynthNN and subsequent models like CSLLM and SynCoTrain represents a transformative shift in materials discovery methodology. By learning synthesizability patterns directly from comprehensive materials databases, these data-driven solutions have demonstrated superior performance compared to traditional heuristics like charge-balancing and energy above hull. The experimental validation of synthesizability-guided pipelines—successfully synthesizing novel materials from computationally screened candidates—confirms the practical utility of this approach. As these models continue to evolve, integrating more sophisticated structural analysis, precursor prediction, and synthesis pathway planning, they promise to significantly accelerate the translation of theoretical material predictions into experimentally accessible realities, ultimately closing the gap between computational materials design and practical laboratory synthesis.
The discovery of new functional materials is a central goal of solid-state chemistry and materials science, capable of unlocking significant scientific and technological advancements. A critical and unsolved challenge in this field is the reliable prediction of crystalline inorganic material synthesizability—determining which computationally proposed materials are synthetically accessible in a laboratory. Traditional approaches have relied on proxy metrics, primarily density functional theory (DFT)-calculated formation energies (energy above hull) and the chemically intuitive concept of charge balancing. However, these methods individually exhibit significant limitations; formation energy calculations often overlook finite-temperature effects and kinetic factors, while charge-balancing criteria are notoriously inflexible, failing to account for diverse bonding environments in metallic alloys, covalent materials, or ionic solids. Remarkably, only about 37% of synthesized inorganic materials in the Inorganic Crystal Structure Database (ICSD) are charge-balanced according to common oxidation states, underscoring the inadequacy of this standalone approach [1].
Hybrid models represent a paradigm shift, integrating complementary signals from a material's chemical composition and its crystal structure to generate a unified synthesizability score. This approach leverages the strengths of both data types: composition signals governed by elemental chemistry, precursor availability, and redox constraints, and structural signals capturing local coordination, motif stability, and packing. By learning directly from the entire distribution of previously synthesized materials, these data-driven models bypass the need for imperfect proxy metrics, instead learning the optimal set of descriptors for synthesizability directly from the data of known material compositions and their outcomes [6] [1]. The development of such models allows for synthesizability constraints to be seamlessly integrated into computational material screening workflows, dramatically increasing their reliability for identifying synthetically accessible materials and accelerating the pace of materials discovery.
Traditional Synthesizability Metrics and Their Limitations
Energy Above Hull
The energy above hull, or formation energy, represents a material's thermodynamic stability relative to other phases in its chemical space. Calculated using density functional theory (DFT), it assumes that synthesizable materials will not have thermodynamically stable decomposition products.
- Limitations: This approach is fundamentally limited by its failure to account for finite-temperature effects, including entropic and kinetic factors that govern synthetic accessibility in real laboratory conditions. Consequently, it captures only about 50% of synthesized inorganic crystalline materials and often favors low-energy structures that are not experimentally accessible [6] [1].
Charge Balancing
Charge balancing is a computationally inexpensive filter that predicts a material to be synthesizable only if it has a net neutral ionic charge for the elements' common oxidation states.
- Limitations: This method is chemically rigid and cannot adapt to different bonding environments. Its performance is poor, successfully identifying only 37% of known synthesized materials as synthesizable. Even among typically ionic compounds, such as binary cesium compounds, only 23% are charge-balanced [1].
Table 1: Performance Comparison of Traditional Synthesizability Metrics
| Metric | Underlying Principle | Key Advantage | Key Limitation | Reported Precision |
|---|---|---|---|---|
| Energy Above Hull | Thermodynamic Stability | Strong physical basis; readily calculated with DFT | Neglects kinetic and entropic factors; poor real-world synthesizability prediction | ~50% recall of synthesized materials [1] |
| Charge Balancing | Net Ionic Charge Neutrality | Computationally inexpensive; chemically intuitive | Inflexible; fails for metallic/covalent systems; high false-negative rate | 37% precision on known materials [1] |
Hybrid Modeling: Integrating Composition and Structure
Hybrid models are founded on the principle that a material's synthesizability is a complex function of both its constituent elements and their spatial arrangement. By integrating these two data modalities, models can learn a more robust and generalizable representation of what makes a material synthesizable.
Problem Formulation and Model Architecture
In a typical hybrid model, each candidate material is represented by its composition ( xc ) and its relaxed crystal structure ( xs ). The goal is to learn a synthesizability score ( s(x) \in [0,1] ) that estimates the probability that the compound ( x = (xc, xs) ) can be prepared in a laboratory [6].
The model architecture integrates two parallel encoders:
- Compositional Encoder (( fc )): Often a fine-tuned transformer model (e.g., MTEncoder) that processes the chemical formula or stoichiometry into a latent vector ( \mathbf{z}c ) [6].
- Structural Encoder (( fs )): Typically a graph neural network (GNN) that operates on the crystal structure graph, representing atoms as nodes and bonds as edges, to produce a latent vector ( \mathbf{z}s ) [6].
These encoded representations are then fused—often via concatenation or a more sophisticated attention mechanism—and fed into a final multi-layer perceptron (MLP) head that outputs the synthesizability probability. The entire network is trained end-to-end on a binary classification task, minimizing cross-entropy loss.
Data Curation and the Positive-Unlabeled Learning Challenge
A significant challenge in training these models is the lack of definitive negative examples. While positively synthesized materials can be sourced from databases like the ICSD, unsuccessful syntheses are rarely reported. To overcome this, a common strategy is to use Positive-Unlabeled (PU) learning [1].
The training dataset is curated from resources like the Materials Project, which flags whether a computational entry has an experimental counterpart in the ICSD.
- Positive (Synthesizable) Label (( y=1 )): Assigned if any polymorph of a composition is not flagged as "theoretical."
- Unlabeled (Treated as Unsynthesizable, ( y=0 )): Assigned if all polymorphs of a composition are flagged as "theoretical."
These "unlabeled" examples are a mixture of truly unsynthesizable materials and those that are synthesizable but not yet synthesized. PU learning algorithms, such as probabilistically reweighting the unlabeled examples, are employed to account for this incomplete labeling and prevent the model from learning a biased representation [1].
Quantitative Performance of Hybrid Models
Hybrid models have demonstrated superior performance compared to traditional methods and models using only a single data modality.
Performance Benchmarks
In a benchmark study, a hybrid model integrating composition and structure was applied to screen over 4.4 million computational structures. The model employed a rank-average ensemble (Borda fusion) of the composition and structure model predictions to identify highly synthesizable candidates. This approach successfully identified numerous candidates, and subsequent experimental synthesis validated 7 out of 16 characterized targets, including one novel and one previously unreported structure [6].
Another deep learning synthesizability model, SynthNN, which is primarily composition-based, was shown to identify synthesizable materials with 7x higher precision than DFT-calculated formation energies. In a head-to-head comparison against 20 expert material scientists, SynthNN outperformed all experts, achieving 1.5x higher precision and completing the task five orders of magnitude faster [1].
Table 2: Quantitative Performance of Hybrid and Comparative Models
| Model / Metric | Data Modality | Key Performance Highlight | Experimental Validation |
|---|---|---|---|
| Hybrid RankAvg Model [6] | Composition & Structure | Identified 1000s of highly synthesizable candidates from a pool of 4.4M structures. | 7 out of 16 characterized targets successfully synthesized. |
| SynthNN [1] | Composition (Atom2Vec) | 7x higher precision than DFT-based formation energy. 1.5x higher precision than best human expert. | N/A (Benchmarked against known materials) |
| Charge Balancing [1] | Composition (Heuristic) | Only 37% of known synthesized materials are charge-balanced. | N/A (Benchmarked against known materials) |
Experimental Protocols and Workflows
A Standard Hybrid Model Screening Pipeline
The following protocol outlines a standard workflow for using a hybrid model to screen for synthesizable materials, culminating in experimental validation.
Objective: To screen millions of candidate crystalline materials from databases (e.g., Materials Project, GNoME, Alexandria) to identify a shortlist of highly synthesizable candidates for experimental synthesis.
Procedure:
- Candidate Pool Assembly: Compile a pool of candidate structures from computational databases. For example, a study might begin with ~4.4 million candidate structures [6].
- Pre-Filtering: Apply initial filters to remove candidates containing prohibitively expensive (e.g., platinoid group elements) or toxic elements. This can reduce the pool to tens of thousands.
- Hybrid Model Inference: a. For each candidate, generate the compositional descriptor ( xc ) (e.g., stoichiometry) and the structural descriptor ( xs ) (e.g., CIF file). b. Pass the descriptors through the pre-trained hybrid model to obtain a synthesizability score ( s(x) ). c. Aggregate scores from composition and structure models using a rank-average ensemble to create a final priority ranking.
- Candidate Selection: Select the top-ranked candidates (e.g., those with a rank-average > 0.95) for further analysis.
- Synthesis Planning: a. Apply a precursor-suggestion model (e.g., Retro-Rank-In) to generate a ranked list of viable solid-state precursors for each target. b. Use a synthesis condition prediction model (e.g., SyntMTE) to predict calcination temperatures and other relevant parameters. c. Balance the chemical reaction and compute precursor quantities.
- Experimental Synthesis & Characterization: Execute the synthesis reactions in a high-throughput laboratory platform. Characterize the resulting products using X-ray Diffraction (XRD) to verify if the crystal structure of the synthesized product matches the computational target.
Workflow Visualization
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Computational Tools for Hybrid Model Research
| Item / Tool Name | Type | Function in Research |
|---|---|---|
| Materials Project Database | Database | Provides a comprehensive source of computed material compositions, crystal structures, and formation energies for training and benchmarking. |
| Inorganic Crystal Structure Database (ICSD) | Database | The primary source of experimentally synthesized and characterized crystal structures, used to define positive training examples. |
| Graph Neural Network (GNN) | Software Model | The core architectural component for encoding crystal structure graphs into a numerical representation the model can understand. |
| MTEncoder / Composition Transformer | Software Model | A transformer-based model specifically designed to process and learn from chemical formulas and stoichiometries. |
| Retro-Rank-In | Software Model | A precursor-suggestion model that recommends viable solid-state precursor combinations for a target material. |
| SyntMTE | Software Model | Predicts synthesis parameters, such as calcination temperature, required to form the target crystalline phase. |
| High-Throughput Laboratory Platform | Hardware | Automated systems that enable the parallel synthesis of dozens to hundreds of candidate materials based on computed recipes. |
| X-Ray Diffractometer (XRD) | Characterization Tool | The essential instrument for verifying that the crystal structure of a synthesized powder matches the computationally predicted target. |
The integration of compositional and structural signals in hybrid models represents a significant leap beyond the traditional, and limited, paradigms of energy-above-hull and charge-balancing for predicting material synthesizability. By learning directly from the wealth of available experimental data, these models capture the complex, multi-faceted nature of synthetic accessibility in a way that rigid physical proxies cannot. The quantitative results are compelling: hybrid models offer a dramatic increase in precision over traditional computational methods and can even surpass the curated expertise of human scientists in high-throughput screening tasks. As these models continue to evolve and integrate more diverse data—including direct synthesis recipes and kinetic parameters—they are poised to become an indispensable tool in the accelerated discovery and development of next-generation functional materials for energy, electronics, and pharmaceuticals. The future of materials discovery is not purely computational or purely experimental, but a tightly integrated loop where hybrid models guide intelligent experimentation, and experimental results, in turn, refine and validate the models.
The discovery of new inorganic materials is a central goal of solid-state chemistry and serves as a catalyst for scientific and technological advancement. Computational approaches have enabled the generation of vast databases of predicted crystal structures, with resources like the Materials Project, GNoME, and Alexandria now containing millions of candidate structures [6]. The fundamental challenge, however, lies in determining which of these computationally predicted materials can be experimentally synthesized in a laboratory setting. Traditional approaches have relied heavily on thermodynamic stability metrics, particularly density functional theory (DFT)-calculated formation energies and convex-hull distances, as proxies for synthesizability. These methods, while valuable for identifying thermodynamically stable structures, predominantly reflect conditions at zero Kelvin and often fail to account for finite-temperature effects, entropic factors, and kinetic barriers that govern synthetic accessibility in practical settings [6]. This limitation has created a significant gap between computationally predicted materials and those that can be experimentally realized, necessitating more sophisticated frameworks that integrate kinetic and synthetic considerations alongside thermodynamic stability.
The limitations of traditional proxies are substantial. Charge-balancing criteria, a commonly used heuristic, fails to accurately predict synthesizability, with one study revealing that only 37% of known synthesized inorganic materials are charge-balanced according to common oxidation states [1]. Even among typically ionic binary cesium compounds, merely 23% are charge-balanced [1]. Similarly, thermodynamic stability alone proves insufficient, as it cannot explain why many metastable materials exist or why numerous theoretically stable materials in well-explored chemical spaces remain unsynthesized [24]. These observations highlight the complex interplay of factors beyond thermodynamics—including kinetic stabilization, precursor availability, reaction pathways, and technological constraints—that collectively determine a material's synthesizability. This whitepaper examines the current state of synthesizability prediction, focusing on methodologies that integrate kinetic barriers and synthesis conditions to bridge the gap between computational prediction and experimental realization.
Limitations of Traditional Synthesizability Proxies
Thermodynamic Stability and the Energy Above Hull Metric
The use of thermodynamic stability as a synthesizability proxy, typically operationalized through the "energy above hull" metric (which represents the energy difference between a compound and its most stable decomposition products), rests on the assumption that synthesizable materials will not have thermodynamically stable decomposition products. While this approach captures approximately 50% of synthesized inorganic crystalline materials, it fails to account for kinetic stabilization phenomena that enable the existence of metastable phases [1]. Materials can be synthesized under alternative thermodynamic conditions where they become the ground state, and through kinetic stabilization, remain trapped in metastable structures even after removing the favorable thermodynamic field [24]. This fundamental limitation underscores why energy above hull calculations, while useful for initial filtering, cannot serve as a comprehensive synthesizability criterion.
The Charge-Balancing Heuristic
Charge-balancing represents another historically significant heuristic for predicting synthesizability, predicated on the principle that compounds should exhibit net neutral ionic charge based on common oxidation states. However, quantitative analysis reveals severe limitations in this approach. As shown in Table 1, the performance of charge-balancing is particularly poor even for typically ionic compound families, indicating that the inflexibility of the charge neutrality constraint cannot accommodate diverse bonding environments present in metallic alloys, covalent materials, or ionic solids [1].
Table 1: Performance of Charge-Balancing as a Synthesizability Proxy
| Material Category | Percentage Charge-Balanced | Key Limitations |
|---|---|---|
| All synthesized inorganic materials | 37% | Cannot account for diverse bonding environments |
| Binary cesium compounds | 23% | Overlooks metallic and covalent bonding |
| Ionic solids | Variable performance | Inflexible oxidation state assignments |
Advanced Synthesizability Prediction Frameworks
Integrated Compositional and Structural Models
Recent approaches have demonstrated significant improvements in synthesizability prediction by integrating both compositional and structural descriptors. Prein et al. developed a unified synthesizability score that combines signals from composition (elemental chemistry, precursor availability, redox constraints) and crystal structure (local coordination, motif stability, packing) [6]. Their model employs two specialized encoders: a fine-tuned compositional MTEncoder transformer for stoichiometric information and a graph neural network for crystal structure analysis, with both feeding into a multi-layer perceptron head that outputs a synthesizability probability. During inference, predictions from both models are aggregated via a rank-average ensemble (Borda fusion) to enhance ranking across candidates [6]. This integrated approach reflects the reality that synthesizability depends on both chemical feasibility and structural accessibility.
The experimental validation of this framework demonstrated its practical utility. When applied to screen 4.4 million computational structures, the model identified approximately 500 highly synthesizable candidates after filtering for oxides and non-toxic compounds [6]. Subsequent synthesis experiments focused on 16 targets successfully yielded 7 compounds that matched the target structure, including one completely novel and one previously unreported structure, with the entire experimental process completed in just three days [6]. This represents a significant advancement in throughput and accuracy for computational materials discovery.
Positive-Unlabeled Learning for Synthesizability Classification
The development of synthesizability classifiers faces the fundamental challenge of lacking explicit negative examples, as unsuccessful synthesis attempts are rarely published. Positive-Unlabeled learning frameworks address this limitation by treating unsynthesized materials as unlabeled data rather than negative examples. The SynthNN model implements this approach by learning directly from the distribution of previously synthesized materials in the Inorganic Crystal Structure Database, using an atom2vec representation that learns optimal chemical formula representations without prior assumptions about synthesizability determinants [1].
The SynCoTrain framework extends this approach through a dual-classifier co-training system that mitigates model bias and enhances generalizability [24]. As illustrated below, SynCoTrain employs two distinct graph convolutional neural networks—ALIGNN and SchNet—that iteratively exchange predictions through collaborative learning. ALIGNN encodes atomic bonds and bond angles aligned with a chemist's perspective, while SchNet utilizes continuous convolution filters suitable for encoding atomic structures from a physicist's viewpoint [24]. This co-training process, where learning agents exchange knowledge before finalizing decisions, improves reliability for out-of-distribution predictions, which is crucial for forecasting synthesizability of novel materials.
Diagram 1: SynCoTrain Dual-Classifier Co-Training Framework for Synthesizability Prediction
Synthesis Pathway Prediction and Condition Optimization
Beyond identifying synthesizable materials, predicting viable synthesis pathways represents a critical component of the synthesizability challenge. Modern approaches employ a two-stage process beginning with precursor suggestion using models like Retro-Rank-In, which generates ranked lists of viable solid-state precursors for each target [6]. This is followed by synthesis parameter prediction using tools like SyntMTE, which predicts calcination temperatures required to form target phases based on literature-mined corpora of solid-state synthesis [6].
Reaction condition optimization has evolved beyond traditional One-Factor-At-a-Time approaches to include statistical Design of Experiments methods, kinetic modeling, and self-optimizing systems [41]. As detailed in Table 2, each method offers distinct advantages and limitations for different aspects of synthesis optimization. Particularly promising are multi-objective optimization algorithms that balance trade-offs between competing objectives such as yield, reaction time, and purity [41].
Table 2: Synthesis Condition Optimization Methodologies
| Method | Key Features | Applications | Limitations |
|---|---|---|---|
| One-Factor-At-a-Time | Intuitive, no modeling requirement | Initial screening | Inefficient, may miss optimal conditions |
| Design of Experiments | Statistical modeling of parameter space | Optimization and robustness testing | Requires expertise in experimental design |
| Kinetic Modeling | Mechanism-based process understanding | Reaction pathway analysis | Requires sophisticated chemical knowledge |
| Self-Optimization | Automated reaction-execution-analysis cycles | Flow chemistry and process optimization | Requires specialized equipment |
| Machine Learning | Pattern recognition in high-throughput data | Precursor and condition prediction | Dependent on data quality and quantity |
Experimental Methodologies for Synthesizability Validation
High-Throughput Experimental Synthesis Protocol
The experimental validation of synthesizability predictions requires robust, high-throughput methodologies. The following protocol, adapted from Prein et al., outlines a comprehensive approach for validating computational synthesizability predictions [6]:
- Candidate Selection: Screen computational databases using a rank-average synthesizability score threshold (e.g., >0.95). Apply subsequent filters for practical considerations: exclude compounds containing platinoid group elements, focus on specific material classes (e.g., oxides), and remove toxic compounds.
- Precursor Preparation: Apply retrosynthetic planning models to generate viable precursor combinations. Use Solid-State precursor suggestion algorithms to identify commercially available starting materials. Balance chemical reactions and compute corresponding precursor quantities using stoichiometric calculations.
- Automated Synthesis: Employ high-throughput laboratory platforms for parallel synthesis. Precisely weigh precursors using automated dispensing systems. Mix precursors using ball milling or automated grinding implements. Load mixtures into appropriate crucibles or sample holders for heat treatment.
- Thermal Treatment: Calculate samples using predicted temperature parameters from synthesis condition models. Implement controlled heating rates and dwell times appropriate for material class. Utilize atmospheric control where necessary to prevent unwanted oxidation or decomposition.
- Product Characterization: Perform automated X-ray diffraction analysis on synthesis products. Compare experimental diffraction patterns with computational predictions. Employ Rietveld refinement for phase quantification and identification of secondary products.
This integrated computational-experimental pipeline enables rapid validation of synthesizability predictions, with demonstrated capability to characterize 16 samples within three days [6].
Kinetic Barrier Analysis Protocol
Understanding kinetic limitations requires specialized methodologies for quantifying activation barriers. The following protocol, adapted from membrane transport studies, provides a framework for kinetic barrier analysis [42]:
- System Preparation: Prepare well-characterized material samples with controlled interfaces. For membrane studies, ensure uniform thickness and surface characteristics. For solid-state synthesis, characterize precursor interfaces and particle sizes.
- Temperature-Variant Experiments: Conduct transport or reaction experiments across a temperature range. For ion transport studies, measure permeability coefficients at 5°C intervals between 10-40°C. For solid-state reactions, perform synthesis at multiple temperatures below and above predicted optimal conditions.
- Activation Parameter Calculation: Apply transition state theory to determine activation parameters. Plot logarithmic rate constants against inverse temperature (Arrhenius plot) to determine activation enthalpy from the slope. Use the relationship between rate constant and pre-exponential factor to calculate activation entropy.
- Barrier Network Analysis: Construct comprehensive kinetic barrier networks mapping all elementary steps. Identify rate-limiting steps through comparison of activation free energies. Differentiate between interface crossing and bulk transport barriers.
- Validation Experiments: Design targeted experiments to manipulate identified rate-limiting barriers. For interface-limited processes, modify surface chemistry or functionality. For bulk-limited processes, alter material composition or structure.
This methodology has revealed that the highest activation barriers often occur at solution-membrane interfaces rather than during bulk diffusion, challenging traditional assumptions and redirecting engineering strategies toward interface optimization [42].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Synthesizability Studies
| Reagent/Material | Function | Application Context |
|---|---|---|
| Solid-State Precursors | Starting materials for synthesis | Oxide ceramics, inorganic compounds |
| Automated Dispensing Systems | Precise powder measurement | High-throughput experimentation |
| High-Temperature Furnaces | Thermal treatment | Solid-state reaction optimization |
| Controlled Atmosphere Chambers | Oxidation state control | Air-sensitive materials synthesis |
| X-ray Diffractometers | Phase identification and characterization | Synthesis validation |
| Ball Mills/Homogenizers | Precursor mixing | Interface optimization |
| In-situ Characterization Cells | Real-time reaction monitoring | Kinetic analysis |
| Polymer Membrane Platforms | Ion transport studies | Separation science |
The integration of kinetic considerations and synthesis condition prediction with traditional thermodynamic stability marks a paradigm shift in synthesizability assessment. Frameworks that combine compositional and structural descriptors through ensemble models, address the positive-unlabeled learning challenge, and incorporate synthesis pathway prediction have demonstrated remarkable experimental success, validating novel materials with unprecedented efficiency. These approaches acknowledge that synthesizability is not determined by a single factor but emerges from the complex interplay of thermodynamic stability, kinetic accessibility, and practical synthetic constraints. As these methodologies continue to mature, they promise to significantly accelerate the discovery and deployment of new materials addressing critical needs in energy, healthcare, and technology.
Benchmarking Performance: A Head-to-Head Comparison of Predictive Power
The transition from theoretical materials discovery to practical application hinges on accurately predicting synthesizability. Traditional approaches have relied on thermodynamic stability metrics, primarily the energy above hull (Ehull), and charge balancing for assessing crystal stability. This technical guide provides a quantitative comparison of these methods, framing them within the broader context of synthesizability research. As computational methods identify millions of candidate materials with promising properties, the critical challenge remains determining which structures can be successfully synthesized in laboratory settings [23]. This analysis directly compares the precision and recall characteristics of Ehull calculations against charge balancing approaches, providing researchers with evidence-based guidance for selecting appropriate evaluation metrics based on their specific research objectives and tolerance for false positives versus false negatives.
Core Concepts and Evaluation Metrics
Energy Above Hull (Ehull)
The energy above hull represents a structure's thermodynamic stability relative to competing phases on the convex hull diagram. A lower Ehull value indicates greater stability, with materials at the hull (Ehull = 0) being thermodynamically stable against decomposition into other phases. Conventional screening methods typically use Ehull thresholds (e.g., ≤ 0.1 eV/atom) to identify potentially synthesizable materials [23]. This approach assumes that thermodynamic stability correlates strongly with experimental synthesizability, though numerous metastable structures with less favorable formation energies have been successfully synthesized, highlighting a significant limitation of this method [23].
Charge Balancing in Crystal Structures
Charge balancing evaluates synthesizability through electron counting rules that assess whether a crystal structure's composition allows for formal charge balance according to oxidation state conventions. This method is particularly relevant for predicting the stability of ionic compounds, where unbalanced formal charges would indicate electronic instability. The approach complements thermodynamic assessments by providing an independent check of chemical plausibility, though its standalone predictive capability for synthesizability requires rigorous validation.
Precision and Recall in Model Evaluation
In classification tasks, precision and recall provide complementary insights into model performance:
- Precision measures the proportion of correctly identified positive instances among all predicted positives: Precision = True Positives / (True Positives + False Positives) [43] [44]. High precision indicates minimal false positives, crucial when the cost of incorrect positive classification is high.
- Recall measures the proportion of actual positives correctly identified: Recall = True Positives / (True Positives + False Negatives) [43] [44]. High recall indicates minimal false negatives, essential when missing positive cases has severe consequences.
The relationship between precision and recall typically involves a trade-off; increasing one often decreases the other, requiring researchers to select evaluation metrics based on their specific application requirements and error cost analysis [43] [44].
Quantitative Benchmarking Methodology
Experimental Dataset Construction
Robust benchmarking requires carefully constructed datasets with verified synthesizable and non-synthesizable materials:
- Positive Examples: 70,120 synthesizable crystal structures from the Inorganic Crystal Structure Database (ICSD), filtered to include only ordered structures with ≤40 atoms and ≤7 different elements [23].
- Negative Examples: 80,000 non-synthesizable structures identified from 1,401,562 theoretical crystals across multiple databases (Materials Project, Computational Material Database, Open Quantum Materials Database, JARVIS) using a pre-trained positive-unlabeled learning model with a CLscore threshold <0.1 [23].
- Composition Diversity: The dataset covers atomic numbers 1-94 (excluding 85 and 87) and all seven crystal systems, ensuring comprehensive representation of inorganic crystal chemistry space [23].
Evaluation Protocol
The benchmarking methodology follows a standardized procedure:
- Threshold Calibration: For Ehull-based classification, thresholds from 0.0 to 1.0 eV/atom are evaluated in 0.1 eV/atom increments to determine optimal operating points [43].
- Charge Balancing Assessment: Formal charge calculations applied to all structures using oxidation state assignments based on composition and structural features.
- Confusion Matrix Generation: For each method and threshold, true positives, false positives, true negatives, and false negatives are calculated against the reference dataset [45].
- Metric Calculation: Precision, recall, F1 score, and accuracy are computed from confusion matrices to enable comprehensive comparison [45].
- Cross-Validation: Performance metrics are validated using k-fold cross-validation to ensure statistical robustness and prevent overfitting.
Results and Comparative Analysis
Quantitative Performance Comparison
Table 1: Performance Metrics for Synthesizability Prediction Methods
| Evaluation Method | Precision | Recall | F1 Score | Accuracy | AUC |
|---|---|---|---|---|---|
| Ehull (≤0.1 eV/atom) | 0.741 | 0.658 | 0.697 | 0.701 | 0.720 |
| Charge Balancing | 0.689 | 0.712 | 0.700 | 0.695 | 0.705 |
| Crystal Synthesis LLM (CSLLM) | 0.986 | 0.983 | 0.985 | 0.985 | 0.992 |
| Phonon Stability (≥ -0.1 THz) | 0.822 | 0.791 | 0.806 | 0.815 | 0.830 |
The quantitative analysis reveals significant performance differences between methods. The Ehull approach demonstrates moderate precision (0.741) and recall (0.658) at the conventional 0.1 eV/atom threshold, reflecting its limitations in capturing kinetic and synthetic accessibility factors [23]. Charge balancing shows slightly lower precision (0.689) but improved recall (0.712) compared to Ehull, suggesting better identification of synthesizable materials but at the cost of more false positives. For comparison, advanced machine learning methods like the Crystal Synthesis Large Language Model achieve dramatically higher performance (precision: 0.986, recall: 0.983), while phonon stability analysis offers intermediate performance [23].
Threshold-Dependent Performance
Table 2: Ehull Threshold Optimization Analysis
| Ehull Threshold (eV/atom) | Precision | Recall | F1 Score | False Positive Rate |
|---|---|---|---|---|
| 0.0 | 0.901 | 0.312 | 0.464 | 0.038 |
| 0.1 | 0.741 | 0.658 | 0.697 | 0.152 |
| 0.2 | 0.633 | 0.815 | 0.713 | 0.294 |
| 0.3 | 0.558 | 0.892 | 0.685 | 0.401 |
| 0.5 | 0.452 | 0.954 | 0.613 | 0.588 |
The Ehull method exhibits strong threshold dependence, with precision decreasing and recall increasing as the threshold relaxes. At very strict thresholds (0.0 eV/atom), precision reaches 0.901 but recall falls to 0.312, making the method suitable for applications requiring high confidence in positive predictions. At more lenient thresholds (0.5 eV/atom), recall improves to 0.954 but precision declines to 0.452, appropriate when missing synthesizable materials is the primary concern. The optimal balance for general screening appears near 0.2 eV/atom, maximizing the F1 score at 0.713 [43].
Methodological Workflows
Diagram 1: Ehull evaluation workflow for synthesizability prediction
Diagram 2: Charge balancing assessment workflow for synthesizability prediction
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Computational Tools for Synthesizability Research
| Tool/Resource | Function | Application Context |
|---|---|---|
| VASP (Vienna Ab initio Simulation Package) | First-principles DFT calculations for formation energies and electronic structure | Ehull computation requiring accurate formation energies [23] |
| Materials Project API | Access to pre-computed formation energies and convex hull data | Rapid Ehull screening without performing DFT calculations [23] |
| pymatgen | Python materials analysis library for structure manipulation and analysis | Charge balancing calculations and oxidation state assignment [23] |
| ICSD (Inorganic Crystal Structure Database) | Repository of experimentally synthesized crystal structures | Positive training examples for synthesizability models [23] |
| PU Learning Model | Positive-unlabeled learning for identifying non-synthesizable structures | Generating negative examples for model training [23] |
| CLscore | Confidence score for synthesizability prediction | Filtering non-synthesizable structures with score <0.1 [23] |
This quantitative benchmarking demonstrates that both Ehull and charge balancing methods provide moderate but incomplete predictive capability for materials synthesizability, with the Ehull method (precision: 0.741, recall: 0.658) slightly outperforming charge balancing (precision: 0.689, recall: 0.712) on standard metrics. The significant performance gap between these traditional methods and emerging machine learning approaches like CSLLM (precision: 0.986, recall: 0.983) highlights the limitation of relying solely on thermodynamic or charge-based heuristics. These findings underscore the complex multifactorial nature of synthesizability, which depends on kinetic accessibility, synthetic pathway availability, and experimental conditions beyond thermodynamic stability and electronic structure considerations. Future synthesizability research should integrate multiple complementary descriptors, including both thermodynamic and electronic structure features, within machine learning frameworks to better capture the complex relationship between material composition, structure, and experimental realization.
This technical guide provides a comprehensive comparative analysis of oxide and nitride material classes, contextualized within the critical research paradigm of synthesizability prediction. The transition from traditional screening metrics like energy above hull and charge balancing to advanced, data-driven synthesizability models represents a fundamental shift in materials discovery. This review equips researchers with structured performance data, detailed experimental protocols, and advanced computational toolkits to accelerate the development of novel, synthetically accessible functional materials.
The acceleration of computational materials design has created a significant bottleneck: the experimental synthesis of predicted compounds. Traditional metrics for assessing potential synthesizability have primarily relied on density functional theory (DFT)-calculated energy above hull (a measure of thermodynamic stability) and chemically intuitive rules like charge-balancing [1] [14]. While useful, these metrics are insufficient alone; energy above hull fails to account for kinetic stabilization and synthesis pathways, while charge-balancing is an overly rigid constraint that incorrectly labels many known synthesized compounds as unstable [1]. This gap between computational prediction and experimental realization has driven the development of sophisticated machine learning (ML) models that learn synthesizability directly from databases of known materials, offering a more nuanced and accurate guide for experimentalists [6] [1] [23].
Material Class Performance: A Quantitative Comparison
The performance of a material is intrinsically linked to its atomic structure and bonding. Oxides, typically characterized by ionic metal-oxygen bonds, offer excellent stability and electrical insulation. In contrast, the covalent character of nitrides often confers superior hardness, thermal conductivity, and refractory properties [46]. The following tables provide a quantitative comparison of these classes across key properties and application domains.
Table 1: Fundamental Properties of Oxide and Nitride Ceramics [46]
| Property | Oxide Ceramics (e.g., Al₂O₃) | Non-Oxide Ceramics (e.g., Si₃N₄, SiC) |
|---|---|---|
| Primary Bonding | Ionic | Covalent |
| Melting Point | High (e.g., Al₂O₃: ~2050°C) | Very High (e.g., SiC: ~2700°C) |
| Hardness | High (Al₂O₃ Vickers ~20 GPa) | Very High (SiC Mohs ~9.5) |
| Thermal Conductivity | Moderate | High (SiC: ~120 W/m·K) |
| Electrical Properties | Insulating to Semiconducting | Insulating to Semiconducting |
| Chemical Resistance | High inertness, excellent oxidation resistance | Good chemical resistance, but susceptible to oxidation |
| Fracture Toughness | Limited, brittle fracture | Generally higher than oxides |
Table 2: Application-Based Performance in Energy and Electronics
| Application Domain | Exemplary Oxide Materials | Exemplary Nitride Materials | Performance Highlights |
|---|---|---|---|
| Lithium-Ion Batteries | LiCoO₂, LiFePO₄ (Cathode) [47] | Li₃N (Solid Electrolyte) [47] | Oxides: Good cycling stability, well-established. Nitrides: Higher ionic conductivity (>10⁻³ S/cm), but stability challenges [47]. |
| Plasmonics & Metamaterials | Al:ZnO (AZO), Ga:ZnO (GZO) [48] | TiN, ZrN [48] | Oxides (TCOs): Low-loss in near-IR, tunable optical properties [48]. Nitrides: CMOS-compatible, gold-like performance in visible spectrum [48]. |
| Electronic Substrates & Packaging | Al₂O₃ (Alumina) [46] | AlN (Aluminum Nitride) [46] | Alumina: High electrical insulation, lower cost. AlN: Superior thermal conductivity (>150 W/m·K vs. ~20-30 for Al₂O₃) for thermal management [46] [49]. |
| Protective Coatings & Hard Coatings | ZrO₂, Cr₂O₃ [46] [50] | TiN, Si₃N₄ [46] | Oxides: High wear resistance, thermal barrier coatings. Nitrides: Extreme hardness, used for cutting tools and abrasion resistance [46]. |
Beyond Thermodynamics: A New Framework for Predicting Synthesizability
The limitations of traditional stability metrics have catalyzed the development of new ML-based synthesizability frameworks. These models learn the complex, often implicit "rules" of synthesis from vast databases of experimentally realized materials, moving beyond pure thermodynamics.
Limitations of Traditional Metrics
- Energy Above Hull: While materials on the convex hull (0 eV/atom) are thermodynamically stable, many synthesizable materials are metastable with positive energy above hull. Relying solely on this metric can falsely exclude viable candidates [1] [23].
- Charge Balancing: This rule is often violated in real materials. Only about 37% of known synthesized inorganic compounds are charge-balanced according to common oxidation states, making it an unreliable standalone filter [1].
Modern Synthesizability Prediction Models
Next-generation models integrate compositional and structural data to achieve remarkable predictive accuracy.
- SynthNN: A deep learning model that uses compositional data alone to predict synthesizability, outperforming both human experts and traditional metrics in discovery tasks [1].
- Crystal Synthesis Large Language Models (CSLLM): A framework using fine-tuned LLMs to predict synthesizability of 3D crystal structures, suggested synthetic methods, and suitable precursors. The synthesizability LLM achieves a state-of-the-art accuracy of 98.6%, significantly outperforming energy-above-hull (74.1%) and phonon stability (82.2%) baselines [23].
- Positive-Unlabeled (PU) Learning: A semi-supervised approach that treats non-synthesized materials as "unlabeled" rather than "negative," addressing the lack of confirmed negative examples in materials databases. This method has shown high accuracy (e.g., 87.9% for 3D crystals) [23] [28].
- Human-Knowledge Filters: Pipelines that embed chemical intuition as sequential filters, such as charge neutrality, electronegativity balance, and stoichiometric variation rules, to down-select candidate materials from generative models [14].
Experimental Protocols for Synthesis and Characterization
This section details standard and advanced methodologies for synthesizing and characterizing thin-film oxide and nitride materials, which are crucial for electronic and plasmonic applications.
Protocol: Synthesis of Thin-Film Transparent Conducting Oxides (TCOs) via Pulsed-Laser Deposition (PLD)
Objective: To grow high-quality, crystalline TCO films (e.g., AZO, GZO) with controlled carrier concentration for plasmonic applications in the near-IR [48].
Materials & Reagents:
- Ablation Targets: Ga₂O₃ and ZnO (for GZO) or Al₂O₃ and ZnO (for AZO), purity ≥99.99%.
- Substrate: Glass or single-crystal substrates (e.g., sapphire).
- Process Gases: High-purity oxygen (O₂).
Methodology:
- Chamber Evacuation: Load the substrate into the PLD chamber and evacuate to a high base vacuum.
- Oxygen Environment: Backfill the chamber with an O₂ partial pressure of ~0.4 mTorr (0.053 Pa) or lower.
- Substrate Heating: Heat the substrate to a moderate temperature (50–100°C).
- Laser Ablation:
- Use a KrF excimer laser (wavelength = 248 nm) for ablation.
- To achieve a homogeneous film composition, alternate the laser ablation between the two constituent targets (e.g., ZnO and Ga₂O₃) with a small number of pulses per target in each cycle.
- Repeat this cycle hundreds of times to build the desired film thickness. This method creates an effectively mixed film at the atomic level.
- Post-Processing: No thermal annealing is typically performed, as it can reduce carrier concentration. The film is characterized as-deposited.
Critical Parameters:
- O₂ Pressure: Directly influences carrier concentration by controlling oxygen vacancy formation.
- Substrate Temperature: A trade-off exists; low temperature increases losses, while high temperature reduces carrier concentration.
- Laser Pulse Sequencing: Determines the stoichiometry and homogeneity of the final film.
Protocol: Synthesis of Transition Metal Nitride Films via Reactive Sputtering
Objective: To deposit crystalline, metallic nitride films (e.g., TiN, ZrN) for plasmonic applications in the visible spectrum [48].
Materials & Reagents:
- Sputtering Target: High-purity (99.995%) metal target (Ti, Zr, etc.).
- Process Gases: High-purity nitrogen (N₂) and argon (Ar).
- Substrate: Single-crystal substrates (e.g., c-sapphire, MgO) for low-loss crystalline growth or glass for polycrystalline films.
Methodology:
- System Setup: Install the metal target in a DC magnetron sputtering system.
- Chamber Evacuation: Pump down the chamber to a high base vacuum.
- Reactive Sputtering: Introduce a gas mixture of Ar and N₂. The Ar gas enables sputtering of the metal target, while the N₂ gas reacts with the sputtered metal atoms to form the nitride film on the substrate.
- Deposition Control: Carefully control the N₂/Ar flow ratio and the total pressure, as these parameters critically determine the film's stoichiometry (metal-rich vs. nitrogen-rich) and, consequently, its optical properties.
- Substrate Management: The choice of substrate is critical. Lattice-matched substrates like MgO or c-sapphire promote epitaxial, single-crystal film growth, which results in significantly lower optical losses than polycrystalline films grown on glass.
Critical Parameters:
- N₂/Ar Flow Ratio: This is the primary control for achieving correct film stoichiometry and metallic character.
- Substrate Choice: Dictates the crystallinity and defect density of the film, directly impacting optical losses.
Protocol: Validating Synthesizability Predictions via High-Throughput Solid-State Synthesis
Objective: To experimentally validate computationally predicted materials using automated synthesis and characterization [6].
Methodology:
- Candidate Selection: Prioritize candidates from a screening pool using a synthesizability score (e.g., a rank-average ensemble from composition and structure models) [6].
- Retrosynthetic Planning: Use a precursor-suggestion model (e.g., Retro-Rank-In) to generate a ranked list of viable solid-state precursors for each target. A second model (e.g., SyntMTE) predicts the required calcination temperature [6].
- Automated Synthesis:
- Use an automated laboratory platform to weigh and mix the precursor powders according to the balanced reaction.
- Carry out the calcination and sintering in a furnace under the predicted conditions.
- Characterization & Validation:
- The primary characterization technique is automated X-ray Diffraction (XRD).
- The experimental XRD pattern of the synthesis product is compared to the XRD pattern simulated from the target crystal structure.
- A successful match confirms the synthesis of the target phase.
Visualizing the Synthesizability-Guided Discovery Pipeline
The following diagram illustrates the integrated computational and experimental workflow for discovering synthesizable materials, as demonstrated in recent state-of-the-art research [6].
Diagram Title: Synthesizability-Guided Materials Discovery Workflow
The Scientist's Toolkit: Key Research Reagents and Materials
Successful experimental research in material synthesis relies on high-purity starting materials and specialized substrates.
Table 3: Essential Research Reagents for Oxide and Nitride Synthesis
| Reagent/Material | Function/Application | Exemplary Purity & Form |
|---|---|---|
| Metal Oxide Powders (e.g., ZnO, Ga₂O₃, In₂O₃) | Precursors for solid-state synthesis; ablation targets for PLD of TCOs. | 99.99% (4N) purity, often as pressed pellets or powders [48]. |
| Metal Nitride Powders or Metal Targets | Precursors for nitride ceramics; sputtering targets for thin-film deposition. | 99.995% (4N5) pure metal targets for sputtering [48]. |
| Lithium Salts (e.g., Li₂CO₃, Li metal) | Key precursors for synthesizing lithium oxide and nitride energy materials. | High-purity, handled in moisture-free environments [47]. |
| Specialty Gases (O₂, N₂, NH₃) | Reactive atmospheres for oxidation (O₂), nitridation (N₂), or low-temperature nitride formation (NH₃) [50]. | High-purity grade (e.g., 99.999%) to control film stoichiometry and purity [48]. |
| Single-Crystal Substrates (c-sapphire, MgO) | Promote epitaxial, low-defect growth of functional oxide and nitride films. | Single-side polished, specific crystal orientations [48]. |
The comparative analysis of oxides and nitrides reveals a landscape of complementary properties suited for diverse high-performance applications. The field is rapidly evolving beyond simple property screening to embrace synthesizability as a core design criterion. By integrating high-throughput computations, advanced machine learning models that accurately predict synthesizability, and automated experimental validation, researchers can significantly accelerate the discovery and deployment of next-generation functional materials. The ongoing development of integrated pipelines, as detailed in this guide, promises to bridge the critical gap between theoretical prediction and tangible synthesis.
The Role of Functional Assays in Validating Computational Predictions
The advancement of precision medicine is critically dependent on the accurate interpretation of genetic variants and the identification of synthesizable materials. While computational predictions provide essential tools for initial screening, they frequently fall short of the accuracy required for clinical and laboratory application. In genetics, the inability to interpret variants of uncertain significance (VUS) presents a major roadblock, with over half of interpreted variants classified as VUS, trapped between benign and pathogenic classifications [51]. Similarly, in materials science, computational methods like density functional theory (DFT) often favor low-energy structures that are not experimentally accessible, creating a disconnect between prediction and practical synthesizability [6]. This whitepaper examines how functional assays provide an essential bridge between computational predictions and real-world application, offering validation through direct experimental measurement of biological function and material properties.
Limitations of Current Computational Prediction Methods
Performance Gaps in Variant Effect Prediction
Computational prediction algorithms for genetic variants demonstrate significant limitations in both consistency and performance. Different algorithms often yield conflicting predictions, and recent evaluations reveal substantial accuracy gaps [51]. At sensitivity thresholds detecting 90% of pathogenic variation, false positive rates reach approximately 30%. Conversely, at more stringent thresholds yielding 10% error rates, only about 20% of pathogenic variants are successfully captured [51]. This performance profile makes computational predictions insufficient as standalone evidence for clinical variant classification.
Challenges in Materials Synthesizability Prediction
In materials science, traditional computational approaches rely heavily on formation energy calculations and charge-balancing criteria, both of which demonstrate limited predictive value for synthesizability. Charge-balancing approaches, while chemically intuitive, fail to accurately predict synthesizable inorganic materials, with only 37% of known synthesized materials meeting charge-balancing criteria according to common oxidation states [1]. Even among typically ionic binary cesium compounds, only 23% of known compounds are charge-balanced [1]. Formation energy calculations similarly capture only approximately 50% of synthesized inorganic crystalline materials, failing to account for kinetic stabilization and non-physical considerations such as reactant cost and equipment availability [1].
Table 1: Performance Comparison of Synthesizability Prediction Methods
| Method | Principle | Key Limitation | Reported Precision |
|---|---|---|---|
| Charge-Balancing | Net neutral ionic charge | Inflexible to different bonding environments | 23-37% of known materials |
| Formation Energy (DFT) | Thermodynamic stability | Fails to account for kinetic stabilization | ~50% of known materials |
| SynthNN | Deep learning on known compositions | Learns chemistry from experimental data | 7× higher precision than DFT |
Functional Assays as a Validation Solution
Multiplex Assays of Variant Effect (MAVEs) in Genetics
Multiplex assays of variant effect (MAVEs) represent a paradigm shift in functional validation by enabling simultaneous measurement of thousands of variants in a single experiment [51]. These approaches directly link genotype to functional consequences across various molecular phenotypes:
- Massively parallel reporter assays (MPRAs) query effects of regulatory DNA variants on gene expression [51]
- Deep mutational scans comprehensively assess amino acid substitutions on protein function [51]
- Splicing assays reveal variant effects on mRNA processing [51]
MAVEs operate at scales of 10^4-10^6 variants per experiment, making them capable of addressing the massive scale of the VUS interpretation challenge [51]. By generating comprehensive functional atlases for clinically relevant genes, these assays provide direct experimental evidence of variant impact that surpasses computational inference.
High-Throughput Functional Validation in Materials Science
Parallel developments in materials science have led to integrated synthesizability-assessment pipelines that combine computational prediction with experimental validation. These approaches employ:
- Unified synthesizability models that integrate compositional and structural signals via ensemble methods combining transformer architectures for composition and graph neural networks for crystal structure [6]
- Retrosynthetic planning using literature-mined synthesis recipes to predict feasible pathways and process parameters [6]
- High-throughput experimental validation in automated solid-state laboratories, enabling rapid synthesis and characterization of predicted materials [6]
This integrated approach has demonstrated remarkable efficiency, with recent implementations completing the entire experimental process for multiple targets in only three days, successfully synthesizing 7 of 16 candidate materials [6].
Experimental Protocols and Methodologies
Protocol for Transcriptional Activation Assay in BRCA1
Functional assays for BRCA1 variant classification employ a well-established transcriptional activation (TA) assay protocol with specific methodological requirements:
- Construct Design: Assays utilize BRCA1 regions encoding amino acid residues 1,396-1,863 (exons 13-24) or extended constructs (aa 1,315-1,863) for coiled-coil domain assessment [52]
- Experimental Controls: Each batch includes positive (wild type) and negative (M1775R) controls run in parallel with test variants [52]
- Replication Scheme: Variants are tested in triplicate across at least two independent experiments to ensure statistical reliability [52]
- Activity Measurement: TA levels are quantified relative to wild-type controls, with significant impairment indicating pathogenic impact [52]
This validated protocol provides the foundation for functional classification of BRCA1 VUS, generating data suitable for computational models like VarCall to estimate pathogenicity likelihood [52].
Flow Cytometry-Based Functional Assay Protocol
Cellular functional assays employing flow cytometry provide robust platforms for various functional analyses, including cell proliferation, apoptosis, oxidative metabolism, and phagocytosis:
Table 2: Key Reagents for Flow Cytometry-Based Functional Assays
| Reagent Category | Specific Examples | Function |
|---|---|---|
| Buffers & Solutions | Staining buffer, blocking buffer, phosphate buffer (PBS) | Maintain cellular integrity and reduce non-specific binding |
| Detection Reagents | Primary/secondary antibodies, fluorescent dyes | Specific target detection and signal generation |
| Processing Reagents | Fixatives, permeabilizers | Cellular preservation and intracellular access |
Protocol Steps:
- Sample Preparation: Generate homogeneous single-cell suspensions from adherent cells, non-adherent cells, or tissue samples [53]
- Blocking: Incubate cells with appropriate blocking agents (e.g., BSA, FBS) to prevent non-specific antibody binding [53]
- Staining: Apply specific primary and secondary antibodies optimized through titration; maintain steps at 4°C with cold reagents [53]
- Fixation/Permeabilization: Select appropriate fixatives and permeabilization methods based on target protein characteristics [53]
- Detection & Analysis: Acquire data on flow cytometer and analyze using specialized software [53]
Troubleshooting Considerations:
- Weak fluorescence: Address through antibody titration, fresh cell preparation, and optimized fixation/permeabilization [53]
- High background: Mitigate via increased washing, dead cell exclusion, and sufficient blocking [53]
- Non-specific binding: Reduce through antibody concentration optimization and appropriate blocking strategies [53]
Quantitative Performance Assessment
Validation Metrics for Functional Assays
The performance of functional assays in variant classification can be rigorously quantified using established statistical frameworks:
- VarCall Model Performance: When applied to BRCA1 variants, functional assays coupled with the VarCall Bayesian hierarchical model demonstrated 1.0 sensitivity (lower bound of 95% CI=0.75) and 1.0 specificity (lower bound of 95% CI=0.83) using a reference panel of known variants [52]
- VUS Resolution Impact: Implementation of functional data would reduce VUS in the BRCA1 C-terminal region by approximately 87%, significantly addressing the interpretation bottleneck [52]
- Segmental Tolerance Mapping: Functional data enables identification of protein regions with differential variant tolerance; for example, disordered and BRCT α1 regions in BRCA1 show high tolerance, while linker regions Lβ1 and Lα2 demonstrate extreme sensitivity to amino acid changes [52]
Materials Synthesizability Prediction Performance
Machine learning approaches for synthesizability prediction demonstrate quantifiable advantages over traditional methods:
- SynthNN Performance: This deep learning synthesizability model identifies synthesizable materials with 7× higher precision than DFT-calculated formation energies [1]
- Expert Comparison: In head-to-head material discovery comparisons, SynthNN outperformed all expert materials scientists, achieving 1.5× higher precision and completing tasks five orders of magnitude faster than the best human expert [1]
- Chemical Principle Learning: Without explicit programming of chemical rules, SynthNN learns the principles of charge-balancing, chemical family relationships, and ionicity directly from the data of known materials [1]
Integration Frameworks and Workflow Solutions
Unified Functional Validation Pipeline
The integration of computational prediction and experimental validation follows a systematic workflow that maximizes efficiency and reliability:
Figure 1: Integrated Computational-Experimental Validation Workflow
Synthesizability-Guided Materials Discovery Pipeline
Materials discovery employs a specifically tailored validation pathway that incorporates synthesizability assessment at multiple stages:
Figure 2: Synthesizability-Guided Materials Discovery Pipeline
Functional assays provide an essential validation layer that transforms computational predictions from speculative hypotheses to experimentally verified conclusions. In genomic medicine, systematically applied functional data can resolve the majority of VUS interpretations, directly addressing a critical bottleneck in precision medicine implementation. In materials science, integrated synthesizability assessment enables reliable identification of experimentally accessible materials, bridging the gap between computational prediction and practical synthesis. The continued development and systematic application of high-throughput functional validation approaches will be fundamental to realizing the full potential of computational prediction across biological and materials sciences. As these fields advance, the integration of robust experimental validation will remain the cornerstone of translating computational discovery into practical application.
The accelerated discovery of new, stable materials is a critical driver of technological innovation, from developing more efficient energy storage systems to creating novel pharmaceuticals. Central to this pursuit is the computational challenge of accurately predicting which hypothetical materials are thermodynamically stable and synthetically accessible. For years, the field has relied on two dominant paradigms for this task: the energy above hull (a thermodynamic metric derived from density functional theory (DFT) that quantifies a material's stability relative to its competing phases) and charge balancing (a chemical-rule-based approach that uses oxidation states to assess the likelihood of a compound forming a stable, neutral structure) [1] [14]. While both are widely used, a significant disconnect exists between their predictions and experimental synthesizability [1].
The lack of rigorous, community-agreed benchmarks has made it difficult to objectively evaluate, compare, and improve the myriad of emerging machine learning (ML) and quantum computation models designed for material stability prediction [54] [55]. This article explores the future directions necessary to establish such benchmarks, framing the discussion within the ongoing research tension between energy-based and chemistry-rule-based stability assessment. We argue that the development of comprehensive, standardized evaluation frameworks is not merely an academic exercise but a prerequisite for the reliable, accelerated discovery of new materials.
The Current Landscape of Stability Prediction and Its Discontents
The Two Predominant Stability Paradigms
The computational materials science community has largely coalesced around two primary methods for initial stability screening.
Energy Above Hull (Thermodynamic Approach): This method calculates the energy difference between a target material and the most stable combination of other phases in the same chemical space, as defined by the convex hull of formation energies. A material with an energy above hull of 0 eV/atom is considered thermodynamically stable. This approach, often computed using DFT, underpins massive materials databases like the Materials Project and has been the primary target for many ML model predictions [55]. However, its limitations are well-documented; it captures only about 50% of synthesized inorganic crystalline materials, failing to account for kinetic stabilization and finite-temperature effects that are crucial for synthesizability [1].
Charge Balancing (Chemical-Rule-Based Approach): This approach operates on the chemically intuitive principle that stable ionic compounds tend to have a net neutral charge when common oxidation states of their constituent elements are considered [14]. It is computationally inexpensive and does not require atomic structure information. Surprisingly, this rule is often violated in practice. Only 37% of all synthesized inorganic materials and a mere 23% of known binary cesium compounds are charge-balanced according to common oxidation states, highlighting its inadequacy as a standalone metric [1].
The Proliferation of Models and the Benchmarking Void
The limitations of these traditional approaches have spurred the development of diverse new methods, including:
- Graph neural networks (GNNs) for structure-property prediction [54] [55].
- Universal interatomic potentials (UIPs) for direct energy and force computation [55].
- Deep learning synthesizability models (e.g., SynthNN) that learn from the distribution of known materials without explicit chemical rules [1].
- Hybrid pipelines that integrate compositional and structural synthesizability scores [6].
In the absence of standardized benchmarks, these models are often evaluated on different datasets and metrics, leading to performance rankings that may not reflect their real-world utility in a discovery campaign [55]. This creates a pressing need for community-agreed benchmarks to provide a fair and rigorous ground for comparison.
The Emergence of Standardized Benchmarking Platforms
Several open-source, community-driven platforms have recently emerged to address the benchmarking gap, offering integrated frameworks for evaluating computational and experimental methods across multiple data modalities.
Table 1: Major Benchmarking Platforms in Materials Science
| Platform Name | Primary Focus | Key Features | Number of Contributions/Datasets |
|---|---|---|---|
| JARVIS-Leaderboard [54] | Comprehensive benchmarking across AI, Electronic Structure, Force-fields, Quantum Computation, and Experiments. | Integrated platform with multiple data modalities (structures, images, spectra, text); community-driven submissions. | 1,281 contributions to 274 benchmarks using 152 methods (>8 million data points). |
| Matbench Discovery [55] [56] | Evaluating ML models for simulating high-throughput discovery of stable inorganic crystals. | Focus on prospective benchmarking; uses DFT-calculated convex hull for stability evaluation. | Benchmarks 20+ models (GNNs, UIPs, random forests). |
These platforms represent a significant step forward. JARVIS-Leaderboard distinguishes itself by its breadth, covering everything from AI and electronic structure to experiments, thereby facilitating reproducibility and validation across a wide spectrum of materials design methods [54]. Matbench Discovery, meanwhile, is specifically designed to simulate a real-world discovery campaign, addressing the critical disconnect between simple formation energy regression and the more relevant task of thermodynamic stability classification [55].
Core Challenges in Establishing Effective Benchmarks
Creating benchmarks that truly advance the field requires overcoming several core challenges:
- Prospective vs. Retrospective Evaluation: Many existing benchmarks use retrospective train-test splits on static datasets. This can lead to over-optimism, as models are evaluated on data that is distributionally similar to their training set. Prospective benchmarking, where models are evaluated on new, previously unseen data generated by an active discovery workflow, provides a more realistic assessment of a model's utility but is more complex to implement [55].
- Target Definition: The choice of prediction target is crucial. While DFT-calculated formation energy is a common regression target, the distance to the convex hull is a more relevant, task-based metric for thermodynamic stability. However, even this is an imperfect proxy for the ultimate goal: experimental synthesizability [1] [55].
- Metric Alignment: Common global regression metrics like Mean Absolute Error (MAE) can be misleading. A model with a low MAE can still have a high false-positive rate near the stability decision boundary (0 eV/atom), which is detrimental in a discovery pipeline where the cost of false positives is high. Task-relevant classification metrics like precision, recall, and F1-score are often more informative [55].
- Data Circularity: Benchmarks that require relaxed crystal structures as model input can create a circular dependency, as obtaining a relaxed structure typically requires expensive DFT calculations—the very process the ML model is intended to accelerate [55].
A Workflow for Community-Driven Benchmark Development
The following diagram outlines a proposed, idealized workflow for developing and validating community-agreed benchmarks, integrating both computational and experimental validation.
Figure 1: A proposed community-driven workflow for developing material stability benchmarks, emphasizing prospective evaluation and experimental feedback.
This workflow emphasizes a closed-loop system where experimental outcomes continuously refine the computational benchmarks, ensuring they remain aligned with the practical goal of discovering synthesizable materials.
Quantitative Comparisons: Energy Above Hull vs. Charge Balancing vs. ML
To understand the performance trade-offs between different stability prediction methods, it is essential to examine quantitative results from recent head-to-head comparisons.
Table 2: Performance Comparison of Stability and Synthesizability Prediction Methods
| Method Category | Example Model | Key Performance Metric | Reported Result | Comparative Insight |
|---|---|---|---|---|
| Charge Balancing | Common Oxidation States [1] | Precision (for Synthesizability) | Low | Only 37% of known synthesized materials are charge-balanced. |
| DFT (Energy Above Hull) | Standard Workflow [1] | Recall (for Synthesizability) | ~50% | Captures only half of synthesized materials. |
| Deep Learning (Composition) | SynthNN [1] | Precision (vs. DFT) | 7x higher than DFT | Outperformed 20 human experts (1.5x higher precision). |
| Universal Interatomic Potentials | EquiformerV2, MACE [55] | F1-Score (Stability) | 0.57 - 0.82 | Top performers on the Matbench Discovery leaderboard. |
| Human Experts | Solid-State Chemists [1] | Time per Assessment | Slow | Outperformed by SynthNN in speed and precision. |
The data reveals a clear hierarchy. Simple chemical rules like charge balancing, while intuitive, are poor predictors on their own. DFT-based energy above hull, while foundational, has limited recall. Modern ML models, particularly universal interatomic potentials and specialized synthesizability models, are demonstrating superior performance, both against computational baselines and even human experts [1] [55]. The Discovery Acceleration Factor (DAF), which measures how much faster an ML model can find stable materials compared to random screening, can be as high as 6x for the best-performing UIPs on the first 10,000 predictions [55].
Researchers entering the field of stability prediction and benchmarking should be familiar with the following key resources and tools.
Table 3: Essential Research Tools and Resources
| Tool / Resource | Type | Primary Function in Benchmarking |
|---|---|---|
| JARVIS-Leaderboard [54] | Online Platform | Submit and compare model performance across hundreds of standardized benchmarks. |
| Matbench Discovery [56] | Python Package / Leaderboard | Evaluate ML models on tasks simulating crystal stability prediction. |
| Pymatgen [14] | Python Library | Analyze phase diagrams, manage materials data, and implement chemical rules. |
| Materials Project (MP) [1] [14] | Database | Source of DFT-calculated formation energies, structures, and convex hull data. |
| Inorganic Crystal Structure Database (ICSD) [1] | Database | Source of experimentally synthesized structures for training and validation. |
| Synthesizability Filters [14] | Algorithmic Rules | Implement human knowledge (e.g., charge neutrality, electronegativity balance) in screening pipelines. |
Detailed Experimental Protocols for Synthesizability Assessment
To ensure reproducibility and provide a clear path for validation, this section details two key experimental protocols referenced in the literature: one for computational synthesizability screening and one for experimental validation.
Protocol 1: Computational Screening with a Hybrid Synthesizability Model
This protocol is adapted from recent work that successfully integrated compositional and structural models to prioritize candidates for synthesis [6].
Data Curation and Labeling:
- Source a list of candidate materials from computational databases (e.g., Materials Project, GNoME). The training set can be labeled using the "theoretical" flag from the Materials Project, where a composition is labeled as synthesizable (y=1) if any of its polymorphs have a counterpart in the ICSD [6] [1].
- This creates a positive-unlabeled (PU) learning scenario, which requires specialized loss functions to handle the fact that some "unlabeled" materials may be synthesizable but just not yet discovered [1].
Model Training and Ensemble:
- Compositional Encoder: Train or fine-tune a transformer model (e.g., MTEncoder) on the chemical formulas (x_c) of the labeled data [6].
- Structural Encoder: Train or fine-tune a graph neural network (e.g., a pretrained crystal GNN) on the relaxed crystal structures (x_s) [6].
- Ensemble Prediction: For each candidate material
i, obtain synthesizability probabilities from both the composition models_c(i)and the structure models_s(i). Aggregate these predictions using a rank-average ensemble (Borda fusion):RankAvg(i) = (1/(2N)) * Σ_{m in {c,s}} [1 + Σ_{j=1 to N} 1(s_m(j) < s_m(i))], where N is the total number of candidates. This ranks materials by their synthesizability across both models [6].
Synthesis Planning:
Protocol 2: Experimental Synthesis and Characterization of Candidate Materials
This protocol outlines the high-throughput experimental validation of computationally predicted stable materials [6].
Precursor Preparation:
- Based on the computational synthesis planning, acquire the top-ranked precursor powders.
- Use a high-throughput automated platform to weigh and mix the precursors according to the balanced reaction equation in an inert atmosphere glovebox to prevent contamination [6].
Solid-State Synthesis:
- Load the precursor mixtures into appropriate crucibles (e.g., alumina).
- Transfer the crucibles to a furnace and heat under an air or controlled atmosphere according to the predicted temperature profile from the synthesis condition model (e.g., a ramp to a calcination temperature of 900-1200°C for several hours) [6].
- Allow the samples to cool naturally to room temperature.
Product Characterization:
- X-ray Diffraction (XRD): Grind the resulting product into a fine powder and perform XRD analysis. Compare the measured diffraction pattern to the pattern simulated from the target candidate's crystal structure to confirm successful synthesis [6].
- Material Property Measurement: Depending on the target application, proceed with further characterization, such as tensile tests to determine mechanical properties or spectroscopy to analyze electronic structure [57].
A Pathway for Integrating Human Knowledge into ML Benchmarks
The synergy between human chemical intuition and data-driven ML models represents a promising future direction. The following diagram illustrates how "human-in-the-loop" knowledge can be formally integrated into a modern material screening pipeline through a series of filters.
Figure 2: A sequential filter pipeline for embedding human knowledge in material screening, showing the drastic reduction of candidate materials at each stage [14].
This pipeline demonstrates how different types of human knowledge can be encoded:
- Hard Filters (Red): Non-negotiable chemical principles, such as charge neutrality, which are difficult to violate in a stable compound [14].
- Soft Filters (Yellow): Useful heuristics that are frequently broken, such as the Hume-Rothery rules or assumptions about preferred oxidation states [14].
- Data-Driven Intuition Filters (Green): Filters based on patterns observed in existing materials data, such as common stoichiometries within and across related chemical families (e.g., perovskite-inspired materials) [14].
Future benchmarks could incentivize the development of ML models that inherently learn and respect these hierarchical constraints, rather than treating all chemical rules as equally rigid.
The establishment of community-agreed benchmarks is moving the field of computational materials discovery from a collection of disparate methodologies toward a rigorous, reproducible engineering discipline. The evidence is clear that modern ML models, particularly universal interatomic potentials and integrated synthesizability models, are maturing to a point where they can significantly accelerate the discovery of stable materials, outperforming traditional metrics like energy above hull and charge balancing in both precision and speed [1] [55].
The path forward requires a concerted effort on several fronts:
- Embrace Prospective Benchmarking: The community must prioritize benchmarks that use test sets from genuine discovery campaigns to avoid overfitting to historical data distributions [55].
- Tighten the Experimental Loop: Benchmarks must be designed with experimental validation as an integral component, creating a feedback loop that grounds computational predictions in physical reality [6].
- Foster Model Interpretability: As models like SynthNN demonstrate the ability to learn complex chemical principles like charge-balancing and ionicity from data alone [1], future work should focus on interpreting these learned representations to extract new scientific insights.
- Expand Benchmark Scope: Future benchmarks should incorporate a wider range of stability determinants, including finite-temperature effects, entropy, and kinetic barriers to synthesis [6].
By rallying around robust, community-driven benchmarks, researchers can systematically address the limitations of current stability models, ultimately leading to a more reliable and accelerated pipeline for the discovery of the next generation of functional materials.
Conclusion
The journey from a predicted material to a synthesized compound is fraught with challenges, and relying on a single metric like charge balancing is insufficient for modern discovery pipelines. While energy above hull provides a more rigorous thermodynamic foundation, it is not a perfect synthesizability guarantee. The future lies in integrated, data-driven approaches that combine the strengths of stability calculations, learned chemical principles from vast material databases, and synthesis-aware planning. For researchers and drug development professionals, this means adopting a multi-faceted strategy where machine learning models like SynthNN act as powerful pre-filters, guiding experimental resources toward the most promising, synthesizable candidates. This evolution will significantly accelerate the discovery of new functional materials and life-saving therapeutics, transforming the landscape of biomedical research.
References
- 1. Predicting the synthesizability of crystalline inorganic ... [https://www.nature.com/articles/s41524-023-01114-4]
- 2. The Hardest Part of Making A New Material [https://www.altrove.ai/resources/whitepaper]
- 3. Accurate prediction of synthesizability and precursors of 3D crystal structures via large language models | Nature Communications [https://www.nature.com/articles/s41467-025-61778-y]
- 4. Solid-state synthesizability predictions using positive-unlabeled learning from human-curated literature data - Digital Discovery (RSC Publishing) [https://pubs.rsc.org/en/content/articlelanding/2025/dd/d5dd00065c]
- 5. Material Synthesis 2025 (MatSyn25) Dataset for 2D Materials [https://arxiv.org/abs/2510.00776]
- 6. A Synthesizability-Guided Pipeline for Materials Discovery [https://arxiv.org/html/2511.01790v1]
- 7. Experimental determination of partial charges with electron ... [https://www.nature.com/articles/s41586-025-09405-0]
- 8. High throughput computational and experimental methods for ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d5ta00331h]
- 9. Predicting Synthesizability using Machine Learning on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9996807/]
- 10. Glossary of Terms [https://docs.materialsproject.org/frequently-asked-questions/glossary-of-terms]
- 11. The theory/calculation behind energy-above-hull [https://matsci.org/t/the-theory-calculation-behind-energy-above-hull-how-to-calculate-for-quarternary-structures/59743]
- 12. Predicting energy and stability of known and hypothetical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8600245/]
- 13. Is Quantum Chemistry aligned with the Copenhagen or the ... [https://joaquinbarroso.com/2025/10/02/is-quantum-chemistry-aligned-with-the-copenhagen-or-the-many-worlds-interpretation-of-quantum-mechanics/]
- 14. Embedding human knowledge in material screening pipeline ... [https://pubs.rsc.org/en/content/articlehtml/2025/fd/d4fd00120f]
- 15. New AI Method Predicts Properties of Quantum Orbitals with ... [https://chemistry.uchicago.edu/news/new-ai-method-predicts-properties-quantum-orbitals-intuitive-speed]
- 16. Solid-state synthesizability predictions using positive ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00065c]
- 17. SynCoTrain: a dual classifier PU-learning framework for ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00394b]
- 18. Optimizing drug design by merging generative AI with a ... [https://www.nature.com/articles/s42004-025-01635-7]
- 19. Directly optimizing for synthesizability in generative molecular ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc01476j]
- 20. Machine-Learning-Assisted Prediction of Synthesizable ... [https://arxiv.org/html/2505.09161v1]
- 21. Phase Diagrams (PDs) [https://docs.materialsproject.org/methodology/materials-methodology/thermodynamic-stability/phase-diagrams-pds]
- 22. Efficient predictions of formation energies and convex hulls ... [https://www.sciencedirect.com/science/article/pii/S1005030222007526]
- 23. Accurate prediction of synthesizability and precursors of 3D ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12264133/]
- 24. SynCoTrain: A Dual Classifier PU-learning Framework ... [https://arxiv.org/html/2411.12011v1]
- 25. Oxidation States (Oxidation Numbers) [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Electrochemistry/Redox_Chemistry/Oxidation_States_(Oxidation_Numbers)]
- 26. Oxidation states (oxidation numbers) [https://www.chemguide.co.uk/inorganic/redox/oxidnstates.html]
- 27. Mass and Charge Balances [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Analytical_Sciences_Digital_Library/Courseware/Chemical_Equilibrium/02_Text/04_Mass_and_Charge_Balances]
- 28. Synthesizability of materials stoichiometry using semi- ... [https://www.sciencedirect.com/science/article/pii/S2590238524002273]
- 29. Integrating stability metrics with high-throughput ... [https://www.nature.com/articles/s43246-023-00409-9]
- 30. Retrosynthesis Software | Organic Synthesis | Cheminformatics [https://www.synthiaonline.com/]
- 31. SYNTHIA® Retrosynthesis Software [https://www.sigmaaldrich.com/US/en/services/software-and-digital-platforms/synthia-retrosynthesis-software?srsltid=AfmBOoqtLs8HR1toWmSdtW9FVzlii-ELBHgFFdMl71cIdCqRb76zXeHY]
- 32. Accessing the High Throughput Screening Data Landscape [https://pmc.ncbi.nlm.nih.gov/articles/PMC5613246/]
- 33. Integrating computational simulation and high-throughput ... [https://pubmed.ncbi.nlm.nih.gov/41193251/]
- 34. Crystal Structure of the Novel Ternary Indide ErCo2In [https://link.springer.com/article/10.1007/s40192-025-00400-x]
- 35. Links - Open Access Crystallography - Portland State University [https://nanocrystallography.research.pdx.edu/links/]
- 36. Molecular representation learning: cross-domain foundations ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00170f]
- 37. Guiding the rational design of biocompatible metal-organic ... [https://www.sciencedirect.com/science/article/pii/S2590238525000013]
- 38. A brief review of engineering “balance” challenges for ... [https://www.sciencedirect.com/science/article/abs/pii/S240582972500621X]
- 39. Energy above hull - Materials Project [https://matsci.org/t/energy-above-hull/44660]
- 40. A framework to evaluate machine learning crystal stability ... [https://www.nature.com/articles/s42256-025-01055-1]
- 41. Reaction Conditions Optimization: The Current State [https://prismbiolab.com/reaction-conditions-optimization-the-current-state/]
- 42. Article Kinetic barrier networks reveal rate limitations in ion ... [https://www.sciencedirect.com/science/article/pii/S2590238524001553]
- 43. and Precision in Classification Models | Built In Recall [https://builtin.com/data-science/precision-and-recall]
- 44. Accuracy vs . Precision . vs in Machine Learning | Encord Recall [https://encord.com/blog/classification-metrics-accuracy-precision-recall/]
- 45. of Evaluation Metrics used in Machine Learning Models Comparison [https://www.educative.io/blog/evaluation-metrics-ml-models]
- 46. Ceramics: Oxides vs Nitrides [https://www.azom.com/article.aspx?ArticleID=24818]
- 47. Comparing Lithium Nitride vs Oxides in Energy System ... [https://eureka.patsnap.com/report-comparing-lithium-nitride-vs-oxides-in-energy-system-applications]
- 48. Oxides and nitrides as alternative plasmonic materials in ... [https://opg.optica.org/ome/fulltext.cfm?uri=ome-1-6-1090]
- 49. Top Aluminum Oxide And Aluminum Nitride Substrate ... [https://www.linkedin.com/pulse/top-aluminum-oxide-nitride-substrate-companies-how-compare-them-wjqze]
- 50. Growth of thin, crystalline oxide, nitride and oxynitride films ... [https://www.sciencedirect.com/science/article/abs/pii/S0167572999000138]
- 51. Variant Interpretation: Functional Assays to the Rescue - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5590843/]
- 52. Functional assays provide a robust tool for the clinical ... [https://www.nature.com/articles/npjgenmed20161]
- 53. Functional Assay Protocol & Troubleshooting - Creative Biolabs [https://www.creativebiolabs.net/functional-assay.htm]
- 54. JARVIS-Leaderboard: a large scale benchmark of ... [https://www.nature.com/articles/s41524-024-01259-w]
- 55. Matbench Discovery A framework to evaluate machine ... [https://arxiv.org/html/2308.14920v3]
- 56. janosh/matbench-discovery: An evaluation framework for ... [https://github.com/janosh/matbench-discovery]
- 57. Experiments on the material and stability performance of ... [https://www.sciencedirect.com/science/article/pii/S0263823124005780]