Beyond Classical Theory: Non-Classical Nucleation Pathways in Inorganic Materials and Their Biomedical Implications
This article explores the paradigm shift from Classical Nucleation Theory (CNT) to non-classical pathways in inorganic and soft materials, a field with profound implications for drug development and biomedical research.
Beyond Classical Theory: Non-Classical Nucleation Pathways in Inorganic Materials and Their Biomedical Implications
Abstract
This article explores the paradigm shift from Classical Nucleation Theory (CNT) to non-classical pathways in inorganic and soft materials, a field with profound implications for drug development and biomedical research. We provide a foundational exploration of key mechanisms like pre-nucleation clusters and particle attachment. The review then details advanced methodological approaches for observing these dynamics and offers strategies for troubleshooting and optimizing crystallization processes. Finally, we present a comparative analysis validating non-classical against classical pathways, highlighting their direct impact on material properties and catalytic performance for researchers and scientists in the field.
Deconstructing the Basics: What Are Non-Classical Nucleation Pathways?
The Limits of Classical Nucleation Theory (CNT)
Classical Nucleation Theory (CNT) has long served as the foundational model for describing the initial stages of crystallization, positing that nuclei become stable after reaching a critical size through the energetically favorable balance between the volume free energy of a new phase and the penalty of creating a new interface [1]. This critical nucleus then grows via the sequential, monomer-by-monomer addition of individual atoms or molecules. Despite its widespread application and notable successes, such as effectively predicting heterogeneous nucleation kinetics even on chemically non-uniform surfaces [2], CNT relies on several restrictive assumptions. These include idealized spherical-cap geometries for nuclei and sharp liquid-solid interfaces. However, due to their atomic and dynamic nature, nucleation processes are inherently difficult to observe experimentally, and a growing body of research across diverse materials systems—from metals and colloids to soft organic materials and two-dimensional semiconductors—reveals complex nucleation behaviors that deviate fundamentally from this classical picture [1] [3] [4]. These deviations, collectively termed non-classical nucleation pathways, demonstrate that atomic and molecular systems often circumvent the high energy barriers predicted by CNT through alternative mechanisms, thereby establishing the practical and theoretical limits of the classical framework.
Key Limitations of Classical Nucleation Theory
The inadequacies of CNT manifest across multiple domains of materials research. A primary limitation is its failure to account for the prevalence and stability of precursor phases and amorphous intermediates. In the homogeneous nucleation of body-centered cubic (BCC) phase in face-centered cubic (FCC) iron, molecular dynamics simulations demonstrate that the system avoids the high energy barrier for homogeneous nucleation by opting for alternative processes such as the coalescence of subcritical clusters and stepwise nucleation [1]. Similarly, during the synthesis of calcium silicate hydrate (C-S-H), the most important hydrate in cement, observations confirm a two-step, non-classical process where discrete globules appear as a metastable precursor before transforming into foil-like C-S-H [5].
Furthermore, CNT is fundamentally challenged by observations of extremely large critical nuclei, a phenomenon incongruent with its thermodynamic formulations. During the vapor-liquid-solid (VLS) growth of monolayer tungsten disulfide (WS₂), in-situ monitoring revealed critical nuclei sizes as large as 38.7 µm, which is orders of magnitude larger than the calculated value for classical nucleation and cannot be explained by monomer addition alone [6]. This finding directly contradicts the CNT prediction of a well-defined, nanoscale critical size.
Finally, CNT oversimplifies crystal growth by focusing on monomer attachment, while non-classical pathways involve more complex multi-stage growth processes. In binary colloidal crystals, growth proceeds via three simultaneous mechanisms: the classical addition of free monomers, the capture and absorption of surrounding amorphous blobs, and the oriented attachment of other crystals [3]. This complexity, observed in systems where interactions can be finely tuned, underscores the inability of the classical model to describe the rich spectrum of crystallization behaviors.
Table 1: Documented Critical Nucleus Sizes Challenging CNT Predictions
| Material System | Observed Critical Nucleus Size | Classical Prediction | Experimental Technique |
|---|---|---|---|
| Monolayer WS₂ [6] | ~38.7 µm | ~1.63 nm | In-situ monitoring chemical vapor deposition |
| BCC phase in FCC Iron [1] | Not specified; involves subcritical clusters | High energy barrier for homogeneous nucleation | Molecular Dynamics (MD) simulations |
| Binary Colloidal Crystals [3] | Forms within amorphous blobs | Direct monomer-by-monomer attachment | Confocal microscopy, Bright-field microscopy |
Established Non-Classical Nucleation Mechanisms
Two-Step Nucleation via Metastable Precursors
The two-step nucleation mechanism is a well-documented non-classical pathway where a dense, often amorphous, metastable phase condenses from the solution or vapor first, followed by the nucleation of the crystalline phase within this precursor. This mechanism is prevalent across vastly different material systems. In binary colloidal systems comprising oppositely charged particles, the process begins with the rapid formation of "particle blobs"—a condensed, liquid-like phase. Crystal nucleation then initiates within these blobs, with the crystallization front becoming visibly distinguishable as it propagates through the amorphous aggregate [3]. This mechanism has also been directly observed in the VLS growth of WS₂, where liquid-phase precursors coalesce on the substrate, and solid nucleation occurs inside these metastable clusters [6].
Particle-Based Crystallization: Coalescence and Oriented Attachment
In contrast to the monomer-by-monomer addition of CNT, non-classical pathways often involve clusters or nanoparticles as primary building blocks. Coalescence of subcritical clusters is a key mechanism by which systems circumvent high classical energy barriers, as identified in FCC iron [1]. A more complex manifestation is oriented attachment, where small, pre-formed crystals come together and fuse in a specific, orientation-dependent manner to form a larger, single crystal. This process is distinct from random aggregation, as the particles align along their crystallographic axes before merging, thereby maintaining a common orientation across the newly formed structure. In colloidal systems, the contact region between attaching crystals often undergoes melting and subsequent re-crystallization, which facilitates perfect alignment and eliminates the initial seam between the structures [3].
The Role of Seeds and Interfaces in Redirecting Pathways
The introduction of crystalline seeds can fundamentally reshape nucleation mechanisms, sometimes converting non-classical pathways into classical ones. Molecular dynamics simulations of zeolite synthesis reveal that crystalline seeds can bypass amorphous intermediates, promoting a classical, monomer-by-monomer crystallization pathway. This creates a complex reaction network where the interplay between the thermodynamic stability and kinetic favorability of intermediate interfacial polymorphs dictates the final nucleation outcome. The synthesis environment is critical; at moderate supersaturation, seeds promote classical nucleation, whereas high supersaturation or the presence of aggregate-based reactants favors non-classical pathways even in the presence of a seed [7].
Table 2: Key Non-Classical Nucleation Mechanisms and Their Characteristics
| Mechanism | Key Feature | Example System | Impact on Crystallization |
|---|---|---|---|
| Two-Step Nucleation [3] [6] [5] | A metastable amorphous intermediate condenses before crystallizing. | Binary colloids, WS₂, C-S-H | Alters kinetics; can lead to different polymorphs. |
| Cluster Coalescence [1] | Subcritical clusters merge to form a stable nucleus. | FCC Iron | Circumvents high energy barriers of classical homogeneous nucleation. |
| Oriented Attachment [3] | Crystals fuse along common crystallographic axes. | Binary Colloidal Crystals | Enables rapid crystal growth while maintaining single-crystalline order. |
| Seed-Mediated Switching [7] | A crystalline seed can convert a non-classical pathway into a classical one. | Zeolites | Allows control over polymorphism and nucleation kinetics. |
Quantitative Experimental Methodologies and Protocols
Molecular Dynamics (MD) Simulations for Atomic-Scale Insights
Objective: To probe the atomic-scale mechanisms and thermodynamics of nucleation in model systems, such as the homogeneous nucleation of BCC phase in FCC iron [1] or heterogeneous nucleation on patterned surfaces [2].
Protocol:
- System Setup: A simulation box is set up with periodic boundaries. For homogeneous nucleation, a pure system of atoms (e.g., iron) is used. For heterogeneous nucleation, the box includes a supercooled liquid confined within a slit pore formed by a nucleating substrate and a repulsive wall [2].
- Interaction Potentials: Particles interact via defined potentials. The Lennard-Jones potential is a common choice for model atomic liquids [2]. The parameters (e.g., energy depth
ε, particle sizeσ) for interactions between different particle types (liquid A, liquiphilic wall B, liquiphobic wall C) are specified to mimic the desired chemical heterogeneity. - Simulation Execution: Newton's equations of motion are integrated using algorithms like velocity Verlet with a small, reduced time step (e.g.,
δt* = 2.5 × 10⁻³). Simulations are run at specific reduced temperatures (kT/εAA) to study supercooled states [2]. - Enhanced Sampling: To overcome the rarity of nucleation events, advanced sampling techniques like Jumpy Forward Flux Sampling (jFFS) are employed. This method biases the simulation to efficiently traverse the nucleation barrier and collect statistics on nucleation pathways and rates [2].
- Analysis: The trajectories are analyzed to identify the formation of critical nuclei, characterize their structure (e.g., using bond-order parameters), calculate free energy barriers, and measure nucleation rates.
Direct Observation viaIn-SituMonitoring and Microscopy
Objective: To directly visualize the nucleation and growth dynamics in real-time, providing spatial and temporal resolution of the process, as demonstrated in colloidal systems [3] and CVD-grown TMDs [6].
Protocol for Colloidal Systems [3]:
- Sample Preparation: Positively and negatively charged colloidal particles are prepared in a salt solution with a precisely controlled concentration. The two groups are mixed in an approximately 1:1 stoichiometric ratio.
- Initiation and Imaging: The mixture is immediately transferred to an observation cell. Time-lapse images are captured using bright-field microscopy to track the entire process. For 3D structural characterization, confocal microscopy is used with refractive index-matched particles.
- Interaction Strength Control (Continuous Dialysis): To dynamically control particle interactions, the observation cell is connected to a deionized water reservoir. As salt diffuses out, the Debye length (
λD) increases, gradually increasing the interaction strength and initiating crystallization in a controlled manner. This allows mapping of crystallization outcomes (classical, two-step, aggregation) against a continuous range of interaction strengths in a single experiment [3]. - Post-Processing: Samples may be quenched at different stages for high-resolution imaging using Scanning Electron Microscopy (SEM) to obtain snapshots at single-particle resolution.
Protocol for 2D Material CVD [6]:
- Reaction Setup: Salt-assisted chemical vapor deposition is performed in a custom reactor equipped with optical imaging capabilities.
- In-Situ Imaging: The substrate is imaged optically during growth in real-time, capturing hundreds of pictures per second.
- Automated Image Analysis: An automated system extracts specific regions from all optical images based on predefined hue, saturation, value (HSV) color index thresholds. This allows independent tracking of monolayer and multilayer regions and the extraction of physical parameters like incubation time (
Δt) and growth speed (vg). - Nucleation Analysis: The initial nucleation phase is investigated by extracting and highlighting the edges of the monolayer color region, revealing the dynamics of liquid precursor particles and the emergence of solid nuclei.
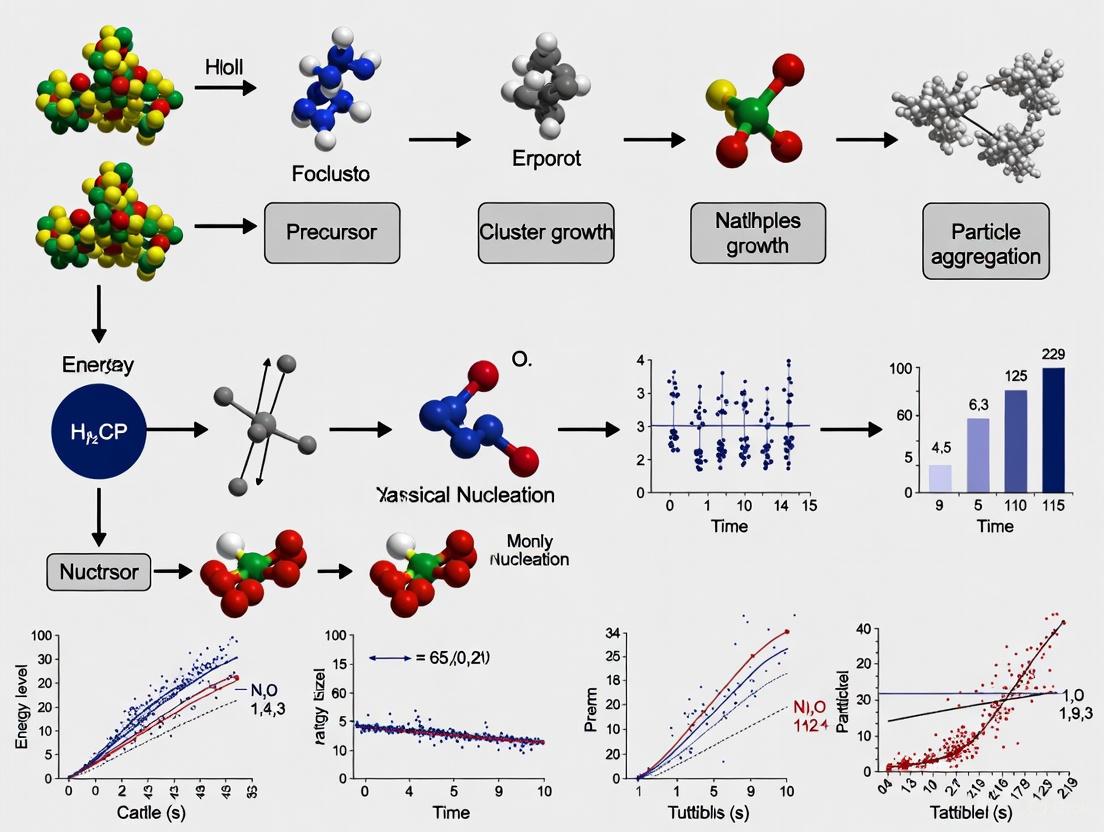
Visualization of Nucleation Pathways
The following diagram synthesizes the competing nucleation pathways discussed, highlighting the decision points between classical and non-classical routes.
Diagram 1: A flowchart comparing classical and non-classical nucleation pathways, showing key system conditions that influence the mechanism.
The experimental workflow for investigating these pathways, particularly in colloidal systems, involves precise control and multiple observation techniques, as shown below.
Diagram 2: Experimental workflow for studying non-classical crystallization in binary colloidal systems.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagents and Materials for Non-Classical Nucleation Studies
| Reagent / Material | Function in Experiment | Example Application |
|---|---|---|
| Oppositely-Charged Colloidal Particles [3] | Serve as model "ions" to directly observe assembly and phase behavior under tunable electrostatic interactions. | Binary colloidal crystal formation. |
| Polymer Brush Coatings [3] | Provide steric repulsion to prevent irreversible aggregation, allowing study of equilibrium structures. | Tuning interparticle potential in colloidal models. |
| Salt Solutions (e.g., NaCl) [3] | Control the Debye screening length (λD), thereby tuning the strength and range of electrostatic attractions. | Mapping crystallization pathways vs. interaction strength. |
| Crystalline Seeds (e.g., CHA zeolite) [7] | Provide a templating surface to study heterogeneous nucleation and its effect on mechanism (classical vs. non-classical). | Zeolite synthesis; polymorph selection studies. |
| Lennard-Jones (LJ) Potential Model [2] | A computationally efficient pair potential to simulate van der Waals interactions in model atomic liquids. | Molecular dynamics studies of nucleation. |
| Metal Oxide Precursors (e.g., WO₃) [6] | The solid source of metal atoms in vapor-phase synthesis of two-dimensional materials. | VLS growth of WS₂ monolayers. |
| Alkali Metal Salt Assistants [6] | Enhance vaporization of metal oxide sources and lower energy barriers, promoting specific growth modes (e.g., VLS). | Salt-assisted CVD of TMDs like MoS₂ and WS₂. |
Non-classical nucleation represents a paradigm shift in our understanding of crystallization processes, moving beyond the traditional model of simple monomer-by-monomer addition to encompass more complex, multi-step pathways. Within inorganic materials research, these pathways frequently involve stable intermediate phases such as pre-nucleation clusters, dense liquid droplets, and amorphous precursors that precede the formation of stable crystalline phases. Recent advances in characterization techniques and computational modeling have revealed that these mechanisms are not merely exceptions but rather common phenomena that fundamentally influence polymorph selection, crystal morphology, and material properties. This whitepaper delineates the principal non-classical mechanisms, supported by quantitative experimental data and detailed methodologies, providing researchers with a framework for understanding and controlling crystallization in materials synthesis and drug development.
Classical Nucleation Theory (CNT) has long served as the foundational model for describing crystallization, positing that ions, atoms, or molecules directly assemble into crystalline structures through stochastic collisions, with the free energy landscape governed by a balance between bulk energy gain and surface energy penalty. However, extensive experimental and computational evidence across diverse material systems now demonstrates that nucleation frequently proceeds through more complex multi-step pathways involving metastable intermediate states.
In non-classical nucleation, the system circumvents the high energy barrier of direct crystallization by first forming intermediate phases, a process with profound implications for controlling material structure and function. For researchers in inorganic materials and pharmaceutical development, understanding these pathways enables precise manipulation of crystallization to achieve desired polymorphic forms, crystal sizes, and morphological characteristics—critical factors in material performance and drug bioavailability.
Key Non-Classical Nucleation Mechanisms
The Two-Step Nucleation Mechanism
The two-step mechanism initially involves the formation of a dense, liquid-like or amorphous intermediate phase from a supersaturated solution, followed by nucleation of the crystalline phase within this precursor medium. This pathway has been observed across diverse systems from proteins to colloidal crystals.
In binary colloidal systems acting as model ions, charged particles first condense into metastable amorphous blobs from the gas phase. Crystal nucleation then initiates within these dense liquid precursors, with the crystallization front propagating through the blob until it becomes fully crystalline [3]. The amorphous blobs serve as a reservoir of particles for subsequent crystal growth, facilitating mechanisms such as Ostwald ripening and direct blob absorption.
Pre-nucleation Clusters and Multistep Pathways
An alternative non-classical pathway involves the formation of stable pre-nucleation clusters (PNCs) in solution prior to the emergence of a separate phase. These nanoscale clusters represent a distinct thermodynamic state that exists in equilibrium with free ions or molecules, acting as building blocks for subsequent nucleation.
In the vapor-liquid-solid (VLS) growth of monolayer tungsten disulfide (WS₂), in-situ monitoring revealed the formation of metastable clusters through the aggregation of droplets, with solid WS₂ nucleation occurring inside these clusters [6]. This mechanism explains observations of very large critical nuclei (up to 38.7 µm) and distinct slow-to-rapid growth transitions that defy classical nucleation theory predictions.
Particle Attachment and Coalescence Pathways
Non-classical nucleation also encompasses pathways where larger particles directly attach to form crystalline structures. This includes mechanisms such as oriented attachment, where crystals fuse along specific crystallographic directions, and random aggregation followed by reorganization.
During the growth of binary colloidal crystals, small crystals can fuse in an orientation-dependent manner to form larger single crystals [3]. This oriented attachment process frequently involves melting and recrystallization at the contact interface, effectively eliminating the seam between the original structures and resulting in a flawless crystalline lattice.
Table 1: Quantitative Characteristics of Non-Classical Nucleation Pathways
| Nucleation Mechanism | Intermediate Phase | Critical Size/Energy Factors | Material Systems Observed |
|---|---|---|---|
| Two-Step Nucleation | Dense liquid droplets, amorphous blobs | Large critical nuclei (µm scale); Reduced energy barrier within dense phase | Binary colloidal crystals [3], Proteins, Minerals |
| Pre-nucleation Clusters | Stable clusters in equilibrium with solution | Cluster stability depends on solution chemistry; Multi-step energy landscape | Calcium carbonate, Iron oxides, Organic molecules [8] |
| Particle Attachment | Nanoparticles, oligomers | Oriented attachment requires crystallographic alignment; Interface elimination | ZnO nanoparticles [9], WS₂ [6], Metal oxides |
| Coalescence of Subcritical Clusters | Amorphous or crystalline nanoclusters | Lower energy barrier vs. classical nucleation; Stepwise growth | FCC iron BCC nucleation [1], Metallic alloys |
Experimental Methodologies and Protocols
Advanced Characterization Techniques
Direct observation of non-classical nucleation pathways requires specialized characterization methods capable of probing nanoscale phenomena in real time under relevant conditions.
Liquid-Cell Transmission Electron Microscopy (LC-TEM): This technique enables direct visualization of nucleation and growth processes in solution at near-atomic resolution. For observing two-step nucleation in colloidal systems, samples are prepared in specialized liquid cells with electron-transparent windows, allowing temporal resolution sufficient to capture phase transitions [10].
In-situ Monitoring Chemical Vapor Deposition: For vapor-phase synthesis of materials like transition metal dichalcogenides, customized CVD systems with optical access permit real-time observation of nucleation events [6]. Automated image analysis of recorded videos using HSV (hue, saturation, value) color thresholding allows precise tracking of precursor dynamics and crystal growth rates.
Cryogenic Transmission Electron Microscopy (Cryo-TEM): By rapidly vitrifying solutions, cryo-TEM captures transient intermediate phases in their native state without crystallization artifacts. This is particularly valuable for identifying amorphous precursors and pre-nucleation clusters in beam-sensitive materials [10].
Atomic Force Microscopy (AFM): High-resolution AFM provides topographical and mechanical property information of emerging nuclei and intermediate phases at solid-liquid interfaces, enabling differentiation between amorphous and crystalline regions during early nucleation stages [10].
Computational and Modeling Approaches
Computational methods provide complementary atomistic insights into nucleation mechanisms that are challenging to observe experimentally.
Machine-Learning Interaction Potentials (MLIP): For accurate modeling of complex systems like zinc oxide, MLIPs combining short-range interactions with long-range electrostatics (PLIP+Q method) enable large-scale molecular dynamics simulations with near-density functional theory accuracy [9]. These potentials correctly reproduce surface energies and polymorph stability, essential for studying nanoparticle nucleation.
Rare-Event Sampling Techniques: Methods like metadynamics and forward flux sampling overcome the timescale limitations of brute-force molecular dynamics, allowing efficient exploration of nucleation pathways and free energy landscapes [9].
Data-Driven Structural Analysis: Gaussian mixture models and other clustering algorithms applied to simulation trajectories enable automatic identification and classification of local structural environments during phase transitions, revealing complex structural evolution in polymorphic systems [9].
Table 2: Experimental Protocols for Investigating Non-Classical Pathways
| Technique | Key Protocol Parameters | Intermediate Phases Identifiable | Limitations and Considerations |
|---|---|---|---|
| Liquid-Cell TEM | Electron dose rate: <10-20 e⁻/Ųs; Solution thickness: <1µm; Frame rate: 1-100 fps | Amorphous precursors, Dense liquid phases, Early crystalline nuclei | Potential electron beam effects; Limited temporal resolution for very fast processes |
| In-situ CVD Monitoring | Temperature: 500-1000°C; Pressure: 1-760 Torr; Imaging rate: 1-30 fps; HSV color thresholding | Liquid precursor droplets, Metastable clusters, Growth dynamics | Limited to vapor-phase processes; Optical resolution limits (~200 nm) |
| Cryo-TEM | Vitrification rate: >10⁴ K/s; Solution temperature: -170°C; Electron dose: <5 e⁻/Ų | Pre-nucleation clusters, Amorphous nanoparticles, Early crystalline phases | Statistically limited sampling; Potential vitrification artifacts |
| Machine-Learning MD | System size: 500-10,000 atoms; Simulation time: ns-µs; Temperature range: 300-2000K | Polymorph transitions, Cluster coalescence, Surface-mediated nucleation | Accuracy depends on training data; Computational cost for large systems |
Signaling Pathways and Process Relationships
The following diagram illustrates the complex relationships and decision points in non-classical nucleation pathways, integrating multiple mechanisms into a unified framework:
Non-Classical Nucleation Pathway Relationships
The experimental workflow for investigating these pathways involves multiple complementary approaches, as illustrated below:
Experimental Workflow for Pathway Investigation
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Experimental Materials
| Reagent/Material | Function in Non-Classical Studies | Example Applications |
|---|---|---|
| Oppositely-charged colloidal particles | Model ions for studying interaction-dependent nucleation pathways; Enable direct visualization of assembly processes | Binary ionic colloidal crystals [3] |
| Salt solutions for dialysis | Precise control of Debye screening length and interaction strength between charged particles; Enable spatiotemporal control of crystallization | Continuous dialysis experiments [3] |
| Metal oxide precursors (WO₃, MoO₃) | Source materials for vapor-liquid-solid growth of transition metal dichalcogenides; Form liquid intermediate phases | WS₂ and MoS₂ synthesis [6] |
| Alkali metal salts (NaCl, KI) | Catalysts in salt-assisted growth; Lower melting points of metal oxides and enhance vaporization | Large-scale TMD growth [6] |
| Polymer brush-coated particles | Provide steric stabilization while allowing electrostatic interactions; Enable tuning of interaction potentials | Binary colloidal crystal assembly [3] |
| Machine-learning potential training sets | Reference data for developing accurate force fields; Include bulk, surface, and nanoparticle configurations | ZnO nucleation simulations [9] |
Implications for Materials Research and Drug Development
The recognition of non-classical nucleation pathways has profound implications for controlling crystallization in applied contexts. In pharmaceutical development, understanding and controlling polymorphic outcomes through intermediate phases is crucial for ensuring drug efficacy, stability, and bioavailability. For inorganic materials synthesis, directing nucleation pathways enables precise control over nanoparticle size, morphology, and crystal phase—parameters that fundamentally determine optical, electronic, and catalytic properties.
The ability to manipulate interaction strengths through solution conditions, as demonstrated in continuous dialysis experiments with colloidal crystals [3], provides a powerful strategy for directing crystallization along specific pathways. Similarly, in silico screening of nucleation barriers using machine-learning potentials offers a computational framework for predicting polymorphic outcomes before resource-intensive experimental work.
Non-classical nucleation pathways represent a fundamental advance in our understanding of crystallization processes, revealing complex multi-step mechanisms that diverge significantly from classical models. The experimental and computational methodologies detailed in this whitepaper provide researchers with powerful tools for investigating these pathways across diverse material systems. By understanding and controlling these processes, scientists can achieve unprecedented precision in materials synthesis and pharmaceutical development, leveraging intermediate phases and particle-based assembly to engineer materials with tailored structures and properties.
The paradigm of crystallization has undergone a fundamental shift with the recognition of non-classical nucleation pathways. Among these, the formation of stable pre-nucleation clusters (PNCs) represents a significant departure from classical nucleation theory (CNT). This whitepaper examines the current understanding of PNCs as solute-based precursors that define early-stage phase separation in mineral and organic systems. Evidence from quantitative models, experimental characterizations, and computational studies reveals that PNCs serve as direct precursors to liquid-liquid phase separation (LLPS), forming dense liquid droplets that eventually solidify into amorphous or crystalline phases. The implications of this mechanism span biomineralization, pharmaceutical development, and advanced materials design, offering novel strategies for controlling crystallization processes through the pre-nucleation stage.
Classical Nucleation Theory (CNT), formulated in the early 20th century, has long served as the fundamental framework for understanding crystallization from solution [11]. CNT posits that nucleation occurs through stochastic fluctuations where ions, atoms, or molecules assemble into unstable critical nuclei that either dissolve or progress to macroscopic crystals based on a balance between bulk and surface energies [12]. This model assumes that (1) nascent nuclei possess the same structure as the macroscopic bulk material, and (2) an interfacial tension exists between the nucleus and solution equivalent to that of a macroscopic interface—the "capillary assumption" [11].
However, numerous observations in biomineralization and biomimetic systems challenge CNT's fundamental premises [11]. Research on calcium carbonate, calcium phosphate, and other systems consistently reveals the existence of stable solute precursors prior to the formation of detectable solid phases [12]. These findings have catalyzed the development of non-classical nucleation concepts, particularly the prenucleation cluster (PNC) pathway [11]. Unlike the unstable, transient clusters envisioned in CNT, PNCs represent thermodynamically stable associations of ions that exist in undersaturated, saturated, and supersaturated solutions [11] [12]. They are not considered distinct particles with a phase interface but rather dynamic solute species with "molecular" character [11]. The PNC pathway provides a mechanistic foundation for understanding previously enigmatic phenomena, including polymer-induced liquid precursors (PILPs), amorphous precursor phases, and mesocrystal formation [11].
The Mechanistic Framework of the PNC Pathway
From Ion Association to Phase Separation
The PNC pathway redefines the initial stages of phase separation. Rather than proceeding directly from ions to solid nuclei, the pathway involves a sequential process where ions first form stable PNCs in homogeneous solution [11]. These clusters subsequently undergo a nanoscopic phase separation event, leading to the formation of dense, liquid-like droplets via liquid-liquid phase separation (LLPS) [13] [12].
A quantitative model developed for calcium carbonate demonstrates that ion association thermodynamics within the homogeneous phase determine the liquid-liquid miscibility gap [13]. In this framework, macroscopically accessible ion association constants define the stability limits for the system. The model successfully predicts both the binodal limit (where phase separation occurs with an energy barrier) and the spinodal limit (where phase separation is barrier-less) based on the thermodynamics of pre-nucleation cluster formation [13]. The mechanism explains that as the dynamics of ion coordination within PNCs decrease upon crossing the liquid-liquid binodal, these solute clusters can transform into phase-separated nanodroplets [13].
Table 1: Key Transitions in the Non-Classical Nucleation Pathway via PNCs
| Stage | Description | Governing Principle | Resulting Structure |
|---|---|---|---|
| 1. Ion Association | Ions form stable complexes in solution | Thermodynamics of ion association & hydration | Prenucleation Clusters (PNCs) in homogeneous solution |
| 2. Liquid-Liquid Phase Separation | PNCs demix from the solution | Crossing of liquid-liquid binodal limit; Decreased coordination dynamics | Dense liquid nanodroplets |
| 3. Solidification | Liquid precursors dehydrate and solidify | Isomorphic transition or surface-induced nucleation | Amorphous nanoparticles with proto-structure |
| 4. Crystallization | Amorphous phase transforms to crystal | Dissolution-reprecipitation or solid-state transformation | Crystalline material |
Distinguishing PNCs from Classical and Other Non-Classical Concepts
The PNC concept fundamentally differs from both classical nuclei and other proposed precursors. Unlike classical critical nuclei, PNCs are thermodynamically stable (not metastable) and exist without a defined phase interface [11]. They are solutes, not particles, and their structures likely do not resemble the final crystalline bulk material [11].
PNCs also differ from the unstable fluctuations in spinodal decomposition. While spinodal decomposition occurs through barrier-less phase separation from the unstable region of phase diagrams, the PNC pathway is based on stable populations of ion associates that serve as precursors to the new phase [13]. The PNC model thus reconciles elements of both binodal and spinodal decomposition by providing a molecular explanation for nanoscopic phase separation [12].
The relationship between PNCs and the widely studied Polymer-Induced Liquid Precursors (PILPs) is particularly noteworthy. Research indicates that liquid precursors observed in PILP systems likely represent polymer-stabilized states of inherently existing liquid phases, rather than being exclusively induced by polymers [13] [14]. The liquid precursors observed in purely inorganic calcium carbonate systems substantiate this interpretation [13].
Experimental Evidence and System Diversity
Calcium Carbonate: The Model System
Calcium carbonate represents the most extensively studied system for PNCs and LLPS, serving as a foundational model for understanding non-classical nucleation pathways [14]. Multiple experimental approaches have verified the existence and role of PNCs in CaCO₃ crystallization:
- Potentiometric Titrations: These measurements allow quantitative determination of the ion activity product (IAP) defining liquid-liquid binodal limits across temperature ranges (15-45°C), revealing the solubilities of initially formed amorphous calcium carbonates (ACCs) [13].
- Stopped-Flow ATR-FTIR Spectroscopy: Kinetic studies using this technique monitor the evolution of carbonate and water vibrational bands after mixing calcium and carbonate solutions, providing time-resolved evidence of phase separation [13]. The kinetics of carbonate ν2 vibrational band transitions show distinct time constants that reach a minimum at the spinodal limit identified potentiometrically, confirming the locus of fastest phase separation [13].
- Cryogenic Transmission Electron Microscopy (Cryo-TEM): Direct imaging of reactive mixtures prior to crystallization consistently reveals "liquid-like" or "emulsion-like" structures, strongly suggesting liquid-phase intermediates before solidification [14].
The evidence shows that amorphous calcium carbonates formed via this pathway have variable solubilities depending on their formation conditions, with the highest possible solubility representing the liquid-liquid spinodal limit [13]. This variability reconciles previously inconsistent literature values for ACC solubility [13].
Extension to Other Mineral and Organic Systems
Beyond calcium carbonate, PNCs and LLPS have been documented across diverse mineral systems, though with varying degrees of experimental confidence [14]:
- Calcium Phosphates: Studies of biomineralization-relevant systems like apatite suggest supportive evidence for liquid-like precursors, though their granular structure is not systematically assigned to colloidal liquids [14].
- Metal Oxalates: Cerium oxalate exhibits very high confidence characteristics for LLPS, with liquid-like morphologies in bulk and porous matrices observed through SEM and cryo-TEM, along with droplet coalescence documented via liquid-phase TEM [14].
- Metal Nanoparticles: Various metallic systems show very high confidence evidence for liquid-like behavior, including dynamic observations via liquid-phase TEM [14].
- Pharmaceutical Compounds: The antiepileptic drug carbamazepine demonstrates LLPS during crystallization, forming amorphous dense liquid clusters (ADLCs) as intermediates in a two-step nucleation process [15]. Micro-droplet precipitation studies reveal that carbamazepine can undergo either a one-step liquid-to-amorphous-solid transition or a two-step liquid-to-crystalline-solid transition, both passing through liquid-to-dense-liquid phase separation [15].
Table 2: Experimental Evidence for PNCs and LLPS Across Different Systems
| System | Supporting Techniques | Confidence Level | Key Observations |
|---|---|---|---|
| Calcium Carbonate | Potentiometry, ATR-FTIR, Cryo-TEM, NMR, MD simulations | Very High | Stable PNCs, liquid-like droplets, variable ACC solubility |
| Cerium Oxalate | SEM, Cryo-TEM, LP-TEM | Very High | Droplet coalescence, liquid morphologies in confined spaces |
| Metal Nanoparticles | Cryo-TEM, AFM, LP-TEM | Very High | Liquid-like dynamics, soft droplets on substrates |
| Apatite | SEM, Cryo-TEM, LP-TEM | Supportive | Liquid-like morphology, dense liquid phases observed |
| Carbamazepine | Micro-droplet assays, polarized microscopy | Supportive | Amorphous dense liquid clusters, solvent-dependent pathways |
| Barium Sulfate | TEM after ethanol quenching | Suggestive | Liquid-like morphologies after fixation |
| Calcium-Silicate-Hydrate | Molecular dynamics simulations | Suggestive | Formation of aggregates from primary particles |
Quantitative Models and Theoretical Advances
Thermodynamic Formulation
The quantitative model for LLPS based on PNCs represents a significant theoretical advance. According to this framework, the spinodal limit (IAPspinodal) can be predicted from the ion association constant Kcluster governing PNC formation [13]:
IAP(spinodal) = [K(cluster)]⁻² [13]
The corresponding binodal limit (IAP_binodal) is accessible through a relationship that incorporates the solubility of the resulting crystalline polymorphs [13]:
IAP(binodal) = A(polymorph) × K_sp(polymorph) × lnK(cluster) [13]
where A(polymorph) is a constant and K_sp(polymorph) is the solubility product of the different polymorphs (calcite, aragonite, or vaterite for CaCO₃) [13]. This model defines a lower-critical solution temperature for the liquid-liquid miscibility gap and accounts for liquid-liquid amorphous polymorphism, potentially explaining mechanisms of polymorph selection [13].
Computational Insights
Computer simulations provide crucial atomistic insights into PNC formation and behavior:
- Enhanced Sampling Techniques: Molecular dynamics simulations with enhanced sampling explore the configurational landscape of ion association, often initializing systems at artificially high ion concentrations to improve sampling efficiency before reconstructing free energy profiles [14].
- Forcefield Development: Simulations employing forcefields like COMPASS (Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies) help describe interactions between molecules in solution, such as between carbamazepine and methanol/water mixtures [15].
- Primary Particle Aggregation: Studies of calcium-silicate-hydrate formation reveal how initial primary particles assemble into stable aggregates, with calcium accelerating this assemblage while releasing water molecules [16].
Experimental Methodologies and Protocols
Potentiometric Titration for Binodal and Spinodal Limits
Objective: Quantitatively determine the liquid-liquid binodal and spinodal limits by measuring the solubility of initially formed amorphous calcium carbonate across different mixing conditions [13].
Procedure:
- Prepare dilute calcium chloride solution (e.g., 10 mM) and carbonate buffer at preset pH (e.g., pH 9.00 for proto-calcite ACC, pH 10.0 for proto-vaterite ACC) [13] [11].
- Titrate calcium solution into carbonate buffer at a constant, slow rate (e.g., 10 μL/min) while maintaining constant pH via simultaneous NaOH addition [11].
- Record calcium potential throughout titration to determine the ion activity product (IAP) at the point of amorphous phase formation [13].
- Repeat at varying temperatures (15-45°C) to establish temperature dependence of binodal limits [13].
- For spinodal determination, implement direct rapid mixing of concentrated solutions (≥100 mM) with concurrent IAP measurements to identify the maximum ACC solubility representing the spinodal limit [13].
Key Parameters:
- Constant pH maintenance via titrant addition
- Controlled addition rate (varied to probe metastable zone)
- Temperature control and variation
- IAP calculation from potential measurements
Stopped-Flow ATR-FTIR Kinetic Analysis
Objective: Independently validate phase separation kinetics and identify spinodal limit through time-resolved vibrational spectroscopy [13].
Procedure:
- Prepare concentrated calcium and carbonate solutions for rapid mixing.
- Utilize stopped-flow apparatus for rapid mixing (millisecond timescale) with ATR-FTIR detection.
- Monitor evolution of characteristic carbonate vibrational bands (particularly ν2 band) and water bands after mixing.
- Collect time-resolved spectra at defined intervals following mixing.
- Fit normalized carbonate ν2 band time transients to generic kinetic models to obtain time constants.
- Identify minimum in time constants corresponding to spinodal limit where phase separation kinetics are fastest [13].
Key Parameters:
- Mixing speed and efficiency
- Spectral acquisition rate
- Carbonate ν2 band monitoring (~873 cm⁻¹)
- Time constant extraction from kinetic fits
Micro-Droplet Platform for Pharmaceutical Compounds
Objective: Statistically analyze phase transitions and capture early-stage crystallization mechanisms of organic drugs like carbamazepine [15].
Procedure:
- Fabricate microfluidic droplet device using polydimethylsiloxane (PDMS) with flow-focusing geometry and 100 μm channel depth [15].
- Prepare drug solutions in varying solvent compositions (e.g., methanol/water ratios for carbamazepine) [15].
- Generate monodisperse micro-droplets using continuous phase (e.g., fluorinated oil with surfactant) and dispersed phase (drug solution) [15].
- Collect droplets onto coated cover glass for microscopic observation.
- Use polarized microscopy to track phase transitions within individual droplets over time.
- Statistically analyze droplet populations for phase transition characteristics (size, number, timing of dense liquid clusters) [15].
Key Parameters:
- Solvent composition ratios
- Droplet size and uniformity
- Statistical sample size (50-100 droplets recommended)
- Image analysis using tools like ImageJ
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for PNC and LLPS Research
| Category/Item | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| Calcium Sources | Calcium chloride (CaCl₂), Calcium hydroxide (Ca(OH)₂) | Provides Ca²⁺ ions for carbonate, phosphate, silicate systems | Purity critical to avoid heterogeneous nucleation |
| Anion Sources | Sodium carbonate (Na₂CO₃), Ammonium carbonate ((NH₄)₂CO₃), Dimethyl carbonate | Provides carbonate ions through direct addition or slow decomposition | Ammonium diffusion technique enables slow pH increase |
| Buffer Systems | Carbonate buffers, Bicarbonate buffers | Maintains constant pH during titration experiments | Critical for controlling ACC proto-structure (pc-ACC vs pv-ACC) |
| Polymers/Additives | Poly(acrylic acid), Poly(aspartic acid), Poly(ethylene glycol) | Stabilizes liquid precursors, enables PILP formation | Historically "induced" liquid precursors now understood as stabilized |
| Solvent Systems | Methanol/Water mixtures, Ethanol for quenching | Controls solubility, nucleation pathway; quenches intermediates | Solvent composition critical for organic drugs like carbamazepine |
| Microfluidic Components | PDMS chips, Fluorinated oils, Surfactants | Creates micro-droplet reactors for statistical analysis | Enables study of homogeneous nucleation in isolated environments |
| Characterization Tools | Potentiometric titrators, ATR-FTIR with stopped-flow, Cryo-TEM | Detects and characterizes PNCs and liquid precursors | Cryo-TEM essential for direct imaging of liquid-like intermediates |
Implications for Materials Research and Pharmaceutical Development
The recognition of PNCs and associated LLPS pathways has profound implications across multiple scientific disciplines:
Advanced Materials Design
Understanding PNCs enables novel approaches to materials synthesis with controlled architectures and properties. The stabilization of liquid precursors allows molding of minerals into complex non-equilibrium shapes, replication of organic matrix templates, and creation of composite materials with tailored hierarchical structures [11]. This capability is particularly valuable for biomimetic materials that seek to replicate the sophisticated structures found in biological minerals like nacre, bone, and teeth [11].
Pharmaceutical Science and Drug Development
For pharmaceutical compounds like carbamazepine, the identification of LLPS pathways enables new strategies for producing amorphous drug forms with enhanced solubility and bioavailability [15]. The micro-droplet platform provides a high-throughput analytical tool for studying amorphous process development, potentially leading to improved formulations for poorly soluble drugs [15]. Understanding and controlling the liquid-to-amorphous-solid transition versus liquid-to-crystalline-solid transition represents a significant opportunity in pharmaceutical manufacturing.
Industrial Crystallization Processes
In industrial contexts, controlling crystallization through PNC manipulation offers routes to optimize pigment and filler properties, improve purification processes, and design novel functional materials [11]. The potential development of nanostructured construction materials through controlled non-classical nucleation pathways could enable revolutionary advances in architectural materials [11].
The paradigm of pre-nucleation clusters as stable liquid-like intermediates has fundamentally transformed our understanding of crystallization pathways. The PNC concept provides a physical chemical foundation for numerous observed non-classical phenomena, including liquid precursors, amorphous intermediates, and complex crystal morphologies. Rather than representing a singular exception, the PNC pathway appears to be a widespread mechanism across diverse mineral and organic systems.
Future research frontiers include more rigorous demonstration of true liquid character across different mineral systems, systematic exploration of structure and dynamics down to atomic and sub-millisecond scales, and integrated experimental-theoretical approaches that capture both thermodynamic and kinetic factors [14]. The development of in situ characterization techniques with enhanced temporal and spatial resolution will be crucial for capturing the rapid dynamics of PNC formation and transformation.
As the field advances, the deliberate manipulation of PNCs and their subsequent transformation pathways promises unprecedented control over crystallization outcomes, enabling next-generation materials with tailored properties and enhanced performance across materials science, pharmaceuticals, and industrial manufacturing.
The Dense Liquid Phase and Two-Step Nucleation Mechanisms
The formation of crystals from solution has traditionally been understood through the framework of Classical Nucleation Theory (CNT), which posits that solute molecules directly assemble into crystalline embryos. According to this century-old theory, crystalline clusters form through the simultaneous processes of densification and structural ordering, with the free energy barrier to nucleation dominated by the interplay between bulk energy gain and surface energy cost [17] [18]. However, an increasing body of experimental and computational evidence now challenges this direct pathway, suggesting instead that crystallization often proceeds through intermediate metastable states [19]. This whitepaper examines the paradigm of two-step nucleation mechanisms, with particular focus on the role of the dense liquid phase as a precursor to crystal formation in organic materials research.
The limitations of CNT have become increasingly apparent, particularly for complex molecular systems. CNT predicts nucleation rates that are often many orders of magnitude lower than those observed experimentally, suggesting that the theory may not fully capture the operative mechanisms in many crystallizing systems [18]. Furthermore, CNT's capillarity approximation, which treats crystal embryos as miniature versions of the bulk crystal with identical properties, fails to account for the complex intermediate states that now appear fundamental to crystallization pathways for a wide range of materials including proteins, small organic molecules, biominerals, and colloids [17] [18] [19].
Theoretical Framework: From Classical to Non-Classical Pathways
Fundamentals of Classical Nucleation Theory
The thermodynamic foundation of CNT, originally developed by Gibbs for liquid droplets and later adapted for crystals, describes the free energy change (ΔG(n)) associated with forming a crystalline cluster of n molecules as:
ΔG(n) = -nΔμ + 6a²n²⁄³α [18]
Where Δμ represents the difference in chemical potential between the solute and crystal (the driving force for crystallization), a is the molecular size, and α is the surface free energy. This relationship produces a free energy maximum at the critical nucleus size n*, where:
n* = 64Ω²α³/Δμ³ and ΔG* = 32Ω²α³/Δμ² = ½n*Δμ [18]
The nucleation rate J, representing the number of nuclei forming per unit volume per unit time, follows an Arrhenius-type dependence on this barrier:
J = νZnexp(-ΔG/kBT) [18]
Where ν* is the attachment frequency of monomers to the nucleus, Z is the Zeldovich factor accounting for the width of the free energy barrier, n is the molecular number density, kB is Boltzmann's constant, and T is temperature [18].
The Two-Step Nucleation Mechanism
In contrast to the direct assembly pathway of CNT, the two-step mechanism proposes that crystalline nuclei form within pre-existing metastable clusters of dense liquid [18] [19]. This process separates the densification and structural ordering processes into distinct stages, significantly reducing the nucleation barrier compared to the direct route [19]. The first step involves the formation of liquid-like clusters several hundred nanometers in size through concentration fluctuations, while the second step entails the emergence of crystalline order within these dense, liquid environments where the surface free energy penalty for creating a crystal-liquid interface is substantially lower than at the crystal-dilute solution interface [18].
The applicability of this mechanism has been demonstrated across diverse systems, including proteins, small organic molecules, colloids, polymers, and biominerals [18]. This universality suggests that two-step nucleation may represent a fundamental pathway for crystallization in solution, particularly for complex molecular systems where the energy landscape between dissolved and crystalline states contains multiple minima.
Table 1: Comparison of Classical and Two-Step Nucleation Mechanisms
| Feature | Classical Nucleation Theory | Two-Step Mechanism |
|---|---|---|
| Pathway | Direct assembly from solution | Nucleation via dense liquid intermediate |
| Order Parameters | Primarily cluster size | Concentration + structural order |
| Critical Nucleus | Defined by size n* | Defined by size and internal order |
| Energy Landscape | Single barrier | Multiple minima and barriers |
| Intermediate States | None | Metastable dense liquid clusters |
| Interface | Sharp crystal-solution boundary | Diffuse liquid-solution and crystal-liquid interfaces |
The Solution-Crystal Spinodal
At high supersaturations typical of many crystallizing systems, the nucleation barrier described by CNT can become negligible, leading to a spinodal-like decomposition regime where the generation of crystal embryos occurs without a significant activation barrier [18]. This solution-crystal spinodal represents a fundamental shift in nucleation behavior, with important implications for polymorph selection and the response to heterogeneous substrates. In this regime, the thermodynamic driving force for crystallization becomes sufficiently large to overcome the interfacial energy penalties without requiring a critical nucleus, enabling spontaneous formation of crystalline regions throughout the solution [18].
Experimental Evidence Across Material Systems
Small Molecule Organic Systems and Supercritical Fluids
Recent thermodynamic assessments of two-step nucleation in supercritical CO₂ fluids have revealed that liquid droplets can precipitate at low supersaturations instead of solid particles, with the metastable liquid phase composition showing only slight dependence on pressure [20]. For systems such as {(S)-Naproxen + CO₂} and {(RS)-Ibuprofen + CO₂} binary mixtures at elevated pressures, the mixture can exist in unstable and/or metastable states with respect to both liquid-vapour and solid-vapour equilibrium [20]. Depending on the supersaturation level, such mixtures may first undergo spinodal decomposition into coexisting liquid and vapour phases before transitioning to solid-fluid equilibrium through nucleation and growth processes [20].
Protein Crystallization
Proteins have provided particularly compelling evidence for two-step nucleation mechanisms. Mesoscopic clusters (10⁵-10⁶ monomers) have been observed in both supersaturated and undersaturated protein solutions, existing as dense liquid phases that are stable with respect to the parent liquid but metastable compared to the emerging crystalline phase [19]. These clusters, typically several hundred nanometers in size, serve as preferential sites for crystal nucleation rather than being dead ends in the crystallization pathway [19].
Experimental approaches including static and dynamic light scattering, brownian microscopy, and laser confocal microscopy with differential interference contrast have demonstrated that the presence of these clusters in solution directly correlates with enhanced nucleation rates and non-classical crystal growth mechanisms [19]. When clusters are removed through rigorous filtration, the nucleation of multilayer crystalline islands is significantly suppressed, establishing a causal relationship between cluster presence and crystallization behavior [19].
Molten Salts and Ionic Systems
Even in relatively simple ionic systems like LiF molten salt, molecular dynamics simulations with machine learning interatomic potentials have revealed complex multistage nucleation pathways [21]. Homogeneous crystal nucleation in undercooled LiF melts preferentially initiates from liquid regions exhibiting both slow dynamics and high bond orientational order simultaneously [21]. Surprisingly, the second-shell order of both precritical nuclei and the surface of postcritical nuclei is dominated by hexagonal close packing and body-centered cubic local structure, despite the nucleus core being dominated by face-centered cubic structure corresponding to the stable rocksalt crystal structure [21]. This illustrates how nucleation pathways can proceed through intermediate states with structural characteristics distinct from the final crystalline form, consistent with Ostwald's step rule which predicts that the phase first nucleated is the one closest in free energy to the parent liquid rather than the globally stable phase [21].
Table 2: Experimental Evidence for Two-Step Nucleation Across Material Classes
| Material System | Key Evidence | Experimental Techniques | References |
|---|---|---|---|
| Proteins | Mesoscopic clusters (100+ nm) preceding crystals | Light scattering, confocal microscopy | [19] |
| Small Organic Molecules/SCF | Liquid droplet precipitation at low supersaturation | Thermodynamic analysis, PREOS | [20] |
| Molten Salts (LiF) | Non-equilibrium local ordering in precritical nuclei | ML-enhanced MD simulations | [21] |
| Colloids | Nucleation from regions with higher bond orientational order | Confocal microscopy, simulation | [17] |
| Biominerals | Amorphous precursors to crystalline phases | TEM, cryo-EM | [18] |
Methodological Approaches and Protocols
Computational Simulation Methods
Molecular dynamics (MD) simulations have provided unique insights into the early stages of crystal nucleation, overcoming the exceedingly small time and length scales that make experimental observation challenging [17]. However, conventional MD faces significant limitations in studying nucleation due to the rare event nature of the process, which often occurs on time scales of seconds—far beyond the reach of standard simulations [17]. Recent advances in machine learning interatomic potentials (MLIPs) have enabled microsecond-scale MD simulations with quantum-level accuracy, as demonstrated in studies of LiF molten salt nucleation [21]. These approaches require careful potential development that considers not only the liquid and stable solids but also all metastable crystalline polymorphs that might participate in the crystallization pathway [21].
For the study of LiF nucleation, researchers developed an Atomic Cluster Expansion MLIP trained on density functional theory data using the SCAN functional, demonstrating excellent agreement with experimentally measured properties [21]. Configurations from multiple crystal structures (B1-B4 and H5) along with equilibrium and undercooled liquids were used in training the potential to ensure accurate representation of both stable and metastable states relevant to the nucleation pathway [21].
Experimental Characterization Techniques
The experimental identification of two-step nucleation pathways requires multidisciplinary approaches capable of detecting both the dense liquid intermediate and the subsequent emergence of crystalline order:
Laser Confocal Microscopy with Differential Interference Contrast (LCM-DIM): This technique provides a mesoscopic field-of-view and relatively fast acquisition time, making it ideal for mapping the temporal dependence of crystal-wide surface topography and observing the formation of looped macrosteps indicative of non-classical growth mechanisms [19].
Brownian Microscopy (BM): BM enables direct tracking of mesoscopic clusters in solution, allowing researchers to correlate cluster number density and dynamics with nucleation events [19].
Static and Dynamic Light Scattering: These approaches characterize the size distribution and stability of dense liquid clusters in solution, providing information about cluster formation and evolution under different thermodynamic conditions [19].
Filtration and Cluster Separation: Rigorous filtration (e.g., triple filtration with 0.2-µm cutoff) allows researchers to create cluster-depleted solutions for comparative studies of nucleation behavior with and without the proposed intermediate phase [19].
The following diagram illustrates the experimental workflow for establishing the role of dense liquid clusters in two-step nucleation:
Thermodynamic Computation Methods
For supercritical fluid systems, thermodynamic computations using equations of state like the Peng-Robinson Equation of State (PREOS) can evaluate mixture stability/metastability/instability with respect to both liquid-vapour and solid-vapour equilibrium [20]. These approaches identify pressure and temperature conditions where two-step nucleation mechanisms are likely to occur, with spinodal limits typically lying at higher supersaturations than the conditions where liquid droplets first precipitate [20].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Reagents and Materials for Studying Two-Step Nucleation
| Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Model Systems | Lysozyme, Glucose Isomerase, Insulin | Well-characterized protein systems for method development | Stability, commercial availability, safety |
| Small Molecule APIs | (S)-Naproxen, (RS)-Ibuprofen | Pharmaceutical relevance in supercritical fluid studies | Purification, polymorphism |
| Solvent Systems | Supercritical CO₂, Aqueous buffers | Control of supersaturation and thermodynamic parameters | Pressure tolerance, purity |
| Filtration Materials | 0.2-µm cutoff membranes | Cluster separation and depletion studies | Protein adsorption, compatibility |
| Characterization Standards | Fluorescent dyes, Size standards | Calibration and validation of microscopy methods | Photostability, interference |
| Computational Tools | MLIP frameworks, Enhanced sampling algorithms | Molecular-level insight into nucleation pathways | Computational cost, accuracy |
Implications for Materials Research and Drug Development
Controlling Polymorphism and Crystal Habit
The recognition of two-step nucleation pathways provides powerful new strategies for controlling crystal polymorphism, a critical concern in pharmaceutical development where different polymorphs can exhibit significantly different bioavailability, stability, and processing characteristics [18]. By manipulating solution conditions to favor specific dense liquid intermediates or by operating in the spinodal regime where the nucleation barrier is negligible, researchers can potentially direct crystallization toward desired polymorphic forms while suppressing unwanted alternatives [18]. This approach is particularly valuable for systems exhibiting multiple polymorphs with similar stability, where traditional crystallization control strategies often prove inadequate.
Enhancing Crystal Purity and Perfection
The non-classical growth mechanisms associated with two-step nucleation, particularly the assimilation of dense liquid clusters by growing crystals, can trigger self-purifying cascades that cleanse impurity-poisoned crystal surfaces [19]. This phenomenon offers potential strategies for producing higher purity crystalline materials without additional processing steps, addressing a significant challenge in pharmaceutical manufacturing where impurities can affect drug efficacy and safety [19].
Advanced Process Design and Optimization
Understanding the role of dense liquid intermediates enables more rational design of crystallization processes for organic materials. By identifying the thermodynamic conditions that favor two-step nucleation and the relationship between intermediate states and final crystal properties, researchers can develop targeted approaches for controlling crystal size distribution, morphology, and internal structure [20] [18]. This knowledge is particularly valuable for continuous manufacturing approaches where precise control over nucleation behavior is essential for consistent product quality.
The following diagram illustrates the decision process for exploiting two-step nucleation in materials design:
Future Perspectives and Challenges
While significant progress has been made in understanding two-step nucleation mechanisms, important challenges remain. The quantitative prediction of nucleation rates for specific material systems still presents difficulties, particularly in relating the properties of dense liquid intermediates to subsequent crystallization behavior [17]. The development of more accurate interatomic potentials and enhanced sampling methods will be crucial for moving toward accurate investigations of realistic systems of practical interest [17].
For experimental studies, clear standards and guidelines for characterizing liquid-liquid phase separation and biomolecular condensates are needed to ensure rigorous identification of two-step nucleation processes [22]. The explosion of physiological and pathological contexts involving liquid-liquid phase separation necessitates careful experimental design to establish causal relationships rather than mere correlations [22].
The integration of computational and experimental approaches, particularly through the use of machine-learning-enhanced simulation methods that bridge time and length scales, represents a promising direction for future research [21]. By combining molecular-level insights from simulation with mesoscopic and macroscopic observations from experiment, researchers can develop comprehensive models of two-step nucleation that enable precise control over crystallization processes across diverse material systems.
As these methods continue to evolve, the deliberate exploitation of two-step nucleation pathways will likely become an increasingly powerful strategy for designing and manufacturing crystalline materials with tailored properties for pharmaceutical, electronic, and structural applications.
In the field of materials science, crystallization has traditionally been understood through Classical Nucleation Theory (CNT), which describes a single-step process where atoms or molecules individually add to a growing crystal lattice [23]. However, advancements in observational techniques have revealed that many materials, particularly in biological and synthetic systems, form through non-classical pathways involving the organized assembly of nanoscale building blocks [24] [25]. Among these mechanisms, oriented attachment (OA)—the crystallographically specific fusion of pre-formed nanoparticles—has emerged as a fundamental process leading to the formation of mesocrystals [26].
Mesocrystals, or mesoscopically structured crystals, are a distinct class of materials defined as "superstructures of crystalline nanoparticles with external crystal faces on the scale of some hundred nanometers to micrometers" [27]. They are not single crystals in the conventional sense, but rather architectures where individual nanocrystals are aligned in a common crystallographic orientation [23] [27]. This ordered arrangement occurs over microscopic length scales, creating a material that exhibits collective physical properties often superior to those of both isolated nanoparticles and conventional single crystals [27]. The study of oriented aggregation and mesocrystal formation provides a critical framework for understanding biomineralization processes in nature and for the bottom-up design of advanced functional materials in fields ranging from catalysis to drug development [23] [26].
Fundamental Mechanisms and Theoretical Framework
Classical vs. Non-Classical Crystallization Pathways
The fundamental distinction between classical and non-classical crystallization lies in the nature of the basic building blocks and the assembly process.
- Classical Crystallization: This is a single-step process where spontaneous nucleation initiates the development of single crystals through the sequential addition of individual atoms, ions, or molecules [23]. The free energy of the system increases until a critical nucleus size is exceeded, after which further growth becomes energetically favorable [25].
- Non-Classical Crystallization: This multi-step pathway involves the self-assembly of nanoparticle clusters, often stabilized by additives [23]. These nanoparticles can transform into intermediate amorphous phases or form mesocrystals through mechanisms like oriented attachment [23]. This pathway is predominant in solution crystallization and plays a crucial role in biomineralization, enabling the formation of complex biological structures like bones, teeth, and shells [23].
Table 1: Comparison of Crystallization Pathways
| Feature | Classical Crystallization | Non-Classical Crystallization |
|---|---|---|
| Building Unit | Atoms, ions, molecules | Nanoparticles, pre-nucleation clusters |
| Pathway | Single-step | Multi-step |
| Key Mechanism | Monomer addition | Particle attachment, self-assembly |
| Common Intermediates | None | Amorphous phases, dense liquid droplets |
| Typical Products | Single crystals, polycrystals | Mesocrystals, hierarchically structured materials |
The Oriented Attachment Mechanism
Oriented attachment is a specific non-classical mechanism where crystalline nanoparticles rotate and align themselves to share a common crystallographic orientation before docking together and fusing into a larger single crystal or mesocrystal [26] [25]. The process can be described in a series of key steps, illustrated in the following workflow.
The driving force for OA is the reduction of surface energy. Nanoparticles possess high surface energy, which provides a thermodynamic impetus for their attachment and fusion to minimize the total surface area [26]. The interaction between nanoparticles is often mediated by their surrounding electrical double layers or organic molecules, which can control the approach and final alignment [23] [26]. Imperfect docking events during OA can lead to the formation of mesocrystals, where the coherence of the final structure is maintained but the original nanoparticle boundaries may remain as defects or grain boundaries [26]. Alternatively, perfect lattice fusion can result in a single crystal that shows no trace of its nanoparticle origins [23].
Experimental Evidence and Formation Pathways
Direct Observation in Protein Systems
Recent cryo-transmission electron microscopy (cryoEM) studies of glucose isomerase (GI) protein crystallization have provided direct, molecular-resolution evidence for oriented attachment [26]. In these experiments, faceted nanocrystals approximately 6-7 nm in size formed within minutes of mixing the protein with a precipitating agent (PEG 1000). The key observations were:
- Faceted Nanocrystals: The initial building blocks were well-defined, crystalline nanoparticles with smooth facets, indicating a one-step nucleation mechanism rather than a disordered precursor [26].
- Lattice Merging: Groupings of nanocrystals were observed to merge into a unified lattice with no discernable stacking faults at their junctions, a hallmark of OA [26].
- Pre-contact Alignment: Nanocrystals were seen to achieve co-alignment well before physical contact, satisfying non-trivial symmetry rules in the process [26].
- Mesocrystal Formation: Larger, composite structures (>1 µm) with pronounced fault lines separating homogeneously aligned domains were identified as mesocrystals, as confirmed by selected area electron diffraction [26].
This study highlights the underappreciated role of interactions between crystalline nuclei themselves, which can dominate the crystallization process at high supersaturation and provide a kinetic shortcut to the final crystalline state [26].
Thermodynamic and Kinetic Controls
The theoretical framework for non-classical nucleation, including OA, has been advanced by combining classical density functional theory (cDFT) with stochastic process theory [24]. This approach can predict nucleation pathways based solely on the interaction potential of the system's particles. For crystallization, the theory illustrates that the process often proceeds by a two-step mechanism: the formation of a dense-solution droplet followed by ordering originating at the core of the droplet [24]. This pathway allows the system to circumvent the high energy barrier associated with direct homogeneous nucleation proposed by CNT [1].
The formation of mesocrystals via OA is highly sensitive to synthetic conditions, which dictate the kinetics and thermodynamics of the assembly process. The table below summarizes the impact of key parameters, primarily derived from studies on inorganic systems like metal oxides.
Table 2: Influence of Synthesis Parameters on Mesocrystal Formation
| Parameter | Effect on Mesocrystal Formation | Example |
|---|---|---|
| pH | Determines surface charge of nanoparticles, affecting stability and interaction. Can transition from mesocrystal to classical crystal formation. | dl-alanine forms rough, porous mesocrystals near its isoelectric point (pH 6.1), but transitions to classical crystallization at pH 10 [23]. |
| Temperature | Influences reaction and crystallization rates. Higher temperatures can lead to fusion into single crystals. | Hematite mesocrystal formation is accelerated with temperature increase. Formation is possible from 45°C to 130°C, while >140°C yields single crystals [23]. |
| Additives / Ligands | Direct nanoparticle alignment by modifying surface energy and interaction; crucial for kinetic stabilization. | Surface ligands like acetate and polyacrylate significantly change nanoparticle alignment in mesocrystals, as indicated by SAED patterns [23]. Oxalate enhances hematite mesocrystal formation [23]. |
| Supersaturation | Affects nucleation rate and probability of nanoparticle interactions. | High supersaturation of glucose isomerase led to rapid nucleation, increasing the probability of nanocrystal interactions and OA [26]. |
Characterization Techniques for Mesocrystals
Confirming mesocrystal structure and elucidating their formation mechanism requires a combination of techniques capable of probing order across multiple length scales—from the atomic arrangement within individual nanoparticles to the long-range mesoscale architecture.
Structural and Morphological Analysis
- Electron Microscopy: Scanning Electron Microscopy (SEM) reveals the overall micrometre-scale morphology [23]. Transmission Electron Microscopy (TEM), especially cryo-TEM for beam-sensitive materials, is indispensable for visualizing internal nanostructure and individual nanoparticle building blocks [26]. Selected Area Electron Diffraction (SAED) in TEM is a critical differentiator: a single-crystal-like spot pattern confirms the crystallographic alignment of a mesocrystal, distinguishing it from the ring pattern of a polycrystalline material [23] [27].
- X-ray Scattering: Small-Angle X-ray Scattering (SAXS) probes the nanoscale structure and periodicity arising from the arrangement of nanoparticles within the mesocrystal [23]. Wide-Angle X-ray Scattering (WAXS) provides information about the atomic-scale crystal structure and crystallinity of the primary nanoparticle units [23].
- In situ Techniques: Liquid cell TEM allows for the direct, video-rate observation of dynamic processes like nanoparticle attachment, coalescence, and mesocrystal growth in their native liquid environment [25]. In situ Atomic Force Microscopy (AFM) can image self-assembly and nucleation events at surfaces with nanometre resolution, and Dynamic Force Spectroscopy (DFS) can quantify the molecular-level interactions underlying these processes [25].
The following diagram illustrates a logical workflow for characterizing mesocrystals using these techniques.
Functional Properties and Applications
The unique architecture of mesocrystals—coupling nanoscale properties with microscale order—confers functional advantages that are exploited across various technological domains.
- Catalysis: Mesocrystals often possess high specific surface area and improved charge transport properties. For example, heterostructures of core-shell CoO/ZnO mesocrystals exhibit enhanced catalytic activity at lower temperatures (60°C-140°C), where the mesocrystalline structure itself plays a major role in boosting performance [23].
- Energy Storage: The hierarchical porosity and efficient electron transport pathways are beneficial for supercapacitors and batteries. W₁₈O₄₉ mesocrystals built from ultra-thin nanowires achieved a high average specific capacitance of 579 F g⁻¹ at a scan rate of 20 mV s⁻¹ [23].
- Optoelectronics and Sensing: Mesocrystals can exhibit novel optoelectronic properties. ZnO mesocrystal microspheres with a core-shell structure generated terahertz emission when exposed to a green laser, while Co-doped ZnO mesocrystal nanowall arrays are suitable for sensor applications due to their large surface area [23].
- Biomedical Technologies: Superparamagnetic magnetite (Fe₃O₄) mesocrystals assembled from 6-10 nm nanoparticles retain superparamagnetism—a property of nanoparticles—while exhibiting a much higher saturation magnetization than the individual nanoparticles, making them ideal for bioseparation, drug delivery, and magnetic resonance imaging [27]. This demonstrates how a mesocrystal can achieve physical properties impossible for a single crystal of the same size.
The Scientist's Toolkit: Key Reagents and Materials
The controlled synthesis of mesocrystals via oriented attachment often requires specific reagents to manipulate nanoparticle surfaces and direct their assembly.
Table 3: Research Reagent Solutions for Mesocrystal Synthesis
| Reagent / Material | Function in Mesocrystal Formation | Example Application |
|---|---|---|
| Polyelectrolytes (e.g., Poly-l-arginine, Polyacrylate) | Act as surface ligands to control nanoparticle charge, stability, and interparticle forces; direct alignment and prevent uncontrolled aggregation. | Used to control the size and magnetic properties of magnetite nanoparticles, influencing their assembly into mesocrystals [23]. |
| Precipitating Agents (e.g., PEG 1000) | Induce supersaturation necessary for the initial nucleation of primary nanocrystals in solution. | Used in the crystallization of glucose isomerase to rapidly form the primary nanocrystals that subsequently undergo OA [26]. |
| Ionic Additives (e.g., Mg²⁺, Oxalate) | Modify specific crystal faces through ion adsorption or complexation, influencing growth rates and nanoparticle morphology to favor oriented attachment. | Oxalate additives enhance the oriented attachment of particles during the formation of hematite mesocrystals [23]. |
| Mineral Precursors (e.g., Zinc Acetate, Iron Salts) | Provide the source of metal ions for the formation of the primary inorganic nanocrystal building blocks (e.g., ZnO, Fe₃O₄). | Fundamental starting material for the synthesis of ZnO and iron oxide mesocrystals [23] [9]. |
Oriented aggregation and mesocrystal formation represent a paradigm shift in our understanding of crystallization, moving beyond the classical atom-by-atom model to a particle-mediated assembly process. This non-classical pathway, driven by the oriented attachment of nanocrystals and finely controlled by synthetic parameters like pH, temperature, and additives, enables the creation of hierarchically structured materials with distinctive functional properties. As advanced in situ characterization techniques and computational models continue to unravel the complexities of these mechanisms, the ability to rationally design mesocrystals with tailored properties for specific applications in catalysis, energy, biomedicine, and sensing will be greatly enhanced. This field stands as a cornerstone of the broader thesis on non-classical nucleation, highlighting how nature's strategies for building complex materials can be understood and harnessed for technological innovation.
Observing the Unseeable: Techniques and Applications in Material Science
In Situ Liquid-Phase Electron Microscopy (LPEM) for Direct Imaging
In situ Liquid-Phase Electron Microscopy (LPEM) represents a transformative advancement in electron microscopy, enabling the direct observation of dynamic processes in liquids at nanoscale resolution. This technique has become indispensable for studying non-classical crystallisation (NCC) pathways in organic materials, particularly for pharmaceutical applications where understanding nucleation events is crucial for controlling polymorph selection and drug efficacy [28]. Unlike Classical Nucleation Theory (CNT), which posits direct organization of atoms or molecules into a critical nucleus, NCC pathways involve intermediate precursor particles such as pre-nucleation clusters (PNCs) and dense liquid phases (DLP) that subsequently develop into crystalline entities [28]. LPEM provides unprecedented temporal and spatial resolution to visualize these transient stages in native liquid environments, offering direct evidence that has previously been inaccessible through conventional ex situ techniques.
The capability to probe early-stage nucleation events is especially valuable for active pharmaceutical ingredients (APIs) like flufenamic acid (FFA), where different crystalline forms (polymorphs) exhibit varying solubility and bioavailability properties [28]. By capturing the precise nucleation events that determine these characteristics, LPEM enables researchers to potentially control and direct crystallization pathways toward more efficacious drug forms, revolutionizing pharmaceutical development and continuous manufacturing processes.
Technical Foundations of LPEM
Instrumentation and Imaging Modes
LPEM instrumentation utilizes specialized liquid cells that encapsulate the sample and liquid medium between thin, electron-transparent membranes (typically silicon nitride) while maintaining the high vacuum requirements of the electron microscope [29]. These environmental chambers incorporate microfluidic channels for introducing and exchanging solutions during imaging, with precisely controlled fluid path lengths (often 50-100 nm) achieved through spacer technology [29].
Two primary imaging modes are employed in LPEM:
- Scanning Transmission Electron Microscopy (STEM): Particularly high-angle annular dark-field (HAADF) STEM, provides superior contrast for biological and organic materials because the signal intensity is approximately correlated with the atomic number squared, making it especially valuable for imaging low-electron-density organic molecules in their liquid surroundings [29].
- Conventional Transmission Electron Microscopy (TEM): Used for specific applications, though with lower contrast for biological and organic samples compared to STEM [29].
The spatial resolution achievable with LPEM depends on multiple factors, including fluid path length, membrane thickness, and electron dose. Under optimal conditions with spherical aberration correction, resolutions of 2.1 Å have been demonstrated for nanoparticles adhered to membrane surfaces, though resolution is typically lower for particles in the bulk liquid or attached to the lower window due to electron probe broadening [29].
Beam-Sample Interactions and Radiolysis Considerations
A critical consideration in LPEM experiments is the interaction between the electron beam and the liquid medium, which induces radiolysis – the dissociation of solvent molecules into reactive species [28]. While traditionally viewed as an undesirable artifact, this phenomenon can be strategically exploited to induce nucleation in undersaturated solutions by altering the local chemical environment to lower energy barriers [28].
For organic molecules like flufenamic acid in ethanol, beam energies exceeding 150 e⁻/Ų/s have been successfully used to initiate nucleation, with radiolysis products potentially catalyzing early-stage nucleation events and accelerating timescales compared to conventional saturated solution experiments [28]. Effective LPEM experimentation requires careful management of electron dose through low-dose imaging techniques, limited exposure times, or continuous flow systems that replenish the solution during observation [28].
Table 1: Key Technical Specifications and Experimental Parameters in LPEM
| Parameter | Typical Range | Impact on Experiment |
|---|---|---|
| Fluid Path Length | 50-1000 nm | Thinner layers improve resolution but limit field of view |
| Silicon Nitride Membrane Thickness | 30-50 nm | Thinner membranes improve signal-to-noise ratio |
| Electron Dose Rate | 10-150 e⁻/Ų/s | Higher doses induce radiolysis-driven nucleation |
| Temporal Resolution | Milliseconds to seconds | Faster capture enables dynamic process imaging |
| Spatial Resolution | <2 nm to ~30 nm | Varies with sample position in liquid cell |
LPEM Experimental Protocol for Studying Non-Classical Nucleation
This section details a standardized methodology for investigating non-classical nucleation pathways of organic materials using LPEM, based on established protocols for pharmaceutical compounds [28] [29].
Sample Preparation
Solution Preparation: Dissolve the organic compound (e.g., flufenamic acid) in an appropriate solvent (e.g., ethanol) at a concentration of 50 mM. For compounds with poor solubility, consider slight heating or sonication to achieve complete dissolution [28].
Liquid Cell Assembly:
- Clean the silicon nitride membrane chips (typically 50 nm thick) with an acetone wash to ensure pristine surfaces [29].
- Prime the microfluidic lines of the continuous flow liquid stage with distilled water to remove air bubbles and particulates [29].
- Deposit 1 μL of the prepared sample solution directly onto the lower silicon nitride window [29].
- Carefully position the upper window, ensuring the membranes face each other with gold spacers (50-100 nm tall) dictating the fluid path length [29].
- Assemble the holder tip and verify vacuum integrity before microscope insertion [29].
Data Acquisition and Imaging
Microscope Alignment: Align the STEM with a spherical aberration corrector using a standard calibration sample (e.g., Platinum/Iridium) to achieve optimal resolution [29].
Imaging Parameters:
- Set the electron probe current density to approximately 30 pA [29].
- Configure dwell times of 2-10 μs per pixel for 512×512 pixel images, resulting in acquisition times of 524 ms to 2.62 s per frame [29].
- Utilize simultaneous bright-field and dark-field detector collection where available [29].
- For nucleation induction, condense the electron beam using the monochromator to increase electron flux in the illuminated region [28].
Continuous Flow Conditions: Implement constant solution flow through the liquid cell to mitigate radiolysis effects and maintain consistent solute concentration during extended observations [28].
Data Analysis and Interpretation
Image Processing: Analyze acquired sequences using specialized software to track particle dynamics, nucleation events, and morphological transformations.
Identification of Non-Classical Intermediates:
- Recognize pre-nucleation clusters as fluctuating, low-contrast entities preceding solid phase appearance [28].
- Identify dense liquid phases as transient droplets that subsequently solidify into crystalline entities [28].
- Document the evolution of these intermediates through Ostwald's rule of stages, potentially observing metastable polymorphs before final stable crystal formation [28].
Key Findings in Non-Classical Nucleation Revealed by LPEM
Direct Observation of Intermediate Stages
LPEM has provided direct visual evidence of non-classical crystallization pathways in small organic molecules. In flufenamic acid, high temporospatial imaging in native organic solvent environments revealed a pathway where pre-nucleation clusters form first, followed by features exhibiting two-step nucleation [28]. This work demonstrated that nucleation pathways are likely an amalgamation of multiple existing non-classical theories rather than following a single universal mechanism [28].
The observed phenomena include:
- Dense Liquid Phase (DLP) Formation: Liquid droplet intermediates appear before solidifying into crystalline entities, serving as an overlapping detail between PNC pathway and two-step nucleation [28].
- Pre-Nucleation Clusters (PNCs): Stable clusters of molecules form in solution prior to the appearance of detectable solid phases, contradicting classical nucleation theory [28].
- Ostwald's Rule of Stages (OSR) Manifestation: Systems transition through less stable states before achieving the final lowest energy crystalline form, though not necessarily passing through all possible intermediate stages [28].
Pharmaceutical Application Case Study: Flufenamic Acid
LPEM experiments with flufenamic acid (FFA), a non-steroidal anti-inflammatory drug, have provided particularly valuable insights:
- Crystallization was induced in an undersaturated 50 mM FFA/EtOH solution through controlled radiolysis, producing hexagonal FFA crystals indicative of form I polymorph used in commercial formulations [28].
- The experiments captured nanoscale early-stage crystallization events, revealing how intermediate pre-crystalline stages evolve into final crystalline entities [28].
- Evidence suggested that water introduction, intended as a troubleshooting step for fluid flow issues, could potentially act as an antisolvent, altering saturation conditions, though this was not conclusively demonstrated [28].
Table 2: Non-Classical Crystallization Pathways Observed via LPEM
| NCC Pathway | Key Characteristics | Observation in LPEM |
|---|---|---|
| Pre-Nucleation Cluster (PNC) | Stable molecular clusters preceding solid formation | Fluctuating, low-contrast entities prior to crystal appearance |
| Two-Step Nucleation | Dense liquid phase intermediate before crystallization | Liquid droplets solidifying into crystalline entities |
| Ostwald's Rule of Stages | Transition through metastable polymorphs | Sequential appearance of different crystalline structures |
| Amalgamated Pathways | Combination of multiple NCC mechanisms | PNC pathway followed by two-step nucleation features |
Essential Research Reagents and Materials
Successful LPEM experiments require specialized materials and instrumentation. The following table details key components of the "LPEM Research Toolkit" for studying non-classical nucleation in organic materials.
Table 3: Essential Research Reagents and Equipment for LPEM Studies
| Component | Specifications | Function/Role |
|---|---|---|
| Liquid Cell Holder | Continuous flow design with microfluidic capabilities | Maintains liquid environment in high vacuum of EM |
| Silicon Nitride Membranes | 50 nm thickness, electron-transparent | Encapsulates liquid while allowing electron transmission |
| Gold Spacers | 50-100 nm height | Controls fluid path length between membranes |
| Organic Solvents | HPLC grade (e.g., ethanol, acetonitrile) | Dissolves organic compounds of interest |
| Pharmaceutical Compounds | High purity (e.g., flufenamic acid) | Target molecules for nucleation studies |
| Aberration-Corrected STEM | JEM-2100F/Cs or equivalent with CEOS corrector | High-resolution imaging of low-contrast materials |
| Continuous Flow System | Syringe pumps with PEEK tubing | Maintains fresh solution, mitigates radiolysis effects |
Advanced Applications and Future Directions
The application of LPEM continues to expand beyond fundamental nucleation studies into more complex systems and technical developments. Emerging areas include:
Biological and Polymeric Systems
LPEM techniques are being adapted for imaging macromolecular protein complexes in fully hydrated environments without staining or chemical fixation [29]. This has enabled visualization of structures like ferritin molecules (approximately 12 nm diameter) with sufficient resolution to distinguish the protein shell from the mineral core [29]. Similar approaches show promise for studying polymeric nanostructures, self-assembling systems, and other soft materials that undergo dynamic transformations in liquid environments.
Technical Innovations and Data Integration
The field is rapidly advancing through several key technological developments:
- Advanced Data Analysis: Artificial intelligence and machine learning approaches are being integrated into LPEM workflows to enhance feature recognition, particle tracking, and dynamic process analysis [30].
- Correlative Techniques: Combination of LPEM with complementary characterization methods such as optical microscopy, X-ray spectroscopy, and mass spectrometry provides multidimensional insights into nucleation phenomena [30].
- Data Integration Platforms: New software tools like Nexus are emerging to synchronize, navigate, and visualize in situ experiment data, enabling researchers to uncover correlations that isolated datasets cannot reveal [31].
In situ Liquid-Phase Electron Microscopy has fundamentally advanced our understanding of non-classical nucleation pathways in organic materials by providing direct nanoscale visualization of previously theoretical intermediate stages. The technique's ability to capture dynamic crystallization events in native liquid environments has demonstrated that nucleation mechanisms often involve amalgamations of pre-nucleation clusters, dense liquid phases, and multi-step pathways rather than following singular theoretical models.
For pharmaceutical research and development, these insights offer unprecedented opportunities to control crystallization outcomes, potentially enabling access to previously inaccessible polymorphs with enhanced therapeutic properties. As LPEM technology continues to evolve through improved liquid cell designs, reduced beam effects, advanced data analysis algorithms, and correlative imaging approaches, its impact on materials science, chemistry, and drug development will undoubtedly expand, solidifying its role as an essential tool for unraveling complex dynamic processes at the nanoscale.
Cryo-TEM and Advanced Light Scattering Techniques
The investigation of non-classical nucleation pathways represents a paradigm shift in inorganic materials research, moving beyond traditional models to encompass complex processes involving pre-nucleation clusters, amorphous precursors, and particle-attachment mechanisms [32]. Understanding these pathways is crucial for gaining fundamental insight into morphology evolution and for the rational design of complex functional materials [33] [32]. This technical guide explores the synergistic application of Cryogenic Transmission Electron Microscopy (cryo-TEM) and advanced light scattering techniques, which together provide a powerful analytical toolkit for elucidating these intricate formation mechanisms directly in solution. Cryo-TEM offers unparalleled nanoscale to near-atomic resolution imaging of transient structures within vitrified solutions [33] [32], while dynamic light scattering (DLS) delivers vital hydrodynamic size and population dynamics information in their native state [34]. When used correlatively, these methods bridge critical gaps in spatial and temporal resolution, enabling researchers to construct a more complete picture of non-classical nucleation and growth phenomena that are central to modern materials chemistry and drug development.
Fundamental Principles of the Techniques
Cryogenic Transmission Electron Microscopy (Cryo-TEM)
Cryo-TEM involves the rapid vitrification of a thin liquid film containing the sample of interest by plunging it into a cryogen (typically ethane or propane) at liquid nitrogen temperatures [33]. This process, known as plunge-freezing, arrests the solution-state structures within a thin layer of amorphous ice, preserving them in a near-native state [32]. The vitrified samples are then transferred to a TEM equipped with a cryo-holder maintained at cryogenic temperatures (typically below -170°C) for imaging [33].
The key advantages of cryo-TEM for studying non-classical nucleation pathways include:
- Minimized Beam Damage: Cooling specimens to cryogenic temperatures significantly reduces structural damage caused by the electron beam through knock-on displacement, radiolysis, or thermal effects, which is particularly crucial for imaging beam-sensitive organic, inorganic, or hybrid materials [33].
- Preservation of Solution-State Structures: By vitrifying the solution, transient and hydrated species—such as pre-nucleation clusters, amorphous precursor particles, and intermediate assemblies—can be immobilized and visualized, overcoming the limitations of conventional dry-state TEM [32].
- High-Resolution Capability: Cryo-TEM enables high-resolution imaging (sub-nanometer to atomic-scale) of these fragile structures, which often cannot be achieved otherwise [33].
Dynamic Light Scattering (DLS)
Dynamic Light Scattering (also known as Photon Correlation Spectroscopy) measures the Brownian motion of particles in suspension by analyzing the fluctuations in the intensity of scattered light from a laser beam [34]. The diffusion coefficient (D) is derived from these fluctuations through an autocorrelation function, and the hydrodynamic diameter (dh) is subsequently calculated using the Stokes-Einstein equation:
d_h = k_B T / 3 π η D
where kB is Boltzmann's constant, T is the absolute temperature, and η is the solvent viscosity [34].
DLS provides population-averaged hydrodynamic size distributions and is exceptionally sensitive to the presence of large aggregates or small amounts of oligomeric species, making it ideal for monitoring the early stages of nucleation and assembly processes in solution [34].
Correlative Workflow for Investigating Non-Classical Nucleation
The power of these techniques is maximized when used within a correlative workflow. The following diagram illustrates the integrated experimental pathway for studying non-classical nucleation.
Experimental Protocols
Cryo-TEM Sample Preparation and Imaging Protocol
Objective: To preserve and visualize transient species and intermediate structures present in a solution during inorganic nucleation processes.
Materials:
- Holey Carbon TEM Grids (e.g., Quantifoil, C-flat)
- Plunge Freezer (e.g., Vitrobot, EM GP)
- Liquid nitrogen and cryogen (ethane or propane)
- Cryo-TEM holder and transfer station
- TEM/STEM with cryo-capabilities
Procedure:
- Grid Preparation: Glow-discharge TEM grids to render them hydrophilic, ensuring uniform vitreous ice formation [33].
- Sample Application: Apply 3-5 µL of the reaction solution to the grid. The solution composition (e.g., solvent, concentration of precursors) should mimic the actual synthesis conditions as closely as possible [32].
- Blotting and Vitrification: Blot the grid with filter paper for a defined time (typically 1-10 seconds) to create a thin liquid film, then rapidly plunge it into the cryogen. Optimal blotting time and humidity are empirically determined to achieve ice of suitable thickness (ideally < 1 µm) [33] [32].
- Cryo-Transfer: Transfer the vitrified grid under liquid nitrogen to the cryo-TEM holder, ensuring the sample remains below -170°C at all times to prevent devitrification [33].
- Imaging and Analysis:
- Perform initial screening at low magnification (e.g., 5,000-20,000x) to assess ice quality and locate regions of interest.
- Acquire high-resolution TEM (HRTEM) or STEM images using low-dose techniques to minimize electron beam radiation damage [33].
- For elemental analysis, acquire cryo-Energy Dispersive X-ray Spectroscopy (cryo-EDS) maps or cryo-Electron Energy Loss Spectroscopy (cryo-EELS) spectra from specific regions of interest [33].
- For 3D structural analysis, collect a tilt series for cryo-Electron Tomography (cryo-ET) [32].
Dynamic Light Scattering Measurement Protocol
Objective: To determine the hydrodynamic size distribution and monitor the temporal evolution of species in solution during nucleation.
Materials:
- DLS Instrument (e.g., Malvern Zetasizer, Wyatt DynaPro)
- Clean, disposable cuvettes (e.g., polystyrene, quartz)
- Temperature-controlled sample chamber
Procedure:
- Sample Clarification: Filter the reaction solution through a 0.1 or 0.2 µm syringe filter (compatible with the solvent) directly into the DLS cuvette to remove dust particles that can dominate the scattering signal [34].
- Instrument Equilibration: Allow the sample to equilibrate in the instrument's temperature-controlled chamber for a set time (e.g., 2-5 minutes) to ensure thermal stability [34].
- Measurement Setup:
- Set the measurement angle (commonly 90° or 173° backscatter).
- Define the measurement duration and number of runs.
- Set the temperature for the experiment, ensuring it matches the reaction conditions of interest.
- Data Acquisition:
- Perform measurements in triplicate or more to ensure reproducibility.
- For kinetic studies, perform sequential measurements over time to track the evolution of particle size.
- Data Analysis:
- Always inspect the autocorrelation function and the baseline for quality. A poor-quality fit or unstable baseline indicates unreliable data [34].
- Report the hydrodynamic diameter (Z-average) and the polydispersity index (PDI) from the cumulants analysis.
- Critically evaluate size distribution plots (intensity-weighted, volume-weighted, and number-weighted). The intensity-weighted distribution is most sensitive to the presence of large aggregates, which can be crucial for identifying the onset of nucleation [34].
Quantitative Data Comparison and Interpretation
Comparison of Technique Capabilities
Table 1: Key characteristics of cryo-TEM and DLS for studying non-classical nucleation.
| Parameter | Cryo-TEM | Dynamic Light Scattering (DLS) |
|---|---|---|
| Spatial Resolution | Sub-nanometer to atomic-scale [33] | N/A (Indirect measurement) |
| Size Information | Direct particle size and morphology [32] | Hydrodynamic diameter (dh) [34] |
| Sample State | Vitrified solution (snapshot) | Native solution (real-time) |
| Chemical/Elemental | Possible with cryo-EDS/EELS [33] | No |
| Temporal Resolution | Seconds to minutes (manual sampling) [32] | Milliseconds (for measurement); suitable for kinetics [34] |
| Key Strengths | Direct visualization of morphology, crystallinity, and coexistence of phases [32] | Rapid assessment of size distribution and solution stability; ideal for monitoring kinetics [34] |
| Primary Limitations | Sample preparation artifacts; electron beam sensitivity; statistical representation [33] | Provides only an indirect measure of size; highly sensitive to aggregates/dust; low resolution for polydisperse systems [34] |
Interpreting Correlative Data: A Roadmap
A critical, combined interpretation of DLS and cryo-TEM data is essential to avoid common pitfalls.
Table 2: Guide to interpreting correlative DLS and cryo-TEM data.
| Observation | Potential Interpretation | Further Investigation |
|---|---|---|
| DLS shows large dh; Cryo-TEM shows small, discrete particles. | The system is highly agglomerated in solution, or the DLS measurement is dominated by a small population of large aggregates [34]. Cryo-TEM may miss loosely-bound aggregates that break apart during blotting. | Check DLS correlation function and size distribution for multiple populations. Use cryo-TEM to search for rare, large species. Consider sample dilution for DLS. |
| DLS shows a single peak; Cryo-TEM reveals multiple distinct morphologies. | Different morphological species (e.g., spheres, rods) have similar hydrodynamic volumes, or one population dominates the scattering intensity due to its larger size [34]. | Analyze the relative proportions of species in cryo-TEM images statistically. Use additional techniques like SAXS to deconvolute populations. |
| DLS size increases over time; Cryo-TEM shows amorphous nanoparticles. | Consistent with non-classical particle-attachment growth or aggregation of amorphous precursors [32]. | Perform time-resolved cryo-TEM sampling. Use cryo-ET to visualize connectivity between particles. |
| DLS indicates stable population; Cryo-TEM shows crystalline structures. | Crystallization may occur locally on the grid after blotting or due to electron beam-induced effects [33] [32]. | Compare cryo-TEM results from grids frozen at different times after blotting. Use low-dose imaging protocols to minimize beam effects. |
Essential Research Reagent Solutions
Table 3: Key reagents and materials for cryo-TEM and DLS studies of inorganic nucleation.
| Reagent/Material | Function/Application | Technical Notes |
|---|---|---|
| Holey Carbon TEM Grids | Support for vitrified ice film. | Grid type (hole size, shape) and hydrophilicity (via glow discharge) are critical for uniform ice thickness [33]. |
| Cryogen (e.g., Ethane) | Medium for rapid vitrification. | High cooling rate prevents water crystallization, forming amorphous ice that preserves native structures [32]. |
| Precursor Solutions | Starting materials for nucleation reactions. | Concentration, pH, ionic strength, and solvent must be carefully controlled to match synthesis conditions [32]. |
| Syringe Filters (0.1-0.2 µm) | Clarification of DLS samples. | Essential for removing dust, which causes spurious scattering and can be misinterpreted as a particle population [34]. |
| Stable Buffer/Solvent Systems | Maintaining solution conditions. | Prevents unwanted changes in pH or ionic strength that could trigger or alter nucleation during measurement [34] [32]. |
The correlative application of cryo-TEM and dynamic light scattering provides a formidable platform for demystifying non-classical nucleation pathways in inorganic materials. While cryo-TEM delivers definitive, high-resolution snapshots of transient intermediate structures—from pre-nucleation clusters to amorphous precursors and crystalline phases—DLS offers indispensable, real-time insights into hydrodynamic size evolution and population dynamics in the native solution state [34] [32]. By following the rigorous protocols and data interpretation frameworks outlined in this guide, researchers can bridge the gap between indirect scattering data and direct visual evidence. This synergistic approach is poised to accelerate the rational design of next-generation functional materials, from tailored nanoparticles for drug delivery to complex hybrid materials inspired by natural biomineralization processes.
Crystallization stands as a fundamental process in materials science, traditionally described by classical theories involving monomer-by-monomer addition to growing crystal surfaces. However, the emergence of non-classical crystallization pathways has fundamentally challenged this paradigm, revealing complex particle-mediated mechanisms that operate across multiple length scales. In zeolites and framework materials, these non-classical pathways involving amorphous intermediates, nanoparticle attachment, and mesoscopic precursors significantly influence crystallization kinetics, final crystal properties, and ultimately, their catalytic performance [3] [4] [35]. Understanding and controlling these pathways is therefore not merely an academic pursuit but a crucial requirement for advancing materials design for applications ranging from industrial catalysis to pharmaceutical development.
This case study examines the mechanistic principles governing non-classical crystallization in zeolitic and framework materials, supported by quantitative data, experimental methodologies, and visualizations of the complex reaction networks involved. The insights gathered provide a framework for rationally designing synthesis protocols that leverage non-classical pathways to achieve materials with tailored properties.
Core Principles and Mechanisms
Non-classical crystallization in zeolites encompasses several distinct but interrelated pathways that diverge from classical stepwise growth:
Particle Attachment Mechanisms: Crystallization proceeds through the assembly of pre-structured nanoparticles or subcolloidal units rather than individual ions or molecules. This includes processes such as oriented attachment, where crystalline nanoparticles fuse along common crystallographic axes [3], and non-oriented aggregation of amorphous precursors that subsequently undergo internal reorganization to form crystalline phases [36] [37].
Two-Step Nucleation Models: A prevalent non-classical mechanism involves initial condensation of solutes into dense liquid-like phases or metastable amorphous intermediates, within which crystalline nucleation subsequently occurs [3] [35]. This pathway significantly reduces the kinetic barrier to nucleation compared to direct crystallization from solution as predicted by Classical Nucleation Theory [35].
Interplay with Classical Pathways: Most real synthetic systems do not follow a single pathway exclusively. Research on MFI zeolite crystallization demonstrates that classical and non-classical pathways often coexist and intertwine throughout the crystallization process [36]. The dominant pathway can shift based on synthesis conditions, with the non-classical pathway frequently predominating in early stages [36].
The following diagram illustrates the key stages and decision points in these competing crystallization pathways:
Quantitative Analysis of Crystallization Parameters
The transition between classical and non-classical crystallization pathways can be systematically controlled by modulating key synthesis parameters. Research on MFI zeolite synthesis demonstrates that varying H₂O/SiO₂ and ethanol/SiO₂ ratios directly influences the dominant crystallization mechanism [36].
Table 1: Effect of Synthesis Parameters on Crystallization Pathways in MFI Zeolite
| Synthesis Parameter | Parameter Range | Effect on Crystallization Pathway | Impact on Final Product |
|---|---|---|---|
| H₂O/SiO₂ Ratio | Reduction from standard ratio | Favors non-classical pathway | Higher aspect ratio nanosheets |
| Ethanol/SiO₂ Ratio | Increase from standard ratio | Promotes classical mechanism | More isotropic nanoparticles |
| Crystallization Stage | Early stages | Non-classical pathway predominates | Determines initial morphology |
| Crystallization Stage | Later stages | Pathways intertwine | Complex morphological evolution |
Furthermore, the presence of crystalline seeds dramatically alters nucleation mechanisms by providing heterogeneous surfaces that can bypass amorphous intermediate stages:
Table 2: Impact of Crystalline Seeds on Nucleation Mechanisms
| Synthesis Condition | Nucleation Mechanism | Key Characteristics | Applicable Systems |
|---|---|---|---|
| Moderate Supersaturation with seeds | Classical nucleation | Monomer-by-monomer addition, bypasses amorphous intermediates | Zeolites, biominerals, pharmaceuticals |
| High Supersaturation with seeds | Non-classical nucleation | Amorphous intermediates persist despite seeds | Zeolite synthesis under aggressive conditions |
| Aggregate-based Reactants | Non-classical nucleation | Particle attachment dominates even with seeds | Systems with pre-formed nanoparticles |
Experimental Protocols and Methodologies
Distinguishing Crystallization Pathways in MFI Zeolite
Objective: To distinguish and directly quantify contributions of classical and non-classical crystallization pathways in MFI zeolite synthesis by varying H₂O/SiO₂ and ethanol/SiO₂ ratios [36].
Materials:
- Tetraethyl orthosilicate (TEOS, 99%) as silica source
- Tetrapropylammonium hydroxide (TPAOH, 25 wt%) as structure-directing agent
- Ethanol (≥99.7%) as solvent
- Deionized water
- Aluminum isopropoxide (for ZSM-5 synthesis)
- Dialysis tube (Molecular weight cutoff: 3.5 kDa)
Procedure:
- Sol Preparation: Prepare sol composition with X SiO₂: 0.39 TPAOH: 13.21 H₂O: 4X ethanol, where X = 1.0–1.9 for H₂O/SiO₂ variation studies.
- Hydrolysis: Add TPAOH solution to TEOS and stir rapidly at room temperature for 24 hours to ensure complete hydrolysis.
- Microwave-Assisted Crystallization: Transfer mixture to microwave reactor for two-step heating:
- Heat at 90°C for 90 minutes
- Immediately heat at 130°C, sampling at intervals up to 600 minutes
- Particle Size Monitoring: Track particle size evolution using Dynamic Light Scattering (DLS).
- Sample Purification: Once system reaches inflection point indicated by DLS, separate solid samples during linear growth stage using two-step dialysis:
- Dialyze against 6 mmol L⁻¹ TPAOH aqueous solution for 24 hours
- Transfer to deionized water for 48 hours, replacing water every 12 hours
- Product Isolation: Purified sol is freeze-dried at -50°C, then calcined at 550°C to remove structure-directing agent.
Analysis:
- Monitor crystallization kinetics using characteristic S-shaped crystallization curves
- Analyze particle morphology evolution via electron microscopy
- Correlate pathway dominance with catalytic performance in furfuryl alcohol etherification
Polymer-Directed Non-Classical Crystallization of TS-1 Zeolite
Objective: To switch TS-1 zeolite crystallization from classical to non-classical mechanism using polyacrylamide (PAM) to accelerate nucleation and enrich active Ti sites [38].
Materials:
- Tetraethyl orthosilicate (TEOS) as silica source
- Tetrabutyl orthotitanate (TBOT) as titanium source
- Tetrapropylammonium hydroxide (TPAOH) as structure-directing agent
- Polyacrylamide (PAM) as crystallization pathway modifier
Procedure:
- Precursor Evolution Control: Utilize specific interactions between PAM and Si/Ti species to promote assembly of colloidal precursors containing ordered structural fragments.
- Two-Step Crystallization: Employ PAM to control precursor structure evolution through a two-step crystallization process.
- Stabilization: PAM stabilizes Ti species in the precursors, preventing titanium leaching and ensuring incorporation into the framework.
- Crystallization Monitoring: Use advanced characterization techniques to track crystallization process and structural evolution.
Analysis:
- Compare crystallization time with conventional methods (1.5-fold shortening expected)
- Analyze Ti content in final TS-1 product (target Si/Ti = 29)
- Evaluate catalytic performance in oxidative reactions
The experimental workflow for controlling crystallization pathways through synthetic parameters is illustrated below:
The Scientist's Toolkit: Essential Research Reagents
Successful investigation of non-classical crystallization pathways requires specific reagents and materials tailored to modulate and characterize these complex processes:
Table 3: Essential Research Reagents for Non-Classical Crystallization Studies
| Reagent/Material | Function in Crystallization Studies | Example Application |
|---|---|---|
| Tetraethyl Orthosilicate (TEOS) | High-purity silica source for clear sol synthesis | MFI, MEL, and EUO zeolite synthesis [36] [39] |
| Structure-Directing Agents (TPAOH, TBAOH) | Template specific zeolite frameworks and influence precursor assembly | MFI (TPAOH) and MEL (TBAOH) structure direction [37] |
| Polyacrylamide (PAM) | Polymer modifier that switches crystallization from classical to non-classical pathway | TS-1 synthesis with enhanced Ti incorporation [38] |
| Heteroatom Sources (Al, Ti, Sn, Zn precursors) | Incorporate framework heteroatoms to tune acidity and catalytic properties | Creating heteroatom subcrystals with tailored active sites [37] |
| Crystalline Seeds | Promote heterogeneous nucleation, converting non-classical to classical pathways | Accelerating nucleation and controlling polymorph selection [40] [41] |
| Alkali Metal Salts (NaOH, Na₂CO₃, NaHCO₃) | Modulate crystallization kinetics and pathway selection | Morphology control in MWW zeolite synthesis [42] |
Implications for Materials Design and Catalytic Performance
The deliberate selection of crystallization pathways directly impacts the functional properties of resulting zeolites and framework materials, with significant implications for catalytic applications:
Acidic Properties Preservation: Studies on ZSM-5 zeolites revealed that shifting the crystallization pathway from non-classical to classical mechanisms had minimal effect on acidic properties but directly impacted catalytic performance [36]. The catalytic activity in furfuryl alcohol etherification correlated with the contribution of the classical pathway, with higher contributions leading to enhanced catalytic activity [36].
Active Site Accessibility: Heteroatom-containing zeolite subcrystals synthesized via non-classical pathways demonstrate high active site accessibility and can serve as versatile building blocks for customized zeolite synthesis [37]. These materials show diverse catalytic performance in reactions such as olefin epoxidation and macromolecular cracking, tailored to meet specific reaction requirements [37].
Morphology-Property Relationships: The crystallization pathway directly influences crystal morphology and aspect ratio. Non-classical pathways often yield high-aspect-ratio nanosheets due to anisotropic growth rates, while classical pathways favor more isotropic nanoparticles [36] [42]. These morphological differences subsequently impact diffusion pathways, accessibility to active sites, and ultimately catalytic efficiency in target applications.
Non-classical crystallization pathways represent a fundamental shift in our understanding of zeolite and framework material formation, moving beyond simple monomer addition models to embrace the complexity of particle-mediated growth, amorphous intermediates, and multi-step nucleation processes. Through careful manipulation of synthesis parameters including H₂O/SiO₂ ratios, polymer additives, crystalline seeds, and heteroatom incorporation, researchers can now exert unprecedented control over these pathways to engineer materials with tailored properties. The continued elucidation of these mechanisms promises to advance the rational design of next-generation catalysts and functional materials, bridging the gap between fundamental crystallization science and practical application demands.
Crystallization is a fundamental process in pharmaceutical development, dictating the critical physicochemical properties of active pharmaceutical ingredients (APIs). While classical crystallization theories have long provided the foundational framework for understanding this process, emerging research on non-classical nucleation pathways reveals substantially more complex crystallization trajectories involving intermediate phases and particle attachment mechanisms. This technical guide examines the crystallization of Flufenamic Acid (FFA), a highly polymorphic nonsteroidal anti-inflammatory drug, as a model system for exploring these non-classical pathways. With nine known polymorphic forms, FFA presents a compelling case study for understanding how crystallization mechanisms impact solid-state properties, stability, and ultimately, drug product performance. This review integrates recent advances in direct observation techniques and analytical methodologies, framing FFA crystallization within the broader context of non-classical crystallization theory to provide pharmaceutical scientists with both theoretical insights and practical experimental guidance.
Flufenamic Acid as a Model System
Polymorphic Landscape and Pharmaceutical Relevance
Flufenamic acid (2-[3-(trifluoromethyl)amino]benzoic acid) is a BCS Class II drug with low solubility and high permeability, making it an ideal candidate for crystallization studies aimed at bioavailability enhancement [43]. According to the Cambridge Structural Database, nine FFA polymorphs have been discovered, with eight structurally characterized forms [43]. These polymorphic systems share common structural features, including strong intramolecular O···H–N hydrogen bonds that hold the phenyl ring carboxylic group and bridging amino group coplanar, while forming dimers through O···H–O hydrogen bonds between adjacent molecules in the crystal packing [43].
The significant interest in FFA polymorphism stems from its direct impact on pharmaceutical processing and performance. Different polymorphic forms can vary considerably in their dissolution rates, compaction behavior, and physical stability, necessitating rigorous control over crystallization outcomes [44]. This complexity is further enhanced by FFA's ability to form liquid crystalline phases and hollow crystal structures, offering unique opportunities for material design [44].
Non-Classical Crystallization Pathways in FFA
Direct observation of FFA crystallization using Liquid Phase Electron Microscopy (LPEM) has provided compelling evidence for non-classical crystallization pathways. Research by Cookman et al. demonstrated that FFA in organic solvents follows a pathway involving Pre-Nucleation Clusters (PNCs) and features consistent with two-step nucleation [45]. This pathway deviates fundamentally from classical models where molecules directly incorporate into growing crystals through monomer-by-monomer addition.
The LPEM observations revealed that FFA crystallization initiates with the formation of dense liquid-like regions containing molecular clusters, which subsequently reorganize into crystalline structures. This mechanism represents an amalgamation of multiple non-classical theories and highlights the importance of intermediate phases in determining final crystal forms [45]. The discovery of these pathways in FFA establishes it as a valuable model system for studying non-classical crystallization in small organic molecules, with implications for pharmaceutical development more broadly.
Crystal Defects and Physical Reactivity
Defect-Mediated Polymorphic Transformations
The relationship between crystal defects and physical reactivity represents a crucial aspect of FFA crystallization behavior. Li et al. demonstrated that crystal defect density directly correlates with the reactivity of physical transformation in FFA polymorphs [46] [47]. Their research established that larger FFA Form I crystals transform to Form III faster than smaller crystals from the same batches, contradicting conventional expectations that smaller crystals might be more reactive due to higher surface area alone [46].
Atomic Force Microscopy (AFM) studies of the major (1 0 0) face of FFA Form I crystals revealed that larger crystals exhibited higher surface defect density, observable as etching pits after treatment with n-pentane [46] [47]. This correlation between crystal size, defect density, and transformation reactivity provides important insights for controlling solid-form transitions in pharmaceutical systems. The findings support the hypothesis that larger crystals accumulate more defects during growth, creating preferential sites for nucleation of new polymorphic forms [47].
Hollow Crystal Formation Through Polymorphic Transformation
FFA's crystallization behavior further demonstrates its complex solid-state chemistry through the formation of hollow crystals. Bhargavi et al. reported that FFA can generate hollow crystals during polymorphic transformation from Form I to Form III in ethanol/ethyl acetate solvent systems [44]. This phenomenon was attributed to FFA's lamellar liquid crystalline properties, which facilitate the development of hollow structures during crystal growth and transformation.
Table 1: Pharmaceutical Performance of FFA Hollow Crystals Compared to Conventional Forms
| Form | Dissolution Performance | Tablet Hardness | Key Characteristics |
|---|---|---|---|
| Hollow Crystals (Form III) | Higher dissolution rate | Greater hardness at all compression pressures | Higher surface area, hollow structure |
| Conventional Form I | Lower dissolution rate | Lower hardness | Standard crystal habit |
| Form III without hollow structure | Intermediate dissolution rate | Intermediate hardness | Same polymorph, different habit |
The hollow crystals demonstrated superior pharmaceutical performance, exhibiting both enhanced dissolution rates and improved compressibility compared to plain FFA Form I and non-hollow Form III crystals [44]. This highlights how control over crystal habit and morphology, in addition to polymorphic form, can significantly impact critical quality attributes of pharmaceutical materials.
Experimental Methodologies for Studying FFA Crystallization
Advanced Characterization Techniques
The complex crystallization behavior of FFA necessitates sophisticated characterization approaches to elucidate transformation pathways and mechanisms:
Hyphenated XRD-DSC Analysis: Simultaneous X-ray powder diffraction and differential scanning calorimetry provides correlated structural and thermal information during polymorphic transitions. This technique has been applied to study heat-induced crystallization of FFA from amorphous solid dispersions, revealing how polymer additives stabilize metastable forms [43]. The experimental setup typically uses synchrotron X-ray sources with a modified DSC cell, collecting diffraction patterns every 1°C during heating ramps.
Liquid Phase Electron Microscopy (LPEM): This technique enables high temporospatial imaging of FFA crystallization in native organic solvent environments, allowing direct observation of pre-nucleation clusters and intermediate stages [45]. LPEM addresses the significant challenge of distinguishing low-electron density organic molecules from their liquid surroundings, providing unprecedented insight into early crystallization events.
Atomic Force Microscopy (AFM): Surface defect characterization of FFA crystals using AFM has proven invaluable for understanding structure-reactivity relationships. After selective etching with n-pentane, AFM can identify and quantify defect density through observation of etching pits on major crystal faces [46] [47].
Fourier Transform Infrared Spectroscopy (FTIR): Used to investigate specific intermolecular interactions between FFA and polymeric additives in amorphous solid dispersions, providing insights into stabilization mechanisms for metastable forms [43].
Diagram Title: Experimental Workflow for FFA Crystallization Studies
Polymer Stabilization of Metastable Forms
Amorphous solid dispersions (ASDs) represent a crucial formulation approach for poorly soluble APIs like FFA, with polymeric additives playing a significant role in directing polymorphic outcomes. Research using hyphenated XRD-DSC has demonstrated that polymers including ethyl cellulose (EC) and various grades of hydroxypropylmethylcellulose (HPMC) can stabilize FFA Form IV by inhibiting transition to Form I during heating [43].
The stabilization mechanism appears to involve a combination of intermolecular interactions (particularly hydrogen bonding) and viscosity effects. Interestingly, the chemical substitution pattern of HPMC polymers (HPMC 2208 vs. HPMC 2910) plays a more significant role in directing polymorphic transitions than polymer viscosity [43]. Increasing polymer content in ASDs further inhibits polymorphic transitions, with drug/polymer ratios of 1:5 w/w resulting in FFA remaining amorphous during heating [43].
Table 2: Key Reagents and Materials for FFA Crystallization Studies
| Reagent/Material | Function/Application | Experimental Context |
|---|---|---|
| Flufenamic Acid (FFA) | Model compound | Polymorphic studies, defect analysis, hollow crystal formation |
| HPMC 2208 (4000 cp, 100000 cp) | Polymer additive | Stabilization of metastable forms in ASDs |
| HPMC 2910 (6 cp) | Polymer additive | Polymorphic direction in ASDs |
| Ethyl Cellulose (4 cp) | Polymer additive | Stabilization of Form IV in ASDs |
| Toluene | Crystallization solvent | Controlled crystal growth for defect studies |
| Ethanol/Ethyl Acetate | Solvent system | Hollow crystal formation |
Non-Classical Crystallization Pathways: Broader Implications
Theoretical Framework and Mechanistic Insights
The non-classical crystallization pathways observed in FFA align with broader discoveries across materials systems. These pathways typically involve intermediate stages such as dense liquid phases, pre-nucleation clusters, and particle-based assembly, contrasting sharply with classical monomer-by-monomer addition [4] [45]. For soft and organic materials, these non-classical trajectories substantially alter crystallization kinetics and outcomes, providing new opportunities for material design [4].
Recent research on binary colloidal systems has further elucidated the mechanisms governing non-classical crystallization, revealing multi-step processes where metastable amorphous blobs condense before evolving into crystalline structures [3]. These findings parallel observations in FFA systems and provide a conceptual framework for understanding the complex crystallization behavior of pharmaceutical materials.
Liquid Crystal Phases and Intermediate States
The discovery of liquid crystal phases during crystallization extends beyond organic molecules to include even simple ionic compounds. Recent studies of sodium halides have revealed the formation of liquid crystal intermediate phases composed of contact ion pairs prior to crystal nucleation [48]. This observation establishes a new theoretical framework for crystal nucleation and growth, with significant implications for understanding similar intermediate phases in organic systems like FFA.
FFA's documented lamellar liquid crystalline properties [44] and its formation of hollow crystals suggest that liquid crystal phases may play important roles in its polymorphic transformations and crystal habit modification. This connection between liquid crystalline behavior and non-classical crystallization pathways represents a promising area for future pharmaceutical research.
Diagram Title: Classical vs. Non-Classical Crystallization Pathways
The case study of flufenamic acid crystallization provides compelling evidence for the importance of non-classical pathways in pharmaceutical crystal formation. The observed behaviors—including defect-mediated transformations, pre-nucleation clusters, hollow crystal formation, and polymer-directed polymorphism—collectively demonstrate the limitations of classical crystallization theories and the need for more sophisticated models that account for intermediate phases and complex assembly mechanisms.
For pharmaceutical scientists, these insights offer new opportunities for controlling crystal form and habit through manipulation of crystallization pathways rather than simply optimizing final conditions. The ability to stabilize metastable forms, generate unique crystal morphologies, and direct polymorphic outcomes through understanding of non-classical mechanisms represents a significant advance in pharmaceutical materials design.
Future research directions should focus on extending these findings to other challenging pharmaceutical systems, developing predictive models for non-classical crystallization outcomes, and translating pathway control strategies to manufacturing processes. As direct observation techniques continue to improve, particularly for beam-sensitive organic materials, our understanding of these complex crystallization trajectories will undoubtedly expand, enabling more precise control over solid-state properties and performance of pharmaceutical materials.
The integration of two-dimensional transition metal dichalcogenides (2D TMDs) into next-generation electronic and optoelectronic devices necessitates the development of scalable, controlled synthesis methods. While chemical vapor deposition (CVD) has emerged as a promising technique, conventional vapor-solid (VS) growth pathways often face limitations in growth rate and crystal quality. The vapor-liquid-solid (VLS) mechanism, particularly when enhanced with promoter agents, represents a significant advancement in the synthesis of high-quality, large-area TMD monolayers. This case study examines the VLS growth of 2D TMDs within the broader context of non-classical nucleation pathways in organic materials research, highlighting how these mechanisms enable precise atomic-level manufacturing of quantum materials.
Fundamental Principles of VLS Growth for TMDs
Classical vs. Non-Classical Nucleation Pathways
Traditional crystal growth follows classical nucleation theory, where atoms or molecules directly attach to a growing crystal surface in a monomer-by-monomer addition process. In contrast, non-classical nucleation involves more complex pathways, often proceeding through intermediate metastable phases that subsequently reorganize into crystalline structures [4]. The VLS growth of TMDs exemplifies this non-classical approach, as it proceeds through a liquid-phase intermediate that facilitates rapid mass transport and lowers energy barriers for crystallization [6].
The VLS mechanism diverges fundamentally from VS growth by introducing a liquid precursor phase that mediates between the vapor sources and the solid crystalline film. This liquid phase acts as a catalytic medium that enhances precursor adsorption, decomposition, and reorganization, ultimately leading to the precipitation of 2D crystals at the liquid-substrate interface [49]. The presence of this intermediate phase enables growth rates at least two orders of magnitude higher than conventional VS growth [6].
Role of Promoters in VLS Growth
Alkali metal salts (e.g., NaCl, KCl) and alkali chalcogenides play crucial roles in facilitating VLS growth by forming low-melting-point intermediates. These promoters function through several mechanisms:
Vaporization Enhancement: Promoters react with metal oxide precursors (e.g., WO₃, MoO₃) to form volatile intermediates such as tungsten oxychlorides (WOₓClᵧ), significantly increasing metal precursor vapor pressure and transport efficiency [49] [6].
Liquid Phase Formation: Alkali metals combine with chalcogens to form alkali chalcogenides (Ak₂Xₐ, where Ak = Na, K and X = S, Se, Te), which subsequently react with metal oxides to create molten alkali tungstates (Ak₂WO₄) or molybdates. These molten salts serve as liquid media for precursor dissolution and diffusion [49].
Reaction Pathway Alteration: Promoters enable alternative reaction pathways with lower activation energies. For instance, alkali chalcogenides provide a lower energy barrier for reaction compared to direct vapor-phase chalcogen reactions [49].
Table 1: Common Promoters and Their Functions in VLS Growth of TMDs
| Promoter | Intermediate Phases Formed | Primary Function | Compatible TMDs |
|---|---|---|---|
| NaCl/KCl | WOₓClᵧ, Na₂WO₄/K₂WO₄ | Lowers melting point of metal precursors, creates liquid phase | WS₂, WSe₂, MoS₂, MoSe₂ |
| LiBH₄ | LixMTe₂ | Solid-state lithiation for exfoliation | MoTe₂, WTe₂, NbTe₂ |
| Alkali Chalcogenides (Na₂S, K₂Se) | Ak₂Xₐ, Ak₂WO₄ | Direct liquid intermediate formation, enables clean transfer | WSe₂, WS₂, WTe₂ |
Experimental Observation of Non-Classical Nucleation in TMDs
In-situ Monitoring of Nucleation Dynamics
Direct observation of the initial nucleation stage in tungsten disulfide (WS₂) growth through in-situ monitoring CVD has revealed distinctive non-classical behavior [6]. This approach has identified critical phenomena that diverge from classical nucleation theory:
Formation of Metastable Clusters: The process begins with the appearance and movement of liquid-phase precursor particles (∼1-5 µm) on the substrate surface, which aggregate to form larger metastable clusters [6].
Large Critical Nuclei Size: The critical nucleus size (r) observed in WS₂ VLS growth reaches approximately 38.7 µm, vastly exceeding theoretical predictions for classical nucleation (typically 1-10 nm) and values reported for other systems like WSe₂ (r = 1.63 ± 0.21 nm) [6].
Two-Step Nucleation Pathway: Analysis of growth dynamics supports a two-step nucleation mechanism where metastable liquid droplets first condense from the vapor phase, followed by solid crystal nucleation within these clusters [6].
The extraordinarily large critical nucleus size observed in VLS-grown WS₂ provides compelling evidence for non-classical nucleation pathways, as this dimension cannot be explained by classical nucleation theory alone [6]. This phenomenon aligns with observations in other material systems where precursor cluster size directly influences critical nucleus dimensions.
Growth Dynamics Analysis
Quantitative analysis of WS₂ growth reveals distinct kinetic phases. The transition from slow to rapid growth coincides with changes in crystal anisotropy, quantitatively verified through phase field simulations combined with Bayesian inference [6]. The incubation time for metastable cluster formation follows a traditional time-temperature transformation diagram, while the subsequent crystal growth phase proceeds through multiple simultaneous mechanisms:
- Direct monomer addition from vapor phase to crystal
- Capture and absorption of surrounding amorphous blobs
- Oriented attachment of crystalline domains with mutual alignment [3]
Diagram 1: Non-classical nucleation pathway in TMD VLS growth. The process proceeds through liquid intermediate and metastable cluster phases before crystallizing.
Advanced VLS Growth Strategies and Mechanisms
Alkali Chalcogenide-Assisted VLS Growth
Recent advances have demonstrated the efficacy of engineered chalcogen-based intermediates in VLS growth. The alkali chalcogenide-assisted approach enables universal growth of high-quality tungsten-based TMDs (WS₂, WSe₂, WTe₂) through distinct reaction pathways [49]:
Route 1 (Vapor-Solid-Solid): Gaseous tungsten oxychlorides (WOₓClᵧ) form through reactions between WO₃ and alkali chlorides, then react with hydrogen chalcogenides (H₂X) to yield WX₂.
Route 2 (Vapor-Liquid-Solid): Excess chalcogen reacts with alkali chlorides to generate alkali chalcogenides (Ak₂Xₐ), which subsequently react with WO₃ to form alkali tungstates (Ak₂WO₄). These molten tungstates serve as liquid intermediates for VLS growth.
Route 3 (Enhanced VLS): Mixing excess chalcogen powders with WO₃ and alkali halides enables growth of continuous few-layer WX₂ films (1.4-5.15 nm) with full coverage over ∼1 cm² areas [49].
This strategy produces TMDs with ultraclean surfaces, attributed to a salt-like alkali-chalcogen interfacial layer that enables support-free film delamination and transfer [49].
Growth Condition Optimization
Precise control over VLS growth parameters enables tailored synthesis of TMDs with specific thicknesses and properties:
Metal/Chalcogen Ratio: A U-type relationship exists between TMD layer number and the ratio of metal to chalcogen precursor, enabling ultrafine thickness control [50].
Interaction Strength Modulation: In related colloidal model systems, crystal formation pathways are highly sensitive to interaction strength, progressing through windows of stability, classical crystallization, two-step crystallization, and random aggregation as interaction strength increases [3].
Temperature Profile: Optimal growth occurs with specific temperature zones for precursor vaporization (∼700-900°C) and substrate deposition (∼650-800°C), with precise thermal profiles influencing nucleation density and crystal size [51].
Table 2: Optimal Growth Parameters for VLS Synthesis of Selected TMDs
| Material | Promoter System | Growth Temperature | Metal/Chalcogen Ratio | Characteristic Features |
|---|---|---|---|---|
| WS₂ | NaCl, KCl | 700-900°C | Tunable for layer control | Millimeter-scale single crystals, large critical nuclei (~40 µm) |
| WSe₂ | KCl, K₂Seₐ | ~700°C | Lower for bilayer | Ultraclean surfaces, support-free delamination |
| WTe₂ | NaCl/KCl + excess Te | 700°C | Chalcogen-rich | Type-II Weyl semimetal, quantum spin Hall effect |
| MoS₂ | NaCl, KI | 650-850°C | U-type relationship | Layer-by-layer sulfurization, rhomboidal flakes |
Characterization and Transfer Techniques
Interfacial Characterization
Advanced characterization techniques confirm the presence and composition of interfacial layers critical to VLS growth:
Auger Electron Spectroscopy (AES): Clear compositional contrast reveals residual potassium selenide (K₂Seₓ) at the WSe₂/substrate interface, evidenced by K KLL (254 eV) and Se LMM (1312 eV) transitions [49].
Electron Microscopy: Atomic-resolution Z-contrast STEM imaging confirms single-crystalline structure with periodic W atom arrangement (0.27 nm spacing) in WS₂ [6].
Raman Spectroscopy: Characteristic vibrational modes (e.g., A₁g at 247.3 cm⁻¹ for WSe₂; multiple modes between 89-212 cm⁻¹ for WTe₂) verify crystallinity and phase purity of as-grown and transferred films [49].
Water-Assisted Transfer Method
The water-soluble nature of alkali chalcogenide interfacial layers enables a novel transfer strategy that eliminates polymer residues and etching damage [49]:
- Immersion: Submersion of as-grown samples in deionized water
- Capillary Infiltration: Water penetration at the film/substrate interface via capillary action
- Dissolution: Alkali chalcogenide layer dissolution releases the TMD film
- Transfer: Direct manipulation onto target substrates
- Drying: Ambient drying completes the process
This support-free approach preserves the structural and electronic integrity of transferred films, enabling ultraclean surfaces for probing quantum phenomena [49].
Research Reagent Solutions
Table 3: Essential Research Reagents for VLS Growth of 2D TMDs
| Reagent Category | Specific Examples | Function in VLS Growth | Key Considerations |
|---|---|---|---|
| Metal Precursors | WO₃, MoO₃, MoCl₅ | Source of transition metal atoms | Purity affects crystal quality; halides offer lower melting points |
| Chalcogen Sources | S, Se, Te powders | Source of chalcogen atoms | Volatility control critical for stoichiometry |
| Alkali Promoters | NaCl, KCl, LiBH₄ | Form low-melting-point intermediates; enhance vaporization | Ratio to metal precursors tunes growth mode (VSS vs VLS) |
| Alkali Chalcogenides | Na₂S, K₂Se, Na₂Te | Direct liquid intermediate formation | Enable clean transfer; lower reaction energy barriers |
| Growth Substrates | SiO₂/Si, sapphire | Support for epitaxial growth | Surface energy and crystallinity influence nucleation |
| Carrier/Reducing Gases | Ar, H₂, N₂ | Atmosphere control; precursor transport | H₂ acts as reducing agent in selenization/tellurization |
The VLS growth mechanism represents a paradigm shift in 2D TMD synthesis, enabling unprecedented control over crystal size, thickness, and quality. The non-classical nucleation pathways observed in these systems—characterized by large critical nuclei, metastable intermediate phases, and two-step crystallization—provide fundamental insights that extend beyond TMDs to broader crystal growth science.
Future research directions should focus on:
- Real-time Interaction Control: Developing dynamic regulation of particle interaction strengths through continuous parameter modulation (e.g., salt concentration gradients) [3]
- Multimodal Characterization: Correlating in-situ growth monitoring with atomic-scale structural and electronic characterization
- Pathway Engineering: Deliberately steering crystallization through classical or non-classical routes by precisely controlling thermodynamic parameters
- Industrial Scaling: Translating laboratory VLS growth to wafer-scale manufacturing processes compatible with existing semiconductor fabrication
The integration of VLS growth with promoter-assisted CVD creates a powerful synthesis platform that aligns with the broader thesis of non-classical pathway engineering in advanced materials research. As understanding of these mechanisms deepens, so too will the capability to design and fabricate quantum materials with atomic-level precision for next-generation technologies.
Controlling the Pathway: Strategies for Optimization and Problem-Solving
The precise control of synthesis conditions, particularly solvent ratios, is a critical frontier in advanced materials science. Moving beyond traditional empirical approaches, modern research leverages these parameters to actively steer nucleation and growth along non-classical pathways, thereby dictating the final material's physicochemical properties. This paradigm is central to designing functional organic and inorganic materials with tailored characteristics for applications ranging from drug delivery to catalysis. Non-classical nucleation, which often involves stable intermediate phases like dense liquid droplets or mesoscopic clusters, provides a versatile framework for such control [52] [53]. Within this context, the ratio of water (H2O) and ethanol to silica (SiO2) precursors emerges as a powerful, tunable variable. This whitepaper delves into the quantitative impact of these ratios, offering a detailed technical guide for researchers and drug development professionals aiming to harness these principles for predictable nanomaterial synthesis.
Quantitative Impact of Ethanol-to-Water Ratio on Silica-Based Materials
The ethanol-to-water ratio in a synthesis solution is a decisive factor influencing the morphology, particle size, and porosity of resulting silica-based materials. These parameters, in turn, directly govern the material's performance in applications such as controlled drug release.
A pivotal study on silica-carbon core–shell (SiO2@C) nanoparticles systematically investigated the effect of the ethanol-to-water ratio [54]. The findings demonstrate that this ratio directly controls the material's architecture and its subsequent functionality as a nanocarrier.
Table 1: Effect of Ethanol-to-Water Ratio on SiO2@C Core-Shell Properties
| Ethanol-to-Water Ratio | Particle Morphology & Size Distribution | Key Material Properties | Impact on Thymol Release & Antibacterial Activity |
|---|---|---|---|
| 20:80 (E20W80) | Spherical core-shell nanostructures with a uniform size distribution [54] | High porous carbon content; Large pore volume [54] | Highest thymol release profile; Prolonged antibacterial activity against S. aureus (40% release over 48 h) [54] |
| Other Tested Ratios | Non-uniform shapes and/or broader size distributions [54] | Lower porous carbon content and smaller pore volume [54] | Lower and less sustained thymol release profiles [54] |
The superior performance of the E20W80 material is linked to the physical adsorption of thymol onto the carbon shell, a mechanism confirmed by Density Functional Theory (DFT) studies [54]. This physisorption mode is advantageous for prolonged release, as it avoids strong chemical bonds that can hinder the active ingredient's release. Consequently, optimizing the solvent ratio directly enabled the development of a nanocarrier with sustained release capabilities [54].
Experimental Protocols for Solvent Ratio Modulation
Protocol: Synthesis of SiO2@C Core-Shell Nanoparticles with Tunable Ethanol-to-Water Ratio
This protocol is adapted from research on producing silica-carbon core–shell materials for prolonged antibacterial activity [54].
- 1. Objective: To synthesize SiO2@C core–shell nanoparticles with controlled morphology and pore structure by modulating the ethanol-to-water ratio, for use as a nanocarrier for active ingredients like thymol.
- 2. Materials:
- Silica precursor (e.g., tetraethyl orthosilicate, TEOS).
- Carbon source.
- Ethanol (absolute).
- Deionized water.
- Catalyst (e.g., ammonia solution).
- Thymol (for drug loading experiments).
- 3. Methodology:
- Solution Preparation: Prepare a series of precursor solutions with varying ethanol-to-water volume ratios, with the optimal condition identified at 20:80 (v/v) ethanol-to-water [54].
- Silica Core Formation: Incorporate the silica precursor into the solvent mixture under controlled stirring and temperature to form the porous silica (SiO2) nanoparticle core.
- Carbon Shell Coating: Introduce the carbon source to the reaction mixture to facilitate the formation of a uniform carbon shell around the silica core.
- Purification: Isolate the resulting SiO2@C core–shell nanoparticles via centrifugation, followed by washing and drying.
- Characterization: Subject the materials to characterization using techniques such as:
- Drug Loading and Release (for application validation):
- Load thymol into the optimized SiO2@C material.
- Conduct in vitro release studies in a suitable buffer.
- Evaluate prolonged antibacterial performance against a relevant model organism like Staphylococcus aureus [54].
Protocol: Investigating Non-Classical Nucleation in Solvent Systems
This generalized protocol outlines the approach for studying non-classical pathways, relevant to understanding solvent-mediated nucleation.
- 1. Objective: To identify and characterize intermediate phases in non-classical nucleation pathways within solvent systems.
- 2. Materials: Model solutes (e.g., sodium halides, hematin, colloidal particles), organic solvents, modifiers (e.g., antimalarials).
- 3. Methodology:
- Solution Preparation: Prepare growth solutions in organic solvents (e.g., octanol saturated with buffer) at defined supersaturations [48] [52].
- Nucleation Monitoring: Use Dynamic Light Scattering (DLS) to track the evolution of the autocorrelation function (g_{2}(\tau)), which reveals the formation of mesoscopic clusters and the onset of crystallization [52].
- Pathway Modulation: Introduce modifiers (e.g., antimalarial compounds) to the system to observe their impact on cluster population and nucleation kinetics (suppression, enhancement, or no effect) [52].
- Direct Observation: Employ techniques like atomic force microscopy (AFM), bright-field microscopy, and 3D confocal microscopy to directly image intermediate phases such as amorphous blobs or liquid crystal phases [52] [53].
- Theoretical Validation: Use Molecular Dynamics (MD) simulations and Density Functional Theory (DFT) to model interactions between solute molecules, solvents, and modifiers, validating the observed mechanisms [54] [52] [53].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Research Reagents and Materials for Controlled Synthesis
| Reagent/Material | Function in Synthesis & Nucleation Studies |
|---|---|
| Ethanol & Water Solvents | Primary media for modulating precursor solubility, reaction kinetics, and final material morphology (e.g., achieving uniform spherical nanoparticles) [54]. |
| Silica Precursors (e.g., TEOS) | Molecular building blocks for the formation of the silica (SiO2) nanoparticle core [54]. |
| Carbon Sources | Precursors for the formation of a porous carbon shell around the silica core, critical for adsorption and release [54]. |
| Sodium Halides (NaCl, NaBr, NaI) | Model ionic compounds for studying non-classical vs. classical nucleation pathways in evaporating microdroplets [48]. |
| Hematin | An organic biocrystal model for studying non-classical nucleation and its suppression by modifiers (e.g., antimalarials) in organic solvents [52]. |
| Charged Colloidal Particles | Model "ions" for direct observation of non-classical, two-step crystallization and growth mechanisms via microscopy [53]. |
| Nucleation Modifiers (e.g., Pyronaridine, Heme-Artesunate) | Compounds used to probe and control nucleation pathways by interacting with solute monomers, dimers, and mesoscopic clusters [52]. |
Visualization of Synthesis Workflow and Nucleation Pathways
Synthesis Workflow and Nucleation Pathways
Modifier Impact on Non-Classical Nucleation
The strategic modulation of H2O/SiO2 and ethanol/SiO2 ratios provides a scientifically grounded method for controlling the synthesis of functional nanomaterials via non-classical pathways. The optimization of the ethanol-to-water ratio to 20:80 for producing superior SiO2@C core–shell nanocarriers underscores a direct translation from synthetic control to application performance [54]. The broader understanding of non-classical nucleation, facilitated by mesoscopic precursors, offers a powerful rationale for designing crystallization processes that can be selectively suppressed or enhanced by specific modifiers [52]. For drug development professionals, these insights are invaluable. They enable the rational design of drug delivery systems with prolonged release profiles and open new avenues for controlling crystallization in active pharmaceutical ingredients (APIs), thereby impacting drug efficacy, stability, and bioavailability.
The Challenge of Polymorphism and Ostwald's Rule of Stages
In the field of organic materials research, controlling the solid form of active pharmaceutical ingredients (APIs) is a fundamental challenge with direct implications for drug efficacy, stability, and manufacturability. Polymorphism, the ability of a compound to crystallize into multiple distinct crystal structures, presents both an opportunity and a risk in pharmaceutical development [55]. Different polymorphs of the same API can exhibit significantly different physical and chemical properties, including solubility, dissolution rate, bioavailability, and physical stability [56] [57] [58]. Approximately 85% of marketed drugs possess the ability to form multiple crystalline forms, making polymorphism a widespread consideration in drug development [58].
Ostwald's Rule of Stages, a principle formulated by Wilhelm Ostwald in 1897, posits that during crystallization from a high-energy state, a system does not directly transition to the most thermodynamically stable phase but instead progresses through a series of increasingly stable intermediate phases [59]. This rule has long provided a theoretical framework for understanding the appearance of metastable polymorphs during crystallization processes. However, recent research on non-classical nucleation pathways in organic materials has revealed significant exceptions and complexities to this century-old rule, challenging our fundamental understanding of polymorph formation and transformation [60] [61] [62].
This technical guide examines the current understanding of polymorphism and Ostwald's Rule of Stages within the context of non-classical nucleation pathways, highlighting experimental approaches for controlling polymorphic outcomes and discussing the implications for pharmaceutical development.
Ostwald's Rule of Stages: Foundations and Mechanisms
Ostwald's Rule of Stages represents a foundational concept in crystallization science. The rule states that "in a crystallisation, the system moves to equilibrium from an initial high-energy state through minimal changes in free energy, and thus implies that the least stable polymorph should be the first isolated in any crystallisation" [59]. This progression occurs because metastable phases with free energies closer to the initial state present lower nucleation barriers compared to the thermodynamically stable phase.
The rule has been experimentally demonstrated in various systems. In dipeptide supramolecular polymers, Boc-diphenylalanine (Boc-FF) self-assembly follows a multi-step pathway through metastable phases [62]. The process begins with the formation of amorphous nanospheres, which subsequently ripen and structurally convert into fibrillar species, before ultimately transforming into highly crystalline tubular structures [62]. This sequential transformation occurs because each phase forms in order of increasing stability, consistent with Ostwald's Rule of Stages.
The underlying mechanism can be understood through classical nucleation theory, where the activation energy barrier for nucleation is lower for metastable phases with higher free energy compared to the stable phase. The system thus minimizes the initial kinetic barrier by forming less stable phases first, then progressively transforming toward the thermodynamic minimum through dissolution and recrystallization processes.
Table 1: Key Concepts in Ostwald's Rule of Stages
| Concept | Description | Experimental Evidence |
|---|---|---|
| Stepwise Progression | System transitions through increasingly stable polymorphs | Boc-FF dipeptide: spheres → fibrils → tubes [62] |
| Free Energy Minimization | Each step involves minimal free energy change | Sequential phase transitions with decreasing free energy [62] |
| Metastable Intermediates | Transient polymorphs with limited stability | Paracetamol Form III as intermediate to Form II [59] |
| Kinetic Control | Initial formation governed by nucleation kinetics rather than thermodynamics | Rapid crystallization favors metastable polymorphs [57] |
Non-Classical Nucleation Pathways: Challenging Traditional Views
Recent advances in crystallization research have revealed that non-classical nucleation pathways substantially alter the crystallization kinetics and outcomes of organic materials [4]. Unlike classical nucleation theory, which describes how nuclei form, become stable after reaching a critical size, and then enlarge through monomer attachment, non-classical pathways involve more complex mechanisms such as metastable structure-mediated and particle attachment-based processes [4].
Direct observation of non-classical crystallization of inorganic nanomaterials was enabled by liquid-phase in situ electron microscopy. However, it has only been recently that the crystallization dynamics of beam-sensitive soft materials have been directly imaged with sufficient spatial resolution [4]. These observations have uncovered a level of microstructural understanding of defects and interfaces that challenges simplistic interpretations of Ostwald's Rule.
For calcium silicate hydrate (C-S-H), the most important hydrate of cement, research has confirmed that homogeneous nucleation follows a two-step, non-classical process [5]. The process begins with the appearance of discrete globules as a metastable precursor, which then transform into foil-like C-S-H accompanied by changes in crystallinity and structure [5]. This pathway demonstrates that non-classical mechanisms operate not only in inorganic systems but also in complex organic and hybrid materials.
The characteristics of soft materials significantly affect their crystallization pathways and kinetics. The flexibility of organic molecules, the strength and directionality of intermolecular interactions, and the presence of solvents or additives can all promote non-classical pathways that deviate from the predictions of Ostwald's Rule [4].
Diagram 1: Classical vs. non-classical nucleation pathways. The classical pathway (yellow to red to green) follows Ostwald's Rule through distinct stages, while non-classical pathways (blue) may involve pre-nucleation clusters, liquid-liquid separation, and particle attachment.
Experimental Challenges in Polymorph Control
The Ritonavir Case Study
The well-documented case of the antiviral drug Ritonavir exemplifies the significant challenges and risks associated with polymorphism in pharmaceutical development [58]. During initial development, only one crystal form (Form I) was identified. However, two years after the drug was marketed in 1996, a more stable polymorph (Form II) with much lower solubility began precipitating out of the semi-solid formulated product [58]. This unexpected polymorphic transformation necessitated an immediate reformulation and temporary withdrawal of the drug from the market, with estimated losses exceeding US$250 million [58].
Remarkably, 24 years after the appearance of Form II, scientists serendipitously discovered a third polymorph (Form III) while studying the crystal nucleation of amorphous Ritonavir [58]. This discovery highlights the persistent challenge of comprehensively mapping the polymorphic landscape of even extensively studied compounds and demonstrates that our ability to predict and control polymorphism remains incomplete.
Paracetamol Polymorphism
Paracetamol presents another instructive case study in polymorph control. The stable Form I does not compress directly into tablets due to its crystal structure containing puckered hydrogen-bonded sheets that resist slipping under compression [59]. This necessitates an additional wet granulation step during tablet manufacturing. Form II, which comprises flat hydrogen-bonded sheets, is more amenable to direct compression but has proven difficult to isolate reliably [59].
A third polymorph (Form III) was reported over a quarter-century ago but remained largely elusive until researchers developed a method to enforce Ostwald's Rule of Stages by crystallizing through controlled warming of the amorphous glassy state rather than by cooling from solution [59]. In the glassy state, molecular degrees of freedom are suppressed, creating large activation energy barriers to interconversion between polymorphic forms and allowing for the isolation of the metastable Forms III and II.
Table 2: Pharmaceutical Consequences of Polymorphism
| Drug Compound | Polymorphic Forms | Impact on Pharmaceutical Properties |
|---|---|---|
| Ritonavir | Form I (initial), Form II (appeared later), Form III (discovered 24 years post-Form II) | Form II had reduced solubility, compromising bioavailability and necessitating reformulation [58] |
| Paracetamol | Form I (stable), Form II (metastable), Form III (metastable) | Form I requires wet granulation for tableting; Form II is directly compressible [59] |
| Benzamide | Form I (most stable), Form II (least stable), Form III (intermediate) | Different hydrogen bonding patterns and pi-pi interactions affect stability [55] |
| Sulfamerazine | Multiple polymorphs | Metastable forms can be stabilized by structurally related additives [57] |
Mechanochemistry: Direct Challenge to Ostwald's Rule
Recent research in mechanochemistry has provided direct experimental challenges to the universality of Ostwald's Rule of Stages. Mechanochemical synthesis, which utilizes mechanical energy to conduct chemical transformations, has emerged as an efficient, solvent-free route for synthesizing and screening polymorphs of organic solids [60] [61].
In a landmark study on the cocrystallization of nicotinamide and adipic acid, researchers demonstrated that the choice of milling assembly (jar material and ball characteristics) could direct polymorphic outcomes, enabling selective synthesis and even reversible interconversion of cocrystal polymorphs [60] [61]. This finding directly contradicts the expectations of Ostwald's Rule, which would predict a consistent progression from metastable to stable polymorphs regardless of mechanical processing conditions.
The research revealed that:
- Milling in poly(methyl methacrylate) (PMMA) jars preferentially produced the thermodynamically more stable Form I
- Milling in stainless steel (ss) jars under otherwise identical conditions yielded a new metastable polymorph (Form II) [61]
Even more remarkably, the study demonstrated reversible polymorph interconversion:
- Milling Form I in ss jars with ACN liquid additive produced phase-pure Form II within 60 minutes
- Milling Form II in PMMA jars converted it back to Form I [61]
This reversible interconversion, achievable simply by modifying the milling assembly, represents a clear exception to Ostwald's Rule of Stages and suggests that modification of energy input in the mechanochemical system, combined with small energy differences between polymorphs, can enable selective synthesis of either stable or metastable forms [60].
Methodologies for Polymorph Screening and Control
Experimental Approaches
Comprehensive polymorph screening requires multiple complementary approaches to fully characterize the solid form landscape of an API. Current methodologies include:
- Solution Crystallization: Systematic variation of solvent systems, concentrations, and temperature profiles to explore polymorphic space [56] [57]
- Mechanochemical Methods: Neat grinding, liquid-assisted grinding (LAG), and polymer-assisted grinding (POLAG) to access polymorphs not readily obtained from solution [61]
- Melt Crystallization: Cooling from the molten state, particularly useful for compounds that degrade in solution [59]
- Glass Crystallization: Controlled warming of the amorphous glassy state to isolate metastable polymorphs by suppressing molecular degrees of freedom [59]
Advanced characterization techniques are essential for identifying and differentiating polymorphs:
- X-ray Powder Diffraction (XRPD): Distinguishes polymorphs based on unique diffraction patterns [61] [55]
- Solid-State Nuclear Magnetic Resonance (ssNMR): Detects differences in molecular environment and conformation [59]
- Differential Scanning Calorimetry (DSC): Reveals thermal transitions and relative stabilities [59]
- Vibrational Spectroscopy (IR, Raman, terahertz): Sensitive to hydrogen bonding patterns and intermolecular interactions [55]
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for Polymorph Research
| Material/Equipment | Function in Polymorph Research | Application Example |
|---|---|---|
| PMMA Milling Jars | Enables selective formation of stable polymorphs in mechanochemistry | Production of Form I nicotinamide-adipic acid cocrystal [61] |
| Stainless Steel Milling Jars | Facilitates formation of metastable polymorphs through efficient energy transfer | Selective synthesis of Form II nicotinamide-adipic acid cocrystal [61] |
| Hot-Stage Microscope | Direct observation of polymorphic transitions in real-time | Monitoring paracetamol form transformations [59] |
| Liquid Additives (ACN, MeOH, H₂O) | Controls polymorphic outcome in liquid-assisted grinding | Varying η parameter (liquid-to-solid ratio) in LAG [61] |
| Amorphous Precursors | Starting point for glass crystallization to access metastable forms | Isolation of paracetamol Forms III and II [59] |
Diagram 2: Workflow for comprehensive polymorph screening and selection. The multi-technique approach ensures thorough mapping of the solid-form landscape and evidence-based selection of the optimal polymorph for development.
The challenge of polymorphism and the limitations of Ostwald's Rule of Stages highlight the complex interplay between thermodynamics and kinetics in crystalline materials formation. While Ostwald's Rule provides a valuable framework for understanding sequential phase transformations in many systems, contemporary research has revealed numerous exceptions and complexities, particularly in the context of non-classical nucleation pathways and mechanochemical synthesis.
The emergence of non-classical crystallization pathways, involving metastable structure-mediated and particle attachment-based mechanisms, has substantially altered our understanding of crystallization kinetics and outcomes [4]. Furthermore, the demonstration that milling assembly characteristics can direct polymorphic outcomes in mechanochemical reactions presents a direct challenge to the universality of Ostwald's Rule [60] [61].
For pharmaceutical development professionals, these advances underscore the importance of comprehensive polymorph screening employing multiple complementary techniques. The increasing structural complexity and molecular weight of new chemical entities present ongoing challenges for crystallization and polymorph control [58]. A survey of 476 new chemical entities studied between 2016 and 2023 revealed an average of 5.5 crystal forms for free forms and 3.7 for salts, demonstrating the prevalence and significance of polymorphism in pharmaceutical compounds [58].
Future research directions will likely focus on:
- Computational prediction of polymorphic landscapes to guide experimental screening [55]
- Advanced in situ characterization techniques to capture transient polymorphic forms [61]
- Rational design of crystallization processes to target specific polymorphic outcomes
- Understanding the molecular mechanisms governing non-classical nucleation pathways [4]
As research continues to unravel the complexities of polymorphism and crystallization pathways, the scientific community moves closer to predictive control over solid-form selection, potentially mitigating risks while optimizing the performance of organic materials and pharmaceutical products.
Harnessing Non-Classical Pathways for Self-Purification
The pursuit of pure materials is a cornerstone of advanced industries, from pharmaceuticals to semiconductors. Classical Nucleation Theory (CNT) has long provided the foundational model for crystallization, describing a direct, single-step process where solutes form crystalline nuclei through stochastic atomic attachment [10]. However, this framework often fails to predict or explain the complex crystallization behaviors of many functional materials. Over the past two decades, the discovery of non-classical pathways has revolutionized our understanding, revealing multi-step nucleation processes involving stable intermediate phases such as pre-nucleation clusters, dense liquid droplets, and amorphous particles [63] [10]. This paradigm shift opens new avenues for controlling material purity.
This technical guide explores the deliberate exploitation of these non-classical mechanisms to achieve self-purification—a process where inherent crystallization dynamics selectively exclude impurities, leading to higher purity products than predicted by classical equilibrium thermodynamics. By understanding and controlling these pathways, researchers can design synthesis conditions that leverage intermediate phases as selective filters, transforming impurity management from a downstream challenge into an intrinsic design feature of the crystallization process itself.
Theoretical Foundations of Non-Classical Nucleation
Non-classical nucleation encompasses several distinct but interrelated mechanisms where crystallization proceeds through intermediate stages rather than via direct molecular attachment to a growing crystal. The key pathways include:
Two-Step Nucleation Mechanism
This predominant non-classical model involves a dense liquid precursor that forms before crystal nucleation. The process initiates with a supersaturated solution undergoing liquid-liquid phase separation, creating dense, solute-rich droplets within a dilute solution phase. Within these metastable droplets, nucleation then occurs [63] [15]. This pathway is characterized by a free energy barrier that is significantly lower than that predicted by CNT, making nucleation more probable under conditions where classical nucleation would be impeded.
Particle-Based Crystallization Pathways
In contrast to molecular addition, particle attachment mechanisms involve the assembly of nano-sized building blocks, including:
- Oriented Attachment: Crystalline nanoparticles align and fuse along specific crystallographic planes [63].
- Mesocrystal Formation: Nanoparticles assemble into a superstructure with long-range order through intermediary phases like amorphous layers [63].
- Ostwald Ripening: A thermodynamically-driven process where larger crystals grow at the expense of smaller, less stable ones, which can be harnessed for purification [63].
Table 1: Key Non-Classical Nucleation Pathways and Their Characteristics
| Nucleation Pathway | Key Intermediate | Driving Force | Purification Potential |
|---|---|---|---|
| Two-Step Nucleation | Dense liquid droplets | Lowered interfacial energy | High - Selective solute partitioning |
| Pre-Nucleation Clusters | Stable solute clusters | Dynamic equilibria | Medium - Impurity exclusion by cluster geometry |
| Oriented Attachment | Nanoparticles | Surface energy minimization | Variable - Dependent on nanoparticle surface chemistry |
| Amorphous Precursor | Metastable amorphous particles | Solubility differential | High - Impurity trapping in amorphous phase |
Experimental Evidence Across Material Systems
Pharmaceutical Compounds: Carbamazepine
The antiepileptic drug carbamazepine exemplifies how non-classical pathways can be harnessed in organic systems. Research utilizing a micro-droplet precipitation system has demonstrated that carbamazepine undergoes a liquid-liquid phase separation before crystallizing, forming amorphous dense liquid clusters (ADLCs) as intermediates [15]. The transition between one-step (direct to amorphous solid) and two-step (through crystalline solid) pathways can be controlled by varying solvent composition, specifically the methanol-to-water ratio [15].
This solvent-dependent pathway selection directly impacts the resulting material's properties. By manipulating these intermediates, researchers can steer the system toward amorphous forms with enhanced solubility—a crucial factor for improving bioavailability of poorly soluble drugs—while potentially excluding molecular impurities that differentially partition between the dense liquid phase and the bulk solution [15].
Zeolites: MFI System Crystallization Pathways
In inorganic framework materials like zeolites, non-classical pathways dominate. Studies of MFI zeolite crystallization have demonstrated that both classical (molecular addition) and non-classical (particle attachment) pathways typically coexist and intertwine [36]. The relative contribution of each pathway can be modulated by simple synthesis parameters:
- Reducing H₂O/SiO₂ ratio promotes non-classical pathways [36]
- Increasing ethanol/SiO₂ ratio favors classical mechanisms [36]
This pathway control has direct implications for catalytic performance. ZSM-5 zeolites prepared with a higher contribution of the classical pathway demonstrated enhanced catalytic activity in furfuryl alcohol etherification, illustrating how crystallization mechanism influences final material function [36].
Two-Dimensional Materials: Tungsten Disulfide
The growth of monolayer transition metal dichalcogenides like WS₂ occurs via vapor-liquid-solid (VLS) mechanisms with clear non-classical characteristics. In-situ monitoring of chemical vapor deposition has revealed very large critical nuclei (up to 38.7 µm) that contradict classical nucleation theory predictions [6]. This observation aligns with the two-step nucleation model where metastable liquid precursors form before solid nucleation.
The VLS growth of WS₂ demonstrates how non-classical pathways can be harnessed for large-scale, high-quality crystal production. The liquid precursor acts as a selective medium where proper atomic arrangement occurs before integration into the crystal lattice, potentially excluding structural defects and impurities [6].
Table 2: Quantitative Parameters for Pathway Control in Different Material Systems
| Material System | Control Parameter | Effect on Pathway | Resulting Material Characteristic |
|---|---|---|---|
| Carbamazepine | Methanol/Water ratio | Determines 1-step vs. 2-step transition | Amorphous vs. crystalline form; Solubility enhancement |
| MFI Zeolites | H₂O/SiO₂ ratio | Lower ratio favors non-classical pathway | Alters crystal morphology and catalytic activity |
| MFI Zeolites | Ethanol/SiO₂ ratio | Higher ratio promotes classical pathway | Increases catalytic activity in etherification |
| WS₂ Monolayers | Precursor supersaturation | Controls critical nucleus size | Domain size and crystal quality |
| ZnO Nanoparticles | Supercooling degree | High supercooling: multi-step with metastable phase | Polymorph selection between WRZ and BCT structures |
Methodologies for Studying Non-Classical Pathways
Advanced Characterization Techniques
Direct observation of non-classical nucleation requires specialized characterization methods that can capture transient intermediates at relevant length and time scales:
- Liquid-Phase Transmission Electron Microscopy: Enables direct visualization of nucleation events in liquid environments, though requires careful management of electron beam effects on beam-sensitive soft materials [63] [10].
- Cryogenic Transmission Electron Microscopy (cryo-TEM): Vitrifies samples to preserve transient intermediate structures, successfully revealing amorphous precursors in carbamazepine crystallization [15].
- Atomic Force Microscopy (AFM): Provides high-resolution topographic imaging of growing crystal surfaces and attached particles [10].
- In-situ Monitoring Chemical Vapor Deposition: Allows real-time observation of phase transitions during materials growth, as demonstrated in WS₂ studies [6].
- Dynamic Light Scattering (DLS): Tracks particle size evolution during zeolite crystallization, helping distinguish between classical and non-classical pathways [36].
Computational and Data Analysis Approaches
Complementary to experimental techniques, computational methods provide atomic-level insights:
- Molecular Dynamics (MD) Simulations: Reveal atomic-scale mechanisms, such as coalescence of subcritical clusters and stepwise nucleation in iron, demonstrating how systems circumvent high energy barriers predicted by classical theory [1].
- Machine-Learning Interaction Potentials (MLIP): Enable accurate modeling of complex systems like ZnO nanoparticles, capturing the competition between different polymorphs during nucleation [9].
- Automated Image Analysis: Processes large datasets from in-situ monitoring experiments, enabling statistical analysis of nucleation events [6].
- High-Throughput Experimental Databases: Resources like the HTEM Database provide large, diverse datasets for data-driven discovery of crystallization trends [64] [65].
Experimental Protocols for Pathway Investigation
Micro-Droplet System for Pharmaceutical Compounds
This protocol, adapted from carbamazepine studies [15], enables high-throughput investigation of non-classical nucleation in organic systems:
Materials Requirements:
- Polydimethylsiloxane (PDMS) microfluidic devices with channel depth of 100 μm
- Fluorinated oil (e.g., Fluorient FC-40) as continuous phase
- 008-Fluorosurfactant for droplet stabilization
- Analytical grade solute (e.g., carbamazepine) and solvents
Procedure:
- Device Preparation: Treat microfluidic channels with aquapel for 10 seconds, followed by nitrogen drying. Incubate with FC-40 oil for 15 minutes prior to experimentation.
- Solution Preparation: Prepare saturated drug solutions in organic solvent (e.g., methanol), with variation in anti-solvent (water) ratio from 0-30% v/v.
- Droplet Generation: Inject solutions into microfluidic device to generate monodisperse droplets (50-100 μm diameter). Collect droplets onto cover glass with FC-40 overlay.
- Observation: Transfer samples to polarized optical microscope stage. Record crystallization process at 1 frame/second for quantitative analysis.
- Image Analysis: Use ImageJ or similar software to quantify droplet size, cluster formation dynamics, and phase transition timing.
Key Measurements:
- Statistical analysis of 50-100 droplets per condition
- Size distribution of dense liquid clusters
- Incubation time for phase transitions
- Correlation of solvent composition with nucleation pathway
Clear Solution Synthesis for Zeolite Pathway Analysis
This protocol, derived from MFI zeolite studies [36], allows systematic investigation of classical and non-classical pathway contributions:
Synthesis Formulation:
- Molar composition: X SiO₂: 0.39 TPAOH: 13.21 H₂O: Y ethanol
- Vary X (1.0-1.9) to modulate H₂O/SiO₂ ratio
- Vary Y (0-10) to modulate ethanol/SiO₂ ratio
Hydrothermal Treatment:
- Solution Preparation: Hydrolyze tetraethyl orthosilicate (TEOS) in tetrapropylammonium hydroxide (TPAOH) solution with 24-hour stirring at room temperature.
- Two-Stage Crystallization:
- Stage 1: Heat at 90°C for 90 minutes under microwave irradiation
- Stage 2: Immediately heat at 130°C, sampling at intervals up to 600 minutes
- Pathway Monitoring: Use dynamic light scattering (DLS) to track particle size evolution and identify crystallization inflection points.
Sample Purification and Analysis:
- Dialysis Purification: Use molecular weight cutoff 3.5 kDa dialysis tubes in two-stage process:
- 24 hours in 6 mmol/L TPAOH aqueous solution
- 48 hours in deionized water (changed every 12 hours)
- Product Isolation: Freeze-dry purified sol at -50°C, then calcine at 550°C to remove structure-directing agent.
- Pathway Quantification: Correlate synthesis parameters with crystallization curves, particle morphology, and catalytic performance to determine classical/non-classical contributions.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Non-Classical Nucleation Studies
| Reagent/Material | Function in Experiments | Example Application |
|---|---|---|
| Polydimethylsiloxane (PDMS) | Microfluidic device fabrication | Creating droplet reactors for pharmaceutical nucleation studies [15] |
| Fluorinated Oils (FC-40) | Continuous phase for droplet generation | Immiscible medium for micro-droplet experiments [15] |
| 008-Fluorosurfactant | Droplet stabilization | Preventing coalescence in emulsion-based systems [15] |
| Tetraalkylammonium Hydroxides (TPAOH) | Structure-directing agents | Zeolite crystallization with controlled porosity [36] |
| Tetraethyl Orthosilicate (TEOS) | Silicon source | Precursor for silicalite-1 and ZSM-5 zeolite synthesis [36] |
| Deuterated Solvents (D₂O, CDCl₃) | NMR spectroscopy | Tracking solution speciation and cluster formation [36] |
| Salt Additives (e.g., NaCl, KI) | Precursor vaporization modifiers | Enhancing VLS growth in 2D TMD synthesis [6] |
Pathway Visualization and Workflow Diagrams
Diagram 1: Non-classical nucleation pathways can be directed toward pure crystal formation through specific experimental controls, in contrast to classical pathways that often result in impurity incorporation.
Diagram 2: An integrated experimental-computational workflow enables comprehensive analysis of non-classical pathways, combining synthesis parameter variation with multiple characterization techniques and computational modeling.
The strategic application of non-classical nucleation pathways represents a paradigm shift in purity control for advanced materials. By moving beyond classical models, researchers can design crystallization processes that actively promote self-purification through several key mechanisms:
- Pathway Selection: Deliberately favor non-classical routes through control parameters like solvent composition, template ratios, and supersaturation levels.
- Intermediate Exploitation: Leverage dense liquid phases and amorphous precursors as selective filters that exclude impurities based on differential partitioning.
- Multi-scale Characterization: Employ integrated analytical approaches spanning from atomic-scale simulations to statistical analysis of droplet-based experiments.
- System-Specific Optimization: Tailor approaches to material class—whether pharmaceutical compounds, zeolites, or 2D materials—based on their specific non-classical behaviors.
The experimental protocols and characterization methodologies outlined in this guide provide a foundation for systematically implementing these strategies. As research continues to reveal the rich complexity of nucleation phenomena, the deliberate harnessing of non-classical pathways will increasingly enable the production of materials with purities and properties that exceed the limitations of classical crystallization approaches.
Mitigating Impurity Poisoning and Defect Formation
The pursuit of high-purity materials and compounds is a cornerstone of advanced research and industrial production across fields ranging from pharmaceuticals to semiconductor manufacturing. Traditional classical nucleation theory, which describes the formation of stable nuclei through monomer attachment, often fails to fully capture the complex reality of crystallization and synthesis processes. The emergence of non-classical nucleation pathways, involving particle attachment and metastable intermediates, substantially alters crystallization kinetics and outcomes, often introducing unique defect structures and impurity incorporation sites [4]. Within this sophisticated framework, impurity poisoning and defect formation are not mere anomalies but inherent challenges that can systematically compromise material integrity, therapeutic efficacy, and functional performance.
This technical guide examines defect mitigation strategies through the lens of non-classical crystallization phenomena, providing researchers with both theoretical foundations and practical methodologies to address these critical issues. By understanding the interplay between impurity behavior, nucleation pathways, and defect formation, scientists can develop more robust prevention and control strategies tailored to their specific material systems, whether working with organic crystalline pharmaceuticals, high-manganese steels, silicon wafers, or specialized chemical intermediates.
Quantitative Landscape of Defect Induction and Mitigation
Systematic measurement of defect induction rates provides crucial data for evaluating mitigation strategy effectiveness. The following tables summarize key quantitative relationships between process parameters, impurity levels, and resulting defect formation across multiple material systems.
Table 1: Defect Induction Rates in AI-Generated Code Relative to Prompt Quality
| Prompt Normativity Level | Description | Code Security Defect Rate | Effective Mitigation Strategies |
|---|---|---|---|
| L0 | High-quality, well-structured prompts | Lowest observed defect rates | Structured validation protocols |
| L1 | Moderate quality with minor inconsistencies | 2.1x increase over L0 | Basic prompt optimization |
| L2 | Significant formulation issues | 3.8x increase over L0 | Chain-of-Thought prompting |
| L3 | Poorly structured, ambiguous prompts | 5.6x increase over L0 | Self-Correction techniques |
Research demonstrates a clear correlation between decreasing prompt normativity and increasing security vulnerabilities in AI-generated code, with defect induction rates rising markedly as prompt quality deteriorates [66]. Advanced mitigation strategies, particularly Chain-of-Thought and Self-Correction prompting techniques, have proven effective at substantially improving code safety across all prompt quality levels.
Table 2: Defect Formation and Mitigation in Material Systems
| Material System | Primary Defect Types | Key Mitigation Approach | Efficacy Measurement |
|---|---|---|---|
| High-Manganese Steels (DED-LB) | Oxide formation, severe cracking | Melt pool size reduction | Reproducible mechanical properties achieved |
| UMG-Si Wafers | Oxygen precipitates, metal impurities | Tabula rasa, gettering, hydrogenation | Carrier lifetime improvement >40% |
| Vinyl Acetate Monomer | Catalyst poisoning, product discoloration | Advanced catalyst systems, purification | 3-7% production cost reduction |
| Pharmaceutical Products | Process-related, degradation impurities | ICH guideline implementation | Threshold-based quality control |
In material science applications, defect formation mechanisms vary significantly by system but share common themes of impurity incorporation and microstructural imperfections [67] [68] [69]. Successful mitigation strategies typically involve process parameter optimization, impurity gettering, and advanced purification techniques.
Experimental Protocols for Defect Analysis and Mitigation
Pharmaceutical Impurity Identification and Qualification
The identification and qualification of unexpected impurities in pharmaceutical products follows a rigorous analytical workflow based on ICH Q3A(R2) and Q3B(R2) guidelines [70] [71]. The protocol begins with detection using chromatographic techniques (LC or GC), with impurities breaching reporting thresholds (typically 0.05-0.1%) requiring identification. Mass spectrometry provides structural information through fragmentation patterns, while Nuclear Magnetic Resonance (NMR) spectroscopy offers complementary structural elucidation capabilities. For impurities exceeding qualification thresholds (typically 0.15-0.2%), biological safety must be established through toxicological assessment.
Key methodological considerations:
- Sample Preparation: Employ stability-indicating methods that preserve impurity profiles
- Method Validation: Demonstrate specificity, accuracy, precision, and detection limits
- Forced Degradation Studies: Stress samples under exaggerated conditions (heat, light, pH, oxidation) to predict long-term stability issues
- Structural Elucidation: Combine multiple spectroscopic techniques for unambiguous identification
The experimental workflow emphasizes threshold-based decision points, with analytical strategies tailored to impurity abundance and phase of drug development. Early-phase investigations focus on major impurities, while later phases require comprehensive impurity profiling with rigorous safety qualification [71].
Defect Prevention in Directed Energy Deposition of Metallic Alloys
Research on high-manganese steels (HMnS) processed via laser beam directed energy deposition (DED-LB) provides a protocol for mitigating defect formation in additive manufacturing [67]. The methodology couples finite element method (FEM) simulations with experimental validation to identify and address defect formation mechanisms.
Experimental sequence:
- Melt Pool Modeling: Simulate thermal profiles and solidification behavior using FEM with varying laser spot diameters (0.66mm vs. 3mm)
- Sample Fabrication: Manufacture X30Mn23 steel specimens with 0-1 wt% in-situ alloyed Al using Ar-gas atomized powders (<90μm)
- Microstructural Characterization: Employ OM, SEM, EDS, EBSD, and APT to quantify oxide formation, precipitate distribution, and crystallographic textures
- Mechanical Testing: Evaluate tensile properties to correlate defect structures with performance metrics
Critical parameters:
- Laser spot diameter (primary influence on melt pool size)
- Solidification rate (higher with smaller beam diameter)
- Shielding gas coverage and purity
- Powder feedstock quality and particle size distribution
This integrated approach successfully demonstrated that reduced melt pool size significantly decreases oxide formation by minimizing atmosphere interaction, ultimately preventing the severe cracking associated with oxide precipitates [67].
Visualization of Defect Mitigation Pathways
Pharmaceutical Impurity Management Workflow
Diagram 1: Pharmaceutical impurity decision pathway
Material Defect Prevention Strategy
Diagram 2: Material defect prevention strategy
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Defect Mitigation Studies
| Reagent/Material | Function in Defect Studies | Application Context |
|---|---|---|
| Gold-Palladium Catalysts | Enhanced selectivity in VAM production | Chemical manufacturing defect prevention [69] |
| Hydrogenation Agents | Passivation of defects and impurities in UMG-Si wafers | Semiconductor material processing [68] |
| Elemental Aluminum Powder | In-situ alloying for SFE control in HMnS | Additive manufacturing defect prevention [67] |
| Ar-Gas Atomized Powders | Consistent feedstock for DED-LB processes | Metal additive manufacturing [67] |
| Chromatography Reference Standards | Impurity identification and quantification | Pharmaceutical impurity profiling [70] [71] |
| Gettering Agents (Phosphorus, Aluminum) | Impurity sequestration in silicon | UMG-Si wafer quality enhancement [68] |
| Antimicrobial Surface Treatments | Biocontamination control in cleanrooms | Semiconductor manufacturing [72] |
These specialized reagents and materials enable researchers to implement targeted defect mitigation strategies across diverse material systems. Proper selection and application of these tools is critical for controlling nucleation pathways, minimizing impurity incorporation, and achieving consistent material properties.
Defect formation and impurity poisoning present multifaceted challenges that require integrated mitigation approaches spanning material design, process optimization, and analytical validation. The non-classical nucleation pathways prevalent in organic and soft materials introduce unique defect formation mechanisms that demand specialized characterization and intervention strategies. By leveraging the protocols, visualization frameworks, and toolkit components presented in this technical guide, researchers can systematically address defect-related challenges across diverse applications from pharmaceutical development to advanced material manufacturing. The continued refinement of these mitigation approaches will enable more reliable production of high-quality materials with tailored properties and enhanced performance characteristics.
Exploiting Metastable Intermediates for Novel Material Design
The pursuit of novel functional materials is increasingly leveraging non-classical nucleation pathways that proceed through metastable intermediates. These kinetically favored, higher-energy states offer a rich design space for creating materials with properties inaccessible through traditional thermodynamic synthesis. This whitepaper provides an in-depth technical examination of strategies for exploiting these transient states, covering advanced computational generative models, innovative experimental trapping techniques, and low-temperature synthesis methodologies. By integrating these approaches, researchers can systematically navigate the energy landscape to discover and stabilize novel inorganic materials with targeted properties for applications ranging from energy storage to drug development.
In the context of inorganic materials research, metastability describes an intermediate energetic state within a dynamical system other than the system's state of least energy. A metastable state persists for a finite lifetime, with all state-describing parameters maintaining stationary values until sufficient external energy triggers a transition to a more stable configuration [73]. This phenomenon is ubiquitous across materials systems, from polymorphic crystals like diamond (metastable at standard temperature and pressure) to self-assembling molecular systems that evolve through transient intermediates before reaching their thermodynamic end states [73].
The strategic importance of metastable intermediates lies in their position within non-classical nucleation pathways. Traditional materials synthesis typically targets the most thermodynamically stable structures, severely limiting the accessible compositional and structural space. In contrast, deliberately targeting and stabilizing metastable intermediates enables access to a vastly expanded range of functional materials with tailored electronic, mechanical, and chemical properties. This paradigm shift requires sophisticated approaches to both predict these transient states and develop methodologies to capture them for technological applications.
Theoretical Foundations of Metastable Intermediates
Energetic Landscapes and Kinetic Persistence
Metastable states inhabit local minima in the energy landscape, separated from the global minimum by activation energy barriers that prevent immediate rearrangement to the most stable configuration. This phenomenon is described as kinetic stability or kinetic persistence - the system remains "stuck" in a thermodynamic trough not due to thermodynamic preference, but because the kinetics of atomic rearrangement are insufficient to overcome the energy barrier [73]. The lifetime of a metastable state depends on the height of this barrier; while some persist for microseconds, others like silica glass have lifetimes estimated at 10^98 years [73].
Classification of Metastable Materials Systems
Metastability manifests across multiple classes of inorganic materials, each with distinct characteristics and stabilization mechanisms:
- Polymorphic Crystals: Different crystalline arrangements of the same composition, such as anatase TiO₂ (metastable) versus rutile TiO₂ (stable), or diamond versus graphite [73].
- Martensitic Phases: Metastable phases critical for controlling material properties like the hardness of steel [73].
- Self-Assembling Supramolecular Systems: Molecular aggregates that evolve through kinetically trapped intermediates like nanoribbons and nanohelices before forming thermodynamic tubular structures [74].
- Glasses and Amorphous Solids: Non-crystalline solids with lifetimes sufficient for practical applications [73].
Computational Approaches for Metastable Material Discovery
Generative artificial intelligence represents a transformative approach for discovering metastable materials by directly proposing candidate structures with targeted properties.
Generative AI and Baselines for Material Discovery
Recent benchmarking studies have established that while traditional methods like data-driven ion exchange of known compounds remain highly effective at generating stable materials that closely resemble known structures, generative models excel at proposing novel structural frameworks [75]. When sufficient training data exists, these models can effectively target specific properties such as electronic band gap and bulk modulus. A critical enhancement for both approaches is implementing post-generation screening using pre-trained machine learning models and universal interatomic potentials, which substantially improves success rates while maintaining computational efficiency [75].
MatterGen: A Diffusion-Based Foundation Model
MatterGen represents a significant advancement in generative models for materials design. This diffusion-based model generates stable, diverse inorganic materials across the periodic table and can be fine-tuned to steer generation toward specific property constraints [76]. The model introduces several technical innovations:
- A customized diffusion process that generates crystal structures by gradually refining atom types, coordinates, and the periodic lattice
- Adapter modules for fine-tuning on desired chemical composition, symmetry, and scalar properties
- Training on diverse datasets (Alex-MP-20 with 607,683 structures) to ensure broad coverage [76]
Table 1: Performance Comparison of Generative Models for Material Discovery
| Model | Stable, Unique, New (SUN) Materials | Average RMSD to DFT-relaxed (Å) | Novelty Rate | Property Targeting |
|---|---|---|---|---|
| MatterGen | 75-78% (within 0.1 eV/atom of convex hull) | <0.076 | 61% new structures | Chemistry, symmetry, mechanical, electronic, magnetic properties |
| CDVAE | Substantially lower than MatterGen | ~10× higher than MatterGen | Lower novelty rate | Limited property set |
| DiffCSP | Substantially lower than MatterGen | ~10× higher than MatterGen | Lower novelty rate | Limited property set |
| Ion Exchange | High stability | N/A | Lower structural novelty | Limited to chemical derivatives |
Generative AI Agents for Guided Exploration
MatAgent represents an alternative approach that harnesses the reasoning capabilities of large language models (LLMs) for materials discovery. This system combines a diffusion-based generative model for crystal structure estimation with predictive models for property evaluation, using iterative, feedback-driven guidance to steer exploration toward user-defined targets [77]. Integrated with cognitive tools including short-term memory, long-term memory, and a comprehensive materials knowledge base, MatAgent emulates human expert reasoning to expand the accessible compositional space while maintaining high compositional validity, uniqueness, and novelty [77].
Experimental Methodologies for Trapping Metastable Intermediates
Photopolymerization Trapping Strategy
A highly efficient method for trapping metastable intermediates in self-assembly processes employs a photopolymerization strategy. This approach uses diacetylene-functionalized building blocks that undergo UV-promoted polymerization to effectively "freeze" intermediate structures before they reorganize into thermodynamic products [74].
Experimental Protocol:
- Prepare a clear solution of chiral perylene-diimide (0.3 mM) in chloroform and mix with ethanol (final concentration 27 μM)
- Heat the mixture to 65°C to ensure complete dissolution
- Cool the solution to 15°C in a temperature-controlled cell to initiate self-assembly
- Monitor formation of metastable intermediates (nanoribbons, nanocoils, nanohelices) via TEM and CD spectroscopy
- Expose intermediates to UV irradiation (254 nm) to initiate polymerization of diacetylene units
- Characterize trapped structures using AFM, XRD, and UV-visible spectroscopy [74]
This method successfully trapped nanoribbons, nanocoils, and nanohelices that normally transform into thermodynamic nanotubes within hours. The photopolymerization occurs with minimal rearrangement of building blocks, preserving the original morphologies of the metastable intermediates [74].
Low-Temperature Synthesis Routes
Low-temperature synthesis methods (<150°C) provide powerful alternatives for accessing metastable materials by leveraging three primary strategies:
- Dative Bonding Approaches: Utilizing weakly bonded precursors (e.g., F₃B-NMe₃) that undergo clean heterolytic bond cleavage to generate reactive synthons without high-temperature conditions [78]
- Ring Strain-Driven Synthesis: Employing strained ring systems that release reactive fragments upon thermolysis or photolysis (e.g., [Me₂Si]₆ eliminating Me₂Si) [78]
- Functional Group Metathesis: Generating reactive intermediates through exchange reactions at room temperature (e.g., producing crystalline Sn from Sn(II) alkoxide and HBPin) [78]
These approaches enable deposition of materials like germanium and indium phosphide at dramatically reduced temperatures while accessing metastable polymorphs not obtainable through traditional high-temperature methods.
Integrated Workflow for Metastable Material Exploration
The strategic exploitation of metastable intermediates requires combining computational prediction with experimental validation in an iterative discovery cycle. The diagram below visualizes this integrated approach.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents for Metastable Intermediate Research
| Reagent/Material | Function | Application Example |
|---|---|---|
| Diacetylene-functionalized building blocks | Photopolymerizable units for trapping intermediates | Self-assembly systems (e.g., chiral PDI) [74] |
| Weak dative bond precursors | Low-temperature source of reactive synthons | F₃B-NMe₃ for main group materials deposition [78] |
| Strained ring systems | Ring strain-driven release of reactive fragments | [Me₂Si]₆ as Me₂Si source [78] |
| Functional group metathesis reagents | Room-temperature generation of reactive intermediates | HBPin for Sn(0) generation from Sn(II) alkoxides [78] |
| Universal interatomic potentials | Machine learning force fields for stability screening | Post-generation filtering of generative model outputs [75] |
| Adapter modules | Fine-tuning generative models for property constraints | MatterGen for targeted property generation [76] |
The strategic exploitation of metastable intermediates represents a paradigm shift in inorganic materials design, moving beyond thermodynamic limitations to access a vastly expanded universe of functional materials. By integrating generative AI prediction with advanced trapping methodologies and low-temperature synthesis, researchers can systematically navigate complex energy landscapes to discover and stabilize materials with precisely tailored properties. As these computational and experimental techniques continue to mature, they promise to accelerate the discovery of next-generation materials for energy storage, catalysis, electronics, and pharmaceutical applications. The future of materials design lies not merely in discovering what nature prefers, but in mastering the full spectrum of possibilities along non-classical nucleation pathways.
Classical vs. Non-Classical: Direct Comparisons and Experimental Validation
The synthesis of zeolites, microporous crystalline oxides central to catalysis and separation processes, has traditionally been viewed through the lens of classical crystallization theory [79]. However, a paradigm shift is underway, recognizing the critical role of non-classical pathways in determining the physicochemical properties of these materials. Within the broader thesis of advanced inorganic materials research, understanding and distinguishing between these dual crystallization pathways—classical and non-classical—is not merely an academic exercise but a fundamental requirement for engineering zeolites with tailored functionality [80] [81]. Classical crystal growth proceeds primarily via the addition of single ions or molecules to a growing crystal surface, resulting in dense morphologies with smooth surfaces [81]. In contrast, non-classical crystallization, often referred to as the particle attachment mechanism, involves the assembly of amorphous or crystalline nanoparticles into larger architectures, frequently yielding hierarchical structures with enhanced porosity and surface accessibility [80].
The imperative to quantify contributions from these pathways stems from their direct impact on catalytic efficiency. Properties such as crystal morphology, mesoporosity development, and the distribution of active sites are profoundly influenced by the crystallization mechanism [80] [82]. For instance, zeolites synthesized via non-classical pathways often exhibit superior performance in reactions involving bulky molecules due to improved mass transport. Consequently, this guide provides researchers with the methodological framework and analytical tools necessary to experimentally distinguish, quantify, and control these pathways, thereby enabling the rational design of next-generation zeolitic materials.
Fundamental Mechanisms: Classical versus Non-Classical Crystallization
Defining the Crystallization Pathways
The journey from amorphous precursor gels to crystalline zeolites can follow distinct mechanistic routes, each with characteristic intermediates and kinetic profiles.
Classical Crystallization Pathway: This mechanism is characterized by a monomer-by-monomer addition process. It begins with soluble species (monomers) in the synthesis gel that nucleate spontaneously. Growth then continues through the direct attachment of these monomers to the crystal surface, leading to the formation of crystals with defined, often smooth, facets and dense morphology [81]. The process is typically described by standard nucleation and growth models, and the resulting crystals are usually compact and micron-sized, as seen in the initial synthesis of MCM-22P without additives [80].
Non-Classical Crystallization Pathway: This pathway proceeds via a particle-based assembly process. Instead of monomers, the system involves the formation of amorphous nanoparticles or pre-structured precursors that aggregate and undergo a gradual solid-state transformation into crystalline material [81]. This mechanism, also described as a "non-classical (i.e., CPA, particle attachment)" route, often results in hierarchical structures, such as the "wool ball" or "ellipsoid" morphologies observed in MWW zeolites synthesized with specific Na salts [80]. These structures typically possess higher external surface areas and improved diffusivity for reactant molecules.
Key Differentiating Factors and Implications
The choice of crystallization pathway has profound implications for the final material's properties. The table below summarizes the core differences and their direct consequences for catalytic application.
Table 1: Fundamental Differences between Classical and Non-Classical Crystallization Pathways
| Aspect | Classical Pathway | Non-Classical Pathway |
|---|---|---|
| Primary Mechanism | Addition of ions/molecular species (monomers) [81] | Attachment and reorganization of particles (nanoprecursors) [80] [81] |
| Common Intermediates | Soluble silicate/aluminate species | Amorphous worm-like particles (WLPs), colloidal precursors [81] |
| Typical Crystal Morphology | Dense, defined facets (e.g., dense hexagon) [80] | Hierarchical, open structures (e.g., ellipsoid, wool ball) [80] |
| Surface Texture | Smooth surfaces [81] | Rough surfaces, composite structures [81] |
| Induction Period | Often longer, dependent on monomer supersaturation | Can be shorter, directed by precursor aggregation [81] |
| Impact on Catalysis | Potential diffusion limitations in micropores | Enhanced accessibility and often a higher density of accessible active sites [81] |
Quantitative Methodologies for Pathway Analysis
Distinguishing between crystallization pathways requires a combination of kinetic, morphological, and structural analyses. The following experimental protocols and quantitative measures are essential for accurate characterization.
Kinetic Analysis of Crystallization
Monitoring the crystallization process over time is a primary method for identifying the operative pathway. This involves isolating solid products at regular intervals during hydrothermal synthesis for analysis.
Experimental Protocol:
- Synthesis Setup: Prepare the zeolite synthesis gel according to the desired composition (e.g., for TS-1: tetraethyl orthosilicate as a silicon source, tetrabutyl orthotitanate as a titanium source, and tetrapropylammonium hydroxide as a structure-directing agent) [81].
- Hydrothermal Treatment: Carry out crystallization in a stirred autoclave at a controlled temperature (e.g., 80°C for the first step and 170°C for the second step in a two-step process).
- Time-Resolved Sampling: At predetermined time intervals, extract a portion of the gel from the autoclave, quench the reaction rapidly, and separate the solid product via centrifugation.
- Crystallinity Measurement: Analyze each solid sample using Powder X-ray Diffraction (PXRD). The relative crystallinity is calculated by comparing the intensity of key characteristic diffraction peaks (e.g., the peak at ~23° for MFI topology) to that of a fully crystalline reference sample [81].
Data Interpretation:
- The induction time is estimated from the initial emergence of characteristic diffraction peaks.
- A significantly shortened induction period and total crystallization time upon adding a modifier (e.g., a polymer) strongly suggests a shift towards a non-classical pathway. For example, polyacrylamide (PAM) reduced the induction time for TS-1 from 6 hours to 2 hours at 120°C, accelerating nucleation by stabilizing colloidal precursors [81].
Advanced Microscopy and Structural Characterization
Direct observation of intermediates is the most conclusive way to identify a non-classical pathway.
Experimental Protocol:
- Sample Preparation: For Transmission Electron Microscopy (TEM), disperse the solid samples (especially those from early crystallization times) in ethanol via ultrasonication and deposit them on a carbon-coated copper grid.
- TEM and SAED Imaging: Analyze the samples using TEM to observe morphology. Perform Selected Area Electron Diffraction (SAED) on different regions of the particles.
- Dynamic Light Scattering (DLS): Analyze the supernatant of the synthesis gel after centrifugation to detect the presence and size distribution of sub-50 nm colloidal particles [81].
Data Interpretation:
- The presence of amorphous worm-like particles that later transform into crystals is a hallmark of non-classical crystallization. For instance, in the PAM-directed synthesis of TS-1, amorphous WLPs of about 200 nm were observed before crystallization, with smaller particles attached to their surfaces [81].
- SAED patterns showing diffuse rings (indicating amorphousness) in these intermediates, which sharpen into diffraction spots over time, confirm a solid-state transformation.
- DLS data showing a population of small, stable nanoparticles in the supernatant supports a non-classical pathway dominated by particle attachment [81].
Probing the Pathway with Inorganic Salts
The use of simple inorganic salts provides a powerful, surfactant-free method to manipulate crystallization pathways and offers insights into the mechanism through their impact on induction time and morphology.
Experimental Protocol:
- Additive Screening: Conduct a series of syntheses of a zeolite like MWW, keeping all parameters constant except for the addition of different sodium salts (e.g., NaOH, Na₂CO₃, NaHCO₃ at various molar ratios relative to SiO₂) [80].
- Characterization: Determine the crystallization kinetics as in Section 3.1 and the final morphology via Scanning Electron Microscopy (SEM).
Quantitative Data and Interpretation:
- The induction time is highly dependent on the anion of the sodium salt. For MWW synthesis, the induction time followed the order: NaHCO₃ (36 h) < Na₂CO₃ (72 h) = NaOH (72 h) [80].
- Different salts lead to distinct morphologies: NaOH produces an ellipsoid shape, Na₂CO₃ a wool ball shape, and NaHCO₃ a uniform hexagon morphology [80].
- This demonstrates that the salt anion can switch the pathway. NaHCO₃ favors a classical route, while NaOH and Na₂CO₃ lead to crystallization through a network of hydrogel via the non-classical pathway [80].
Table 2: Quantitative Impact of Inorganic Salts on MWW Zeolite Crystallization Pathways [80]
| Additive | Induction Time (h) | Proposed Crystallization Pathway | Resulting Crystal Morphology |
|---|---|---|---|
| None | Not specified | Classical | Dense Hexagon |
| NaOH | 72 | Non-classical | Ellipsoid with central hole |
| Na₂CO₃ | 72 | Non-classical | Wool Ball |
| NaHCO₃ | 36 | Classical | Uniform Hexagon |
Visualizing Workflows and Pathway Logic
The following diagrams, generated using Graphviz DOT language, illustrate the core experimental and analytical workflows for studying zeolite crystallization pathways.
Experimental Workflow for Pathway Analysis
Decision Logic for Identifying Crystallization Pathways
The Scientist's Toolkit: Essential Research Reagents and Materials
Controlling zeolite crystallization pathways requires a precise set of chemical reagents and analytical tools. The following table catalogues the key materials used in the featured experiments and their specific functions in directing synthesis.
Table 3: Research Reagent Solutions for Zeolite Synthesis and Pathway Analysis
| Reagent/Material | Function in Synthesis | Role in Pathway Control | Example Use Case |
|---|---|---|---|
| Organic Structure-Directing Agents (SDAs) (e.g., 1-Butyl-2,3-dimethyl-1H-imidazol-3-ium hydroxide, Tetrapropylammonium hydroxide) | Directs the formation of a specific zeolite topology by fitting within the developing pores [80] [79]. | Primary template around which the framework assembles; its hydrophobicity and size can influence the energy landscape of nucleation. | Synthesizing MCM-22P (MWW) and TS-1 (MFI) zeolites [80] [81]. |
| Dual SDAs (e.g., Pyrrolidine + Isopropylamine) | Two amines work cooperatively to direct structure formation [82]. | Widens the synthesis composition window, alters acid site distribution, and can speed up crystallization, potentially influencing the nucleation pathway [82]. | Synthesis of ZSM-23 zeolite with improved properties for n-hexadecane hydroisomerization [82]. |
| Polymer Modifiers (e.g., Polyacrylamide - PAM) | Acts as a crystal growth modifier (CGM) [81]. | Switches pathway from classical to non-classical via specific interactions with Si/Ti species, stabilizing colloidal precursors and accelerating nucleation [81]. | Producing TS-1 with enriched Ti content and hierarchical features [81]. |
| Inorganic Salts (e.g., NaOH, Na₂CO₃, NaHCO₃) | Provides alkali metal cations (Na⁺) and adjusts pH and supersaturation in the gel [80]. | Anion choice can dictate the crystallization pathway (classical vs. non-classical) and dramatically alter final crystal morphology without surfactants [80]. | Morphology regulation of MWW zeolite (ellipsoid, wool ball, hexagon) [80]. |
| Hydrofluoric Acid (HF) | Mineralizing agent in certain syntheses, particularly for all-silica or high-silica zeolites. | Influences the solubility of silica species and the kinetics of the synthesis, thereby affecting the nucleation and growth mechanism. | Used in the synthesis of pure silica MWW zeolites with Na salts [80]. |
The ability to distinguish and quantify contributions from classical and non-classical pathways is a cornerstone of modern zeolite design. By employing the kinetic, microscopic, and synthetic tools detailed in this guide—from time-resolved PXRD and TEM to the strategic use of polymer modifiers and inorganic salts—researchers can move beyond empirical formulations. This methodological framework allows for the deliberate steering of crystallization towards pathways that yield materials with optimized hierarchical structures, enriched active sites, and superior catalytic performance, thereby advancing the broader field of inorganic materials research.
Comparative Analysis of Nucleation Kinetics and Growth Rates
Nucleation and crystal growth are fundamental processes in materials science, dictating the critical structural and functional properties of a wide range of inorganic materials and pharmaceuticals. The controlled formation of crystals is paramount for applications spanning from the design of high-performance flame retardants and battery electrodes to the precise formulation of active pharmaceutical ingredients (APIs). For decades, the Classical Nucleation Theory (CNT) has provided the foundational framework for understanding these initial stages of phase formation. However, recent advances have revealed that non-classical nucleation pathways, involving multi-step mechanisms and metastable intermediates, are more prevalent than previously recognized, particularly in inorganic systems [1] [9].
This whitepaper provides a comparative analysis of nucleation kinetics and growth rates, situating its discussion within the evolving context of non-classical pathways. Aimed at researchers and drug development professionals, it synthesizes recent theoretical and experimental findings to serve as a technical guide for understanding and controlling crystallization processes. By integrating quantitative data, standardized protocols, and conceptual visualizations, this document aims to bridge the gap between fundamental mechanistic studies and industrial process optimization.
Theoretical Framework: Classical vs. Non-Classical Nucleation
The journey from a dissolved solute to a stable crystal begins with nucleation. This section contrasts the established classical view with emerging non-classical perspectives.
Classical Nucleation Theory (CNT)
CNT describes nucleation as a single-step, stochastic process where solute molecules in a supersaturated solution aggregate to form a stable nucleus of a critical size. The formation of this nucleus is governed by a trade-off between the bulk free energy (which favors growth) and the surface free energy (which opposes it). The key kinetic parameter is the nucleation rate (J), which quantifies the number of stable nuclei formed per unit volume per unit time and is described by the fundamental equation:
J = kn exp(-ΔG / RT) [83]
Here, k_n is the nucleation rate kinetic constant, ΔG is the Gibbs free energy of nucleation, R is the gas constant, and T is the temperature. The Gibbs free energy barrier is influenced by the supersaturation of the system; higher supersaturation lowers the energy barrier and increases the nucleation rate. This model allows for the calculation of critical parameters such as the surface free energy and the radius of the critical nucleus from experimental metastable zone width (MSZW) data [83].
Non-Classical Nucleation Pathways
Contrary to the direct path of CNT, non-classical nucleation proposes alternative, often more complex, routes to a stable crystalline phase. Atomic-scale studies, particularly through molecular dynamics simulations, have revealed that systems can circumvent the high energy barrier of classical homogeneous nucleation through mechanisms such as:
- Two-Step Nucleation: In this pathway, the formation of a stable crystal is preceded by an intermediate metastable phase. For example, in the crystallization of zinc oxide (ZnO) from a nano-droplet melt, the system can first form a dense liquid droplet or an amorphous precursor, which then reorganizes into a crystalline structure. This pathway can lead to a narrower particle size distribution due to a "catching-up" behavior during growth [84] [9].
- Coalescence of Subcritical Clusters: Molecular dynamics simulations of iron have shown that instead of a single cluster growing to a critical size, multiple subcritical clusters can form and coalesce to bypass the classical energy barrier [1].
- Polymorphic Competition: In nanocrystal formation, different crystal structures (polymorphs) can compete during the nucleation stage. The predominance of one polymorph over another depends on synthesis conditions, such as the degree of supercooling. For instance, in ZnO nanoparticles, a competition between the wurtzite (WRZ) and body-centered tetragonal (BCT) phases is observed, with the nucleation pathway (classical vs. multi-step) varying with temperature [9].
These non-classical mechanisms significantly expand the toolbox for controlling material properties, as the pathway taken can directly influence the final crystal size, size distribution, and polymorphic form.
Quantitative Kinetics: A Comparative Data Analysis
The following tables consolidate key kinetic parameters from recent studies across various materials, including inorganic salts, APIs, and a model biomolecule, to allow for direct comparison.
Table 1: Comparative Nucleation Kinetics for Selected Materials [83]
| Material | Solvent System | Nucleation Rate Constant, kn (molecules m⁻³ s⁻¹) | Gibbs Free Energy of Nucleation, ΔG (kJ mol⁻¹) | Surface Free Energy, γ (mJ m⁻²) | Critical Nucleus Radius, r* (nm) |
|---|---|---|---|---|---|
| APIs (e.g., Paracetamol) | Various (Water, Ethanol) | 10²⁰ – 10²⁴ | 4 – 49 | 1.3 – 2.1 | 0.6 – 1.5 |
| Lysozyme | NaCl Solution | ~10³⁴ | 87 | 3.3 | 1.2 |
| Glycine (Amino Acid) | Water | - | 28 | 1.6 | 0.9 |
| Inorganic Salts (e.g., KNO₃, NH₄Cl) | Water | - | 5 – 18 | 0.9 – 1.6 | 0.5 – 1.1 |
Table 2: Experimentally Determined Nucleation and Growth Kinetics for Magnesium Hydroxide [85]
| Process Parameter | Nucleation Rate Order | Growth Rate Order | Observations |
|---|---|---|---|
| Mg²⁺ Concentration | 0.48 ± 0.07 | 0.09 ± 0.01 | Nucleation is more sensitive to concentration changes than growth. |
| Current Density | 0.32 ± 0.03 (Stage 1) | 0.15 ± 0.01 | Higher current density promotes nucleation over growth, leading to a "competition." |
| Temperature | Positive Correlation | Positive Correlation | Elevated temperature accelerates both processes. |
Key Insights from Quantitative Data:
- Material-Dependent Energetics: The Gibbs free energy of nucleation (ΔG) varies significantly, from as low as 4 kJ mol⁻¹ for some APIs to 87 kJ mol⁻¹ for lysozyme. This indicates a much higher energy barrier for the nucleation of large, complex molecules [83].
- Process-Dependent Kinetics: The exponents for Mg²⁺ concentration and current density in Mg(OH)₂ crystallization reveal that nucleation is more sensitive to changes in supersaturation than growth is. This differential sensitivity is a critical lever for controlling crystal size distribution; operating at higher driving forces favors the generation of more nuclei, potentially leading to smaller crystals [85].
- Pathway-Dependent Outcomes: The "catching-up" behavior observed in two-step nucleation pathways can result in a narrower particle size distribution compared to the one-step classical pathway. This size-focusing effect is a direct consequence of the nucleation mechanism and offers a strategy for producing more uniform particles [84].
Experimental Protocols for Kinetic Analysis
Standardized and advanced methodologies are crucial for obtaining reliable and comparable kinetic data.
Automated and Standardized Crystallization Kinetics
Objective: To determine secondary nucleation and crystal growth kinetics in a reproducible manner, specifically for strong electrolyte systems like inorganic salts.
Methodology:
- Equipment: Utilize an automated laboratory reactor system (e.g., Technobis Crystalline) equipped with in-situ imaging probes (e.g., particle vision microscope or FBRM).
- Process: Conduct automated antisolvent crystallizations. For example, crystallize potassium chloride or potassium sulfate from water by systematically adding ethanol as an antisolvent.
- Data Collection: The in-situ probes continuously monitor crystal count and size in real-time.
- Kinetic Modeling: Fit the experimental data (crystal size and concentration over time) using a population balance model. The key advancement for inorganic salts is the explicit incorporation of activity coefficients to accurately calculate the thermodynamic driving force (supersaturation) in strong electrolyte systems, which dissociate in solution [86].
Outcome: The model yields quantitative kinetic parameters for nucleation and growth, allowing for the direct comparison of different solutes, such as organic molecules versus inorganic salts.
Determining Nucleation Rates from Metastable Zone Width (MSZW)
Objective: To estimate the nucleation rate (J), kinetic constant (kn), and Gibbs free energy of nucleation (ΔG) using readily measurable MSZW data.
Methodology:
- Experimental Setup: Prepare a saturated solution at a known temperature (T). Using a polythermal method, cool the solution at a defined, constant cooling rate (dT/dt).
- Measurement: Monitor the solution (e.g., via turbidity or visual inspection) to detect the nucleation temperature (Tnuc). The MSZW is defined as ΔTmax = T* - Tnuc.
- Data Processing: For multiple experiments at different cooling rates, calculate the maximum supersaturation at nucleation, ΔCmax, using the solubility curve.
- Model Application: Apply the linearized form of the classical nucleation theory model: ln(ΔCmax / ΔTmax) = ln(kn) - (ΔG / R) * (1 / Tnuc) [83]
- Parameter Extraction: Plot ln(ΔCmax / ΔTmax) versus 1/Tnuc. The slope gives -ΔG/R and the intercept gives ln(kn). Subsequently, surface energy and critical nucleus size can be calculated.
Outcome: This protocol provides a powerful method to extract fundamental nucleation parameters from standard laboratory experiments, validated across APIs, inorganics, and biomolecules [83].
Numerical Simulation of Nucleation Pathways
Objective: To study the atomistic mechanisms and effects of different nucleation pathways (one-step vs. two-step) on particle formation.
Methodology:
- Force Field Development: Construct a machine-learning interaction potential (MLIP) that accurately captures both short-range and long-range atomic interactions. For materials like ZnO, this is critical for correctly modeling surface energies and polymorph stability [9].
- Simulation Setup: Model a system such as a liquid nano-droplet of the material (e.g., 500-1500 atoms of ZnO).
- Sampling: Perform brute-force molecular dynamics simulations at different temperatures (degrees of supercooling). Complement with rare-event sampling techniques (e.g., seeded simulations) to overcome free energy barriers.
- Pathway Analysis: Use data-driven structural analysis (e.g., Gaussian-mixture models) to characterize the local ordering of atoms and identify the emergence of different phases (e.g., liquid, metastable BCT, stable WRZ) [9].
- Population Balance Modeling (PBM): Incorporate the insights from atomistic simulations into a mesoscale PBM. The model should account for pathway-dependent effects, such as different growth rates for droplets and crystals in a two-step mechanism, to predict the final particle size distribution [84].
Outcome: This multi-scale approach provides an atomistic picture of the nucleation process and quantitatively links the chosen nucleation pathway to control over particle characteristics like size and polymorphism.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions and Materials
| Item | Function/Application | Example Use Case |
|---|---|---|
| Technobis Crystalline | Automated reactor system for automated crystallization data collection with in-situ imaging. | Standardized determination of nucleation and growth kinetics for APIs and inorganic salts [86]. |
| In-situ Microscope / FBRM Probe | Real-time monitoring of crystal count and size distribution in a slurry. | Tracking crystal nucleation and growth dynamics without stopping the process [86]. |
| Machine-Learning Interaction Potentials (MLIP) | High-accuracy force fields for molecular dynamics simulations, including long-range interactions. | Atomistic study of nucleation pathways and polymorph competition in nanomaterials like ZnO [9]. |
| In-situ pH Microsensor | Monitoring local pH changes at an electrode surface during electrochemical deposition. | Real-time tracking of OH⁻ generation for studying the nucleation kinetics of metal hydroxides (e.g., Mg(OH)₂) [85]. |
| Population Balance Model (PBM) | Mathematical framework to describe the evolution of a particle population over time. | Predicting crystal size distribution by modeling nucleation, growth, and other rate processes [84]. |
The comparative analysis of nucleation kinetics and growth rates underscores a paradigm shift in materials research, from a purely classical view to one that embraces the complexity and opportunity of non-classical pathways. The quantitative data and standardized protocols presented herein provide researchers with a robust framework for analyzing these processes. Understanding that nucleation can proceed through mechanisms like two-step pathways or cluster coalescence provides new levers for material design—enabling control over polymorphic selection, crystal size distribution, and morphology. Integrating advanced experimental techniques with multi-scale computational models is the path forward. This synergistic approach will ultimately empower scientists to move from empirical optimization to predictive design of crystalline materials, accelerating innovation in pharmaceuticals, advanced inorganic materials, and beyond.
The pursuit of enhanced catalytic performance has driven research into precisely controlling material properties at the nanoscale. This whitepaper examines the profound influence of morphology and defects on the efficacy of inorganic catalysts, framed within the emerging paradigm of non-classical nucleation pathways. Modern synthesis approaches increasingly exploit these pathways, which involve intermediate phases like dense liquid states and nanoparticle attachments, to deliberately craft catalysts with specific surface architectures and defect populations. A growing body of evidence confirms that parameters such as facet expression, oxygen vacancy concentration, and structural ordering are not merely incidental but are critical design parameters that directly govern activity, selectivity, and stability. This guide synthesizes current research to provide a technical framework for understanding these relationships, detailing relevant characterization methodologies, and presenting quantitative performance data, with the ultimate goal of empowering the rational design of next-generation catalytic materials.
Classical nucleation theory (CNT) posits a direct, monomer-by-monomer pathway from a disordered solution to a stable crystalline phase. However, this framework is often insufficient to describe the complex crystallization behaviors observed in many functional inorganic materials. Non-classical nucleation pathways, which include processes like two-step nucleation through metastable intermediate phases and oriented attachment of pre-formed nanoparticles, offer a powerful alternative for material design [4] [3].
These pathways provide unique opportunities to control the final product's morphology and defect landscape. For instance, the crystallization of sodium halides like NaBr and NaI has been shown to proceed through a liquid crystal intermediate phase composed of contact ion pairs, a deviation from the classical pathway followed by NaCl [48]. Similarly, in binary colloidal systems—a model for atomic crystallization—the formation of ionic crystals occurs via the condensation of metastable amorphous blobs, within which nucleation begins [3]. The growth then proceeds through a combination of monomer addition, blob absorption, and oriented attachment. Understanding and controlling these mechanisms allows researchers to deliberately introduce specific crystal defects and morphological features that serve as active sites, thereby directly tailoring the catalytic properties of the resulting material.
The Interplay of Morphology, Defects, and Performance
The Critical Role of Morphology and Facet Engineering
The external morphology of a catalyst nanoparticle determines the specific crystallographic facets exposed at the surface. Different facets possess distinct atomic arrangements and coordination, which directly influence their surface energy and reactivity.
A prime example is found in manganese-ceria (MnCeOx) solid solutions. Studies show that nanorods, which preferentially expose the (111) facets of ceria, consistently outperform nanocubes, which expose (100) facets, in the catalytic reduction of NO with CO [87]. This performance enhancement is directly linked to the higher population density of oxygen vacancies that can be formed on the (111) facets. The morphology thus dictates the type of facet exposed, which in turn determines the density and nature of defects that can be engineered, ultimately governing catalytic activity. This demonstrates a direct morphology-facet-defect-performance relationship.
Defects as Active Sites
Defects are deviations from the perfect crystal lattice and are categorized as either point defects or extended defects [88]. In catalysis, these sites are crucial because they break the periodicity of the lattice, creating localized regions with unsaturated coordination and altered electronic structures. These regions can facilitate the adsorption and activation of reactant molecules.
- Point Defects: These include vacancies (missing atoms), interstitial defects (atoms in non-lattice sites), and substitutional defects (foreign atoms replacing host atoms) [88]. Oxygen vacancies in metal oxides are a quintessential example, often serving as the primary active site for reactions involving oxygen transfer.
- Extended Defects: These include dislocations (line defects) and grain boundaries (interfaces between crystallites) [88]. These extended defects can create strain fields and unique electronic environments that enhance catalytic activity.
The intentional introduction of these defects, known as defect engineering, is a key strategy for developing advanced catalysts [88]. For instance, doping ceria with manganese (Mn) facilitates the formation of surface oxygen vacancies via a pathway described as "Mn—O_L(V_Ö)—Mn—O_L(V_Ö)—Ce" connectivities, where O_L is a lattice oxygen and V_Ö is an oxygen vacancy. This process enhances the reduction of NO according to the steps: CO* + O_L → CO_2* + V_Ö and NO* + V_Ö → N* + O_L [87].
Table 1: Defect Types and Their Catalytic Roles
| Defect Type | Description | Catalytic Role | Example |
|---|---|---|---|
| Vacancy Defect | A missing atom from its lattice site. | Creates unsaturated sites for strong reactant adsorption; alters local electronics. | Oxygen vacancies in MnCeOx for NO reduction [87]. |
| Substitutional Defect | One atom replaced by a different element. | Introduces new electronic states; can facilitate defect formation. | Mn dopants in ceria promoting oxygen vacancy formation [87]. |
| Interstitial Defect | An atom occupies a site between lattice points. | Can create strain and modify diffusion pathways. | -- |
| Grain Boundary | Region where two crystal grains meet. | Acts as a high-energy surface with enhanced reactivity; can impede sintering. | -- |
Quantitative Performance Comparisons
The impact of morphology and defect engineering is quantifiable. Research on MnCeOx solid solutions provides clear data on how morphology-driven defect populations translate to catalytic performance.
Table 2: Quantitative Performance of MnCeOx Catalysts with Different Morphologies [87]
| Catalyst Morphology | Preferentially Exposed Facet | Relative Population of Oxygen Vacancies | Catalytic Performance (NO Reduction with CO) |
|---|---|---|---|
| MnCeOx Nanorods | (111) | Higher | Outperforms nanocube counterpart |
| MnCeOx Nanocubes | (100) | Lower | Lower performance |
The data in Table 2 underscores that the superior catalytic performance of the nanorod morphology is a direct consequence of its higher capacity for generating oxygen vacancies, a key active site for the reaction.
Experimental Methodologies
Synthesis Protocols for Tailored Morphologies and Defects
Controlling nucleation and growth is fundamental to achieving desired morphologies and defect structures.
1. Synthesis of MnCeOx Solid Solutions with Controlled Morphology [87]
- Objective: To prepare MnCeOx nanorods (exposing (111) facets) and nanocubes (exposing (100) facets) to investigate morphology effects on oxygen vacancy formation and catalytic performance in NO reduction by CO.
- Protocol:
- Nanocube Synthesis: Typically involves a hydrothermal method using cerium precursors in a strong alkaline environment, which favors the growth of the (100) facet.
- Nanorod Synthesis: Also employs hydrothermal methods, but with specific structure-directing agents or mineralizers that promote anisotropic growth along certain crystal axes, leading to rod-like morphologies that expose (111) facets.
- Doping: Manganese is incorporated into the ceria lattice during the synthesis via co-precipitation or impregnation, followed by calcination to form a solid solution.
2. Microdroplet Crystallization for Studying Non-Classical Pathways [48]
- Objective: To investigate the crystallization pathways of sodium halides (NaCl, NaBr, NaI) under homogeneous nucleation conditions.
- Protocol:
- Microdroplets of the salt solution are created and observed under controlled evaporation.
- The crystallization process is monitored using optical and computational analyses to detect the formation of any intermediate phases.
- This method revealed that NaBr and NaI form a liquid crystal intermediate phase (a non-classical pathway), while NaCl follows a classical pathway.
3. Continuous Dialysis for Binary Colloidal Crystals [3]
- Objective: To achieve fine spatiotemporal control over interaction strength during the crystallization of binary colloidal crystals.
- Protocol:
- A mixture of oppositely charged colloidal particles is placed in an observation cell connected to a deionized water reservoir.
- As salt diffuses out of the sample into the reservoir, the Debye length (λD) increases, gradually strengthening the electrostatic attraction between particles.
- This slow, controlled change in interaction strength allows for the direct observation of non-classical pathways, such as the formation of amorphous blobs that later crystallize, and helps identify the precise conditions for classical vs. non-classical crystallization.
Essential Research Reagent Solutions
The following table details key materials and their functions in experiments related to morphology and defect-controlled catalysis.
Table 3: Research Reagent Solutions for Catalytic Material Synthesis
| Reagent/Material | Function in Experiment |
|---|---|
| Cerium (Ce) Precursors (e.g., Cerium nitrate) | Primary metal source for constructing the ceria (CeO₂) support lattice. |
| Manganese (Mn) Precursors (e.g., Manganese nitrate) | Dopant source to create substitutional defects and facilitate oxygen vacancy formation in CeO₂. |
| Structure-Directing Agents | Chemicals (e.g., specific surfactants, amines) used to control the growth of specific crystal facets and achieve desired morphologies like nanorods or nanocubes. |
| Oppositely Charged Colloidal Particles | Model "ions" for studying fundamental nucleation and growth pathways in binary ionic crystal systems [3]. |
| Salt Solutions (e.g., NaCl, NaI) | Used to control the ionic strength and Debye screening length (λD) in colloidal systems, thereby tuning particle interaction potentials [3]. |
Characterization and Data Analysis Techniques
Advanced Characterization of Defects
A multi-technique approach is essential to fully characterize the defect structures and morphologies of engineered catalysts.
- Microscopy: Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM) visualize crystal structure at the atomic scale and surface morphology, identifying defects like dislocations and grain boundaries [88].
- Spectroscopy: X-ray Photoelectron Spectroscopy (XPS) detects chemical defects like vacancies and impurities. Raman Spectroscopy is highly sensitive to defects such as oxygen vacancies in oxides. X-ray Absorption Spectroscopy (XAS) probes the local structure and electronic properties of metal centers [88] [87].
- In Situ/Operando Characterization: Techniques like in situ XRD, XAS, and Raman are used to track the dynamic evolution of defects under actual reaction conditions, providing insights into the active state of the catalyst [88].
Presenting Quantitative Comparisons
When presenting quantitative data comparing catalytic performance between different groups (e.g., morphologies), summary tables and comparative graphs are essential.
- Summary Tables: Should include the sample size (n), mean, median, standard deviation, and interquartile range (IQR) for each group. When comparing two groups, the difference between their means should be calculated [89].
- Comparative Graphs: Boxplots are highly effective for comparing the distribution of a quantitative variable (e.g., reaction rate) across different categories (e.g., catalyst type). They visually display the median, quartiles, and potential outliers, facilitating a quick comparison of central tendency and variability [89].
Visualization of Concepts and Workflows
Relationship Between Synthesis and Properties
The following diagram illustrates the logical pathway from synthesis strategy to final catalytic performance, emphasizing the role of non-classical pathways.
Defect Characterization Workflow
This workflow outlines the key techniques used to identify and analyze defects in catalytic materials.
The deliberate engineering of morphology and defects represents a cornerstone of modern catalyst design. By moving beyond classical crystallization models and leveraging non-classical nucleation pathways, researchers can precisely control the formation of active sites at the atomic level. The evidence is clear: the morphology of a catalyst particle dictates the available surface facets, which in turn influences the type and density of defects that can be stabilized. These defects, particularly oxygen vacancies and substitutional dopants, are often the very centers responsible for catalytic activity, as quantitatively demonstrated in systems like MnCeOx.
Future research will likely focus on several key areas:
- Advanced Synthesis: Developing more precise methods to control defect type, density, and location during synthesis.
- Dynamic Observation: Expanding the use of in situ and operando characterization to understand how defects evolve and function under real operating conditions [88].
- Multi-scale Modeling: Integrating computational materials science with experimental work to predict new catalyst compositions and structures with desired properties.
- Exploration of New Materials: Applying these principles to emerging material classes, such as high-entropy intermetallics (HEIs), which combine multimetallic composition with ordered structures, offering unprecedented opportunities for tuning surface environments and catalytic performance [90].
The integration of non-classical crystallization concepts with sophisticated characterization and defect engineering provides a robust framework for designing the high-performance catalysts needed for sustainable energy and chemical processes.
{# The Rheological and Thermodynamic Signatures of Non-Classical Nucleation Mechanisms
Non-classical nucleation mechanisms, which diverge from the established framework of Classical Nucleation Theory (CNT), are increasingly recognized as fundamental pathways in the crystallization of organic, inorganic, and soft materials. This whitepaper delineates the defining rheological and thermodynamic signatures of these pathways, including stable prenucleation clusters, liquid-liquid phase separation, and particle-based assembly. Within the context of organic materials research, particularly for pharmaceuticals, understanding these signatures is paramount for exerting precise control over crystallization outcomes, enabling the design of novel materials with tailored properties and the suppression of pathological crystallization.
Classical Nucleation Theory (CNT) has long provided the foundational framework for understanding the initial formation of a new phase. CNT posits that nucleation is a single-step process where monomers (atoms, ions, or molecules) stochastically assemble to form a critical nucleus that possesses the bulk crystal structure; its stability is governed by a balance between volume free energy gain and surface free energy cost [11]. However, a paradigm shift has occurred over recent decades, with Vekilov (2020) noting that "two-step nucleation is by now ubiquitous and registered cases of classical nucleation are celebrated" [14].
Non-classical nucleation encompasses mechanisms that deviate from CNT's core assumptions. These pathways often involve stable, multi-ion prenucleation clusters (PNCs) that do not have a phase interface, dense liquid precursor phases formed via liquid-liquid phase separation (LLPS), or the assembly of nanoparticles [11] [14]. For researchers working with organic materials, such as active pharmaceutical ingredients (APIs), these pathways are not merely academic curiosities. They directly influence critical material properties, including polymorphism, crystal habit, purity, and bioavailability [28]. This whitepaper synthesizes current evidence to provide a technical guide for identifying and leveraging the unique signatures of non-classical nucleation.
Thermodynamic Signatures of Non-Classical Pathways
The thermodynamics of non-classical nucleation are distinctly different from those described by CNT. These deviations provide the first key signatures for identifying such mechanisms.
The Preamble of Prenucleation Clusters
A key non-classical concept involves stable Prenucleation Clusters (PNCs). These are solute species with "molecular" character in solution that exist before the nucleation of a new, solid phase and are not considered to have a phase interface [11]. Their existence challenges CNT, which assumes that small clusters are thermodynamically unstable and dissolve rapidly.
Experimental evidence for PNCs often comes from potentiometric titrations combined with analytical techniques. For instance, in calcium carbonate systems, the difference between the total concentration of titrated Ca²⁺ and the concentration of free Ca²⁺ measured by an ion-selective electrode defines the concentration of "bound" Ca²⁺. Studies have consistently shown that a significant fraction (65–75%) of calcium is bound in solution before any solid nucleation occurs [91]. Isothermal Titration Calorimetry (ITC) investigations reveal that PNC formation is an endothermic process, indicating that the driving force is entropic, likely due to the release of water molecules upon ion association [11].
Metastable Liquid Precursors and Lowered Energy Barriers
Liquid-Liquid Phase Separation (LLPS) into a dense liquid phase (DLP) is another widespread non-classical pathway. This process acts as a precursor to crystallization, profoundly altering the nucleation landscape.
The thermodynamic signature of this pathway is a dramatically lowered nucleation barrier. In classical nucleation, the formation of a crystal nucleus directly from a dilute solution incurs a high energy cost due to the creation of a solid-solution interface. In a two-step mechanism, nucleation occurs within a dense liquid droplet where the solute concentration is already near-crystalline densities. The effective surface energy (γ) for a crystal nucleus forming within this dense liquid environment is significantly lower—potentially by an order of magnitude—than for a nucleus forming in the bulk solution. Since the nucleation barrier in CNT scales with γ³, this translates to a reduction of the barrier by about 1000-fold, making nucleation feasible at conditions where CNT would predict it to be imperceptibly slow [52].
Table 1: Thermodynamic Signatures of Classical vs. Non-Classical Nucleation
| Feature | Classical Nucleation Theory | Non-Classical Nucleation |
|---|---|---|
| Primary Units | Atoms, ions, molecules | Prenucleation clusters, nanoparticles, dense liquid droplets |
| Cluster Stability | Pre-critical clusters are unstable and transient | Stable Prenucleation Clusters (PNCs) can exist in undersaturated/ saturated solutions |
| Nucleation Pathway | Single-step: solution → critical crystal nucleus | Multi-step (e.g., solution → PNCs / DLP → crystal nucleus) |
| Interfacial Energy | High, based on macroscopic crystal-solution interface | Lowered for nucleation within a dense liquid precursor |
| Energetic Driving Force | Predominantly enthalpic (bulk energy gain) | Can be entropic for PNC formation (e.g., dehydration) |
Rheological and Kinetic Signatures
The presence of metastable precursors and multi-step pathways imparts unique rheological and kinetic behaviors that serve as practical observational signatures.
Coalescence and Liquid-like Behavior
A direct rheological signature of a dense liquid precursor is its liquid-like character, evidenced by the coalescence of droplets. This has been observed in diverse systems:
- In calcium carbonate, cryo-TEM and LP-TEM show emulsion-like structures that coalesce over time [14].
- In the early nucleation stage of monolayer tungsten disulfide (WS₂), in-situ monitoring reveals that particles move on the substrate and increase crystal size through collision and coalescence [6].
- During the formation of cerium oxalate, LP-TEM has directly captured the coalescence of droplet precursors [14].
This behavior is a definitive marker of a liquid state and is incompatible with classical models of atomic attachment.
Anomalous Nucleation Kinetics and Critical Sizes
Non-classical pathways can lead to nucleation kinetics that deviate from CNT predictions. A key signature is the observation of very large critical nucleus sizes. During the VLS growth of WS₂, the observed critical nucleus size was found to be as large as 38.7 µm, which is orders of magnitude larger than the CNT-predicted value of ~1.6 nm for a similar system [6]. This macroscopic critical size is a hallmark of a non-classical, two-step nucleation process where the nucleus forms within a much larger metastable cluster.
Furthermore, the nucleation process often exhibits a distinct slow-to-rapid growth transition. An initial incubation period is followed by a sudden acceleration in growth, which aligns with the time-temperature transformation diagram for metastable cluster formation [6].
Table 2: Experimentally Observed Signatures in Various Material Systems
| Material System | Observed Non-Classical Signature | Experimental Technique |
|---|---|---|
| Flufenamic Acid (Pharmaceutical) | Formation of pre-crystalline stages and dense liquid phases preceding solidification. | Liquid Phase Electron Microscopy (LPEM) [28] |
| β-Hematin (Malaria Pigment) | Nucleation is hosted within mesoscopic solute-rich clusters; can be suppressed or enhanced by modifiers. | Dynamic Light Scattering (DLS), Molecular Simulations [52] |
| Iron (BCC phase in FCC) | Nucleation via coalescence of subcritical clusters and stepwise nucleation, circumventing classical energy barriers. | Molecular Dynamics (MD) Simulations [1] |
| Calcium Silicate Hydrate (C-S-H) | Homogeneous nucleation is a two-step process: discrete globules (precursors) transform into foil-like C-S-H. | TEM, XRD, FT-IR, ²⁹Si NMR [5] |
Experimental Protocols for Detection
Identifying non-classical mechanisms requires a toolkit of in-situ and real-time characterization methods.
In-Situ Monitoring of Chemical Vapor Deposition (CVD)
Objective: To directly observe the early nucleation dynamics of 2D materials, such as transition metal dichalcogenides (TMDs). Methodology:
- A custom CVD system is equipped with optical imaging capabilities to capture the substrate during growth in real-time.
- Images are captured at a high frame rate (e.g., 1 frame per second).
- Automated image analysis is employed, using thresholds for HSV (Hue, Saturation, Value) color indices to independently extract regions corresponding to monolayer and multilayer growth from the optical images.
- The area of the monolayer is plotted as a function of growth time to extract physical parameters like incubation time (Δt) and growth speed (v_g) [6]. Key Insight: This protocol enabled the direct observation of liquid-phase precursor movement and coalescence, leading to the identification of a two-step nucleation mechanism in WS₂ [6].
Liquid Phase Electron Microscopy (LPEM) for Organic Pharmaceuticals
Objective: To visualize the nanoscale early-stage nucleation events of beam-sensitive organic molecules in their native solvent environment. Methodology:
- A solution of the organic molecule (e.g., Flufenamic Acid, FFA) is prepared in a suitable solvent (e.g., ethanol).
- The solution is loaded into a liquid cell holder with silicon nitride windows for TEM.
- The electron beam is used both to image the process and to induce nucleation through controlled radiolysis of the solvent.
- Imaging is performed at high temporal resolution, often using a high electron dose (>150 e⁻/Ų/s) to ensure nucleation occurs within the observation window.
- The resulting video data is analyzed for the appearance and evolution of pre-crystalline features, such as dense liquid phases and their transformation into solids [28]. Key Insight: This protocol provided the first direct observations of a PNC pathway followed by features exhibiting two-step nucleation for a small organic pharmaceutical crystal [28].
The Scientist's Toolkit: Essential Reagents and Materials
Controlling non-classical nucleation often requires specific additives or modifiers that interact with the precursors.
Table 3: Key Research Reagent Solutions for Modulating Non-Classical Nucleation
| Reagent / Modifier | Function in Non-Classical Nucleation | Example System |
|---|---|---|
| Polymeric Additives (e.g., PEG, PAA) | Induce or stabilize polymer-induced liquid precursors (PILPs), enabling non-classical growth of complex morphologies. | Calcium Carbonate [14] [11] |
| Antimalarial Compounds (e.g., Pyronaridine) | Suppress crystal nucleation by interacting with and destabilizing mesoscopic solute-rich clusters that host nucleation. | β-Hematin [52] |
| Alkali Metal Salts | Act as catalysts in salt-assisted growth; lower melting/boiling points of source powders, promoting vapor-liquid-solid (VLS) growth over vapor-solid (VS) growth. | Transition Metal Dichalcogenides (WS₂, MoS₂) [6] |
| Solvent Mixtures (e.g., Octanol with Aqueous Buffer) | Mimic the composition of in vivo environments (e.g., lipid nanospheres in malaria parasites) to study biologically relevant nucleation pathways. | β-Hematin [52] |
Visualization of Experimental Workflows
Integrating the various experimental signatures and methods into a coherent workflow is essential for rigorous analysis.
The recognition of non-classical nucleation mechanisms, identifiable through their distinct rheological and thermodynamic signatures, represents a fundamental advancement in materials science. For researchers focused on organic materials, this paradigm offers powerful levers for control. The ability to manipulate nucleation by targeting precursor clusters or liquid phases—for instance, to suppress pathological crystallization as demonstrated with β-hematin [52] or to access desired polymorphs in pharmaceuticals as explored with flufenamic acid [28]—shifts the process from a statistical phenomenon to a more designable one.
Future research will hinge on the continued development of integrated in-situ methodologies, like LPEM and advanced simulation, to capture these transient events with greater fidelity across a wider range of organic systems. The ultimate challenge and opportunity lie in moving from observation to prediction and precise control, enabling the rational design of crystalline organic materials with pre-defined properties and functions.}
Validation through Bayesian Inference and Quantitative Phase Field Simulation
The pursuit of advanced organic materials, particularly in pharmaceutical development, is increasingly focused on controlling crystallization pathways and resulting polymorphs. Non-classical nucleation pathways, which proceed through intermediate states rather than direct monomer-by-monomer addition, present both challenges and opportunities for controlling material properties [92] [7]. This technical guide establishes an integrated framework combining Bayesian inference for uncertainty quantification and quantitative phase field simulation for microstructure prediction. This synergistic approach provides researchers with a robust methodology for validating hypotheses about complex nucleation mechanisms in organic systems, enabling more reliable prediction and control of crystalline forms essential for drug development.
Theoretical Foundation: Non-Classical Nucleation Pathways
Traditional Classical Nucleation Theory (CNT) provides a phenomenological description based on overcoming a single free energy barrier from a disordered fluid to an ordered crystal [92]. However, this framework often proves insufficient for describing complex crystallization processes in organic and pharmaceutical systems. Non-classical nucleation pathways involve multiple steps separated by several free energy barriers, frequently proceeding through amorphous intermediates or stable pre-nucleation clusters [92] [7].
Recent molecular dynamics simulations reveal that crystalline seeds can fundamentally reshape nucleation mechanisms, potentially converting non-classical pathways with amorphous intermediates into classical, monomer-by-monomer crystallization [7]. The competition between these pathways is governed by the interplay between thermodynamic stability and kinetic favorability of intermediate interfacial polymorphs [7]. Markov State Models applied to heterogeneous ice nucleation have demonstrated that both classical one-step and non-classical two-step nucleation pathways can coexist with comparable fluxes at specific temperatures (e.g., 230 K) [92]. In these systems, disordered mixing of rhombic and hexagonal ice creates favorable configurational entropy that stabilizes critical nuclei in non-classical pathways [92].
Table 1: Key Characteristics of Classical vs. Non-Classical Nucleation Pathways
| Characteristic | Classical Nucleation | Non-Classical Nucleation |
|---|---|---|
| Pathway Steps | Single-step [92] | Multi-step (2+ barriers) [92] |
| Intermediate States | None | Amorphous precursors, pre-nucleation clusters [7] |
| Free Energy Landscape | Single barrier [92] | Multiple barriers [92] |
| Stabilizing Factors | Favorable energetics [92] | Configurational entropy [92] |
| Impact of Seeds | Promotes monomer addition [7] | Can bypass amorphous intermediates [7] |
| Temperature Dependence | Preferred at higher temperatures [92] | Competitive at lower temperatures [92] |
Bayesian Inference for Quantitative Validation
Theoretical Framework
Bayesian methods provide a probabilistic framework for quantifying uncertainty in materials modeling, particularly valuable when dealing with limited experimental data. The core principle involves updating prior beliefs about model parameters (θ) based on observed data (D) to obtain a posterior distribution using Bayes' theorem:
[ P(\theta|D) = \frac{P(D|\theta)P(\theta)}{P(D)} ]
where ( P(\theta|D) ) is the posterior distribution, ( P(D|\theta) ) is the likelihood, ( P(\theta) ) is the prior distribution, and ( P(D) ) is the model evidence [93]. This approach naturally handles both aleatoric uncertainties (inherent stochasticity in data generation) and epistemic uncertainties (from limited data and model imperfections) [93].
Implementation Methods
For materials modeling applications, several Bayesian approaches have been developed:
Bayesian Neural Networks (BNNs) learn probability distributions over network weights rather than point estimates, providing natural uncertainty quantification [93]. Gaussian Processes (GPs) offer a non-parametric Bayesian framework for building surrogate models, with built-in uncertainty estimates [94]. For problems involving both qualitative and quantitative variables, the Latent-Variable Gaussian Process (LVGP) approach maps qualitative factors to underlying numerical latent variables, enabling effective modeling of mixed-variable materials design problems [94].
Table 2: Bayesian Methods for Materials Modeling Validation
| Method | Key Features | Best-Suited Applications | Uncertainty Types Quantified |
|---|---|---|---|
| Bayesian Neural Networks (BNNs) | Probability distributions over weights [93] | Surrogate modeling, structure-property linkages [93] | Aleatoric, Epistemic [93] |
| Gaussian Processes (GPs) | Non-parametric, kernel-based [94] | Expensive simulation surrogates, small datasets [94] | Predicted variance [94] |
| Latent-Variable GP (LVGP) | Maps qualitative factors to latent space [94] | Mixed-variable problems, materials selection [94] | Interpolation uncertainty [94] |
| Markov Chain Monte Carlo (MCMC) | Samples from true posterior [93] | High-accuracy inference, small networks [93] | Full posterior distribution [93] |
| Variational Inference (VI) | Approximate, scalable [93] | Large networks, computational efficiency [93] | Approximate posterior [93] |
Experimental Protocol: Bayesian Force Field Parameterization
Objective: Determine posterior distributions for partial charge parameters in biomolecular force fields using ab initio molecular dynamics (AIMD) reference data [95].
Materials & Data Requirements:
- Reference Data: AIMD trajectories of solvated molecular fragments [95]
- Quantities of Interest (QoIs): Radial distribution functions, hydrogen bond orders, ion-pair distance distributions [95]
- Prior Distribution: Truncated normal prior based on established force field ranges [95]
- Computational Tools: MD simulation software, Bayesian inference framework [95]
Methodology:
- AIMD Reference Generation: Perform ab initio MD simulations of solvated molecular fragments to generate reference structural data [95]
- Surrogate Model Training: Train Local Gaussian Process (LGP) surrogates to map partial charges to QoIs using trial force field MD simulations [95]
- Likelihood Evaluation: Compute likelihood of candidate parameter sets against AIMD reference data using LGP predictions [95]
- Posterior Sampling: Employ Markov Chain Monte Carlo to sample from posterior parameter distribution [95]
- Validation: Assess accuracy using normalized mean absolute error (NMAE) from posterior samples [95]
Key Considerations:
- Apply global constraint on total molecular charge (e.g., scaled by 0.8) [95]
- Include multiple simulation setups: aqueous solute, direct contact ion pairing, solvent-shared ion pairing [95]
- Expected performance: RDF errors <5%, hydrogen bond count deviations <10-20% [95]
Quantitative Phase Field Simulation
Theoretical Background
The phase-field method is a diffuse-interface model for simulating microstructure evolution during phase transformations, particularly valuable for modeling crystallization processes on experimentally relevant length and time scales [96]. Unlike molecular dynamics simulations, which provide atomic-scale insights but limited temporal and spatial scales, phase-field modeling can directly simulate growth rates and growth modes observable in experimental systems [96].
The model describes the system using two primary field variables:
- Phase field (ϕ): Distinguishes between solid (ϕ ≈ -1) and liquid (ϕ ≈ +1) phases [96]
- Concentration field (c): Describes solute distribution [96]
The governing equations for crystal growth are [96]:
[ \frac{\partial \phi}{\partial t} = -M \frac{\delta F}{\delta \phi} ]
[ \frac{\partial c}{\partial t} = \nabla \cdot [D(\phi)\nabla c] + \nabla \cdot [D(\phi)c(1-c)\nabla \phi] ]
where M is mobility, F is free energy functional, and D(ϕ) is diffusion coefficient [96].
Anisotropy and Crystal Growth
For modeling crystalline materials with specific habits, anisotropy must be incorporated through the surface energy term [96]:
[ \epsilon(\theta) = \epsilon\sigma(\theta) ]
with the anisotropy function:
[ \sigma(\theta) = 1 + \delta\cos(j(\theta - \theta_0)) ]
where δ is anisotropy strength, j is anisotropy mode number, θ is the angle between the x-axis and surface normal, and θ₀ indicates high-symmetry direction [96].
Experimental Protocol: NaCl Crystal Growth Simulation
Objective: Simulate growth of cubic NaCl crystals in 2D to investigate concentration-dependent transition from compact to non-compact growth [96].
Simulation Parameters:
- Diffusion constant (D): 1.3 × 10⁻⁹ m²s⁻¹ [96]
- Solid density (ρc): 37.04 mol L⁻¹ (2165 kg m⁻³) [96]
- Equilibrium concentration (Ceq): 5.55 mol L⁻¹ (room temperature) [96]
- Surface energy (S): 0.100 J m⁻² along (100), 0.114 J m⁻² along (111) [96]
- Interface mobility (L): 4.11 × 10⁻³ m³ J⁻¹ s⁻¹ [96]
- Reaction rate constant (kR): 2.33 × 10⁻³ m s⁻¹ [96]
Numerical Implementation:
- Domain Setup: Initialize small crystal seeds with appropriate anisotropy [96]
- Grid Definition: Set grid size Δx < ε (interface width parameter) for accuracy [96]
- Time Stepping: Determine stable time step Δt using instability condition: Δt < (Δx)⁴/(10Dε²) [96]
- Boundary Conditions: Implement appropriate boundary conditions for concentration and phase fields [96]
- Solution Method: Solve coupled phase field and concentration equations using finite difference methods [96]
Validation Metrics:
- Growth speed as function of supersaturation [96]
- Transition from compact to non-compact growth morphology [96]
- Comparison with experimental crystal growth rates [96]
Integrated Framework for Non-Classical Nucleation Analysis
Synergistic Methodology
The integration of Bayesian inference with phase field simulation creates a powerful validation framework for investigating non-classical nucleation pathways. This combined approach enables researchers to:
- Quantify uncertainties in phase field parameters through Bayesian calibration [93]
- Validate model predictions against limited experimental data using probabilistic measures [94]
- Identify dominant nucleation pathways through Bayesian model selection [92]
- Optimize experimental conditions for targeting specific polymorphs [7]
Application to Pharmaceutical Polymorph Selection
For drug development applications, this framework can address the critical challenge of polymorph control. Using Bayesian optimization with mixed-variable modeling (LVGP), researchers can simultaneously optimize qualitative factors (solvent selection, additive types) and quantitative parameters (temperature, concentration, cooling rates) to direct nucleation through specific pathways [94]. Phase field simulations then predict the resulting microstructure and growth morphology under these optimized conditions [96].
Bayesian-Phase Field Experimental Protocol
Objective: Validate non-classical nucleation pathways in organic crystal formation using integrated Bayesian-phase field framework.
Step 1: Bayesian Experimental Design
- Define mixed-variable design space: qualitative factors (solvent type, polymer additives) and quantitative parameters (temperature, concentration) [94]
- Establish priors for phase field parameters based on literature data [96]
- Use Bayesian optimization to identify informative experimental conditions [94]
Step 2: Data Collection & Surrogate Modeling
- Perform limited experiments or molecular simulations at designed conditions [95]
- Measure key observables: nucleation rates, growth morphology, polymorph identity [7]
- Build Gaussian process surrogate models linking parameters to observables [94]
Step 3: Phase Field Model Calibration
- Implement phase field model with anisotropic crystal growth [96]
- Use Bayesian calibration to determine posterior distributions for critical parameters:
- Validate against experimental growth rates and morphologies [96]
Step 4: Pathway Analysis & Prediction
- Apply Bayesian model selection to identify dominant nucleation pathways [92]
- Predict conditions favoring specific polymorphs through non-classical pathways [7]
- Design targeted experiments to validate predictions [94]
Table 3: Research Reagent Solutions for Nucleation Experiments
| Reagent/Condition | Function in Nucleation Studies | Bayesian Treatment |
|---|---|---|
| Solvent Selection | Controls solubility, molecular mobility, and interfacial energy [7] | Qualitative variable in LVGP [94] |
| Polymer Additives | Modifies interface energy, directs polymorph selection [7] | Qualitative variable with latent representation [94] |
| Crystalline Seeds | Promotes heterogeneous nucleation, can bypass amorphous intermediates [7] | Binary qualitative factor [94] |
| Supersaturation (Ω) | Driving force for nucleation, affects pathway competition [96] | Quantitative variable with uncertainty [93] |
| Temperature | Affects kinetic rates and thermodynamic stability [92] | Quantitative variable with prior distribution [93] |
| Impurity Species | Can inhibit or promote specific pathways through surface adsorption [7] | Qualitative mixture variable [94] |
This technical guide establishes a comprehensive framework for validating non-classical nucleation pathways in organic materials through the integration of Bayesian inference and quantitative phase field simulation. The synergistic combination of these approaches enables researchers to address the fundamental challenges in pharmaceutical crystal engineering: quantifying uncertainties in model parameters, validating multiscale simulations against limited experimental data, and predicting conditions that favor specific polymorphic outcomes through controlled nucleation pathways. The provided experimental protocols and visualization frameworks offer researchers practical tools for implementing this integrated approach in drug development and functional materials design. As crystallization research increasingly focuses on pathway engineering rather than simply controlling final outcomes, this Bayesian-phase field framework provides the necessary mathematical foundation for reliable prediction and validation of non-classical nucleation mechanisms in complex organic systems.
Conclusion
The exploration of non-classical nucleation pathways reveals a complex and versatile landscape far beyond the scope of CNT. The synthesis of evidence from inorganic materials, pharmaceuticals, and soft matter confirms that mechanisms involving pre-nucleation clusters, dense liquid intermediates, and particle attachment are not mere exceptions but fundamental processes that can be harnessed. The key takeaway is that controlling the nucleation pathway directly dictates critical material properties, from catalytic activity in zeolites to bioavailability in pharmaceuticals. Future research must focus on refining in situ characterization techniques to unravel the atomic-scale details of these pathways and develop predictive models for pathway selection. For biomedical and clinical research, this promises a new era of rational design for drug polymorphs with optimized efficacy and stability, advanced biomimetic materials, and highly efficient catalytic systems for industrial biotechnology.
References
- 1. Fundamental study of nonclassical nucleation mechanisms ... [https://www.sciencedirect.com/science/article/pii/S1359645422000404]
- 2. Robustness of classical nucleation theory to chemical ... [https://arxiv.org/html/2510.01420v1]
- 3. Direct observation and control of non-classical ... [https://www.nature.com/articles/s41467-025-58959-0]
- 4. Non-Classical Crystallization in Soft and Organic Materials [https://www.pnnl.gov/publications/non-classical-crystallization-soft-and-organic-materials]
- 5. New insights into the non-classical nucleation of C-S-H [https://www.sciencedirect.com/science/article/abs/pii/S0008884623000479]
- 6. Non-classical nucleation in vapor–liquid–solid growth of ... [https://www.nature.com/articles/s41598-021-01666-9]
- 7. Crystallization Seeds Reshape Nucleation Mechanisms [https://chemrxiv.org/engage/chemrxiv/article-details/67abd90e6dde43c90874d79d]
- 8. Crystal Nucleation and Growth of Inorganic Ionic Materials from... [https://pubmed.ncbi.nlm.nih.gov/35678091/]
- 9. Competing nucleation pathways in nanocrystal formation [https://www.nature.com/articles/s41524-024-01371-x]
- 10. Recent experimental explorations of non-classical nucleation [https://pubs.rsc.org/en/content/articlelanding/2020/ce/d0ce00480d]
- 11. Review Prenucleation clusters and non-classical nucleation [https://www.sciencedirect.com/science/article/abs/pii/S1748013211001174]
- 12. Pre-nucleation clusters as solute precursors in crystallisation [https://pubs.rsc.org/en/content/articlehtml/2014/cs/c3cs60451a]
- 13. Stable Prenucleation Calcium Carbonate Clusters Define ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7187218/]
- 14. Liquid–liquid phase separation into reactant-rich precursors ... [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00695c]
- 15. Two-Step Nucleation and Amorphization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389563/]
- 16. Mechanistic insights into calcium-silicate-hydrate ... [https://www.sciencedirect.com/science/article/pii/S000888462500198X]
- 17. Crystal Nucleation in Liquids: Open Questions and Future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4919765/]
- 18. Nucleation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2995260/]
- 19. Role of clusters in nonclassical nucleation and growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3918807/]
- 20. Thermodynamic assessment of two-step nucleation ... [https://www.sciencedirect.com/science/article/pii/S089684462400127X]
- 21. Multistage nucleation pathway in LiF molten salt mirrors the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12207450/]
- 22. Considerations and Challenges in Studying Liquid ... [https://www.sciencedirect.com/science/article/pii/S0092867418316490]
- 23. From formation to functionality: Insights into mesocrystal ... [https://www.sciencedirect.com/science/article/abs/pii/S0022459625001549]
- 24. How crystals form: A theory of nucleation pathways - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6450691/]
- 25. Nucleation and phase transformation pathways in ... [https://www.sciencedirect.com/science/article/abs/pii/S1359029417301334]
- 26. Nucleation of protein mesocrystals via oriented attachment [https://pmc.ncbi.nlm.nih.gov/articles/PMC8222410/]
- 27. Mesocrystals: structural and morphogenetic aspects [https://pubs.rsc.org/en/content/articlehtml/2016/cs/c6cs00208k]
- 28. Non-classical crystallisation pathway directly observed for ... [https://www.nature.com/articles/s41598-020-75937-2]
- 29. Visualizing macromolecular complexes with in situ liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9979698/]
- 30. 2026 Liquid Phase Electron Microscopy Conference GRC [https://www.grc.org/liquid-phase-electron-microscopy-conference/2026/]
- 31. DENSsolutions' Post [https://www.linkedin.com/posts/denssolutions_mm25-mm2025-insitu-activity-7356110683819630592-LZhf]
- 32. CryoTEM as an Advanced Analytical Tool for Materials ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5518272/]
- 33. Unravelling complex mechanisms in materials processes with ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc05188b]
- 34. Dynamic light scattering and transmission electron microscopy ... [https://pubs.rsc.org/en/content/articlehtml/2023/mh/d3mh00717k]
- 35. A nonclassical pathway of β-hematin crystal nucleation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12350662/]
- 36. insights into their contributions and impact on the final ... [https://pubs.rsc.org/en/content/articlehtml/2025/qi/d5qi00224a]
- 37. Synthesis of heteroatom zeolite subcrystals as catalytic ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979725013001]
- 38. Impact of a polymer modifier on directing the non-classical ... [https://pubs.rsc.org/en/content/articlelanding/2022/sc/d2sc04544c]
- 39. Synthesis and crystallization mechanism of EUO zeolite [https://www.sciencedirect.com/science/article/abs/pii/S1387181122002293]
- 40. Crystallization Seeds Reshape Nucleation Mechanisms [https://chemrxiv.org/engage/chemrxiv/article-details/680ba520927d1c2e66c12d42]
- 41. Crystallization Seeds Reshape Nucleation Mechanisms [https://pubmed.ncbi.nlm.nih.gov/40472260/]
- 42. Morphology Regulation of Zeolite MWW via Classical ... [https://www.mdpi.com/1420-3049/29/1/170]
- 43. Monitoring Polymorphic Phase Transitions in Flufenamic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9097533/]
- 44. a case study of flufenamic acid [https://pubs.rsc.org/en/content/articlelanding/2018/ce/c7ce01984j]
- 45. Non-classical crystallisation pathway directly observed for ... [https://pubmed.ncbi.nlm.nih.gov/33154480/]
- 46. Crystal quality and physical reactivity in the case of ... [https://pubmed.ncbi.nlm.nih.gov/20589945/]
- 47. Crystal Quality and Physical Reactivity in the Case of ... [https://www.sciencedirect.com/science/article/abs/pii/S0022354915324874]
- 48. Discovery of a liquid crystal phase of sodium halides via ... [https://www.sciencedirect.com/science/article/pii/S2772834X25000727]
- 49. Alkali chalcogenides-assisted vapor–liquid–solid growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12423382/]
- 50. Universal Precise Growth of 2D Transition-Metal ... [https://pubmed.ncbi.nlm.nih.gov/32648731/]
- 51. Synthesis of 2D transition metal dichalcogenides by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6160381/]
- 52. A nonclassical pathway of β-hematin crystal nucleation ... [https://www.nature.com/articles/s42004-025-01612-0]
- 53. Direct observation and control of non-classical crystallization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12003862/]
- 54. Effect of the ethanol-to-water ratio on the properties of silica ... [https://www.sciencedirect.com/science/article/pii/S0169433223013958]
- 55. Crystal polymorphism [https://en.wikipedia.org/wiki/Crystal_polymorphism]
- 56. The Polymorphism of Drugs: New Approaches to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7022426/]
- 57. Recent advances in drug polymorphs: Aspects of ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517321011261]
- 58. Polymorphism: A Major Risk That Pharma Companies Must ... [https://www.ccdc.cam.ac.uk/discover/blog/polymorphism-a-major-risk-that-pharma-companies-must-mitigate/]
- 59. Isolation of paracetamol forms III and II [https://www.sciencedirect.com/science/article/abs/pii/S0928098707001017]
- 60. Challenging the Ostwald rule of stages in mechanochemical ... [https://pubs.rsc.org/en/content/articlelanding/2020/sc/d0sc03629c]
- 61. Challenging the Ostwald rule of stages in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8162427/]
- 62. Ostwald's rule of stages governs structural transitions and ... [https://www.nature.com/articles/ncomms6219]
- 63. - Non crystallization in soft and... | Nature Reviews classical Materials [https://www.nature.com/articles/s41578-023-00637-y?error=cookies_not_supported&code=6f865b46-6d8f-4224-9724-99c5a35aea61]
- 64. An open experimental database for exploring inorganic ... [https://www.nature.com/articles/sdata201853]
- 65. Automated Data Analysis | Materials Science [https://www.nrel.gov/materials-science/automated-data-analysis]
- 66. [2510.22944] Is Your Prompt Poisoning Code? Defect Induction Rates... [https://arxiv.org/abs/2510.22944]
- 67. Defect formation and prevention in directed energy ... [https://www.sciencedirect.com/science/article/abs/pii/S0921509319314741]
- 68. Frontiers | A review of defect for UMG-Si wafers mitigation strategies [https://www.frontiersin.org/journals/photonics/articles/10.3389/fphot.2023.1331471/full]
- 69. Defect Formation In VAM And Prevention Strategies [https://eureka.patsnap.com/report-defect-formation-in-vam-and-prevention-strategies]
- 70. An unexpected impurity in my drug product, what now? [https://www.nelsonlabs.com/an-unexpected-impurity-in-my-drug-product-what-now/]
- 71. Safety Considerations of Impurities in Pharmaceutical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3909164/]
- 72. Semiconductor Manufacturing: Best Defect Reduction ... [https://future-bridge.us/semiconductor-manufacturing-best-defect-reduction-strategies/]
- 73. Metastability [https://en.wikipedia.org/wiki/Metastability]
- 74. Light-directed trapping of metastable intermediates in a self ... [https://www.nature.com/articles/s41467-020-20172-6]
- 75. Establishing baselines for generative discovery of inorganic ... [https://pubs.rsc.org/en/content/articlehtml/2025/mh/d5mh00010f]
- 76. A generative model for inorganic materials design [https://www.nature.com/articles/s41586-025-08628-5]
- 77. Accelerated Inorganic Materials Design with Generative AI ... [https://arxiv.org/abs/2504.00741]
- 78. Review Main group materials syntheses at low temperatures [https://www.sciencedirect.com/science/article/abs/pii/S2589597425002072]
- 79. Zeolite Synthesis | Mark E. Davis Research Group [https://markdavisgroup.wordpress.com/zeolite-synthesis/]
- 80. Morphology Regulation of Zeolite MWW via Classical/ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10780621/]
- 81. Impact of a polymer modifier on directing the non-classical ... [https://pubs.rsc.org/en/content/articlehtml/2022/sc/d2sc04544c]
- 82. Synthesis of ZSM-23 zeolite with dual structure directing ... [https://www.sciencedirect.com/science/article/abs/pii/S138718111830218X]
- 83. Nucleation rate and Gibbs free energy of nucleation of APIs ... [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00467e]
- 84. Numerical simulation of particle formation processes via a ... [https://www.sciencedirect.com/science/article/pii/S2590123025012034]
- 85. Understanding the Mechanism of Nucleation and Growth ... [https://www.sciencedirect.com/science/article/abs/pii/S0927775725022976]
- 86. Extension to Inorganic Salts [https://www.sciencedirect.com/science/article/pii/S0263876225005064]
- 87. Morphology effects in MnCeOx solid solution-catalyzed NO ... [https://link.springer.com/article/10.1007/s12274-023-5407-6]
- 88. Defect Engineering of Nanomaterials for Catalysis [https://www.mdpi.com/2079-4991/13/6/1116]
- 89. 14 Comparing quantitative data between individuals [https://bookdown.org/pkaldunn/SRM-Textbook/BetweenQuantData.html]
- 90. High-entropy intermetallics: emerging inorganic materials for ... [https://pubs.rsc.org/en/content/articlehtml/2024/sc/d3sc03897a]
- 91. A classical view on nonclassical nucleation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5617248/]
- 92. Temperature-dependent kinetic pathways of heterogeneous ice... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8367957/]
- 93. Bayesian neural networks for uncertainty quantification in ... [https://www.sciencedirect.com/science/article/abs/pii/S0045782521004102]
- 94. Bayesian Optimization for Materials Design with Mixed ... [https://www.nature.com/articles/s41598-020-60652-9]
- 95. Bayesian learning for accurate and robust biomolecular ... [https://arxiv.org/html/2511.05398v1]
- 96. Phase field modelling of crystal growth of NaCl in two ... [https://pubs.rsc.org/en/content/articlehtml/2023/ce/d2ce01640k]