Beyond Classical Theory: Nonclassical Nucleation Mechanisms in Inorganic and Biomimetic Crystals
This article provides a comprehensive analysis of classical and nonclassical nucleation theories, with a specific focus on their implications for inorganic and biomimetic crystals in pharmaceutical and materials science.
Beyond Classical Theory: Nonclassical Nucleation Mechanisms in Inorganic and Biomimetic Crystals
Abstract
This article provides a comprehensive analysis of classical and nonclassical nucleation theories, with a specific focus on their implications for inorganic and biomimetic crystals in pharmaceutical and materials science. It explores the fundamental limitations of Classical Nucleation Theory (CNT) and details the experimental evidence for alternative pathways like two-step nucleation and pre-nucleation clusters. For researchers and drug development professionals, the content covers advanced methodological approaches for observing these phenomena, strategies for troubleshooting and optimizing crystallization processes, and a comparative validation of theoretical models against experimental data. The synthesis aims to equip scientists with the knowledge to control polymorph selection, enhance crystal properties, and develop more effective therapeutics.
Deconstructing Nucleation: From Classical Dogma to Nonclassical Realities
Classical Nucleation Theory (CNT) serves as the primary theoretical framework for quantitatively describing the kinetics of first-order phase transitions, such as the formation of a solid crystal from a supersaturated solution or liquid melt [1]. As the initial step in the spontaneous formation of a new thermodynamic phase, nucleation often dominates the kinetics of the overall transformation process, with nucleation times varying by orders of magnitude from negligible to experimentally unobservable timescales [1]. This whitepaper details the core principles of CNT, focusing on the fundamental concepts of the nucleation energy barrier and critical nuclei formation, with specific application to organic crystal research in pharmaceutical development. The central aim of CNT is to explain and quantify this immense variation in nucleation rates and provide researchers with a predictive model for controlling phase transformations.
Theoretical Foundations of CNT
The development of CNT dates back to the 1930s with foundational work by Becker, Döring, and others, building upon earlier ideas from Gibbs and Volmer and Weber [2]. The theory employs a simplified thermodynamic approach to describe the formation of a stable new phase from a metastable parent phase.
The Free Energy Landscape
CNT conceptualizes nucleation as a process governed by competing energy terms. For the formation of a spherical nucleus of radius (r), the change in free energy (\Delta G) is expressed as:
[ \Delta G = \frac{4}{3}\pi r^3\Delta g_v + 4\pi r^2\sigma ]
Where (\Delta g_v) is the Gibbs free energy change per unit volume (typically negative for a spontaneous process), and (\sigma) is the interfacial surface tension per unit area (always positive) [1]. The first term represents the bulk energy gain from phase transformation, while the second term represents the energy cost of creating a new interface.
Table 1: Components of Nucleation Free Energy
| Term | Mathematical Expression | Energetic Contribution | Dependence on Radius (r) |
|---|---|---|---|
| Volume (Bulk) Term | (\frac{4}{3}\pi r^3\Delta g_v) | Favorable (Negative) | Cubic ((r^3)) |
| Surface Term | (4\pi r^2\sigma) | Unfavorable (Positive) | Square ((r^2)) |
| Total Free Energy ((\Delta G)) | (\frac{4}{3}\pi r^3\Delta g_v + 4\pi r^2\sigma) | Determines Nucleation Viability | Combination |
The critical concept in CNT is that the surface term dominates for small cluster sizes, creating an energy barrier that must be overcome for nucleation to proceed [1]. This relationship produces the characteristic free energy profile shown in the diagram below.
The Critical Radius and Energy Barrier
The critical nucleus radius ((r_c)) occurs where the free energy (\Delta G) reaches its maximum value, found by setting the derivative (d\Delta G/dr = 0):
[ rc = \frac{2\sigma}{|\Delta gv|} ]
Nuclei smaller than (rc) (embryos) are unstable and tend to dissolve, while those larger than (rc) are stable and likely to grow [1]. Substituting (r_c) back into the free energy equation gives the height of the nucleation energy barrier ((\Delta G^*)):
[ \Delta G^* = \frac{16\pi\sigma^3}{3|\Delta g_v|^2} ]
This barrier represents the activation energy that must be supplied by thermodynamic fluctuations for nucleation to occur [1]. The strong dependence on surface tension ((\sigma^3)) highlights its crucial role in determining nucleation kinetics.
For crystallization from solution, the driving force (\Delta g_v) can be expressed in terms of supersaturation ((S)), leading to practical expressions for the critical radius and energy barrier:
[ rc = \frac{2\gamma vm}{kBT \ln S} ] [ \Delta G^* = \frac{16\pi}{3} \frac{\gamma^3 vm^2}{(k_BT \ln S)^2} ]
Where (\gamma) is surface tension, (vm) is molecular volume, (kB) is Boltzmann's constant, (T) is temperature, and (S) is supersaturation ratio [2]. These relationships demonstrate mathematically how increasing supersaturation reduces both the critical size and energy barrier, thereby dramatically enhancing nucleation rates.
Nucleation Kinetics and Rate Prediction
The central result of CNT is the prediction of nucleation rate ((R)), defined as the number of nuclei formed per unit volume per unit time. The CNT expression for (R) is:
[ R = NS Z j \exp\left(-\frac{\Delta G^*}{kB T}\right) ]
Where (N_S) is the number of potential nucleation sites, (Z) is the Zeldovich factor (accounting for non-equilibrium effects), and (j) is the rate at which molecules join the critical nucleus [1]. The exponential term dominates the temperature and supersaturation dependence of the rate, explaining why nucleation can vary by orders of magnitude with small changes in conditions.
Table 2: Parameters in Classical Nucleation Rate Equation
| Parameter | Symbol | Physical Meaning | Typical Range/Value |
|---|---|---|---|
| Nucleation Rate | (R) | Number of nuclei per unit volume per time | Varies widely (e.g., 10⁻⁸³ to >10³ m⁻³s⁻¹) |
| Energy Barrier | (\Delta G^*) | Free energy required to form critical nucleus | ~275 (k_BT) for ice at 19.5°C supercooling [1] |
| Zeldovich Factor | (Z) | Kinetic pre-factor accounting for non-equilibrium | ~10⁻³ (example for ice nucleation) [1] |
| Molecular Flux | (j) | Rate at which molecules join critical nucleus | ~10¹¹ s⁻¹ (example for ice nucleation) [1] |
For researchers in pharmaceutical development, understanding these kinetic parameters is essential for controlling crystal polymorphism, particle size distribution, and ultimately drug bioavailability.
Experimental Validation and Methodologies
Pharmaceutical Precipitation Studies
CNT has been successfully applied to simulate the precipitation of poorly soluble basic compounds in biorelevant media, a critical process in oral drug absorption. In one key study, researchers used infusion-precipitation data to validate CNT parameters for basic drug compounds in fasted state simulated intestinal fluid [3].
The primary nucleation rate per volume was described by:
[ \frac{dN{nc}}{dt} = \beta D{mono}(NA C{aq})^2 \left(\frac{kB T}{\gamma}\right)^{1/2} \ln\left(\frac{C{aq}}{S{aq}}\right) \exp\left(-\frac{16\pi}{3} \frac{\gamma}{(kB T)^3} \frac{vm^2}{\ln(C{aq}/S_{aq})^2}\right) ]
Where (N{nc}) is the number of nuclei, (NA) is Avogadro's number, (C{aq}) is aqueous concentration, (S{aq}) is solubility, (D_{mono}) is monomer diffusion coefficient, and (\beta) is a fitting parameter related to the number of nuclei formed [3].
Table 3: Experimental Parameters from Pharmaceutical Precipitation Study
| Parameter | Symbol | Value Obtained | Methodology |
|---|---|---|---|
| Surface Tension | (\gamma) | 0.0030 N/m | Visual fitting to experimental data at 0.5 mL/min infusion rate [3] |
| Pre-exponential Factor | (\beta) | 1 × 10⁻¹⁸ | Fitting to match precipitation characteristics [3] |
| Critical Supersaturation Ratio | CSSR | 1.7 | Calculated from fitted parameters (GOF = 3.2) [3] |
The methodology involved visually fitting simulation curves to experimental precipitation data at a specific infusion rate (0.5 mL/min), then validating the parameters by predicting precipitation behavior at different infusion rates [3]. This approach successfully simulated key precipitation characteristics, including the increase in precipitation rate and the reduced sensitivity of maximum concentration to increased infusion rate.
The Researcher's Toolkit: Essential Materials and Reagents
Table 4: Key Research Reagent Solutions for CNT Experiments
| Reagent/Material | Function in CNT Studies | Application Example |
|---|---|---|
| Simulated Intestinal Fluids | Biorelevant media for precipitation studies | Predicting in vivo precipitation of poorly soluble drugs [3] |
| Basic Compound Solutions | Model drugs for nucleation kinetics | Studying pH-dependent supersaturation and precipitation [3] |
| Polymer Additives | Nucleation inhibitors/modifiers | Controlling crystallization kinetics and polymorph selection |
| Salt Forms (APIs) | Enhance dissolution and create supersaturation | Rapid dissolution creating metastable zone for nucleation studies [3] |
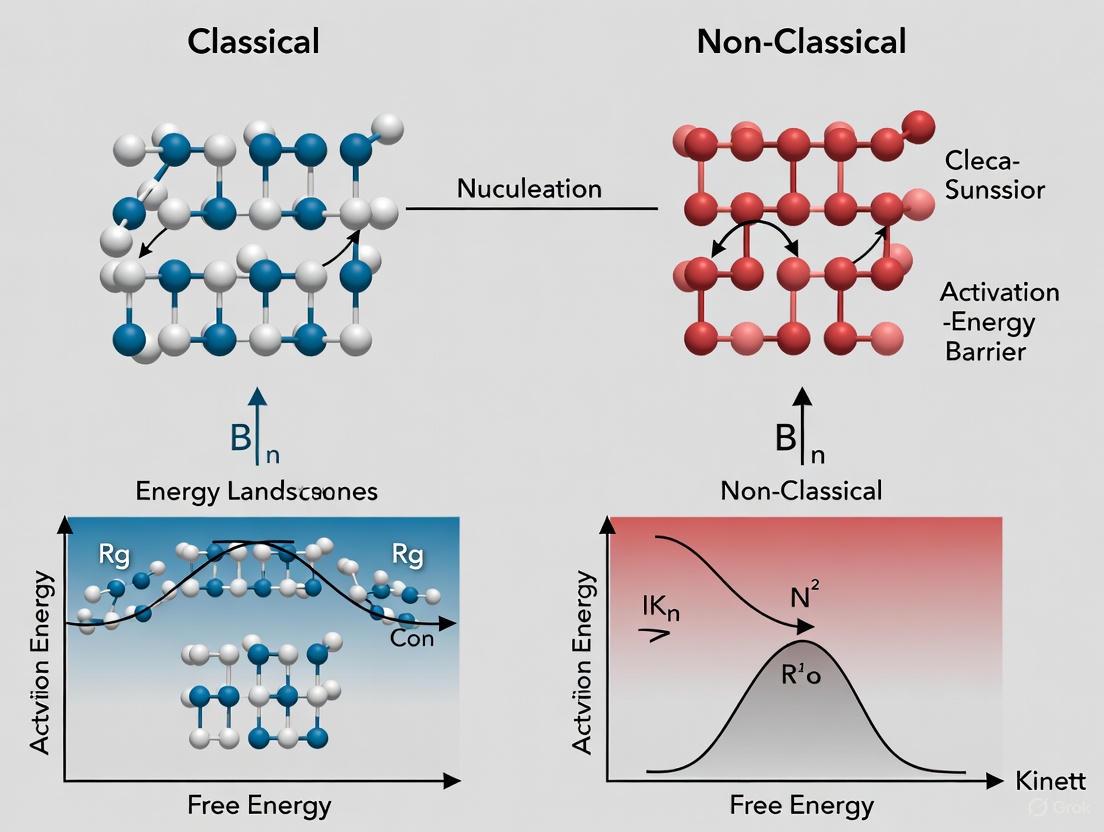
Classical vs. Non-Classical Nucleation Theory
While CNT provides a robust conceptual framework, it incorporates significant simplifications that limit its quantitative accuracy. The "capillary assumption" – treating small nuclei as having the same interfacial properties as macroscopic interfaces – is particularly debated [2]. Consequently, non-classical pathways with lower energy barriers have been identified.
The non-classical pathway, exemplified by the Prenucleation Cluster (PNC) theory, proposes that ions or molecules first form thermodynamically stable, dynamic clusters without a defined phase interface [2]. These PNCs then undergo a structural change to form phase-separated nanodroplets, which aggregate and eventually solidify into crystalline phases [2]. This mechanism fundamentally differs from CNT in that the precursor clusters are stable and form independently of supersaturation level, rather than being unstable entities whose formation probability depends exponentially on supersaturation.
Classical Nucleation Theory remains an essential framework for understanding and predicting nucleation phenomena in organic crystal research, particularly in pharmaceutical development. The concepts of the critical nucleus and energy barrier provide fundamental insights into the kinetic control of crystallization processes. While CNT has limitations in quantitative predictive accuracy, particularly due to its simplified treatment of small clusters, it continues to serve as the foundational model from which more sophisticated theories have evolved. For drug development professionals, CNT offers valuable guidance for controlling polymorphism, particle size, and precipitation behavior – critical factors determining drug product performance and bioavailability.
Classical Nucleation Theory (CNT) has long provided a fundamental framework for understanding the initial stages of phase transformation, positing that a nucleus grows based on a deterministic balance between the volumetric free energy gain of a new phase and the energy cost of creating a new interface. This capillarity approximation assumes that the properties of a microscopic nucleus—such as surface tension and density—are identical to those of the macroscopic bulk phase. While this model offers valuable simplicity and has demonstrated predictive success in many systems, its application to complex, technologically critical materials like carbon nanotubes (CNTs) consistently reveals significant shortcomings. The growth of CNTs, a process central to their application in next-generation electronics, drug delivery, and composite materials, involves intricate atomic-scale dynamics that CNT's macroscopic, thermodynamic view struggles to capture. This whitepaper delineates the specific, quantitative gaps between CNT's predictions and experimental observations in CNT synthesis and behavior, drawing upon recent computational and experimental studies. It further explores the non-classical nucleation mechanisms that more accurately describe these processes, providing researchers with a detailed critique of the classical framework and protocols for its experimental evaluation.
Theoretical Shortcomings of CNT in CNT Nucleation
The fundamental assumptions of CNT create a series of theoretical gaps when applied to the nucleation of carbon nanotubes. These are not minor discrepancies but foundational failures to explain deterministic outcomes.
The Failure of the Capillarity Approximation
The core assumption of CNT is the capillarity approximation, which treats a nascent nucleus as a miniature version of the bulk crystal with identical interfacial and thermodynamic properties. A rigorous falsifiability test of this premise was conducted using a binary mixture of tetravalent patchy particles designed to form three distinct crystalline polymorphs (DC-8, DC-16, DC-24) with identical bulk free energies and interfacial free energies at all state points [4]. Within the capillarity approximation, all three polymorphs should have identical nucleation rates. However, molecular dynamics simulations demonstrated radically different nucleation properties, with the nucleation rates varying significantly between the polymorphs. The study concluded that CNT's primary limitation is its neglect of structural fluctuations within the liquid phase, which pre-emptively templates specific polymorphs [4]. This finding directly undermines the universality of the capillarity approximation for complex crystals.
Inability to Deterministically Predict Chirality
A paramount challenge in CNT synthesis is controlling chirality, as this single parameter determines the nanotube's electronic properties. CNT offers a stochastic view of nucleation and growth, unable to explain the high enantiomeric purity (e.g., 92% for (12,6) and 97% for (14,4)) achieved in experiments [5]. The classical perspective attributes chirality enrichment to edge matching during the growth phase. In contrast, a deterministic nucleation theory demonstrates that chirality is encoded during the nucleation event itself through the selective formation of specific carbon caps on catalyst surfaces [5]. The topology of the initial cap, which incorporates six pentagons into a hexagonal lattice, deterministically defines the chirality vector (n, m) of the resulting nanotube. This cap formation is guided by epitaxial matching to specific catalyst facets, such as the (0 0 12) facet of W₆Co₇ for (12,6) nanotubes, a process outside the scope of CNT's phenomenological description [5] [6].
Table 1: Core Theoretical Gaps Between CNT Predictions and Experimental Observations of CNT Nucleation
| Classical Nucleation Theory (CNT) Prediction | Experimental/Observed Reality in CNT Systems | Theoretical Gap |
|---|---|---|
| Nucleation rate is purely stochastic and determined by macroscopic thermodynamics (capillarity approximation). | Nucleation is guided by atomic-scale pre-ordering in the liquid phase and is often deterministic for specific polymorphs [4]. | Neglect of local structural fluctuations and their templating effect. |
| Chirality is a stochastic outcome, potentially influenced during the growth phase. | Chirality is deterministically encoded during the cap nucleation step via structural matching with the catalyst [5]. | Inability to account for atomic-level catalyst-carbon interactions that define cap topology. |
| The critical nucleus is a simple, monolithic entity with bulk properties. | Nucleation can proceed via non-classical pathways, including the coalescence of sub-critical clusters and stepwise nucleation [7]. | Oversimplified model of the nucleus as a single, well-defined phase. |
| Interfacial free energy is a constant for a given crystal-liquid pair. | The effective interfacial energy is pathway-dependent and influenced by transient, non-equilibrium cluster structures [7] [4]. | Assumption of equilibrium thermodynamics applied to a non-equilibrium process. |
Quantitative Discrepancies in Experimental Data
The theoretical shortcomings of CNT manifest as concrete, quantitative gaps when its predictions are compared with experimental and high-fidelity computational data.
Nucleation Pathways and Energy Barriers
Molecular dynamics simulations of homogeneous nucleation in iron reveal mechanisms that CNT cannot describe. The atomic system avoids the high energy barrier predicted by CNT for homogeneous nucleation by undergoing alternative, nonclassical processes, specifically "coalescence of subcritical clusters" and "stepwise nucleation" [7]. The observation of these pathways indicates that the nucleation process is not a simple, single-step activation event but a more complex evolution involving multiple interacting clusters, a phenomenon not captured by the classical model.
On the atomic scale, Density Functional Theory (DFT) calculations of CNT nucleation on a Ni(1 1 1) surface provide quantitative data on energy barriers that are absent from CNT. These studies examine the formation and diffusion of small carbon clusters (C₂ to C₆), revealing that C₃ exhibits the lowest diffusion barrier and identifying the preferred nucleation pathway in terms of energetics [6]. For instance, the mobility and stability of these small clusters dictate the kinetics of the early nucleation stage, yet CNT possesses no framework for incorporating such atomistic information.
Macroscopic Fiber Properties vs. Individual CNT Performance
The failure to control nucleation and growth translates directly to performance gaps in macroscopic CNT materials. While individual CNTs possess extraordinary theoretical tensile strength and electrical conductivity, CNT fibers (assemblies of many CNTs) often fall short of these ideals. The FCCVD (Floating Catalyst Chemical Vapor Deposition) method, a leading production technique, faces critical challenges in achieving property consistency [8]. The performance of the macroscopic fiber is hampered by weak intertube interactions, misalignment, and structural defects originating from the imperfect nucleation and growth of the constituent nanotubes [8]. This discrepancy between the properties of the individual nucleated product and the macroscopic assembled material is a direct consequence of the inability to perfectly control the underlying nucleation process, a problem CNT does not address.
Table 2: Comparison of CNT Fabrication Methods and Their Limitations Relative to Nucleation Control
| Fabrication Method | Tensile Strength | Electrical Conductivity | Production Scalability | Key Nucleation-Related Challenges |
|---|---|---|---|---|
| Wet Spinning | ★ Low | ★ Low | ★★ Moderate | Poor CNT alignment and weak intertube bonding due to disordered nucleation precursors [8]. |
| Array Spinning | ★★★ High | ★★★ High | ★ Low | Stringent processing conditions limit scale-up; nucleation control is confined to small areas [8]. |
| Floating Catalyst Chemical Vapor Deposition (FCCVD) | ★★★ High | ★★★ High | ★★★ High | Catalyst uniformity and structural defects during nucleation lead to batch-to-batch inconsistency [8]. |
Non-Classical Nucleation Mechanisms
The failures of CNT have spurred the identification and study of non-classical nucleation mechanisms that more accurately describe the formation of CNTs and other complex crystals.
Coalescence and Stepwise Nucleation: As identified in solid-state phase transformations in iron, the "coalescence of subcritical clusters" involves small, stable clusters merging to form a larger, stable nucleus. "Stepwise nucleation" describes a more complex, multi-stage assembly process [7]. These mechanisms allow the system to circumvent the high energy barrier predicted by CNT, explaining nucleation phenomena observed in simulations that classical theory cannot.
Deterministic Cap Formation: Contrary to the stochastic model, evidence shows that the chiral structure of a CNT is determined during the formation of the initial carbon cap on the catalyst surface. The vector sum rule provides a topological framework linking the architecture of the cap—defined by the positions of its six pentagons—to the final chirality (n, m) of the nanotube [5]. The catalyst surface acts as a template, epitaxially matching specific cap structures and thereby deterministically selecting the nucleation outcome.
Multiscale Growth Phenomena: The CNT growth process is inherently multiscale, involving phenomena not captured by a purely thermodynamic theory. As outlined by [9], these include (1) the decomposition of carbon sources on the catalyst, (2) the diffusion of carbon atoms/clusters, (3) the removal of carbon by etching agents, and (4) the integration of carbon atoms into the CNT wall. Each stage involves complex kinetics and diffusion at the atomistic level, which then manifest in the macroscopic structure and quality of the nanotube.
Experimental Protocols for Investigating Nucleation Gaps
To bridge the gap between theory and experiment, researchers can employ the following advanced protocols.
Density Functional Theory (DFT) for Early Nucleation Energetics
Objective: To calculate the reaction energy barriers and identify the preferred nucleation pathway for carbon atoms on a specific catalyst surface (e.g., Ni(1 1 1)) [6].
- Model Setup: Construct a slab model of the catalyst surface (e.g., Ni(1 1 1)) with periodic boundary conditions. Ensure the slab thickness and vacuum space are sufficient to minimize interactions between periodic images.
- Adsorption and Diffusion Calculations:
- Place individual carbon atoms and small clusters (C₂ to C₆) on various high-symmetry surface sites (e.g., fcc, hcp, top, bridge).
- Use DFT to optimize the geometry of each adsorption configuration and calculate the adsorption energy.
- Use the Nudged Elastic Band (NEB) method or dimer method to locate the transition state between stable sites and compute the diffusion energy barrier for each cluster.
- Reaction Pathway Analysis:
- Calculate the formation energies for larger clusters (C₅, C₆) from smaller ones, considering all possible formation scenarios.
- Compare the energetics of different pathways to identify the most thermodynamically and kinetically favorable route for the early nucleation process.
Molecular Dynamics (MD) for Non-Classical Pathways
Objective: To observe and characterize non-classical nucleation mechanisms, such as cluster coalescence, in a simulated material system [7] [4].
- System Design: Choose an appropriate interatomic potential (e.g., for iron) or a patchy particle model (e.g., the N2c8 binary mixture) that exhibits polymorphic crystallization [7] [4].
- Simulation Run: Perform isothermal-isobaric (NPT) or canonical (NVT) ensemble MD simulations at a supercooled state point where nucleation is expected to occur on the simulation timescale.
- Cluster Analysis:
- Use a common neighbor analysis (CNA) or bond order parameter (e.g., Steinhardt parameters) to identify solid-like atoms in the simulation box.
- Implement a clustering algorithm (e.g., Dense Neighborhood) to group solid-like atoms into distinct clusters.
- Track the size, composition, and lifetime of all clusters over the course of the simulation.
- Pathway Identification: Analyze the trajectory to identify events where two or more sub-critical clusters merge (coalesce) to form a stable nucleus, or to observe the stepwise assembly of a nucleus.
In-situ Characterization and CFD Integration
Objective: To correlate synthesis conditions with nucleation outcomes in chemical vapor deposition (CVD) reactors and scale up production [8] [9].
- Reactor Instrumentation: Employ in-situ characterization techniques, such as environmental transmission electron microscopy (ETEM) or Raman spectroscopy, to observe CNT nucleation and growth in real-time within a model CVD reactor.
- Computational Fluid Dynamics (CFD) Modeling:
- Develop a multiphase flow model of the industrial-scale CVD reactor.
- Simulate gas flow patterns, heat transfer, and species transport to identify regions of turbulence, non-uniform temperature, and inconsistent precursor concentration [8].
- Data Integration: Correlate the observed nucleation yield and CNT chirality distribution from (1) with the localized process conditions predicted by the CFD model in (2). This helps identify how macroscopic reactor flow and temperature gradients lead to microscopic nucleation inconsistencies.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for CNT Nucleation Research
| Item | Function in Nucleation Research | Specific Examples / Notes |
|---|---|---|
| Catalyst Precursors | Seed the formation of catalytic nanoparticles for CNT growth. | Ferrocene, acetylferrocene, ferrocene methanol; Bimetallic systems (Fe/Co, Co/Mo); Metal-Organic Frameworks (MOFs) like ZIF-67 [8] [6]. |
| Carbon Sources | Provide the carbon atoms for CNT construction. | Ethanol, methane, ethylene; Multicomponent sources (e.g., ethanol + thiophene); Biomass-derived sources (lignin, tannic acid) [8] [6]. |
| Substrate Materials | Provide a surface for catalyst deposition and CNT growth. | Silicon wafers with oxide layer (SiO₂/Si), Alumina (Al₂O₃), Quartz, Zeolites [10]. |
| Etching Agents / Promoters | Control catalyst activity and remove amorphous carbon. | Sulfur-containing compounds (e.g., thiophene), Hydrogen gas (H₂), Water vapor (H₂O) [8] [9]. |
| Computational Software | Model nucleation energetics and dynamics at the atomic scale. | QuantumATK (for DFT), LAMMPS (for MD), Kinetic Monte Carlo (kMC) platforms [6] [9]. |
The evidence is clear and compelling: Classical Nucleation Theory falls short in explaining the experimental data of carbon nanotube nucleation. Its failures are rooted in fundamental assumptions—the capillarity approximation, the neglect of liquid-phase structure, and the stochastic worldview—that are incompatible with the deterministic, atomic-scale mechanisms governing CNT formation. Quantitative gaps in energy barriers, chirality distributions, and macroscopic fiber properties all trace back to these theoretical limitations. The path forward for researchers lies in embracing and refining non-classical frameworks that incorporate deterministic cap formation, cluster dynamics, and multiscale modeling. Integrating advanced computational methods like DFT and MD with in-situ experimental data and reactor-scale modeling is no longer optional but essential. By moving beyond the classical paradigm, the scientific community can unlock predictable, chirality-controlled synthesis of carbon nanotubes, fully realizing their potential to revolutionize technology from post-Moore's Law electronics to targeted drug delivery systems.
For decades, the prevailing paradigm for crystal formation has been Classical Nucleation Theory (CNT), which describes crystallization as a single-step process where individual atoms or molecules in a supersaturated solution directly assemble into a critical nucleus that then grows into a crystal. This model, while foundational, operates under significant limitations, particularly when the old and new phases differ by multiple order parameters such as density and structure simultaneously [11]. The implicit and explicit assumptions of CNT are often poorly justified in complex systems, leading to its failure in accurately predicting and describing nucleation behavior across a wide range of materials [11]. In contrast, non-classical crystallization (NCC) pathways encompass mechanisms that do not comply with CNT's core criteria, typically involving precursor particles larger than single atoms or molecules [12]. These pathways, particularly those involving prenucleation clusters (PNCs) and two-step nucleation, have emerged as vital frameworks for understanding crystallization in diverse systems ranging from biominerals to pharmaceutical compounds.
The significance of these non-classical pathways extends beyond academic interest. In pharmaceutical development, for instance, where a molecule can exist in multiple crystal structures (polymorphs), understanding and controlling the nucleation pathway is paramount. The crystal structure dictates critical drug properties including solubility and bioavailability, directly impacting drug efficacy and safety [12]. The intermediate stages of pre-crystalline processes can harbor previously inaccessible polymorphs with potentially more desirable properties, opening avenues to direct their formation through pathway engineering [12].
Theoretical Foundations of Non-Classical Nucleation
Prenucleation Clusters (PNCs)
Prenucleation clusters are defined as thermodynamically stable associates of atoms, ions, or molecules that form in solution. They are typically 1–3 nm in size and exist as solutes themselves, forming based on dynamic chemical equilibrium in both undersaturated and supersaturated solution states [13]. Contrary to being mere density fluctuations as envisioned in CNT, PNCs are stable solute species that play a key role as precursors in the nucleation of a second phase from metastable solutions [13].
A critical characteristic of PNCs is their liquid-like nature. Experimental studies on protein systems have revealed that these clusters are not rigid solid particles but dynamic, dense liquid phases. For example, in lysozyme solutions, these protein-rich clusters measure approximately 100 nm in size and contain between 10,000-100,000 molecules, yet occupy less than 10⁻³ of the total solution volume [14]. This dense liquid phase serves as an intermediate that facilitates the subsequent formation of crystalline nuclei.
The Two-Step Nucleation Mechanism
The two-step nucleation mechanism provides a specific pathway wherein PNCs act as the precursor to crystalline phases. This mechanism posits that crystal formation occurs through two distinct stages: first, the formation of a dense, liquid-like intermediate cluster, and second, the nucleation of crystals within these confined environments [14]. The thermodynamic driving force for this process differs fundamentally from CNT.
Table 1: Supersaturation Parameters in Two-Step Nucleation [15]
| Supersaturation Type | Mathematical Definition | Physical Meaning |
|---|---|---|
| For C-phase in O-phase | Δμco ≡ μo - μc | Driving force for direct crystallization from old phase |
| For M-phase in O-phase | Δμmo ≡ μo - μm | Driving force for metastable phase formation |
| For C-phase in M-phase | Δμcm ≡ μm - μc | Driving force for crystallization within metastable phase |
According to the composite-cluster model within CNT extension, two-step nucleation requires that Δμco > 0 and Δμcm > 0 simultaneously [15]. This means both the old phase (solution) must be supersaturated with respect to the crystal phase, and the metastable phase (the dense liquid cluster) must also be supersaturated with respect to the crystal phase. When these conditions are met, the system can bypass the high energy barrier of direct solid formation by first transitioning through the "nearest lying state" as advised by the Ostwald step rule [15].
Figure 1: The Two-Step Nucleation Pathway. The non-classical route proceeds through a dense liquid intermediate (M-phase), contrasting with the direct classical pathway.
Experimental Evidence Across Material Systems
Protein Crystallization
Proteins have served as ideal model systems for studying non-classical pathways due to their large molecule sizes, which facilitate observation. Experimental evidence shows that mesoscopic protein clusters can significantly impact both nucleation and growth stages. When these clusters merge with a growing crystal surface, they lead to a non-classical growth mechanism characterized by instantaneous multilayer formation, also referred to as looped macrosteps or 3D islands [11]. This mechanism differs fundamentally from classical growth via spiral dislocations or 2D nucleation.
Remarkably, these clusters also contribute to crystal perfection. Research has demonstrated that cluster assimilation by a crystal can trigger a self-purifying cascade of impurity-poisoned crystal surfaces [11]. Essentially, the coalescence of a cluster with an impurity-poisoned crystal surface leads to rapid and complete cleansing of the entire surface—a property with significant implications for growing high-quality crystals for pharmaceutical or structural applications.
The physical origin of protein clusters appears linked to protein conformational flexibility. Studies exploring the effect of chaotropic agents like urea and shear stress—both known protein denaturants—found that these treatments strongly decrease cluster size [14]. This supports a mechanism of cluster formation involving partial protein unfolding followed by dimerization. Nuclear magnetic resonance monitoring of amide hydrogen-deuterium exchange has further highlighted that lysozyme conformational flexibility is a prerequisite for forming protein-rich clusters that facilitate crystal nucleation [14].
Pharmaceutical Compounds
Direct observation of non-classical pathways in small organic molecules, particularly active pharmaceutical ingredients (APIs), has historically been challenging due to difficulties in distinguishing these low-electron density entities from their liquid surroundings. However, recent advances in Liquid Phase Electron Microscopy (LPEM) have enabled high temporospatial imaging of nucleation events in native environments.
For the common NSAID flufenamic acid (FFA), LPEM observations in organic solvent have captured intermediate pre-crystalline stages consistent with a PNC pathway followed by features exhibiting two-step nucleation [12]. The observed phenomena suggest that nucleation pathways are likely an amalgamation of multiple existing non-classical theories rather than following a single rigid mechanism. This understanding is particularly valuable for pharmaceutical processing, where continuous manufacturing approaches demand precise control over nucleation events to ensure consistent polymorphic form and product quality [12].
Colloidal and Nanoscale Systems
Ionic colloidal crystals provide excellent model systems for visualizing non-classical pathways due to their size scales, which allow direct observation of assembly mechanisms. Studies with binary mixtures of oppositely charged colloids have revealed a clear two-step process where metastable amorphous blobs first condense from the gas phase before evolving into small binary crystals [16]. These small crystals then grow via multiple simultaneous processes:
- Monomer-by-monomer addition from the bulk solution
- Capture and absorption of surrounding amorphous blobs
- Oriented attachment of other crystals [16]
The interaction strength between particles, often tunable through parameters like salt concentration, plays a crucial role in modulating these pathways. At moderate interaction strengths, two-step crystallization dominates, while classical crystallization occurs only in a narrow window of conditions, and random aggregation prevails at very high interaction strengths [16].
In semiconductor nanocrystal synthesis, such as ZnSe quantum dots, PNCs play a definitive role. These clusters form at specific temperatures and can follow different transformation pathways depending on environmental conditions: they can isomerize into magic-size clusters (MSCs) at lower temperatures or fragment into monomers that feed the nucleation and growth of quantum dots at higher temperatures [17].
Table 2: Experimental Evidence for Non-Classical Pathways Across Material Systems
| Material System | Experimental Technique | Key Findings | Reference |
|---|---|---|---|
| Proteins (Lysozyme, etc.) | Light scattering, Brownian microscopy, Laser confocal microscopy | Liquid-like clusters (100nm, 10⁴-10⁵ molecules) mediate nucleation and growth; Enable self-purification | [11] [14] |
| Pharmaceutical (Flufenamic Acid) | Liquid Phase Electron Microscopy (LPEM) | Direct observation of PNC pathway and two-step nucleation features in organic solvent | [12] |
| Ionic Colloids | Bright-field/confocal microscopy, SEM, Simulations | Two-step process: amorphous blobs → crystals; Growth via monomer addition, blob capture, oriented attachment | [16] |
| Semiconductor (ZnSe) | Optical absorption spectroscopy | PNCs form above 120°C; Transform to MSCs at 25°C or fragment to monomers for QD growth at 220°C | [17] |
Methodologies for Investigating Non-Classical Pathways
Experimental Approaches
A diverse toolkit of characterization techniques has been essential for elucidating the details of non-classical nucleation pathways:
- Light Scattering Techniques: Static and dynamic light scattering provide information on cluster size distributions and stability in solution over time [11].
- Brownian Microscopy (BM): Allows direct tracking of cluster motion and determination of cluster number densities, revealing their temporal evolution and instability [11].
- Laser Confocal Microscopy with Differential Interference Contrast (LCM-DIM): Enables non-invasive, mesoscopic mapping of crystal surface topography with fast acquisition times, ideal for observing looped macrostep formation and other non-classical growth features [11].
- Liquid Phase Electron Microscopy (LPEM): Provides high temporospatial imaging of nucleation events in native liquid environments, though requires careful consideration of radiolysis effects which can be mitigated through low-dose techniques or exploited to induce nucleation [12].
- Confocal Microscopy with Refractive Index Matching: Allows 3D characterization of distinct phases in colloidal systems, confirming the amorphous nature of intermediate blobs and pinpointing nucleation locations [16].
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Non-Classical Crystallization Studies
| Reagent/Material | Function/Application | Example Use |
|---|---|---|
| Model Proteins (Lysozyme, Glucose Isomerase, etc.) | Well-characterized systems for studying protein crystallization fundamentals | Testing cluster roles in nucleation and growth; observing looped macrostep formation [11] |
| Pharmaceutical Compounds (Flufenamic Acid, Ibuprofen) | Study API crystallization pathways and polymorph control | Direct observation of PNCs and intermediate stages via LPEM [12] |
| Oppositely Charged Colloidal Particles | Model "ions" for visualizing assembly pathways | Investigating two-step crystallization and multiple growth mechanisms [16] |
| Chaotropic Agents (Urea) | Perturb protein folding to test cluster formation mechanisms | Demonstrating connection between partial unfolding and cluster size reduction [14] |
| Continuous Dialysis Setup | Precise control over interaction strength via salt concentration | Identifying conditions for classical vs. non-classical crystallization windows [16] |
| H₂O/D₂O Mixtures | Density-matching solvent for gravity-neutral experiments | Confirming non-classical mechanisms are not gravity-dependent [16] |
Technical Protocols for Key Experiments
Establishing Cluster Role in Crystal Growth
Objective: Demonstrate the causal relationship between mesoscopic clusters in solution and non-classical crystal growth mechanisms [11].
Figure 2: Experimental workflow for establishing the causal relationship between solution clusters and crystal growth mechanisms.
Procedure:
- Prepare a supersaturated solution of the target protein (e.g., glucose isomerase).
- Split the solution into two aliquots: one subjected to intensive filtration (0.2-µm cutoff, three times) to remove clusters, and one unfiltered control.
- Monitor cluster number densities in both solutions using Brownian Microscopy (BM), confirming significant reduction in the filtered sample.
- Expose pregrown seed crystals to both solutions and observe the (011) face using Laser Confocal Microscopy with Differential Interference Contrast (LCM-DIM).
- Quantify the nucleation rate of looped macrosteps (Jclust) and correlate with temporal cluster density.
Expected Results: Unfiltered solutions will show instantaneous looped macrostep formation, while filtered solutions exhibit significantly reduced or completely absent 3D growth mechanisms. The temporal dependence of cluster number density should correlate strongly with the nucleation rate of multilayer islands [11].
Direct Observation of Pharmaceutical Nucleation via LPEM
Objective: Capture nanoscale early-stage crystallization events of small organic molecules like flufenamic acid (FFA) in organic solvent [12].
Procedure:
- Prepare a 50 mM solution of FFA in ethanol.
- Load solution into a liquid cell with silicon nitride windows, ensuring proper solution presence between windows.
- Initiate imaging with a transmission electron microscope equipped for liquid phase experiments.
- To induce nucleation, condense the electron beam using the monochromator to increase electron flux, or exploit radiolysis effects by using a high dose (>150 e⁻/Ų/s).
- Capture sequential images of the nucleation events, focusing on early-stage intermediate structures.
- Troubleshoot for common issues including silicon nitride window scarring or absence of solution between windows by introducing water to the syringe pumping system to dislodge particulates.
Expected Results: Observation of intermediate pre-crystalline stages consistent with a PNC pathway, potentially showing features of two-step nucleation. The gathered evidence typically reveals that nucleation pathways are an amalgamation of multiple non-classical theories rather than following a single rigid mechanism [12].
Discussion and Implications
The experimental evidence across diverse systems confirms that non-classical nucleation pathways represent fundamental mechanisms of crystallization rather than rare exceptions. The convergence of findings from protein, pharmaceutical, colloidal, and nanomaterial systems suggests that two-step nucleation through dense liquid phases may be a universal phenomenon under appropriate conditions. The extension of Classical Nucleation Theory to incorporate composite clusters provides a mathematical framework that unifies classical and non-classical approaches, demonstrating that 1S nucleation is merely a limiting case of the more general 2S nucleation process [15].
For pharmaceutical scientists and materials engineers, understanding these pathways enables new strategies for crystal engineering. The ability of clusters to trigger self-purifying cascades on crystal surfaces addresses a fundamental challenge in producing high-purity crystals for pharmaceutical applications [11]. The discovery that intermediate stages may contain transient metastable polymorphs opens possibilities for accessing previously inaccessible crystal forms with potentially superior properties [12].
Future research directions will likely focus on developing more sophisticated control strategies to direct crystallization along specific non-classical pathways. The continuous dialysis approach demonstrated for colloidal systems, which allows fine control over interaction strength in both time and space, represents a promising methodology for such pathway engineering [16]. As observation techniques continue to improve, particularly in situ methods like LPEM, our understanding of the intricate details of these nucleation pathways will further mature, enabling more precise control over crystallization outcomes across scientific and industrial applications.
The Role of Mesoscale Clusters as Ubiquitous Precursors
The longstanding paradigm for crystal formation from solution has been Classical Nucleation Theory (CNT), which posits a one-step process where individual solute molecules (monomers) randomly assemble into a critical nucleus with a long-range ordered crystalline structure [18]. However, advanced analytical techniques have revealed that primary nucleation, particularly for organic molecules, is often far more complex. A growing body of evidence now supports non-classical nucleation theories, which emphasize the crucial role of mesoscale clusters—solute-rich, often liquid-like aggregates typically 50-500 nm in size—as ubiquitous precursors in the crystallization pathway [18] [19]. These clusters challenge traditional understandings of phase transitions and have significant implications for controlling crystallization in fields ranging from pharmaceutical development to materials science.
This technical guide synthesizes current understanding of mesoscale clusters, framing their properties and roles within the broader context of classical versus non-classical nucleation theory. It provides researchers and drug development professionals with a comprehensive overview of the experimental evidence, characterization methodologies, and implications for industrial processes.
Theoretical Framework: Classical vs. Non-Classical Nucleation
The Classical Nucleation Theory (CNT) Model
Classical Nucleation Theory describes nucleation as a continuous, stochastic process where monomers progressively attach to an emerging cluster. The cluster becomes stable and capable of further growth only after reaching a critical size that overcomes the free energy barrier associated with creating a new interface [18]. This model assumes that the nascent nucleus possesses the same internal structure and long-range order as the final macroscopic crystal. CNT has historically dominated the field due to its mathematical tractability, despite persistent difficulties in quantitatively predicting nucleation rates for new systems [18].
The Non-Classical Paradigm and Mesoscale Clusters
Non-classical theories propose alternative pathways where nucleation occurs through intermediate stages that are structurally and energetically distinct from the final crystal. A predominant non-classical model is two-stage nucleation, where a dense, often disordered liquid-like cluster forms first, followed by internal reorganization and crystallization within this cluster [20]. Mesoscale clusters are central to these models, acting as pre-nucleation intermediates that can concentrate solute, lower interfacial energy barriers, and provide a favorable environment for the birth of crystalline order [18] [19].
The diagram below illustrates the key stages in this non-classical pathway, highlighting the role of mesoscale clusters.
Experimental Evidence for Mesoscale Clusters
Indirect Experimental Indications
Early evidence for clusters came from observing their effects on bulk solution properties:
- Gravity and Diffusion: Experiments with aqueous citric acid in columns demonstrated concentration gradients in supersaturated solutions, suggesting the presence of dense, settling clusters [18]. Measurements of diffusivity via Gouy interferometry showed that diffusion coefficients in supersaturated glycine and urea solutions decrease with both increasing concentration and solution age, indicating the formation and growth of clusters over timescales of 10-100 hours [18].
- Shear Flow: Studies using Taylor-Couette flow cells demonstrated that fluid shear rate inversely correlates with nucleation induction time for compounds like glycine and butyl paraben. Proposed mechanisms include shear-promoted aggregation of clusters or attrition effects at different agitation intensities [18].
- Solution History and Filtration: The propensity of a solution to nucleate can depend on its thermal pre-treatment history, with effects lasting hours or even days, suggesting slow cluster reorganization kinetics [18]. Furthermore, nanofiltration (e.g., 0.2 μm filters) has been shown to suppress nucleation in systems like glycine and lysozyme, an effect often attributed to the removal of clusters, though the mechanism may be complex and involve other factors like dissolved gasses [18] [21].
Direct Experimental Detection
Modern analytical techniques now allow direct observation and characterization of mesoscale clusters:
- Light Scattering Techniques: Dynamic Light Scattering (DLS) and Static Light Scattering (SLS) have identified mesoscale clusters in undersaturated and supersaturated solutions of various small organic molecules (e.g., glycine, DL-alanine) and proteins [19]. These studies reveal clusters with colloidal-scale radii (100–150 nm for glycine) that exist in equilibrium with the bulk solution [19].
- Advanced Microscopy: Cryogenic Transmission Electron Microscopy (cryo-TEM) has visualized faceted nanocrystals and their subsequent oriented attachment into larger mesocrystals in protein systems like glucose isomerase (GI), providing direct evidence for non-classical growth pathways [20].
- Spectroscopic and Scattering Methods: Nuclear Magnetic Resonance with Diffusion Ordered Spectroscopy (NMR-DOSY) and Small-Angle X-ray Scattering (SAXS) provide complementary data on cluster size, population, and molecular dynamics. For example, SAXS and DLS confirmed the presence of glycine-rich nanodroplets in solution [19].
Table 1: Summary of Direct Experimental Evidence for Mesoscale Clusters in Various Systems
| System | Experimental Technique | Cluster Size/Characteristics | Key Findings | Reference |
|---|---|---|---|---|
| Glycine (Aqueous) | DLS, NTA, SAXS, NMR-DOSY | Molecular clusters (0.3-0.5 nm); Mesoscale clusters (100-150 nm) | Clusters stable in undersaturated solutions down to 1 mg/g water; Molecular clusters consistent with hydrated dimers. | [19] |
| OTBN (Organic Molecule) | SLS, DFT, NMR | Mesoscale clusters formed at high concentration; π–π stacking dimers key. | Clusters are disordered, liquid-like, and do not grow indefinitely; formation driven by dimerization. | [22] |
| CPEB4 (Protein) | EPR, Light Scattering | 30-300 nm; Core-shell structure. | Clusters are stable and above Critical Aggregation Concentration (CAC); LLPS consists of aggregate clusters. | [23] |
| FUS (Protein) | Light Scattering | Size strongly concentration-dependent. | Clusters are less stable, below CAC; LLPS forms via cluster coalescence. | [23] |
| Glucose Isomerase (Protein) | cryo-TEM | Faceted nanocrystals (∼10 nm scale). | Nucleation via one-step mechanism followed by oriented attachment of nanocrystals into mesocrystals. | [20] |
Formation and Stabilization Mechanisms
The persistence of mesoscale clusters in solution poses a thermodynamic puzzle, as their high surface area should make them unstable. Research points to several stabilization mechanisms:
- Oligomerization as a Precursor: For small organic molecules like OTBN, the formation of stable dimers via specific interactions (e.g., π-π stacking) is a crucial first step. The resulting reaction-diffusion kinetics between monomers and dimers can lead to the formation of stable, solute-rich mesoscale clusters [22]. Density Functional Theory (DFT) calculations and NMR studies confirm that the formation of π-π stacking dimers in solution is thermodynamically favorable and aligns with the building units found in the final crystal structure [22].
- Core-Shell Model for Proteins: For intrinsically disordered proteins like CPEB4 and FUS, a core-shell model is proposed. In this model, proteins can adopt different conformations: a hydrophobic configuration in the cluster core, a water-soluble configuration in the bulk solvent, and an amphiphilic configuration at the cluster interface. The shell of amphiphilic proteins, with hydrophilic residues facing outwards and hydrophobic residues facing the core, acts as a surfactant, stabilizing the entire cluster and defining its size through bending modulus and interfacial tension [23].
- Free Energy Considerations: The exchange of solute between the cluster and the bulk solution is governed not only by concentration gradients but also by the free energy excess per molecule. In some systems, inserting a monomer into a high-concentration cluster requires energy, suggesting that stable clusters must be composed of other species, such as dimers or oligomers, which have a lower chemical potential in the clustered state [22].
Characterization Techniques and Experimental Protocols
A multi-technique approach is essential for comprehensively studying mesoscale clusters. The workflow below outlines a logical progression for cluster investigation, from preparation to advanced analysis.
Detailed Methodologies
Investigating Clusters via Light Scattering and Filtration
- Objective: To detect the presence and size distribution of mesoscale clusters and test their role in nucleation (e.g., Laser-Induced Nucleation) [21].
- Materials:
- Supersaturated glycine solution in water (e.g., 375 g glycine per 1 kg water).
- HPLC vials, syringes, and polyethersulfone (PES) syringe filters of various pore sizes (e.g., 0.2 μm to 5 μm).
- Dynamic Light Scattering (DLS) instrument (e.g., ALV/CGS-3 with λ = 632.8 nm).
- Nd:YAG laser system for NPLIN experiments (e.g., 1064 nm, 6 ns pulses, 10 Hz).
- Procedure:
- Prepare supersaturated solutions at elevated temperature (e.g., 60°C) with stirring.
- Divide the solution into aliquots. Process one as an unfiltered control and others by hot-filtration through different filter pore sizes.
- For each sample (unfiltered and filtered), perform DLS measurements at a 90° scattering angle. Analyze the autocorrelation function using the cumulant method and Stokes-Einstein equation to determine hydrodynamic radius distributions.
- Transfer identical samples to a temperature-controlled stage and irradiate with a laser pulse (e.g., ~3.75 J cm⁻² fluence for 1 minute).
- Monitor vials automatically (e.g., image every 2 minutes) to determine the nucleation induction time, defined as the time between irradiation and the detection of a crystal of a specified size (e.g., 1 mm).
- Plot cumulative probability distributions of induction times and correlate with DLS data to assess the link between clusters and nucleation propensity.
Probing Oligomerization and Cluster Formation with NMR and DFT
- Objective: To understand the molecular-level interactions driving cluster formation, using a small organic molecule (e.g., OTBN) as a model [22].
- Materials:
- OTBN (2-Cyano-4'-methylbiphenyl) solutions in methanol across a concentration range.
- NMR spectrometer for ¹³C and DOSY measurements.
- Computational chemistry software (e.g., Gaussian, Molclus) for DFT calculations.
- Procedure:
- Record ¹³C NMR spectra at different solute concentrations. Monitor the concentration-dependent chemical shift changes, as consistent downfield trends suggest specific, directional molecular associations.
- Use Diffusion-Ordered Spectroscopy (DOSY) to measure the diffusion coefficients of solute species as a function of concentration. A decrease in diffusivity indicates the formation of larger aggregates.
- Employ computational tools (Genmer, DFT optimization) to generate and energetically rank possible dimer configurations. Compare the binding energy of the most stable dimer structure with that of solute-solvent complexes.
- Calculate local electronic structures (e.g., Fukui functions) to identify molecular regions with high nucleophilic or electrophilic character, revealing the driving forces for π-π stacking or other intermolecular interactions.
- Integrate experimental NMR parameters (e.g., reaction rate constants from chemical shifts, diffusion coefficients) into a reaction-diffusion kinetic model to simulate the time evolution and spatial distribution of cluster formation.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagents and Materials for Mesoscale Cluster Research
| Item | Function/Application | Example Usage |
|---|---|---|
| Polyethersulfone (PES) Syringe Filters (0.2 μm) | Removal of mesoscale clusters and/or solid impurities from solution to test their role in nucleation. | Used to suppress laser-induced nucleation of glycine, helping to discriminate between nucleation mechanisms [21]. |
| Deuterated Solvents (e.g., D₂O) | Solvent for NMR spectroscopy to allow locking and shimming without a strong proton signal from the solvent. | Essential for conducting ¹³C NMR and DOSY experiments to study molecular self-assembly in solution [22] [19]. |
| Cryo-EM Grids | Support for vitrified aqueous samples for direct imaging by cryo-TEM. | Used to visualize protein nanocrystals and their oriented attachment in glucose isomerase solutions [20]. |
| Intrinsically Disordered Proteins (e.g., CPEB4, FUS) | Model systems for studying protein cluster formation and Liquid-Liquid Phase Separation (LLPS). | Used to establish core-shell models of cluster stabilization and link cluster properties to the nature of LLPS [23]. |
| Small Organic Molecules (e.g., Glycine, OTBN) | Model solutes for studying cluster formation in small molecule systems relevant to pharmaceuticals. | Glycine is used in DLS, SAXS, and nucleation studies; OTBN is used for combined NMR/DFT studies of dimerization [22] [19] [21]. |
Implications for Pharmaceutical Research and Drug Development
The existence of mesoscale clusters has profound implications for controlling crystallization processes in the pharmaceutical industry, where the solid form of an Active Pharmaceutical Ingredient (API) is critical.
- Polymorphic Control: The pathway-dependent nature of non-classical nucleation, mediated by clusters, can influence the polymorphic outcome. For example, stirring can lead to the nucleation of a metastable glycine polymorph (α) while quiescent conditions favor the stable γ-form, potentially due to different cluster shear histories [18]. Understanding which clusters lead to which polymorph is key to robust manufacturing.
- Process Design and Scalability: The stochastic nature of nucleation and its sensitivity to solution history (e.g., pre-treatment temperature, filtration) can be traced to the slow kinetics of cluster formation and reorganization [18]. Standardizing solution pre-history (e.g., holding at a set temperature for a set time) becomes crucial for reproducible crystallization processes in API manufacturing.
- Targeting Clusters for New Materials: Deliberately inducing cluster formation provides a unique route to produce meso-sized particles or gels. The narrow size distribution of clusters at steady-state makes them attractive as building blocks for novel biomaterials or nano-particle manufacturing [22].
Mesoscale clusters are indeed ubiquitous precursors in crystallization from solution, representing a fundamental shift away from the purely classical nucleation viewpoint. Evidence from diverse systems—small organic molecules, proteins, and inorganic compounds—confirms their role as essential intermediates in non-classical pathways that may involve oligomerization, two-step nucleation, or oriented attachment.
For researchers and drug development professionals, acknowledging the role of these clusters is no longer optional but necessary for achieving predictive control over crystallization. The experimental toolkit, combining light scattering, spectroscopy, microscopy, and computation, allows for a detailed dissection of cluster properties and dynamics. Integrating this knowledge into process design will be pivotal for overcoming long-standing challenges in polymorph selection, crystal engineering, and ensuring the robust and reproducible manufacturing of pharmaceutical products with desired properties.
This technical guide examines the critical roles of interfacial energy and supersaturation as determining factors in nucleation pathway selection between classical and non-classical mechanisms. Within crystalline materials research, particularly for organic crystals and pharmaceutical compounds, the interplay between these thermodynamic and kinetic parameters dictates whether systems follow traditional or alternative crystallization pathways with significant implications for polymorph selection, crystal habit, and final material properties. Through synthesis of current research findings, we demonstrate how targeted manipulation of these drivers enables precise control over nucleation mechanisms, providing researchers with evidence-based strategies for directing crystallization outcomes in both industrial and scientific contexts.
The fundamental processes governing the initial formation of crystalline phases from solution represent a critical determinant of material properties across pharmaceutical, materials science, and chemical engineering disciplines. Traditional understanding of crystallization has been dominated by Classical Nucleation Theory (CNT), which posits a continuous, stochastic process wherein individual atoms or molecules assemble into stable nuclei through a single step of random monomer addition. This perspective assumes the formation of structureless clusters that gradually evolve into ordered crystalline phases, with the free energy barrier dominated by the competition between bulk and surface energy terms.
In contrast, Non-classical Nucleation Theory encompasses multiple alternative pathways that deviate from this conventional model, including oriented attachment of pre-structured clusters, multi-step nucleation through intermediate phases, and pre-nucleation cluster pathways. The selection between these mechanisms is not random but is governed by specific thermodynamic and kinetic parameters, primarily interfacial energy and supersaturation levels, which create distinct energy landscapes that favor particular nucleation routes.
Understanding the interplay between these drivers provides researchers with the ability to predict and control crystallization outcomes, enabling targeted polymorph production and crystal engineering with precision previously unattainable through empirical approaches alone.
The Interfacial Energy Determinant
Fundamental Principles and Theoretical Background
Interfacial energy (σ), representing the excess energy at the boundary between a nascent crystal nucleus and its surrounding solution, constitutes a primary thermodynamic driver in nucleation pathway selection. According to Classical Nucleation Theory, the critical Gibbs free energy requirement for nucleation (ΔG*) is directly proportional to the cube of the interfacial energy, establishing σ as a dominant factor in determining nucleation barriers [24].
The relationship between interfacial energy and material solubility reveals a consistent pattern: highly soluble salts typically exhibit low interfacial energies, while sparingly soluble compounds demonstrate elevated interfacial energies. This correlation has profound implications for nucleation mechanisms, as the interfacial energy directly influences both the thermodynamic feasibility and kinetic rates of nucleation events [24].
Experimental Evidence: Interfacial Energy Directing Nucleation Pathways
Recent investigations have quantified the role of interfacial energy in directing nucleation mechanisms. For low interfacial energy systems (highly soluble salts), research demonstrates reduced nucleation barriers that favor heterogeneous primary nucleation mechanisms, particularly at interfaces such as membrane surfaces. This pathway predominates because the limited relative supersaturation (Δc/c*) can be overcome through the reduced energy barrier provided by the substrate [24].
Conversely, for high interfacial energy systems (less soluble salts), studies reveal that primary nucleation does not occur until Δc/c* exceeds a threshold value of approximately 1. At this supersaturation level, the available excess chemical potential becomes sufficient to favor homogeneous primary nucleation in the bulk solution, effectively mitigating scale formation on membrane surfaces [24].
Table 1: Relationship Between Interfacial Energy, Solubility, and Resulting Nucleation Pathways
| Interfacial Energy Level | Salt Solubility | Relative Supersaturation Threshold | Preferred Nucleation Pathway | Observed Outcome |
|---|---|---|---|---|
| Low | High | Low (<1) | Heterogeneous on substrates | Membrane scaling |
| High | Low | High (≥1) | Homogeneous in bulk | Reduced scaling, bulk crystallization |
Supersaturation as a Kinetic Control Parameter
Supersaturation Fundamentals and Metastable Zone Width
Supersaturation represents the thermodynamic driving force that overcomes the energy barrier for nucleation, defined as the ratio of the actual concentration to the equilibrium concentration (Δc/c*). The metastable zone refers to the supersaturation region between solubility and nucleation thresholds, where spontaneous nucleation does not occur despite the thermodynamically unstable state of the solution [25].
Experimental evidence demonstrates that increasing the concentration rate shortens induction time and raises supersaturation at induction, effectively broadening the metastable zone width. This expanded driving force favors homogeneous primary nucleation pathways by providing sufficient energy to overcome the nucleation barrier without substrate assistance [25].
Strategic Supersaturation Control in Crystallization Processes
Advanced supersaturation control strategies enable precise regulation of nucleation and crystal growth mechanisms following induction. Research shows that membrane area adjustment can modify crystallization kinetics without introducing changes to mass and heat transfer within the boundary layer, providing a targeted approach to supersaturation management [25].
Furthermore, in-line filtration techniques have proven effective for crystal retention within the crystallizer, reducing deposition and enabling consistent supersaturation rates to be sustained. This approach permits longer hold-up times following induction, with population balance studies confirming a reduction in nucleation rate due to solvent desaturation caused by crystal growth, ultimately resulting in larger crystal sizes [25].
Table 2: Supersaturation Control Strategies and Their Effects on Crystallization Outcomes
| Control Strategy | Mechanism of Action | Effect on Nucleation | Effect on Crystal Growth | Overall Outcome |
|---|---|---|---|---|
| Membrane area modulation | Alters concentration kinetics without changing boundary layer transfer | Repositions system within metastable zone; favors homogeneous nucleation at higher supersaturation | Creates discrete supersaturation regions for controlled growth | Reduced scaling, improved crystal size distribution |
| In-line filtration | Retains crystals in bulk solution, reduces membrane deposition | Lowers nucleation rate through sustained desaturation | Promotes growth through extended hold-up times | Larger crystals, improved habit and purity |
| Segregated crystal phase | Develops two discrete regions of supersaturation | Separates nucleation control from growth optimization | Enables independent crystal growth management | Enhanced product quality, tailored crystal properties |
Experimental Methodologies for Pathway Investigation
Non-invasive Characterization Techniques
Cutting-edge investigation of nucleation mechanisms employs non-invasive tools to characterize both surface and bulk nucleation events without disturbing the delicate crystallization process. These methodologies enable real-time monitoring of nucleation induction times, rates, and locations with unprecedented spatial and temporal resolution [24].
Advanced in-situ characterization methods have established direct correlations between nucleation rate and crystal size with interfacial energy parameters, confirming compatibility with established crystallization literature across a comparable range of salt solubilities. These approaches provide quantitative data linking thermodynamic parameters with observable crystallization outcomes [24].
Time-Resolved Cryo-Transmission Electron Microscopy
Revolutionary insights into molecular-scale nucleation mechanisms have emerged through time-resolved cryo-transmission electron microscopy (cryo-TEM), which enables direct imaging of nucleation events at near-molecular resolution. This technique has uncovered specific nucleation pathways leading to multiple crystalline states and gelled states by capturing transient intermediate structures previously undetectable through conventional methods [26].
Application of this methodology to protein crystals (glucose isomerase) has demonstrated that polymorph selection occurs at the earliest stages of structure formation, based on specific building blocks for each space group. This challenges previous conceptions of protein nucleation by revealing that nucleation events are driven by oriented attachments between subcritical clusters that already exhibit crystallinity, rather than proceeding through metastable dense liquid precursors [26].
Machine-Learning Enhanced Molecular Dynamics Simulations
Computational approaches have achieved unprecedented insight into nucleation competitions through machine-learning interaction potentials (MLIP) that accurately capture long-range atomic interactions. These advanced simulation strategies overcome previous limitations in modeling both bulk and surface effects, particularly for complex nanostructured systems [27].
The development of Physical LassoLars Interaction Potential (PLIP) methodology, incorporating both short-range interactions and scaled point charge models for long-range electrostatic forces, has enabled precise modeling of polymorphic competitions in zinc oxide nanoparticles. This approach has revealed temperature-dependent nucleation pathways, with multi-step processes involving metastable crystal phases occurring at high supercooling, while classical nucleation mechanisms dominate at moderate supercooling levels [27].
Pathway Selection in Organic and Pharmaceutical Systems
Polymorph Control in Macromolecular Systems
The competition between possible crystalline structures carries dramatic consequences in pharmaceutical applications, where different polymorphic forms can exhibit varying bioavailability, stability, and toxicity profiles. Research demonstrates that control over polymorphic outcomes requires intervention at the earliest stages of structure formation, as polymorph selection is determined by initial nucleation events rather than subsequent phase transformations [26].
Studies of protein glucose isomerase have revealed that specific building blocks for each space group direct polymorph selection through their assembly characteristics. This understanding enables strategic control over polymorphic outcomes through site-directed mutagenesis to selectively tune intermolecular bonding or through gel seeding techniques that introduce specific nucleation templates [26].
Competing Nucleation Pathways in Nanocrystal Formation
Nanoscale crystallization processes introduce additional complexity to pathway selection, as surface effects expand the structural landscape of possible polymorphic structures. Research on zinc oxide nanoparticles has revealed competitions between homogeneous nucleation in the core and heterogeneous nucleation in the peripheral regions, with the predominant mechanism determined by the degree of supercooling [27].
Notably, computational investigations have demonstrated that different nucleation pathways compete depending on the investigated degree of supercooling, with a multi-step process involving metastable crystal phases favored at high supercooling, while classical nucleation mechanisms prevail at moderate supercooling levels. This temperature-dependent pathway selection has significant implications for controlling nanocrystal properties through thermal management [27].
Advanced Visualization of Nucleation Pathways
Pathway Selection Logic Diagram
Experimental Workflow for Nucleation Studies
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 3: Key Experimental Resources for Nucleation Pathway Studies
| Category | Specific Reagents/Methods | Research Function | Application Context |
|---|---|---|---|
| Model Systems | Calcium sulphate salts | Low solubility, high interfacial energy model | Membrane distillation crystallization [24] |
| Sodium chloride crystals | High solubility, low interfacial energy model | Crystal growth kinetics studies [25] | |
| Glucose isomerase protein | Polymorph competition model system | Pharmaceutical protein crystallization [26] | |
| Zinc oxide nanoparticles | Nanocrystal formation model | Polymorph competition at nanoscale [27] | |
| Characterization Techniques | Non-invasive in-situ monitoring | Induction time and nucleation location tracking | Real-time pathway determination [24] |
| Time-resolved cryo-TEM | Molecular-resolution nucleation imaging | Pathway visualization at molecular scale [26] | |
| Machine-learning interaction potentials (PLIP+Q) | Long-range interaction modeling | Nanoscale nucleation simulation [27] | |
| Gaussian-mixture model clustering | Local ordering characterization | Structural landscape analysis [27] | |
| Control Strategies | Membrane area modulation | Supersaturation kinetics adjustment | Pathway selection without boundary layer disruption [25] |
| In-line filtration | Crystal retention and scaling reduction | Sustained supersaturation management [25] | |
| Site-directed mutagenesis | Intermolecular bonding tuning | Polymorph control via molecular engineering [26] | |
| Gel seeding | Template-directed nucleation | Selective polymorph formation [26] |
The systematic investigation of interfacial energy and supersaturation as governing parameters in nucleation pathway selection provides researchers with a principled framework for controlling crystallization outcomes across diverse material systems. The evidence presented demonstrates that these factors do not operate in isolation but interact complexly to determine the energetic landscape through which nucleation proceeds.
Future research directions will likely focus on the real-time manipulation of these parameters during crystallization processes, enabled by advanced process analytical technologies and machine-learning approaches for dynamic control. The integration of multi-scale modeling—from molecular simulations to process-level optimization—promises to unite fundamental understanding with practical application, particularly in pharmaceutical development where polymorph control remains a critical challenge.
As characterization techniques continue to improve, particularly in the realm of in-situ monitoring with enhanced temporal and spatial resolution, our understanding of the subtle transitions between nucleation pathways will refine further, enabling increasingly precise control over crystalline materials design and production.
Observing the Unseeable: Methodologies and Applications in Material Science
The study of nucleation mechanisms, particularly the debate between classical and non-classical pathways in organic crystal formation, relies heavily on advanced characterization techniques. Classical nucleation theory (CNT) posits a direct, one-step formation of crystalline phases from solution, where atoms or molecules add to a growing nucleus. In contrast, non-classical theory suggests more complex, multi-step pathways involving intermediate phases such as dense liquid droplets or amorphous precursors. Molecular Dynamics (MD), Liquid-Phase Electron Microscopy (LP-EM), and Dynamic Light Scattering (DLS) provide the necessary multi-scale analytical framework to distinguish between these mechanisms by offering insights into temporal and structural evolution from molecular to nanoscale levels. This technical guide examines these core techniques, their methodologies, and their specific applications in modern pharmaceutical and materials research on nucleation phenomena.
Theoretical Framework: Connecting Techniques to Nucleation Theory
The Critical Role of Advanced Characterization
Distinguishing classical from non-classical nucleation pathways requires observing processes across multiple length and time scales. MD simulations model atomic-level interactions and the earliest stages of molecular assembly, providing data on the free energy landscape and critical nucleus formation that are challenging to obtain experimentally. LP-EM enables direct visualization of nucleation events and nanoparticle evolution in their native liquid environment, potentially capturing non-classical intermediate phases. DLS offers solution-based monitoring of hydrodynamic size and population distributions over time, sensitive to the presence of precursor species or aggregation processes indicative of non-classical pathways. The complementary nature of these techniques creates a powerful toolkit for building mechanistic nucleation models.
Table 1: Technique Capabilities in Nucleation Research
| Technique | Spatial Resolution | Temporal Resolution | Key Measurable Parameters | Relevance to Nucleation Studies |
|---|---|---|---|---|
| Molecular Dynamics | Atomic-scale (Å) | Femtoseconds to microseconds | Free energy (ΔG), Critical nucleus radius, Molecular trajectories | Calculates nucleation barriers and critical nucleus sizes; simulates early molecular clustering |
| Liquid-Phase EM | Near-atomic to nanoscale (1-10 Å in situ) | Milliseconds to minutes | Particle size, Morphology, Assembly pathways | Directly visualizes nucleation events and intermediate phases in liquid phase |
| Dynamic Light Scattering | Nanoscale (sub-nm to μm) | Microseconds to hours | Hydrodynamic diameter, Size distribution, Diffusion coefficient | Monitors size evolution and population changes in real-time, detects precursor populations |
Dynamic Light Scattering: Principles and Applications in Nucleation
Fundamental Principles
Dynamic Light Scattering operates by measuring the Brownian motion of particles in suspension. When a monochromatic laser light encounters macromolecules or nanoparticles, the scattered light undergoes intensity fluctuations due to the constant motion of the particles. Analysis of these fluctuations via an autocorrelation function yields the diffusion coefficient (D~t~), which relates to the hydrodynamic diameter (D~h~) through the Stokes-Einstein equation [28] [29]:
D~h~ = k~B~T / 3πηD~t~
Where k~B~ is Boltzmann's constant, T is temperature, and η is solvent viscosity. The hydrodynamic size represents the diameter of a sphere that diffuses identically to the particle being measured, including any solvation layers [29].
Critical Experimental Protocol for Nucleation Studies
Sample Preparation:
- Prepare solutions using high-purity solvents and analytes; filter through appropriate membranes (e.g., 0.02-0.2 μm Anopore syringefilters) to remove dust and pre-existing nuclei
- Use clean, particle-free cuvettes to avoid interference from contaminants
- Optimize concentration to balance signal-to-noise ratio while avoiding multiple scattering (typically 0.1-1 mg/mL for proteins)
Instrument Calibration and Measurement:
- Temperature equilibration is critical (wait 5-15 minutes after loading)
- Set temperature control with accuracy of ±0.1°C due to viscosity dependence
- For nucleation studies, use backscatter detection (e.g., 173°) to minimize multiple scattering
- Set measurement duration to capture sufficient correlation decay (typically 5-10 runs of 10-60 seconds each)
- Monitor correlation function intercept (should be 0.1-1.0; values outside indicate poor signal)
Data Analysis and Interpretation:
- Use cumulants analysis for monomodal distributions (reporting z-average and PDI)
- Apply multiple algorithms (e.g., non-negative least squares, maximum entropy) for polydisperse systems
- For nucleation studies, perform time-resolved measurements to track size evolution
- Employ a dilution series and dynamic Debye plot when concentration effects are suspected [30] [31]
Table 2: DLS Data Interpretation Guide
| Parameter | Definition | Interpretation | Nucleation Context |
|---|---|---|---|
| Z-Average Diameter | Intensity-weighted harmonic mean size | Primary size indicator from cumulants analysis | Average size of nucleating particles |
| Polydispersity Index (PDI) | Width of distribution | <0.05: monodisperse; 0.05-0.08: nearly monodisperse; >0.08: polydisperse | Indicates uniformity of nuclei population |
| Intensity Distribution | Size distribution by scattered intensity | Sensitive to larger particles/aggregates | Detects early aggregate formation |
| Number Distribution | Transformation from intensity | Estimation of particle number; use with caution | Can misrepresent populations in polydisperse systems |
| Correlation Function Intercept | Signal-to-noise ratio | <0.1 indicates poor measurement conditions | Validates data quality for nucleation kinetics |
Applications in Nucleation Research and Limitations
DLS provides crucial evidence for distinguishing nucleation mechanisms through time-resolved size distribution analysis. In non-classical nucleation pathways, DLS can detect the presence of stable pre-nucleation clusters or amorphous intermediates before crystal appearance. For example, in protein crystallization, DLS can identify conditions where proteins form dense liquid phases prior to ordering. The technique's sensitivity to sub-populations makes it valuable for detecting transient species in the early stages of nucleation [28] [31].
Key limitations include:
- Hydrodynamic size interpretation: Differences from TEM sizes arise from solvation, particle shape, and concentration effects rather than just "hydration shell" [31]
- Concentration dependence: Interparticle interactions at high concentrations affect diffusion; requires extrapolation to infinite dilution for accurate sizing [30]
- Size resolution limits: Approximately 0.6 nm for modern instruments with optimal configuration [30]
- Assumption of sphericity: Non-spherical particles require complementary techniques for accurate morphology assessment
Molecular Dynamics Simulations
Principles and Methodologies
Molecular Dynamics simulations calculate the time evolution of a molecular system by numerically solving Newton's equations of motion for all atoms. The forces between atoms are derived from molecular mechanics force fields, which approximate the potential energy surface of the system. Key components include bond stretching, angle bending, torsion terms, and non-bonded interactions (van der Waals and electrostatic). Modern MD implementations can simulate systems containing millions of atoms over microsecond timescales, capturing essential molecular processes involved in nucleation [32].
For nucleation studies, MD provides atomic-resolution insight into the formation of critical nuclei, the structure of the solution-solute interface, and the molecular kinetics of assembly. Advanced sampling techniques such as metadynamics and umbrella sampling enable calculation of free energy barriers, a crucial parameter in nucleation theory [32].
Computational Protocol for Nucleation Studies
System Setup:
- Build initial configuration with solute molecules randomly distributed in solvent box
- Apply periodic boundary conditions to minimize edge effects
- Select appropriate force field (e.g., CHARMM, AMBER, OPLS-AA) with validated parameters for specific molecules
- Set up electrostatic interactions using Particle Mesh Ewald method
Simulation and Analysis:
- Energy minimization to remove steric clashes
- Equilibration in NVT and NPT ensembles (1-10 ns)
- Production run with trajectory saving (nanoseconds to microseconds)
- Implement enhanced sampling for rare events like nucleation
- Analyze using:
- Cluster analysis to identify nascent nuclei
- Steinhardt order parameters to distinguish ordered/disordered clusters
- Committor analysis for true transition states
- Potential of Mean Force calculations for free energy barriers
Applications to Nucleation Theory Validation
MD simulations have provided critical evidence challenging purely classical nucleation models. Studies of calcium carbonate, for instance, revealed stable pre-nucleation clusters that contradict CNT assumptions. Similarly, MD of protein systems has shown that the free energy landscape of early-stage assembly often features multiple minima corresponding to different oligomeric states or polymorphic forms [32].
Recent advancements integrate MD with experimental data through:
- Experimental parameterization of force fields using DLS and other solution-phase data
- Multi-scale modeling approaches that bridge quantum mechanical calculations with coarse-grained MD
- Markov state models to extract kinetic information from multiple trajectories
Liquid-Phase Electron Microscopy
Principles and Technical Considerations
Liquid-Phase Electron Microscopy enables direct observation of nanoscale processes in liquid environments with near-atomic resolution. The technique employs specialized liquid cells with electron-transparent windows (typically silicon nitride) that encapsulate the liquid sample. As the electron beam traverses the sample, scattered electrons form an image that captures dynamic processes in real time. For nucleation studies, LP-EM provides unprecedented direct visualization of the early stages of crystal formation and growth [31].
Experimental Protocol for Nucleation Studies
Liquid Cell Preparation:
- Assemble liquid cell with appropriate window thickness (typically 10-50 nm)
- Load solution containing solute at controlled concentration
- Implement flow cells for continuous replenishment of solution to minimize radiolysis effects
- For nucleation studies, often use mixing cells to initiate crystallization at specific time points
Imaging Parameters:
- Use low electron doses (1-100 e/Ų) to minimize beam effects
- Employ fast imaging to capture dynamic events
- Implement stage heating/cooling for temperature-controlled studies
- Acquire video sequences at 1-30 frames per second depending on process kinetics
Data Analysis:
- Track particle appearance, growth, and dissolution events
- Measure size and morphology evolution
- Correlate with electron diffraction to identify crystalline phases
- Statistical analysis of multiple nucleation events
Applications in Nucleation Mechanism Elucidation
LP-EM has directly revealed non-classical nucleation pathways in various systems. Observations of oriented attachment of nanoparticles, formation of amorphous precursors, and liquid-liquid phase separation preceding crystallization provide visual evidence challenging CNT. In pharmaceutical contexts, LP-EM can track polymorphic transformations and the effect of additives on nucleation pathways at unprecedented spatial and temporal resolution [31].
Key considerations for nucleation studies:
- Electron beam effects can induce secondary nucleation or alter pathways
- Statistical significance requires analysis of multiple events across different regions
- Resolution limitations in liquid (~1-2 nm) may obscure molecular-level details
- Correlation with bulk techniques like DLS is essential to validate observations
Integrated Workflow for Nucleation Studies
The most powerful approach to elucidating nucleation mechanisms combines all three techniques in a complementary workflow. MD simulations generate atomic-level hypotheses about molecular assembly pathways, which can be tested experimentally. DLS provides solution-phase validation of predicted behaviors under realistic conditions. LP-EM offers direct visualization of the nanostructures that form during nucleation, bridging molecular and microscopic scales.
Diagram 1: Integrated technique workflow for nucleation mechanism studies
Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Nucleation Studies
| Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Purified Macromolecules | Lysozyme, Apotransferrin, BSA | Model proteins for nucleation studies | Require characterization of initial oligomeric state by DLS [33] |
| Pharmaceutical Compounds | APIs: Paracetamol, Ibuprofen, Glycine | Study of drug crystallization pathways | Monitor polymorphic transitions via combined DLS/LP-EM [33] |
| Solvent Systems | Aqueous buffers, Organic solvents, Mixed solvents | Control solubility and nucleation kinetics | Viscosity and refractive index critical for DLS interpretation [30] |
| Nucleation Modifiers | Polymers, Additives, Ionic liquids | Alter nucleation pathways and kinetics | Study mechanisms via MD simulation of additive interactions [32] |
| Characterization Standards | Nanosphere size standards, Viscosity standards | Instrument calibration and validation | Essential for quantitative comparison across techniques [31] |
The integration of Molecular Dynamics, Liquid-Phase EM, and Dynamic Light Scattering provides a powerful multi-scale approach to unraveling nucleation mechanisms in organic crystals. MD delivers atomic-level understanding of the free energy landscape and molecular interactions driving nucleation. DLS offers robust solution-phase characterization of size distributions and population dynamics under realistic conditions. LP-EM bridges these scales with direct visualization of nanostructure formation and evolution. Together, these techniques enable researchers to move beyond the classical/non-classical dichotomy toward a more nuanced understanding of nucleation as a system-dependent process with multiple potential pathways. This integrated methodological framework continues to drive advances in pharmaceutical development, materials design, and fundamental physical chemistry.
Nucleation, the initial step in phase transformations, fundamentally determines the microstructure and properties of metallic materials. While Classical Nucleation Theory (CNT) has long provided the foundational framework for understanding this process, recent advanced molecular dynamics simulations have revealed pervasive nonclassical pathways in metallic systems like iron. This whitepaper examines compelling evidence from atomic-scale simulations demonstrating how FCC iron circumvents CNT-predicted energy barriers through mechanisms such as multi-step nucleation and cluster coalescence. These findings necessitate a paradigm shift in our fundamental understanding of crystallization processes in metals, with significant implications for materials design, geophysics, and pharmaceutical development where precise control over crystal structure is critical.
Classical Nucleation Theory (CNT) has served as the predominant theoretical model for describing phase transformations for over a century. CNT posits that nucleation occurs through a single-step process where monomeric units (atoms, ions, or molecules) stochastically assemble to form a nucleus of the new phase. This process must overcome a free-energy barrier arising from the competition between the bulk free energy gain of the new phase and the energy cost of creating the interface between the parent and product phases [34]. The critical nucleus size represents the point where this energy barrier is maximized – smaller clusters tend to dissolve, while larger ones are statistically likely to grow.
Despite its widespread application, CNT faces significant challenges in accurately predicting nucleation behavior across diverse systems. The theory often underestimates nucleation rates, requires unrealistically large undercooling to match experimental observations and struggles to account for complex pathways involving intermediate states. This is particularly evident in iron, where direct nucleation of the stable hexagonal close-packed (HCP) phase under Earth's core conditions was calculated to require approximately 1,000 K of undercooling – a thermodynamically improbable scenario given the core's estimated cooling rate of 100 K/Gyr [35]. This "inner core nucleation paradox" exemplifies the fundamental limitations of the classical model and has motivated the search for alternative nucleation mechanisms.
Nonclassical Nucleation Mechanisms
Nonclassical nucleation encompasses multiple pathways that deviate from the single-step CNT model by involving transient intermediate states. These mechanisms provide lower-energy alternative routes for phase transformation.
Key Nonclassical Pathways in Metallic Systems
Multi-step Nucleation: This pathway involves the formation of one or more metastable intermediate crystalline phases before the appearance of the stable phase. In iron, this commonly manifests as FCC→BCC transformation through an intermediate phase or BCC→HCP transition via a metastable FCC intermediate [36] [35].
Cluster Coalescence: Instead of single-atom addition, this mechanism involves the assembly and merging of pre-existing subcritical clusters that individually would be unstable [7]. These clusters coalesce to form a supercritical nucleus, effectively bypassing the high energy barrier associated with the classical pathway.
Dense Liquid Precursor: Some systems undergo liquid-liquid phase separation before crystallization, forming a dense liquid phase that acts as a precursor for the solid phase [34]. While more commonly reported in biomineralization systems like calcium carbonate, similar mechanisms may operate in metallic systems under specific conditions.
Thermodynamic and Kinetic Advantages
Nonclassical pathways provide significant thermodynamic and kinetic advantages over classical nucleation. The multi-step mechanism reduces the overall energy barrier by proceeding through intermediate phases with lower interfacial energies against the parent phase [35]. Cluster coalescence enables the system to leverage the stability of pre-existing assemblies rather than building the nucleus atom-by-atom. These alternative pathways explain how nucleation can occur under conditions where CNT predicts kinetic inhibition, effectively resolving paradoxes like the inner core nucleation problem.
FCC Iron: A Paradigm Case for Nonclassical Nucleation
Homogeneous Nucleation Mechanisms
Advanced molecular dynamics simulations of homogeneous BCC nucleation in FCC iron have revealed two predominant nonclassical mechanisms that enable the system to circumvent the high energy barrier predicted by CNT [7]. The stepwise nucleation pathway involves a structural transformation through a distinct intermediate phase rather than a direct FCC→BCC transition. Simultaneously, cluster coalescence occurs where discrete subcritical BCC clusters aggregate to form a stable nucleus. These mechanisms collectively enable nucleation under conditions that would be kinetically forbidden according to classical theory.
Heterogeneous Nucleation at Defect Sites
Heterogeneous nucleation at defect sites exhibits even more complex nonclassical behavior. Molecular dynamics simulations of BCC-phase nucleation at FCC-grain-boundary dislocations reveal a pseudo-cylindrical morphology of the nucleus that follows the dislocation network [36]. While the overall energy change as a function of nucleus size conforms to Cahn's classical model with no energy barrier, the atomic-scale pathway shows distinct nonclassical characteristics.
The transformation proceeds through a three-step "FCC→intermediate→BCC" process rather than a direct structural transition. Additionally, researchers observed the aggregation of discrete subnuclei along the dislocation line, which subsequently coalesce into a continuous nucleus. These nonclassical aspects contribute significantly to reducing the energy barrier and stabilizing the BCC nucleus, even when the overall thermodynamics appear classical.
Quantitative Analysis of Nucleation Mechanisms
Table 1: Comparative Analysis of Nucleation Mechanisms in FCC Iron
| Mechanism | Nucleation Pathway | Energy Barrier | Critical Nucleus Size | Experimental Evidence |
|---|---|---|---|---|
| Classical (CNT) | Direct FCC→BCC | High | Large | Not observed in MD simulations |
| Stepwise Nucleation | FCC→intermediate→BCC | Reduced | Variable | MD simulations [7] |
| Cluster Coalescence | Subcritical clusters → BCC | Significantly reduced | Distributed | MD simulations [7] |
| Grain Boundary Assisted | FCC→intermediate→BCC at dislocations | Minimal/None | Pseudo-cylindrical | MD simulations [36] |
Nonclassical Nucleation in Other Metallic Systems
The phenomenon of nonclassical nucleation extends well beyond iron, appearing in diverse metallic systems under various conditions.
BCC to HCP/FCC Transitions in Iron
Under anisotropic compressions at high strain rates, BCC iron undergoes complex phase transformations to HCP and FCC structures that demonstrate strong nonclassical characteristics [37]. The transformation pathway and resulting microstructure show remarkable sensitivity to the strain path and resulting shear deformations. Under 1D-dominated compression (along the [001] direction), HCP nucleation predominates with only limited FCC stacking faults. In contrast, 2D-dominated compression activates more equivalent slip systems and significantly increases the FCC mass fraction, eventually exceeding the HCP phase fraction under certain conditions [37].
This strain-path dependence demonstrates how external mechanical fields can selectively promote different nucleation pathways by altering the relative stability of competing intermediate states. The ability to manipulate phase selection through strain engineering has profound implications for materials processing and design.
The Earth's Inner Core: A Geophysical Manifestation
The formation of Earth's inner core presents a fascinating geophysical example of nonclassical nucleation. The "inner core nucleation paradox" arose when calculations indicated that direct nucleation of stable HCP iron would require approximately 1,000 K of undercooling – impossible given Earth's thermal history [35]. This paradox finds resolution through a two-step nucleation mechanism where the metastable BCC phase nucleates first, followed by transformation to the stable HCP phase.
Using the persistent embryo method and molecular dynamics simulations, researchers demonstrated that the BCC phase has a significantly higher nucleation rate than HCP under inner core conditions [35]. The nucleation barrier for BCC iron is substantially lower than for HCP across all undercooling temperatures studied, despite HCP having a larger bulk driving force. This two-step pathway reduces the required undercooling to physically plausible levels, effectively resolving the nucleation paradox.
Table 2: Nucleation Parameters for Iron Phases at 323 GPa (Inner Core Boundary)
| Phase | Melting Temperature (K) | Nucleation Barrier (eV) | Critical Nucleus Size (atoms) | Relative Nucleation Rate |
|---|---|---|---|---|
| BCC | 5,750 | 65 | ~550 | High |
| HCP | 6,350 | 110 | ~950 | Low |
| Liquid | - | - | - | - |
Comparative Analysis Across Metallic Systems
The prevalence of nonclassical nucleation pathways across metallic systems reveals common principles:
- Multi-step pathways consistently demonstrate lower activation barriers than direct transformations
- Metastable intermediate phases play crucial roles in facilitating nucleation kinetics
- Mechanical stress states significantly influence the preferred nucleation pathway
- Nucleation rates for metastable phases often exceed those for stable phases under realistic conditions
These common features suggest that nonclassical nucleation is not an exception but rather a fundamental characteristic of phase transformations in metallic systems, necessitating a revision of theoretical models and computational approaches.
Methodologies for Investigating Nonclassical Nucleation
Molecular Dynamics Simulation Protocols
Molecular dynamics (MD) simulations have emerged as the primary tool for investigating nonclassical nucleation mechanisms at atomic resolution. The standard protocol involves:
System Preparation: Create initial FCC iron crystal structure with 80×80×80 unit cells (approximately 2 million atoms) oriented along [100], [010], and [001] directions [37]. Employ periodic boundary conditions in all directions to minimize surface effects.
Potential Selection: Utilize embedded-atom-method (EAM) potentials specifically developed for high-pressure iron phases [35]. Critical potential validation includes comparison with ab initio MD results for latent heat, elastic properties, and liquid structure [35].
Strain Application: Apply anisotropic compression with fixed bulk strain rate (typically 1.5×10⁹ s⁻¹) while varying strain ratios along different crystallographic directions to introduce controlled shear components [37].
Trajectory Analysis: Identify phase transformations using multiple complementary methods: radial distribution functions (RDFs), coordination numbers (CNs), and centro-symmetry parameters (CSPs) [37]. Track nucleation events through potential energy, stress tensor components, and local bond order parameters.
Enhanced Sampling Techniques
Conventional MD faces limitations in simulating rare nucleation events. Advanced sampling methods overcome these constraints:
Persistent Embryo Method (PEM): This approach identifies critical nucleus sizes by monitoring plateau regions in nucleus growth curves [35]. The nucleation barrier is calculated as ΔG* = ½|Δμ|N, where Δμ is the bulk free energy difference and N is the critical nucleus size.
Free Energy Calculations: Determine Δμ using the Gibbs-Helmholtz equation combined with MD simulations of solid-liquid coexistence [35]. These calculations enable quantitative comparison of nucleation barriers between competing phases.
Machine Learning Enhancement: Recent advances leverage machine learning to identify collective variables and reaction coordinates that accurately describe complex nucleation pathways [38]. This approach enables more efficient exploration of configuration space and identification of previously unknown intermediate states.
Experimental Validation Methods
While simulation provides atomic-scale insights, experimental validation remains crucial:
Cryogenic Electron Microscopy: Enables direct imaging of transient intermediate phases in materials like calcium carbonate, providing reference frameworks for metallic systems [34].
High-Pressure X-ray Diffraction: Provides experimental determination of phase stability and melting curves under Earth's core conditions [35].
Dynamic Light Scattering: Tracks nucleation and growth kinetics in solution-based systems, revealing size evolution and Ostwald ripening phenomena [34].
Table 3: Key Computational and Experimental Resources for Nucleation Research
| Tool/Resource | Function | Application Example |
|---|---|---|
| EAM Potentials | Describe atomic interactions in metals | Voter-Chen potential for iron phase transitions [37] |
| LAMMPS | Molecular dynamics simulation | Large-scale nucleation simulations [37] |
| Persistent Embryo Method | Enhanced nucleation sampling | Critical nucleus size determination [35] |
| Centro-symmetry Parameter | Crystal structure identification | Distinguishing HCP/FCC/BCC coordination [37] |
| High-Pressure DAC | Experimental pressure application | Iron phase studies under core conditions [35] |
The compelling evidence from FCC iron and other metallic systems demonstrates that nonclassical nucleation mechanisms represent the rule rather than the exception in solid-state phase transformations. The multi-step pathways, cluster coalescence, and intermediate phases observed across diverse systems reveal fundamental limitations of Classical Nucleation Theory and necessitate more sophisticated theoretical frameworks.
These findings have profound implications across multiple disciplines. In geophysics, the two-step nucleation mechanism resolves the long-standing inner core nucleation paradox [35]. In materials science, understanding nonclassical pathways enables better control of microstructure evolution during processing. In pharmaceutical development, similar principles apply to controlling polymorph selection in organic crystals [38].
Future research directions should focus on developing quantitative predictive models that incorporate the complexity of nonclassical pathways, extending investigations to multi-component systems with light elements, and leveraging machine learning approaches to accelerate the discovery of novel nucleation mechanisms [38]. The integration of advanced simulation methods with experimental validation will continue to unravel the complexities of nucleation, enabling precise control of crystalline materials across scientific and industrial applications.
The control of crystal polymorphs—solid materials with identical chemical compositions but different crystal structures—represents a fundamental challenge in materials science and pharmaceutical development. Polymorphs exhibit distinct physical and chemical properties, making their selective formation crucial for product performance and functionality in industrial applications [39] [40]. Despite its importance, the mechanistic understanding of polymorph selection during crystallization has remained incomplete, particularly within the ongoing scientific discourse comparing classical and non-classical nucleation theories.
Classical nucleation theory posits a single-step process where randomly arranged atoms or molecules in a solution spontaneously form stable crystals of the most thermodynamically favorable phase. In contrast, non-classical nucleation theory suggests a multi-step pathway where metastable intermediate phases form first, subsequently transforming into the stable polymorph [41]. This case study examines how recent research utilizing colloidal crystals as model systems has provided unprecedented molecular-level insights into these non-classical pathways, specifically through the technique of heteroepitaxial growth.
Experimental System and Methodology
Colloidal Crystals as a Model System
Colloidal crystals contain suspended, submicron-sized particles arranged in ordered arrays analogous to atomic or molecular crystals. Their primary advantage for crystallization studies lies in the ability to perform in-situ observations with single-particle resolution using conventional microscopy techniques, enabling direct visualization of processes that occur at inaccessible scales in molecular systems [39] [40] [42]. This system bridges the conceptual gap between theoretical models and experimental observation in crystallization research.
Heteroepitaxial Growth Protocol
The research employed heteroepitaxial growth, where a crystalline epitaxial phase grows on a substrate crystal with a different composition or structure [40]. The specific experimental protocol encompassed several critical stages:
- Substrate Fabrication: Thin colloidal crystal films were fabricated using convective assembly with polystyrene particles of specific sizes (1100 nm, 1300 nm) [41] [40].
- Epitaxial Phase Preparation: Polystyrene particles of different sizes (860 nm, 700 nm, or 790 nm) were suspended in solution for the epitaxial growth phase [41].
- Induced Depletion Attraction: Depletion attraction between particles was induced by adding sodium polyacrylate polymers (polymerization degree: 30,000-40,000) to the solution. The polymer concentration (Cp) was carefully controlled as it directly modulates the intensity of particle interactions [41].
- In-situ Observation: The crystallization process was monitored in real-time using optical microscopy, with analysis of particle number densities (ρ) on the substrate and characterization of crystal structures through orientational order parameters (θ6) [41].
Table 1: Key Experimental Components in Colloidal Heteroepitaxy
| Component | Specification | Function/Role |
|---|---|---|
| Substrate Particles | 1100-1300 nm polystyrene | Provides templating surface for epitaxial growth |
| Epitaxial Particles | 700-860 nm polystyrene | Forms polymorphic structures on substrate |
| Depletion Agent | Sodium polyacrylate polymer | Induces attractive interparticle forces |
| Size Ratio | ~0.78 (epitaxial/substrate) | Critical parameter for polymorph formation |
| Primary Analysis | Single-particle resolution imaging | Enables direct observation of nucleation pathways |
Results: Polymorphic Transitions and Nucleation Pathways
Polymorph Formation and Characteristics
The heteroepitaxial growth system produced two distinct polymorphic structures through meticulous control of the particle size ratio between epitaxial and substrate particles at approximately 0.78 [41] [40]:
- α-phase: Characterized as three-dimensional islands with the same in-plane orientation as the substrate. Particles in this phase did not localize to specific positions on the substrate, allowing variable particle distances that decreased with increasing polymer concentration [41].
- β-phase: Appeared as two-dimensional layers with a 30° rotated in-plane orientation relative to the substrate. Particles occupied specific positions (either above substrate particles or between them) with constant particle distances unaffected by polymer concentration changes [41].
The system exhibited enantiotropic behavior, where the relative stability of the polymorphs reversed depending on polymer concentration. The α-phase was more stable at low Cp, while the β-phase dominated at high Cp, with their solubility curves intersecting at intermediate Cp values [41].
Three Types of Polymorphic Transitions
The research identified three distinct polymorphic transition (PT) mechanisms that transformed crystallization into non-classical pathways:
Diagram 1: Polymorphic transition pathways in colloidal heteroepitaxy.
- Type I and II: Solid-State Transitions: These transitions occurred directly within the solid phase, where a cluster of one polymorph transformed into the other without dissolving into solution [41].
- Type III: Solution-Mediated Transition: This unique transition type, rarely observed in hard-sphere systems, involved the dissolution of one polymorph followed by the nucleation of the other from the solution phase [41].
Critical Factors Governing Polymorphic Transitions
The research revealed that polymorph selection depends not solely on traditional thermodynamic stability considerations but significantly on kinetic factors:
- Cluster Stability Dictates Nucleation: The probability of polymorphic transitions during nucleation depended more on the stability of metastable clusters than on bulk phase stability. This finding directly challenges classical nucleation theory, which predicts formation of the most thermodynamically stable phase [41].
- Crystal Size Influences Transitions: The size of polymorphic crystals emerged as a critical parameter governing transitions during growth, with specific size thresholds determining transition probabilities [41].
- Pathway Complexity: The final polymorphic product resulted from the complex interplay of these transition types throughout nucleation and growth stages, illustrating the multi-pathway nature of non-classical crystallization [41] [40].
Table 2: Characteristics of Polymorphic Transitions in Colloidal Heteroepitaxy
| Transition Type | Mechanism | Key Influencing Factors | Observation Frequency |
|---|---|---|---|
| Type I: Solid-State | Direct structural transformation | Cluster stability, orientation | Frequent at specific Cp ranges |
| Type II: Solid-State | Direct structural transformation | Cluster stability, orientation | Frequent at specific Cp ranges |
| Type III: Solution-Mediated | Dissolution followed by nucleation | Relative solubility, polymer concentration | Unique to attractive systems |
| All Types Combined | Multiple pathways | Polymer concentration, particle density | Determines final polymorph distribution |
Polymorph Control Strategies
Manipulation of Experimental Conditions
The research demonstrated that polymorphic outcomes can be controlled through several strategic approaches:
- Polymer Concentration Modulation: By adjusting Cp, researchers could manipulate the relative stability of polymorphs, selecting for either α-phase (low Cp) or β-phase (high Cp) based on the enantiotropic relationship [41].
- Particle Size Ratio Control: Maintaining the critical particle size ratio of approximately 0.78 between epitaxial and substrate particles was essential for forming the distinct polymorphic structures [41] [43].
- Particle Additives: Introducing differently sized particles effectively controlled polymorph formation by exploiting their differential effects on cluster formation, which varied due to structural differences between polymorphs [41] [39].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Colloidal Heteroepitaxy
| Reagent/Material | Function in Experiment | Technical Specifications |
|---|---|---|
| Polystyrene Particles | Primary building blocks for crystal formation | Diameters: 700-1300 nm; precise size matching critical |
| Sodium Polyacrylate | Depletion agent inducing interparticle attraction | Polymerization degree: 30,000-40,000 |
| Size-Tuned Additives | Polymorph control through selective cluster effects | Varied particle sizes to target specific polymorphs |
| Aqueous Dispersion Medium | Solvent for colloidal suspension | Enables Brownian motion and self-assembly |
Implications for Classical vs. Non-Classical Nucleation Theory
This research provides compelling experimental evidence supporting non-classical nucleation theory through several key observations:
- Metastable Intermediates: The consistent observation of polymorphic transitions during nucleation and growth confirms the existence and importance of metastable intermediate phases, a cornerstone of non-classical theory [41] [40].
- Pathway Dependency: The finding that multiple pathways (through different transition types) lead to final polymorphic products contradicts the single-pathway assumption of classical theory [41].
- Kinetic Control Dominance: The demonstrated importance of kinetic factors (cluster stability, crystal size) over purely thermodynamic considerations in polymorph selection further aligns with non-classical perspectives [41] [39] [42].
Diagram 2: Classical versus non-classical nucleation pathways.
The colloidal crystal model demonstrates that non-classical behavior emerges naturally when polymorphic transitions occur during nucleation and growth, transforming what might appear as direct crystallization into a multi-step process with metastable intermediates [41]. This mechanistic understanding provides a foundation for developing more predictive models of crystallization across material systems.
This case study demonstrates that colloidal heteroepitaxy provides a powerful model system for elucidating the complex mechanisms of polymorph selection during crystallization. The research establishes that polymorphic transitions play a decisive role in determining nucleation and growth pathways, ultimately controlling final crystalline products. Rather than following simple thermodynamic predictions, these processes involve intricate interplay between cluster stability, crystal size, and environmental conditions.
The findings significantly advance the understanding of non-classical nucleation phenomena, providing concrete evidence for multi-step pathways with metastable intermediates. From a practical perspective, the identified control strategies—including manipulation of interaction parameters, particle size ratios, and selective additives—offer tangible approaches for polymorph regulation technologies with potential applications in pharmaceutical development and advanced material fabrication [41] [39] [40]. As research continues, these insights bridge fundamental crystallization theory with practical polymorph control, addressing a critical need across multiple scientific and industrial domains.
In the pathophysiology of malaria, the parasite's survival within human erythrocytes hinges on a critical biocrystallization process: the detoxification of heme into crystalline hemozoin [44]. During hemoglobin catabolism, the Plasmodium parasite releases Fe(II) heme, which rapidly oxidizes to toxic Fe(III) hematin. To avoid oxidative damage, the parasite sequesters this hematin into an inert crystalline pigment known as hemozoin [44]. The synthetic analogue of hemozoin, β-hematin, serves as the primary model for in vitro studies of this life-saving crystallization process [45] [44]. For decades, the most successful antimalarial drugs, including quinolines (e.g., chloroquine) and artemisinins, have shared a common mechanism: the inhibition of hematin crystallization [44] [46]. Consequently, understanding and disrupting the nucleation of β-hematin crystals is a cornerstone of antimalarial drug development. This guide examines these disruption mechanisms through the lens of classical and non-classical nucleation theories, providing researchers with the technical foundation and methodologies to advance this crucial biomedical field.
Classical vs. Non-Classical Nucleation Theory in Biocrystallization
The formation of β-hematin crystals can be understood by two competing theoretical frameworks, which inform distinct inhibition strategies.
Classical Nucleation Theory (CNT)
Classical Nucleation Theory (CNT) posits a single-step process where solute molecules (hematin) directly assemble into a stable crystal nucleus once a critical size is surpassed [45] [47]. The theory defines a high energy barrier, ΔG*, which is a function of the surface free energy (γ) at the crystal-solution interface and the supersaturation level [45]. For β-hematin, calculations using CNT predict an immense nucleation barrier—approximately 2500 times the thermal energy (k~B~T) at physiological supersaturations [45]. This presents a thermodynamic paradox, as such a high barrier should render nucleation imperceptibly slow, contradicting observed crystallization within parasites. While CNT suggests inhibition can occur by reducing solute concentration or increasing surface energy, it offers limited and coarse strategies for suppression, primarily through reducing the concentration of available hematin [45].
Non-Classical Nucleation Theory
Non-classical theory, particularly the two-step nucleation pathway, resolves the contradictions of CNT. This mechanism involves the initial formation of metastable, mesoscopic solute-rich clusters (dense-liquid droplets), within which crystal nuclei subsequently form [45] [47]. These clusters, a few tens of nanometers across, concentrate hematin to near-crystalline densities, thereby drastically lowering the free-energy penalty for building a critical nucleus [45]. The surface energy (γ) for a nucleus within a cluster is about 10-fold lower than for one in solution, reducing the nucleation barrier by approximately 1000-fold and making crystallization under physiological conditions feasible [45]. This pathway provides a richer framework for inhibition, as it introduces a vulnerable precursor stage—the mesoscopic cluster—that can be targeted by antimalarial compounds [45].
The following diagram illustrates this non-classical, two-step pathway for β-hematin crystallization and the potential points of inhibition.
Experimental System: A Biomimetic Approach
To yield physiologically relevant data, in vitro experiments must replicate the unique environment of the parasite's digestive vacuole (DV). The consensus from multiple studies is that a lipid-like organic phase is the preferred medium for crystallization.
- The Biomimetic Solvent: The most robust experimental results are obtained using n-octanol saturated with citric buffer at pH 4.8 (CBSO) [45] [44]. This solvent mimics the reported lipid nanospheres or the lipid-rich membrane interface within the DV. Key evidence supports its use:
- Crystal Morphology & Size: β-hematin crystals grown in CBSO exhibit the characteristic morphology and powder X-ray diffraction pattern of natural hemozoin and can reach sizes up to 30 μm, unlike those grown in aqueous buffers [44].
- Solubility & Kinetics: Hematin solubility in CBSO is ~10⁵ times higher than in aqueous buffer at the same pH. Since growth rates scale with solubility, this indicates crystallization from an organic phase is a significantly faster and more plausible detoxification pathway for the parasite [44].
- Inhibitor Solubility: While antimalarials like chloroquine have lower solubility in CBSO than in water, their measured solubility (0.19 mM) remains sufficient for effective inhibition, being twofold higher than hematin's solubility in CBSO [44].
Quantitative Effects of Antimalarials on Nucleation
The non-classical nucleation pathway enables a sophisticated level of control, where different compounds can selectively suppress, enhance, or have no effect on nucleation by modulating the precursor clusters [45]. The following table summarizes the quantified impact of various antimalarials on β-hematin nucleation rates, as determined by dynamic light scattering (DLS) assays in CBSO.
Table 1: Quantified Impact of Antimalarial Compounds on β-Hematin Nucleation [45]
| Antimalarial Compound | Class | Effect on Nucleation Rate | Proposed Molecular Mechanism |
|---|---|---|---|
| Pyronaridine (PY) | Quinoline | ~Full Suppression | Interacts with hematin monomers/dimers to suppress mesoscopic cluster formation [45]. |
| Chloroquine (CQ) | Quinoline | No Significant Effect | Does not significantly perturb nucleation precursor populations [45]. |
| Mefloquine (MQ) | Quinoline | No Significant Effect | Does not significantly perturb nucleation precursor populations [45]. |
| Heme-Artesunate (H-ARS) | Artemisinin adduct | Significant Enhancement | Promotes the formation or stability of mesoscopic clusters that host nucleation [45]. |
Detailed Experimental Protocol: Monitoring Nucleation & Inhibition
This section provides a detailed methodology for assessing β-hematin nucleation and its inhibition, based on the DLS protocols from the search results [45].
Reagent Preparation
- Citric Buffer-Saturated Octanol (CBSO): Saturate n-octanol with 0.1 M citric acid buffer, pH 4.8, by vigorous mixing for 1 hour. Allow phases to separate completely and use the organic (top) layer [44].
- Hematin Stock Solution: Dissolve hematin in CBSO to a concentration above the desired working concentration (e.g., 0.24 mM or 0.48 mM for 2x or 4x supersaturation). Solubility at 22°C is ~0.12 mM [45] [44]. Sonicate and filter (0.2 μm) to ensure complete dissolution and remove pre-existing particulates.
- Inhibitor Stock Solutions: Dissolve antimalarial compounds (e.g., PY, CQ, H-ARS) in CBSO at a concentration suitable for achieving the final desired testing concentration upon dilution in the hematin solution.
Dynamic Light Scattering (DLS) Assay
Objective: To track the real-time nucleation of β-hematin crystals by monitoring the evolution of scattered light intensity.
- Solution Preparation: In a clean DLS cuvette, prepare the working solution by mixing hematin stock solution with CBSO or inhibitor stock solution to achieve the final target hematin and inhibitor concentrations.
- Data Acquisition:
- Place the cuvette in the DLS instrument thermostatted to 22°C.
- Continuously measure the autocorrelation function,
g₂(τ), at short time intervals (e.g., every minute). - In a homogeneous solution with no crystals,
g₂(τ)will show negligible decay. The appearance and growth of a shoulder in theg₂(τ)function indicates the formation of β-hematin crystallites, whose Brownian motion causes intensity fluctuations [45].
- Data Analysis:
- The amplitude (A₁) of the autocorrelation function is proportional to the number and size of the nucleated crystallites.
- Plot A₁ versus time. A faster increase in A₁ indicates a faster nucleation rate. Compare the A₁ vs. time curves for solutions with and without inhibitors to quantify the extent of nucleation suppression or enhancement [45].
The workflow for this experimental protocol is summarized below.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for β-Hematin Nucleation Research
| Reagent / Material | Critical Function & Rationale |
|---|---|
| n-Octanol (saturated with citric buffer, pH 4.8) | Serves as a biomimetic solvent (CBSO) replicating the lipid subphase of the parasite digestive vacuole, essential for physiologically relevant crystal growth and inhibitor action [45] [44]. |
| Hematin (Fe(III)PPIX) | The core solute and crystallizing unit; the substrate for β-hematin formation and the primary target for antimalarial compounds [45] [44]. |
| Quinoline Antimalarials (e.g., Pyronaridine, Chloroquine) | Function as nucleation or growth modifiers. Pyronaridine is a key tool for studying potent nucleation suppression via precursor interaction [45]. |
| Artemisinin-Hematin Adducts (e.g., H-ARS) | Metabolite-based inhibitors that provide insights into non-classical pathways and irreversible growth inhibition at high driving forces [45] [46]. |
| Dynamic Light Scattering (DLS) Instrument | Enables real-time, label-free monitoring of nucleation kinetics by detecting the formation and diffusion of crystal nuclei [45]. |
| Atomic Force Microscopy (AFM) | Provides molecular-level, in-situ visualization of crystal surface evolution, step growth, and inhibitor binding mechanisms [44] [46]. |
Advanced Concepts: Irreversible Inhibition and Future Directions
Moving beyond simple nucleation suppression, recent research reveals cooperative mechanisms that lead to irreversible inhibition of β-hematin crystal growth, even after the inhibitor is removed from the system [46]. This is critically relevant for drug efficacy, given the limited residence time of antimalarials in the body.
- Artemisinin Adducts (H-ARS): At high hematin concentrations, H-ARS incites the copious nucleation of non-extendable nanocrystals. These incorporate into larger crystals, inducing internal lattice strain that permanently impedes further growth [46].
- Pyronaridine (PY): This quinoline drug promotes the formation of step bunches on crystal surfaces, which evolve into abundant dislocations—another source of persistent lattice strain that hinders recovery of growth [46].
These non-classical, irreversible inhibition mechanisms correlate with high-efficacy parasite killing in limited-time pulse studies, highlighting their biomedical significance and opening new avenues for designing next-generation antimalarials [46]. Furthermore, the exploration of metal-based antimalarial complexes (e.g., zinc- or copper-amodiaquine complexes) shows promise, as these compounds can exhibit enhanced inhibition of β-hematin formation and potent activity against resistant parasite strains [48].
The paradigm shift from classical to non-classical nucleation theory has fundamentally advanced the understanding of β-hematin formation. The identification of mesoscopic hematin-rich clusters as nucleation precursors provides a powerful and previously overlooked target for antimalarial intervention [45]. The experimental frameworks and data outlined in this guide provide researchers with the tools to probe these mechanisms further. The future of this field lies in leveraging these insights to design novel inhibitors that specifically target the nucleation precursor phase or exploit cooperative mechanisms to achieve irreversible crystal growth suppression, ultimately leading to more effective treatments for malaria.
Leveraging Solvent Effects and Additives to Direct Crystallization Pathways
The control of crystallization pathways represents a critical frontier in materials science and pharmaceutical development. This process is fundamentally governed by the competition between classical and non-classical nucleation mechanisms. Within the context of organic crystal research, directing this competition through solvent effects and strategic additives allows scientists to selectively target desired polymorphs, control crystal size and shape, and optimize material properties. The traditional view of crystallization, encapsulated by Classical Nucleation Theory (CNT), posits a direct, one-step formation of a stable crystal nucleus from a supersaturated solution. However, growing evidence across multiple systems reveals more complex, non-classical pathways involving metastable intermediates and pre-nucleation clusters [49] [7]. Understanding and leveraging these mechanisms is particularly crucial in the pharmaceutical industry, where over 90% of active pharmaceutical ingredients (APIs) are delivered as crystalline solids, and their solid form dictates critical performance characteristics including solubility, stability, and bioavailability [50]. This guide provides an in-depth technical framework for researchers and drug development professionals to actively direct these pathways, marrying theoretical foundations with practical experimental strategies.
Theoretical Framework: Classical vs. Non-Classical Nucleation
Classical Nucleation Theory (CNT)
Classical Nucleation Theory (CNT) provides the historical baseline for understanding crystallization. It describes a continuous process where molecules in a supersaturated solution spontaneously aggregate. The formation of a nucleus is governed by a free energy balance: the energy gain from the transition to a more stable phase is offset by the energy cost of creating a new surface. This results in an energy barrier that defines the critical nucleus size. A cluster must reach this critical size to become stable and proceed to grow into a macroscopic crystal. CNT assumes this process occurs in a single step, directly from solution to the thermodynamically stable phase. While CNT is powerful, it often fails to fully explain experimental observations, particularly in complex molecular systems where multiple solid forms can coexist.
Non-Classical Nucleation Mechanisms
Non-classical nucleation mechanisms challenge the simplicity of CNT by introducing intermediate steps. These pathways are characterized by the initial formation of metastable species that subsequently transform into the final, stable crystal. Key mechanisms include:
- Pre-nucleation Clusters: The formation of stable, dynamic aggregates in solution prior to the emergence of a definite crystalline structure [49].
- Multistep Nucleation via Metastable Polymorphs: A system first nucleates a metastable polymorph, which then acts as a precursor for the nucleation of the stable polymorph. A seminal study on glycine demonstrated that in aqueous solution with or without NaCl, the metastable β-glycine forms first. The additive (NaCl) then stabilizes this metastable form for several hours, preventing its conversion to the α-glycine, before the final stable γ-glycine nucleates on the surface of the β-glycine crystal [49].
- Coalescence and Stepwise Nucleation: Molecular dynamics simulations of iron have shown that systems can circumvent the high energy barrier of homogeneous CNT through mechanisms like the coalescence of subcritical clusters [7].
The table below summarizes the core distinctions between these two theoretical frameworks.
Table 1: Core Tenets of Classical vs. Non-Classical Nucleation Theory
| Feature | Classical Nucleation Theory (CNT) | Non-Classical Nucleation Theory |
|---|---|---|
| Nucleation Pathway | Single-step, direct | Multi-step, indirect |
| Mechanism | Homogeneous/heterogeneous formation of a critical nucleus | Often involves metastable intermediates (e.g., pre-nucleation clusters, liquid droplets, unstable polymorphs) |
| Governing Principles | Free energy balance between bulk (favorable) and surface (unfavorable) terms | Kinetic trapping, Ostwald's Rule of Stages, surface stabilization |
| Role of Additives | Primarily alter supersaturation or interfacial free energy | Can selectively stabilize or destabilize specific intermediates in the pathway |
| Observable Evidence | Challenging to observe directly | Detection of transient intermediate phases (e.g., via SCNS) [49] |
Visualizing Crystallization Pathways
The following diagram illustrates the key decision points between classical and non-classical pathways and how additives can influence them.
Quantitative Data on Additive and Solvent Effects
Empirical data is crucial for validating theoretical models and designing experiments. The following tables consolidate quantitative findings from key studies on how additives and molecular properties influence crystallization outcomes.
Table 2: Experimental Data on Additive-Induced Polymorphic Selection in Glycine
| System | Proposed Mechanism | Key Finding | Measurement Technique |
|---|---|---|---|
| Glycine in H₂O | Non-classical | β-glycine (metastable) forms first through a non-classical pathway. | Single-Crystal Nucleation Spectroscopy (SCNS) [49] |
| Glycine in H₂O + NaCl | Non-classical & Stabilization | NaCl stabilizes metastable β-glycine for several hours and prevents conversion to α-glycine. Final γ-glycine nucleates on β-glycine surface. | Single-Crystal Nucleation Spectroscopy (SCNS) [49] |
| Generic Protein Crystallization | Surface Entropy Reduction | Low levels of surface side chain entropy and a high fraction of small surface residues (e.g., Gly, Ala) assist crystallization. Replacing large residues (Lys, Glu) with Ala facilitates crystallization. | Gaussian Process Regression (GPR) on 182 proteins [51] |
Table 3: Impact of Key Surface Residues on Protein Crystallization Propensity
| Residue | Role/Property | Impact on Crystallization | Inferred Mechanism |
|---|---|---|---|
| Aromatic (F, Y, W) | Hydrophobic, large, can form π-π interactions | Significant role; small Gaussian Process length scale indicates high importance [51]. | Specific interactions at crystal contacts; hydrophobic patches. |
| Cysteine (C) | Potential for disulfide bonds, specific interactions | Previously undetected significant role identified by GPR model [51]. | May facilitate specific, directional intermolecular contacts. |
| Glutamic Acid (E) | Charged, can form salt bridges | Significant role; importance confirmed in current and earlier studies [51]. | Specific electrostatic interactions at crystal contacts. |
| Small Residues (G, A) | Low side chain entropy, flexible | High fraction assists crystallization; basis for Surface Entropy Reduction (SER) mutagenesis [51]. | Reduces conformational entropy penalty upon incorporation into crystal lattice. |
The Scientist's Toolkit: Research Reagent Solutions
Success in directing crystallization pathways relies on a suite of specialized reagents and tools. The following table details essential materials for conducting experiments in this field.
Table 4: Essential Research Reagents and Materials for Crystallization Studies
| Reagent/Material | Function/Application | Example Use-Case |
|---|---|---|
| Salt Additives (e.g., NaCl) | Modulate electrostatic interactions, stabilize specific crystal faces or metastable polymorphs. | Stabilizing the polar face of β-glycine, directing the pathway towards γ-glycine [49]. |
| High-Throughput Crystallization Screens | Arrays of chemical conditions (e.g., 1,536 conditions) to empirically identify initial crystal hits. | Initial screening of proteins or small molecules to find promising conditions for optimization [51]. |
| Divalent Cations (e.g., Mg²⁺) | Act as electrostatic bridges, particularly for charged molecules like DNA. | Essential for facilitating the interaction between negatively charged DNA and a mica substrate in substrate-assisted growth [52]. |
| Surface Entropy Reduction (SER) Kit | Mutagenesis primers for replacing large, flexible surface residues (Lys, Glu) with small residues (Ala). | Engineering a protein surface to reduce conformational entropy and promote crystal contact formation [51]. |
| Charged Substrates (e.g., Mica) | Provide a catalytic surface for nucleation via electrostatic interactions, lowering the entropic cost. | Used in Substrate-Assisted Growth (SAG) of DNA crystals to achieve precise coverage control [52]. |
Advanced Experimental Protocols
Protocol A: Single-Crystal Nucleation Spectroscopy (SCNS) for Pathway Analysis
Purpose: To investigate the early stages of crystallization and detect transient metastable polymorphs at the single-crystal level [49].
Materials:
- Microscope: In situ Raman microspectroscopy system.
- Sample Chamber: A controlled environment chamber for the solution (e.g., a well or capillary).
- Additives: High-purity salt or organic additives.
- Solution: Supersaturated solution of the target molecule (e.g., glycine).
Procedure:
- Sample Preparation: Prepare a supersaturated aqueous solution of the target molecule (e.g., glycine). For the test condition, add the selected additive (e.g., NaCl) at the desired concentration.
- Loading: Place a small volume (e.g., 5-10 µL) of the solution into the sample chamber of the Raman microscope.
- Data Acquisition: Focus the laser on a specific, microscopically small volume within the solution. Continuously monitor and collect Raman spectra over time at a high frequency (e.g., every few seconds).
- Nucleation Triggering: Allow nucleation to occur spontaneously or induce it through a controlled stimulus (e.g., a temperature jump).
- Spectral Analysis: Analyze the acquired Raman spectra in real-time or post-acquisition. Identify characteristic peaks that are unique to different polymorphs.
- Pathway Mapping: Correlate the temporal sequence of polymorph-specific spectral signatures to reconstruct the crystallization pathway (e.g., appearance of β-glycine peaks followed by their disappearance as γ-glycine peaks emerge).
Protocol B: Substrate-Assisted Growth (SAG) for Quantitative Crystal Growth Analysis
Purpose: To achieve molecular-level control over crystal coverage and study the effects of external parameters on crystal size and distribution, as demonstrated with DNA crystals [52].
Materials:
- Substrate: Negatively charged mica sheet (5 mm x 5 mm).
- Buffer: Physiological buffer containing divalent cations (e.g., Mg²⁺ in Tris-EDTA buffer).
- DNA Solution: Solution of DNA monomers (tiles or single-stranded tiles) at a defined concentration.
- Annealing Equipment: Programmable thermal cycler or heat block.
Procedure:
- Substrate Immersion: Immerse the mica substrate in a 200 µL solution containing the DNA monomers and the Mg²⁺-containing buffer.
- Programmed Annealing: Place the sample in a thermal cycler and execute a controlled cooling ramp. A standard protocol is to cool from 80°C to 25°C in 5°C decrements, holding at each step for 30 minutes (total time ~6 hours).
- Parameter Variation:
- Concentration: Repeat steps 1-2 with a series of monomer concentrations (e.g., 5 nM to 50 nM) to establish a threshold and saturation concentration.
- Temperature/Time: Vary the initial annealing temperature (e.g., 40°C, 60°C, 80°C) or the total annealing time (e.g., 1 h, 3 h, 6 h) to probe thermodynamic and kinetic effects.
- Imaging & Analysis: After annealing, remove the substrate and image the surface using Atomic Force Microscopy (AFM). Quantify the crystal coverage and calculate the average crystal size (Savg) and size distribution using image analysis software.
Workflow for Directing Crystallization Pathways
The following diagram outlines a generalized experimental workflow for investigating and controlling crystallization pathways, integrating the protocols and measurement technologies discussed.
Measurement Technologies for Pathway Characterization
Accurately monitoring crystallization is paramount. The following table compares key technologies used for characterizing crystallization processes, especially in situ.
Table 5: Comparison of Crystal Measurement Technologies (CMTs)
| Technology | Measurement Mode | Primary Information | Key Advantages | Key Challenges |
|---|---|---|---|---|
| Raman Spectroscopy | In situ / On-line | Polymorphic form, molecular vibration signatures | Identifies specific polymorphs, suitable for aqueous solutions, can be quantitative [50]. | Fluorescence interference, weak signal, complex data analysis for overlapping peaks. |
| Focused Beam Reflectance Measurement (FBRM) | In situ | Particle count and chord length distribution (CLD) | Provides real-time particle count and size trends, robust for high-concentration slurries [50]. | Chord length is not a direct measure of particle size; affected by particle shape and orientation. |
| Process Video Microscopy (PVM) | In situ | Direct 2D images of particles | Provides direct visual evidence of crystal shape, size, and presence of agglomerates [50]. | 2D projection, difficult to automate image analysis for overlapping particles. |
| Atomic Force Microscopy (AFM) | Off-line | 3D surface topography at nano-scale | Extremely high resolution, can measure single molecules and crystal layers [52]. | Ex-situ technique, requires sample preparation, small analysis area. |
The journey from a supersaturated solution to a macroscopic crystal is no longer seen as a singular event but as a branched pathway with critical decision points. The framework presented in this guide—grounded in the distinction between classical and non-classical nucleation, and empowered by strategic additive use and advanced measurement technologies—provides researchers with a systematic approach to control this outcome. By understanding the mechanistic role of solvents and additives in stabilizing specific intermediates or altering surface energetics, scientists can deliberately steer crystallization away from default pathways and toward desired polymorphic forms and morphologies. As the field advances, the integration of high-throughput experimentation, real-time analytics, and predictive modeling will further transform crystallization from an empirical art into a precise and rational engineering discipline, accelerating development across pharmaceuticals and advanced materials.
Troubleshooting Crystallization: Strategies for Control and Suppression
Crystal nucleation, the initial and critical step in the formation of solid phases from solution, fundamentally determines the structural, morphological, and functional properties of crystalline materials across industries ranging from pharmaceuticals to advanced materials synthesis. The precise control of nucleation remains a formidable scientific challenge, primarily due to the complex interplay between two core determinants: interfacial energy and kinetic hurdles. Within contemporary research, the understanding of these processes is framed by the competition between long-established Classical Nucleation Theory (CNT) and emerging evidence for non-classical pathways that diverge from its foundational assumptions.
Classical Nucleation Theory posits that nucleation occurs via the direct, step-by-step addition of monomers (individual ions or molecules) to form a nascent crystal cluster. This cluster becomes stable only after surpassing a critical size, beyond which the bulk free energy favoring crystallization overcomes the surface free energy penalty of creating a new interface [53]. The kinetic hurdle in CNT is the energy barrier associated with forming this critical nucleus. In contrast, non-classical crystallization encompasses pathways involving metastable intermediate states, such as dense liquid droplets or amorphous precursors, which can substantially alter crystallization kinetics and outcomes [54] [45]. Diagnosing nucleation problems, therefore, requires a dual-focused investigation into the thermodynamic role of interfacial energy and the kinetic rates governing molecular assembly, all while considering which theoretical framework best describes the system at hand.
Theoretical Foundations: Classical vs. Non-Classical Paradigms
Core Tenets of Classical Nucleation Theory (CNT)
CNT provides a thermodynamic and kinetic framework for understanding the formation of a stable crystal nucleus from a supersaturated solution. The theory hinges on the balance between the volume free energy, which drives the phase transition, and the surface free energy, which resists it.
Thermodynamic Barrier: The free energy change (ΔG) for forming a spherical cluster of radius r is given by: ΔG = -(4/3)πr³Δgv + 4πr²γ where Δgv is the free energy change per unit volume (negative and proportional to the supersaturation, S), and γ is the crystal-liquid interfacial energy [53]. This relationship results in an energy barrier, ΔGc. The critical nucleus size, r*, and the energy barrier are derived as: r* = 2γ / Δgv and ΔGc = 16πγ³ / (3Δgv²) The strong, cubic dependence of the barrier on γ highlights why this parameter is a primary diagnostic for nucleation problems [53].
Kinetic Hurdle and Nucleation Rate: The steady-state nucleation rate J, representing the number of nuclei formed per unit volume per unit time, is expressed in the Arrhenius form: J = A exp(-ΔGc / kB T) where A is a pre-exponential factor accounting for the kinetic frequency of molecular attachment, kB is Boltzmann's constant, and *T* is temperature [53]. The kinetic hurdle is thus governed by both the thermodynamic barrier (ΔGc) and the molecular mobility (captured in A).
Key Principles of Non-Classical Nucleation Theory
Non-classical pathways challenge several assumptions of CNT, particularly the ideas of monomer-based growth and the simultaneous development of density and order.
The Precursor Pathway: A prominent non-classical mechanism is the two-step nucleation process, where the formation of crystalline nuclei is preceded by the creation of metastable, solute-rich clusters. These clusters, which can be dense liquid droplets or amorphous aggregates, act as precursors that lower the kinetic hurdle for nucleation [45]. Within these precursors, the local solute concentration is dramatically higher than in the bulk solution, reducing the interfacial energy penalty (γ) for forming a crystalline nucleus. This can lower the nucleation barrier by orders of magnitude, making nucleation feasible at supersaturations where CNT would predict negligible rates [45].
Particle-Based Assembly: Another non-classical route involves the attachment of pre-existing nanoparticles, rather than individual molecules, to form larger crystals [54]. This pathway can bypass the classical monomer-addition growth model and lead to crystals with complex morphologies and internal structures.
The following diagram illustrates the key stages and decisions in diagnosing a nucleation problem, guiding the researcher through the fundamental questions that distinguish classical from non-classical behavior.
Quantitative Interfacial Energy and Nucleation Kinetics
A critical step in diagnosing nucleation problems is the quantitative determination of key parameters. The following table consolidates experimental data and calculated parameters for various materials, illustrating the direct relationship between interfacial energy, nucleation barrier, and resulting kinetics.
Table 1: Experimentally Determined Nucleation Parameters for Various Compounds
| Compound | Solvent | Interfacial Energy (γ) [mJ/m²] | Nucleation Rate (J) [molecules/m³/s] | Gibbs Free Energy of Nucleation (ΔG) [kJ/mol] | Primary Nucleation Mechanism |
|---|---|---|---|---|---|
| β-hematin | Octanol/Buffer | 25 ± 2 [45] | - | ~10⁻¹⁷ J/particle [45] | Non-classical (within mesoscopic clusters) |
| Lysozyme | Aqueous NaCl | - | 10³⁴ [33] | 87 [33] | Varies (classical & non-classical) |
| Typical APIs | Various | - | 10²⁰ – 10²⁴ [33] | 4 – 49 [33] | Model-dependent |
| Alkali Halides (e.g., NaCl) | Melt | Model-dependent [55] | - | - | Classical (often heterogeneous) |
The data reveals several key insights. First, the interfacial energy is a pivotal variable. For instance, with β-hematin, the classical nucleation barrier calculated using the measured γ=25 mJ/m² is impossibly high (~10⁻¹⁷ J, or about 2500 *kBT), effectively ruling out a classical pathway and forcing consideration of non-classical mechanisms where the effective γ is much lower [45]. Second, the nucleation rates and Gibbs free energies can vary over immense ranges, as seen in the comparison between large biomolecules like lysozyme and smaller active pharmaceutical ingredients (APIs) [33]. This variability underscores the need for system-specific measurement.
The thermodynamic parameter B and the nucleation rate J are powerfully linked to interfacial energy and supersaturation, as described by the equations: B = 4c³γ³v₀² / 27(k_B T)³ J = A exp(-B / ln² S) [53]
These relationships show that a small reduction in γ can lead to a dramatic increase in J due to its cubic dependence in the exponent. This explains why strategies aimed at reducing interfacial energy are so effective in controlling nucleation.
Experimental Protocols for Diagnosis
Determining Metastable Zone Width (MSZW) and Induction Time
The Metastable Zone Width (MSZW) is the region between the solubility curve and the supersaturation level at which spontaneous nucleation occurs. The induction time (t_ind) is the time elapsed between achieving supersaturation and the detectable formation of nuclei. These are critical, experimentally accessible parameters for diagnosing nucleation problems.
Protocol for Polythermal MSZW Measurement:
- Prepare a saturated solution at an elevated temperature and hold it with constant agitation to ensure dissolution.
- Cool the solution at a constant, predefined rate (e.g., 0.1 to 1.0 °C/min).
- Monitor the solution for the first appearance of crystals using an in-situ probe such as focused beam reflectance measurement (FBRM), particle vision microscope (PVM), or simple turbidity measurement.
- Record the temperature at which nucleation is detected (T_nuc). The MSZW is defined as ΔTmax = Tsat - Tnuc, where Tsat is the saturation temperature [33].
- Repeat at different cooling rates to model nucleation kinetics.
Protocol for Isothermal Induction Time Measurement:
- Prepare a supersaturated solution at a temperature where the solute is fully dissolved.
- Rapidly cool or otherwise adjust the solution to a target temperature within the metastable zone.
- Immediately begin monitoring for nucleation using the techniques listed above.
- Record the time from the establishment of supersaturation until the detection of crystals as the induction time, t_ind [56].
- Repeat at varying levels of supersaturation.
Recent advances allow the extraction of key nucleation parameters directly from MSZW data. A 2025 model enables the prediction of the nucleation rate (J) and Gibbs free energy of nucleation (ΔG) using MSZW data obtained at different cooling rates, according to the linearized relationship [33]: ln(ΔCmax / ΔTmax) = ln(kn) - ΔG / (R Tnuc) where kn is the nucleation rate constant, and ΔCmax is the maximum supersaturation at the point of nucleation.
Probing for Non-Classical Precursors
When nucleation behavior deviates significantly from CNT predictions, investigating the presence of non-classical precursors is essential.
Dynamic Light Scattering (DLS) Protocol:
- Prepare a supersaturated solution, ensuring it is free of dust by filtration.
- Place the sample in a DLS instrument and monitor the autocorrelation function g₂(τ) over time at a constant temperature.
- In a classical system, the correlation function may remain flat until a sudden change indicates the appearance of crystals. In a non-classical system, a growing shoulder or a slow decay in g₂(τ) at early times may indicate the presence and evolution of metastable clusters prior to crystallization [45].
- The amplitude A₁ of the autocorrelation function decay is proportional to the number and size of scattering entities (clusters or crystallites), allowing the kinetics of precursor formation and crystallization to be tracked.
Liquid-Phase In-Situ Electron Microscopy: While technically demanding, this technique enables the direct visualization of nucleation events, including the formation of amorphous precursors and particle-attachment pathways, with high spatial resolution [54].
The Scientist's Toolkit: Key Reagents and Methods
The following table summarizes critical reagents, additives, and methodological approaches used in modern nucleation research to diagnose and manipulate interfacial energy and kinetic hurdles.
Table 2: Research Reagent Solutions for Nucleation Control
| Reagent / Method | Function / Purpose | Example Application / Effect |
|---|---|---|
| Micro-/Nanobubbles | Green heterogeneous nucleation templates; reduce effective interfacial energy at gas-liquid interface. | Shortens induction time, promotes nucleation of specific polymorphs, retards calcite growth by up to 53% [56]. |
| Antimalarial Compounds (e.g., Pyronaridine) | Modifiers that interact with solute to suppress non-classical nucleation pathways. | Nearly fully suppresses β-hematin nucleation by disrupting precursor clusters [45]. |
| Polythermal MSZW Analysis | Diagnostic method to determine nucleation kinetics and model parameters like ΔG and J. | Used to predict nucleation rates for APIs and inorganics across different cooling rates [33]. |
| In-Situ Spectroscopy & Scattering | Provides molecular-level insight into nucleation mechanisms and precursor chemistry. | Elucidated the role of TOPO in CsPbBr3 quantum dot formation; revealed continuous growth pathways for iron oxide NCs [57]. |
Diagnosing nucleation problems effectively requires a structured approach that interrogates both the thermodynamic influence of interfacial energy and the nature of the kinetic hurdles involved. The researcher must first establish whether the system's behavior aligns with Classical Nucleation Theory or if it necessitates a non-classical description involving metastable precursors. Quantitative tools, such as the analysis of Metastable Zone Width and induction time, provide a pathway to extract fundamental parameters like nucleation rate and Gibbs free energy. Simultaneously, experimental techniques like DLS are vital for detecting the presence of non-classical intermediates. As research progresses, the integration of advanced in-situ characterization methods with sophisticated theoretical models continues to refine our diagnostic capabilities, enabling more precise control over crystallization outcomes in the synthesis of pharmaceuticals, functional materials, and beyond. The key is to systematically eliminate potential issues—starting with supersaturation, then interfacial energy, then kinetic factors, and finally by exploring non-classical pathways—to identify and address the root cause of any nucleation problem.
Exploiting Solvent Influence to Modulate Nucleation Kinetics and Polymorphic Outcomes
The strategic selection of solvents is a powerful tool for controlling crystallization processes, enabling precise manipulation of nucleation kinetics, crystal growth, and ultimate polymorphic outcomes. Within pharmaceutical and materials science, this control is paramount for ensuring product efficacy, stability, and manufacturability. This guide examines solvent influence through the dual lenses of Classical Nucleation Theory (CNT) and emerging non-classical perspectives, providing a technical framework for exploiting solute-solvent interactions. CNT describes nucleation as the formation of a critical nucleus through stochastic molecular addition, with a rate R expressed as ( R = A \exp(-\Delta G^/k_B T) ), where ( \Delta G^ ) is the free energy barrier for forming a stable nucleus [1]. The nucleation barrier for homogeneous nucleation is given by ( \Delta G^* = \frac{16\pi \gamma{sl}^3}{3|\Delta \mu|^2} ), where ( \gamma{sl} ) is the solid-liquid interfacial tension and ( \Delta \mu ) is the thermodynamic driving force [58]. Solvents directly modulate this process by altering both ( \gamma_{sl} ) and ( \Delta \mu ), thereby influencing the kinetics and mechanism of nucleation. While CNT provides a robust foundational model, its limitations in fully capturing molecular-level heterogeneity have spurred the development of non-classical theories that consider multi-step pathways and pre-nucleation clusters, offering a more nuanced understanding of polymorph selection [58] [59].
Theoretical Framework: Classical vs. Non-Classical Perspectives
Classical Nucleation Theory (CNT) and Solvent Role
Classical Nucleation Theory establishes that the nucleation rate exhibits an exponential dependence on the thermodynamic driving force and the solid-liquid interfacial energy. The core equation for the homogeneous nucleation rate is: [ R = NS Z j \exp\left(-\frac{\Delta G^*}{kB T}\right) ] where ( N_S ) is the number of potential nucleation sites, ( Z ) is the Zeldovich factor, and ( j ) is the monomer attachment rate [1]. The dominant influence of the free energy barrier, ( \Delta G^* ), means that even minor perturbations to this barrier by solvent interactions can alter nucleation rates by orders of magnitude. Solvents impact CNT parameters through several key mechanisms:
- Modification of Interfacial Tension (( \gamma_{sl} )): Preferential adsorption of solvent molecules at specific crystal faces reduces the interfacial tension of those faces, lowering the nucleation barrier ( \Delta G^* ) and accelerating nucleation [60] [1].
- Alteration of Thermodynamic Driving Force (( \Delta \mu )): The effective supersaturation is governed by a solute's activity, not merely its concentration. In strong electrolyte systems or mixed solvents, activity coefficients must be accounted for to accurately estimate the true driving force for nucleation [61].
- Heterogeneous Nucleation: The presence of impurities or interfaces drastically reduces the nucleation barrier. The modified barrier is given by ( \Delta G^_{\text{het}} = f(\theta) \Delta G^_{\text{hom}} ), where ( f(\theta) ) is a factor less than 1 that depends on the contact angle (( \theta )) between the nucleus and the substrate [1]. Recent studies demonstrate CNT's remarkable robustness even on chemically heterogeneous surfaces, where nuclei maintain a fixed contact angle through pinning at patch boundaries [58].
Beyond CNT: Non-Classical Nucleation and Solvent-Induced Pathways
Non-classical nucleation mechanisms challenge the single-step, monomer-by-monomer addition model of CNT. Evidence suggests that solvent-mediated pre-nucleation clusters and two-step nucleation pathways, where a dense liquid phase precedes crystal formation, are critical in many systems. The influence of solvent is profound in these scenarios:
- Pathway Selection: Softening intermolecular potentials through solvent choice can alter critical nucleus composition and introduce distinct nucleation pathways, leading to different polymorphic outcomes without significantly changing the nucleation rate itself [59]. This decoupling of kinetics and pathway is a key concept in non-classical theory.
- Polymorphic Control via Solvent-Template Effects: Strong, selective adsorption of solvent molecules can create a "relay mechanism" for crystal growth, where solvent molecules act as transient templates that direct incoming solute molecules to specific lattice sites, thereby stabilizing metastable polymorphs [60].
The following diagram illustrates the core concepts of how solvents influence nucleation pathways within these theoretical frameworks.
Solvent Influence on Nucleation Pathways
Quantitative Data: Solvent Effects on Nucleation and Growth Kinetics
The following tables consolidate quantitative findings on how solvents and processing conditions impact key crystallization kinetic parameters, drawing from recent experimental studies.
Table 1: Solvent Impact on Nucleation Kinetics for Various Compounds
| Compound | Solvent System | Nucleation Rate Kinetic Constant (k_n) | Gibbs Free Energy of Nucleation (ΔG) [kJ/mol] | Key Finding |
|---|---|---|---|---|
| Lysozyme [33] | NaCl Solution | ~10³⁴ molecules/m³s | 87.0 | Extremely high ΔG for large biomolecules |
| Generic APIs [33] | Various | 10²⁰ – 10²⁴ molecules/m³s | 4 – 49 | Wide variability based on solvent & API structure |
| Potassium Salts [61] | Ethanol-Water Mixtures | N/A | N/A | Ethanol inhibits both nucleation & growth kinetics |
| Lennard-Jones System [58] | Model Atomic Liquid | N/A | N/A | Nucleation rate retains CNT-predicted temperature dependence on heterogeneous surfaces |
Table 2: Solvent-Induced Crystal Habit Modifications and Dissolution Performance
| Drug Compound | Crystallization Solvent | Resulting Crystal Habit | Impact on Dissolution Rate | Postulated Mechanism |
|---|---|---|---|---|
| Tolbutamide [62] | Methanol/Ethanol | Small, circular plates | Increase (86% vs 67% in 60 min) | Increased specific surface area & exposed faces |
| Tolbutamide [62] | Acetone | Needle-like | Decrease (vs pure powder) | Reduced surface area & unfavorable face exposure |
| Mefenamic Acid [62] | Acetone | Needles | Increase (Acetone > Ethanol > IPA) | Higher solubility/wettability from Schiff-base reaction |
| Dipyridamole [62] | Benzene | Rod-shaped | Increase (vs methanol needles) | Preferential face growth & improved wettability |
Experimental Protocols for Kinetic Analysis
Automated, Standardized Kinetic Determination
A modern approach for quantifying secondary nucleation and crystal growth kinetics utilizes automated, small-scale reactors coupled with population balance modeling to minimize material usage and maximize reproducibility [61] [63].
- Equipment Setup: Experiments are performed in an automated reactor system (e.g., Technobis Crystalline) equipped with in-situ imaging probes (e.g., particle vision microscope, PVM) and/or Fourier Transform Infrared (FTIR) spectroscopy for concentration monitoring. The typical suspension volume is 5 mL for batch desupersaturation experiments [61] [63].
- Procedure:
- Saturation & Supersaturation Generation: The solute is dissolved in the chosen solvent or solvent-antisolvent mixture at an elevated temperature to ensure complete dissolution. The solution is then cooled at a controlled rate or with antisolvent addition to generate a defined supersaturation.
- In-situ Monitoring: The crystallization process is monitored in real-time using PVM to track crystal count and size evolution, and FTIR to measure solute concentration in solution.
- Data Processing: Image analysis software processes the PVM data to extract crystal population data (count and size distributions). The desupersaturation curve from FTIR is used to calculate the solution supersaturation profile over time.
- Kinetic Parameter Estimation: A population balance model is fitted to the experimental data (population and concentration) to extract the kinetic parameters for secondary nucleation (( kb ), ( b )) and crystal growth (( kg ), ( g )). For inorganic salts or strong electrolytes, the model must be expanded to use solute activity (calculated from activity coefficients) instead of concentration for accurate supersaturation estimation [61].
- Key Consideration: This methodology's expansion to inorganic salts requires explicit modeling of the role of dissolved salts and mixed aqueous-organic solvents on activity coefficients, which is critical for estimating accurate kinetic constants [61].
Metastable Zone Width (MSZW) Measurement and Analysis
The Metastable Zone Width is a critical parameter defining the supersaturation limit before spontaneous nucleation. Its measurement provides insights into nucleation kinetics.
- Polythermal Method Protocol:
- A saturated solution is prepared at a known saturation temperature (( T^* )).
- The solution is heated to approximately 5°C above ( T^* ) to dissolve all nuclei.
- The solution is cooled at a constant, predefined cooling rate (( R' )), and the nucleation temperature (( T_{\text{nuc}} )) is detected visually or via turbidity probe.
- The MSZW is calculated as ( \Delta T{\text{max}} = T^* - T{\text{nuc}} ) [33].
- Analysis for Nucleation Kinetics: A recently developed model enables the extraction of nucleation rates and Gibbs free energy of nucleation directly from MSZW data obtained at different cooling rates [33]. The model linearizes as: [ \ln(\Delta C{\text{max}} / \Delta T{\text{max}}) = \ln(kn) - \frac{\Delta G}{R T{\text{nuc}}} ] A plot of ( \ln(\Delta C{\text{max}} / \Delta T{\text{max}}) ) vs. ( 1/T{\text{nuc}} ) yields a slope of ( -\Delta G/R ) and an intercept of ( \ln(kn) ), allowing for the calculation of both the nucleation rate constant and the thermodynamic barrier [33].
The workflow for this integrated experimental and modeling approach is detailed below.
Crystallization Kinetics Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Crystallization Studies
| Reagent/Material | Function in Experimentation | Application Context |
|---|---|---|
| Automated Crystallization Platform (e.g., Technobis Crystalline) | Provides automated, standardized temperature control, mixing, and data collection in small volumes (≤ 5 mL), ensuring reproducibility [61] [63]. | High-throughput screening of solvent/antisolvent systems; kinetic parameter estimation. |
| In-Situ Process Analytical Technology (PAT) | Enables real-time monitoring of crystallization processes without manual sampling. | |
| - Particle Vision Microscope (PVM) | Provides real-time images of crystals for particle size and shape (habit) analysis [61] [63]. | Tracking crystal growth and nucleation events. |
| - FTIR/ATR-FTIR Probe | Measures solute concentration in solution in real-time to construct desupersaturation curves [63]. | Determining supersaturation profiles for kinetic modeling. |
| Tailor-Made Additives | Molecules structurally similar to the solute but with altered moieties; selectively adsorb on specific crystal faces to inhibit growth and modify morphology [60]. | Directing crystal habit towards more isometric shapes; investigating growth mechanisms. |
| Molecular Modeling Software (e.g., BIOVIA Materials Studio) | Predicts crystal morphology in vacuum and the interaction energy of solvents/additives with specific crystal faces (e.g., via Modified Attachment Energy model) [64]. | In-silico solvent selection and pre-screening of habit modifiers. |
The deliberate exploitation of solvent influence represents a sophisticated strategy for controlling crystallization outcomes. By leveraging the principles of Classical Nucleation Theory and acknowledging the insights from non-classical pathways, scientists can rationally design crystallization processes. The integration of automated, standardized experimental methods with advanced modeling and in-situ analytics provides a powerful framework for quantifying kinetic parameters and understanding fundamental mechanisms. This approach enables the transition from empirical trial-and-error to a predictive science, facilitating the robust design of crystals with desired physicochemical properties, polymorphic form, and performance characteristics—a critical capability for advanced drug development and materials engineering.
Using Additives and Modifiers to Suppress or Enhance Specific Pathways
The control of crystallization pathways is a cornerstone in the development of advanced materials, active pharmaceutical ingredients (APIs), and functional biomaterials. This process hinges on the strategic use of additives and modifiers to suppress or enhance specific nucleation and growth mechanisms. Within the field of inorganic crystal research, a fundamental tension exists between the long-established Classical Nucleation Theory (CNT) and the more recent non-classical theories, which account for phenomena like pre-nucleation clusters and particle-based crystallization. CNT describes nucleation as a single-step process where atoms or molecules assemble into a critical nucleus with a distinct interface, governed by a competition between bulk free energy gain and surface free energy cost [65]. In contrast, non-classical pathways often involve multi-step processes, where stable pre-nucleation clusters aggregate and reorganize into crystalline phases [66].
The precise application of additives serves as a powerful experimental tool to probe these mechanisms. By altering the thermodynamic and kinetic landscape of crystallization, modifiers can selectively inhibit or promote specific pathways, thereby revealing the underlying principles of crystal formation. This guide provides a technical overview of how additives function, detailing experimental protocols and data analysis methods for researchers and drug development professionals working at the frontier of crystal engineering.
Theoretical Framework: Classical vs. Non-Classical Nucleation
Core Principles and the Role of Additives
Classical Nucleation Theory (CNT) provides a foundational model for predicting nucleation rates. It posits the formation of a new thermodynamic phase through the stochastic assembly of ions or molecules into a critical nucleus. The rate of this process is exponentially dependent on the Gibbs free energy barrier, which is influenced by the interfacial tension between the nucleus and the solution [65]. A key prediction of CNT is that nucleation can be significantly slowed by factors that increase this energy barrier. This model has been successfully applied to a wide range of systems, from simple liquids to complex organic molecules [67] [65].
Non-Classical Nucleation Theory challenges the CNT view by introducing multi-step pathways. A prominent mechanism involves the initial formation of stable pre-nucleation clusters, which are dense liquid phases or amorphous nanoparticles that act as precursors to the final crystalline phase. These clusters can aggregate and undergo internal restructuring before a crystal nucleus emerges [66]. This pathway is particularly relevant for biomineralization systems, where organic polymers often direct crystallization.
Additives and modifiers exert their influence differently within these two frameworks:
- In classical pathways, they typically function by adsorbing to the surface of the nascent crystal, blocking active growth sites such as kinks or steps. This increases the energetic cost of forming a stable nucleus or slows the lateral motion of steps, effectively inhibiting crystallization [68].
- In non-classical pathways, modifiers often interact with metastable precursors, such as pre-nucleation clusters. They can stabilize these intermediates against aggregation or reorganization, or direct their transformation into specific crystalline polymorphs [69] [66].
The following diagram illustrates the sequential stages of both pathways and the points where additives exert influence.
Quantitative Models for Crystal Growth Inhibition
The microkinetic model offers a quantitative framework for understanding growth inhibition, requiring only the adsorption energy of the inhibitor on the crystal step as a key parameter. This model successfully describes calcite growth inhibition by ions like Mg²⁺ and SO₄²⁻, as well as small organic molecules. The model's equations show that inhibition primarily occurs through the blocking of kink nucleation sites on the crystal surface. For instance, the microscopic growth rate ((r)) in the presence of an inhibitor like Mg²⁺ can be expressed as:
[r{\mathrm{Mg}} = \frac{{a \cdot K{\mathrm{IP},\mathrm{CaCO}3}\left[ {\mathrm{Ca}^{2 + }} \right]\left[ {\mathrm{CO}3^{2 - }} \right]}}{{\left( {1 + K{\mathrm{Ca}}\left[ {\mathrm{Ca}^{2 + }} \right] + K{\mathrm{Mg}}\left[ {\mathrm{Mg}^{2 + }} \right]} \right)\left( {1 + K{\mathrm{CO}3}\left[ {\mathrm{CO}3^{2 - }} \right]} \right)}} \ \frac{{kT}}{h}{\mathrm{e}}^{ - \left( {\frac{{\Delta G{\mathrm{IP},\mathrm{CaCO}_3}}}{{RT}}} \right)}]
Where (K_{\mathrm{Mg}}) represents the adsorption equilibrium constant for Mg²⁺, and other (K) terms correspond to other ions [68]. This model demonstrates that even a single, physically meaningful parameter can accurately predict inhibition behavior across diverse chemical systems.
Experimental Protocols for Pathway Analysis
In Situ Monitoring of Heterogeneous Nucleation on Functionalized Surfaces
Objective: To quantify the kinetics of gypsum nucleation on surfaces with different functional groups and hydrophobicity, providing data to distinguish between classical and non-classical pathways [66].
Materials:
- Self-Assembled Monolayers (SAMs): Gold substrates functionalized with −CH₃, −COOH, −SO₃, −NH₃, −OH, and hybrid −NH₂/−COOH groups.
- Precursor Solutions: Aqueous solutions of CaCl₂ and Na₂SO₄, prepared at various saturation indices (σ) from 0.97 to 1.63.
- Flow Cell System: Equipped with an optical microscope for real-time imaging.
- Analysis Software: For counting crystallites and calculating number density over time.
Procedure:
- Surface Characterization:
- Verify SAM quality using Atomic Force Microscopy (AFM) to confirm roughness below 1.3 nm.
- Measure water contact angles to determine surface hydrophilicity/hydrophobicity.
- Analyze surface chemistry using X-ray Photoelectron Spectroscopy (XPS) to confirm functional group presence.
Nucleation Experiment:
- Mount the functionalized substrate in the flow cell.
- Introduce mixed CaCl₂/Na₂SO₄ solution at a constant flow rate (e.g., 30 mL h⁻¹).
- Record optical microscopy images at regular time intervals (e.g., every 30 seconds) for up to 2 hours.
Data Analysis:
- Count the number of gypsum crystallites present in each image frame.
- Plot crystallite number density ((N)) versus time ((t)).
- Calculate the steady-state heterogeneous nucleation rate ((J_0)) from the slope of the linear region of the (N) vs. (t) plot.
- Relate (J0) to the saturation index ((\sigma)) to determine the thermodynamic barrier ((B)) using the relationship derived from CNT: (J0 \propto \exp(-B / \sigma^2)).
Expected Outcomes:
- Hydrophobic surfaces (e.g., −CH₃) will typically exhibit the highest nucleation rates and lowest (B) values, indicating a lower energy barrier.
- Hydrophilic surfaces (e.g., −OH) will show the lowest nucleation rates and highest (B) values.
- Molecular dynamics (MD) simulations can corroborate these findings by showing higher density of ion clusters near hydrophobic surfaces, suggesting a prevalence of classical nucleation, versus ion anchoring on hydrophilic functional groups, suggesting surface-induced non-classical pathways [66].
Determining Metastable Zone Width (MSZW) and Nucleation Thermodynamics
Objective: To measure the Metastable Zone Width (MSZW) for an API or inorganic material and use this data to calculate fundamental nucleation parameters using a modern mathematical model [67].
Materials:
- Analyte Solution: A solution of the target compound (e.g., an API, glycine, lysozyme) in a suitable solvent.
- Crystallization Setup: A jacketed crystallizer vessel connected to a programmable cooling bath, an agitator, and a turbidity probe or focused beam reflectance measurement (FBRM) probe.
- Data Logging System: To record temperature and turbidity/particle count in real time.
Procedure:
- Saturation Temperature Determination:
- Prepare a saturated solution of the analyte at a known elevated temperature.
- Cool the solution slowly until the first crystal appears, noting the temperature. Alternatively, use gravimetric analysis.
MSZW Measurement:
- Heat a clear, undersaturated solution 5-10°C above its saturation temperature to ensure complete dissolution.
- Cool the solution at a constant, controlled cooling rate (e.g., 2, 5, 10 °C/h) under constant agitation.
- Record the temperature at which a sudden change in turbidity or particle count is detected, indicating nucleation. This is the nucleation temperature ((T_n)).
- Repeat for at least three different cooling rates.
Data Analysis using the Vashishtha-Kumar Model:
- The MSZW is the difference between the saturation temperature ((Ts)) and the nucleation temperature ((Tn)).
- Use the model to calculate the nucleation rate ((J)) and Gibbs free energy of nucleation ((\Delta G_{nuc})) directly from the MSZW data obtained at different cooling rates.
- The model avoids the limitations of earlier approaches (Nývlt, Kubota) by directly linking cooling rate to nucleation rate, enabling the prediction of key parameters like surface free energy, critical nucleus size, and number of unit cells.
Expected Outcomes:
- Nucleation rates for APIs typically range from 10²⁰ to 10²⁴ molecules per m³ s, while larger molecules like lysozyme can reach up to 10³⁴ molecules per m³ s [67].
- Gibbs free energy of nucleation typically varies from 4 to 49 kJ mol⁻¹ for most compounds, but can reach 87 kJ mol⁻¹ for large biomolecules [67].
Data Presentation and Analysis
Quantitative Effects of Inorganic and Organic Additives
Table 1: Inhibition mechanisms and adsorption energies for selected calcite growth inhibitors. Data adapted from Nature Communications [68].
| Inhibitor | Class | Primary Mechanism | Key Parameter (Adsorption Energy) | Impact on Growth Rate |
|---|---|---|---|---|
| Mg²⁺ | Inorganic Ion | Kink-site blocking, step adsorption | Fitted adsorption constant ((K_{Mg})) | Strong inhibition; rate decreases with increasing [Mg²⁺] |
| SO₄²⁻ | Inorganic Ion | Kink-site blocking, step adsorption | Fitted adsorption constant ((K{SO4})) | Moderate inhibition |
| Acetate | Small Organic Anion | Solution complexation (reduces free Ca²⁺) | Not primarily adsorption | Weak to moderate inhibition |
| Benzoate | Small Organic Anion | Solution complexation (reduces free Ca²⁺) | Not primarily adsorption | Weak to moderate inhibition |
| Polyacrylic Acid (PAA) | Organic Polymer | Kink-site blocking, surface adsorption | N/A | Strong inhibition; can direct polymorphism |
Table 2: Influence of surface functional groups on gypsum heterogeneous nucleation rates. Data adapted from Nature Communications [66].
| Surface Functional Group | Hydrophobicity (Contact Angle) | Relative Nucleation Rate | Proposed Nucleation Pathway |
|---|---|---|---|
| −CH₃ | High (98.1°) | Highest | Bulk (Classical) Nucleation |
| −Hybrid (NH₂/COOH) | Medium (81.8°) | High | Intermediate |
| −COOH | Medium-High (50.5°) | Medium-High | Surface-Induced (Non-Classical) |
| −SO₃ | Low (32.5°) | Low | Surface-Induced (Non-Classical) |
| −NH₃ | Low | Low | Surface-Induced (Non-Classical) |
| −OH | Low (60.8°) | Lowest | Surface-Induced (Non-Classical) |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents and materials for nucleation pathway experiments.
| Reagent/Material | Function in Experiment | Example Application |
|---|---|---|
| Self-Assembled Monolayers (SAMs) | Provides a well-defined, chemically modified surface to study heterogeneous nucleation mechanisms. | Investigating the effect of surface functionality and hydrophobicity on gypsum scale formation [66]. |
| Mg²⁺, SO₄²⁻ ions | Acts as inorganic inhibitors that adsorb to specific crystal growth sites. | Probing the kink-blocking inhibition mechanism in calcite growth using microkinetic models [68]. |
| Carboxylate-based Polymers (e.g., PAA) | Organic macromolecules that chelate cations and interact with crystal surfaces. | Modifying crystal morphology and polymorphism in MICP/EICP processes [69]. |
| Chitosan | A bio-based polymer additive that provides nucleation sites and enhances aggregation. | Improving the strength and stability of bio-cemented materials in MICP [69]. |
| Sodium Citrate | A chelating agent that complexes calcium ions in solution. | Regulating the availability of free Ca²⁺ ions to control the kinetics of carbonate precipitation [69]. |
| Geochemical Speciation Software (e.g., PHREEQC) | Calculates solution speciation, saturation indices, and ion pair formation. | Essential for accurate interpretation of growth inhibition experiments and microkinetic modeling [68]. |
The targeted use of additives and modifiers provides a powerful methodology for dissecting and controlling crystallization pathways. Experimental data unequivocally shows that the effectiveness of an additive is governed by its specific molecular interaction with either the crystal surface, the solvated ions, or metastable precursors. Hydrophobic surfaces and kink-blocking ions like Mg²⁺ often promote pathways aligned with Classical Nucleation Theory, while hydrophilic functional groups and polymeric additives frequently engage in the complex, multi-stage processes characteristic of non-classical nucleation.
The integration of robust experimental protocols—such as in situ monitoring on functionalized surfaces and MSZW analysis—with advanced modeling techniques like microkinetic models and molecular dynamics simulations, creates a comprehensive framework for crystal engineering. This integrated approach enables researchers to move beyond empirical observations toward a predictive understanding of crystallization, facilitating the rational design of materials with tailored properties for pharmaceutical, industrial, and environmental applications.
The solid form of an Active Pharmaceutical Ingredient (API) is a critical quality attribute that dictates the efficacy, safety, and stability of a drug product. Among the various solid forms, crystalline polymorphs—different crystal structures of the same chemical compound—present both a challenge and an opportunity for pharmaceutical scientists. The phenomenon of "disappearing polymorphs," where a previously accessible crystalline form becomes irreproducible, often due to the emergence of a more stable form, has led to costly product recalls and underscores the necessity of robust polymorph control strategies [70]. The selection and control of a specific polymorph are governed by the intricate mechanisms of crystallization, a process whose understanding has evolved significantly from the oversimplified Classical Nucleation Theory (CNT) to encompass a spectrum of non-classical pathways involving intermediate phases [71] [72]. This whitepaper delves into the theoretical foundations of nucleation, outlines advanced strategies for polymorph control, details essential experimental protocols, and provides a toolkit for researchers, all framed within the context of modern crystallization research.
Theoretical Foundations: From Classical to Non-Classical Crystallization
Classical Nucleation Theory (CNT) and Its Limitations
Classical Nucleation Theory (CNT) has long served as the primary model for understanding crystallization. It posits that nucleation is a one-step process where solute molecules in a supersaturated solution associate stochastically to form a critical nucleus with the same internal structure as the final bulk crystal [72]. The formation of this nucleus is governed by a free energy barrier (ΔG*), which arises from the competition between the lower free energy of the new crystalline volume and the positive surface free energy required to create the crystal-solution interface [72]. A key limitation of CNT is its assumption that the crystalline nucleus appears directly from the molecularly dissolved state. For many organic molecules and APIs, experimental evidence has shown that nucleation rates can be many orders of magnitude lower than CNT predictions, suggesting a more complex reality [72].
Non-Classical Crystallization (NCC) Pathways
Recent advances in direct imaging techniques have revealed that crystallization often proceeds through non-classical pathways that do not comply with CNT. These mechanisms involve precursor particles larger than single molecules, such as nanoparticles or disordered clusters [12].
- Pre-Nucleation Clusters (PNCs) and Two-Step Nucleation: A widely supported non-classical mechanism is the two-step nucleation pathway. In this model, the crystalline nucleus forms inside pre-existing, metastable clusters of dense liquid suspended in the solution [72]. For the API flufenamic acid (FFA), Liquid Phase Electron Microscopy (LPEM) directly captured the intermediate pre-crystalline stages, suggesting a pathway that amalgamates PNCs and two-step nucleation [12].
- Continuum Crystallization Model: The dichotomy of "classical" versus "non-classical" is often insufficient to describe the observed diversity of crystallization mechanisms. A proposed continuum crystallization model posits that the evolution of crystalline order depends on the degree of molecular ordering within the initially formed intermediates and the efficiency of molecular rearrangements within them [71]. For instance, cryo-TEM studies of ibuprofen showed the instantaneous formation of final crystalline order within round-shaped, low-density aggregates—a scenario that is "classical" in its instant order but "non-classical" in its association with intermediate aggregates [71].
Table 1: Key Characteristics of Classical and Non-Classical Nucleation Theories
| Feature | Classical Nucleation Theory (CNT) | Non-Classical Crystallization (NCC) |
|---|---|---|
| Nucleation Pathway | One-step, direct from solution [71] | Multi-step, via intermediate phases [12] [71] |
| Key Intermediate | Critical nucleus (identical to final crystal) | Pre-nucleation clusters (PNCs), dense liquid phase, amorphous nanoparticles [12] [72] |
| Governing Principle | Overcoming a single free energy barrier (ΔG*) [72] | Evolution of order within intermediates; spinodal decomposition at high supersaturation [71] [72] |
| Implication for Polymorphs | Predicts the fastest-nucleating polymorph emerges [72] | Explains kinetic trapping of metastable polymorphs; Ostwald's Rule of Stages [12] |
Strategies and Tools for Polymorph Control
Supersaturation Management and the Metastable Zone
Supersaturation is the fundamental driving force for both nucleation and crystal growth. Effective control of supersaturation is the most powerful lever for directing crystallization along a desired pathway. The Metastable Zone (MSZ), where the solution is supersaturated but spontaneous nucleation is unlikely, is a crucial concept for polymorph control. Operating within the MSZ allows for controlled crystal growth, often initiated by seeding with crystals of the desired polymorph, thereby avoiding the unstable zone where spontaneous and rapid nucleation leads to uncontrolled particle size and potential formation of unwanted polymorphs [73]. Refractive Index (RI) monitoring is a highly effective Process Analytical Technology (PAT) for tracking supersaturation in real-time, providing a selective measurement of the mother liquor concentration even in the presence of solids and gas bubbles [73].
The Role of Solvents, Additives, and Molecular Design
The crystallization environment profoundly impacts polymorphic outcome.
- Solvent-Mediated Phase Transformation (SMPT): The choice of solvent can govern polymorph selection by influencing molecular conformations and intermolecular interactions in solution. A study on Tegoprazan (TPZ) demonstrated that protic solvents like methanol favored the direct crystallization of the stable Polymorph A, while aprotic solvents like acetone promoted the transient formation of metastable Polymorph B, which subsequently converted to form A via an SMPT [70].
- Crystal Engineering and Additives: Crystal engineering leverages intermolecular interactions to design crystallization pathways. This includes using additives that selectively adsorb to specific crystal faces to control habit or using co-crystallization with pharmaceutically acceptable co-formers to stabilize specific crystal forms under milder conditions [74]. Mechanochemistry, which induces crystallization through solid-state grinding, offers a solvent-free, low-energy alternative for polymorph control [74].
Advanced Process Design and Scale-Up
The transition from laboratory discovery to commercial manufacturing requires careful process design.
- Quality by Design (QbD) and PAT: Implementing QbD principles involves defining a Design Space for critical process parameters (e.g., cooling rate, seeding point) that ensure consistent polymorphic outcome. PAT tools, such as in-situ Raman spectroscopy or RI sensors, are essential for monitoring and controlling within this design space [75] [76].
- Continuous Crystallization: Compared to traditional batch processes, continuous crystallization offers improved control over supersaturation, reduced energy consumption due to minimized heating/cooling cycles, and better scalability, all of which contribute to more robust polymorph control [74].
Table 2: Analytical Techniques for Polymorph Characterization and Control
| Technique | Function | Application in Polymorph Control |
|---|---|---|
| Cryogenic Transmission Electron Microscopy (Cryo-TEM) | Direct imaging of nucleation intermediates and early-stage crystals [71] | Elucidates nucleation mechanism (classical vs. non-classical); identifies amorphous precursors and early polymorphic forms. |
| Liquid Phase Electron Microscopy (LPEM) | High temporospatial imaging of crystallization in native liquid environment [12] | Direct observation of nanoscale nucleation events and intermediate pathways for APIs like flufenamic acid. |
| Powder X-Ray Diffraction (PXRD) | Fingerprint analysis of crystalline phases [70] | Definitive identification of polymorphic form; monitoring solvent-mediated phase transformations (SMPTs). |
| Refractive Index (RI) Monitoring | Real-time, in-situ concentration measurement [73] | Tracks supersaturation to identify optimal seeding points and maintain crystallization within the metastable zone. |
| Differential Scanning Calorimetry (DSC) | Thermal analysis of phase transitions [70] | Determines relative thermodynamic stability of polymorphs; identifies melting points and solid-solid transitions. |
Experimental Protocols for Polymorph Investigation
Protocol for Investigating Solvent-Mediated Phase Transformation (SMPT)
Objective: To monitor the kinetic transition of a metastable polymorph to a stable polymorph in a slurry and model the transformation rate.
Materials:
- API solid forms (metastable and stable polymorphs, amorphous form).
- Solvents of varying polarity (e.g., methanol, acetone).
- Laboratory slurry reactor with overhead stirring.
- Powder X-Ray Diffractor (PXRD).
- Vacuum filtration setup.
Method:
- Slurry Preparation: Prepare a slurry by suspending a known mass of the starting solid form (e.g., metastable Polymorph B or amorphous TPZ) in a selected solvent. Maintain isothermal conditions and constant agitation [70].
- Time-Point Sampling: At predetermined time intervals, extract a small aliquot of the slurry.
- Solid Analysis: Immediately vacuum-filter the aliquot to separate the solids. Wash the filter cake with a small amount of the same solvent to remove residual mother liquor and air-dry the sample. Analyze the solid residue using PXRD to identify the crystalline phase [70].
- Kinetic Modeling: Quantify the fraction of the new stable polymorph (α) over time. Fit the data to the Kolmogorov–Johnson–Mehl–Avrami (KJMA) equation: ( \alpha(t) = 1 - \exp(-kt^n) ), where ( k ) is the rate constant and ( n ) is the Avrami exponent, which provides insight into the transformation mechanism [70].
Protocol for Seeded Cooling Crystallization with PAT
Objective: To consistently produce a desired polymorph by controlling supersaturation and nucleation via seeding.
Materials:
- API solution in a suitable solvent.
- Pre-characterized seed crystals of the target polymorph.
- Process refractometer (e.g., Vaisala Polaris PR53AC).
- Jacketed laboratory reactor with temperature control.
Method:
- Solubility Curve Generation: Use the process refractometer to measure the RI of clear API solutions at various temperatures to construct a solubility curve [73].
- Generate Supersaturation: Heat the reactor contents to dissolve the API completely, then initiate a controlled cooling profile.
- Identify Seeding Point: Monitor the RI in real-time. The point at which the solution enters the metastable zone (supersaturated but not nucleated) is indicated by the RI trend deviating from the solubility curve. This is the optimal point for seeding [73].
- Seeding and Growth: Introduce a known amount and size of seed crystals. Continue cooling along a trajectory designed to maintain a constant, moderate level of supersaturation, as guided by the RI trend, to promote growth on the seeds without secondary nucleation [73].
- Isolation and Characterization: Filter the final product and characterize the crystals using PXRD to confirm the target polymorph.
Visualization of a Polymorph Control Workflow
The following diagram outlines a logical workflow for polymorph investigation and control, integrating computational, experimental, and analytical elements.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Polymorph Research
| Item | Function | Application Example |
|---|---|---|
| Solvents (Protic & Aprotic) | Create crystallization environment; influence molecular conformation and hydrogen bonding [70]. | Protic solvents (e.g., methanol) can direct crystallization to the stable polymorph, while aprotic solvents (e.g., acetone) may kinetically trap metastable forms [70]. |
| Co-formers | Molecules used to form co-crystals, altering the API's crystal packing and properties [74]. | Used in crystal engineering to create a stable solid form with improved bioavailability or physical stability. |
| Heterogeneous Substrates | Surfaces that can catalyze nucleation and influence polymorph selection [72]. | Can be used to selectively nucleate a specific polymorph by reducing its nucleation barrier. |
| Characterized Seed Crystals | Provide a controlled surface for crystal growth of the desired polymorph, suppressing primary nucleation [73]. | Added at the optimal supersaturation point in a cooling crystallization to ensure consistent production of the target form. |
| Stable Isotope-Labeled Compounds | Used for advanced nuclear magnetic resonance (NMR) studies, such as NOE-based conformational analysis [70]. | Elucidate the solution-state conformation of flexible API molecules, linking solution structure to solid-form outcome. |
The journey toward robust polymorph control in pharmaceutical API crystallization is a multifaceted endeavor that bridges fundamental science and practical engineering. The paradigm has shifted from the simplistic view of Classical Nucleation Theory to a more nuanced understanding that includes a continuum of nucleation pathways, often involving dense liquid intermediates and amorphous precursors. Leveraging this knowledge through strategic supersaturation control, intelligent solvent selection, and the application of QbD principles and PAT empowers scientists to design reliable crystallization processes. Furthermore, advanced direct-imaging techniques and computational tools provide unprecedented insights into the molecular events that dictate polymorphic outcome. As the pharmaceutical industry continues to embrace continuous manufacturing and green chemistry principles, the lessons learned from polymorph control will be instrumental in developing the next generation of efficient, sustainable, and robust drug manufacturing processes.
Overcoming the Slowness of Biomolecular Crystal Nucleation
The pursuit of high-resolution biomolecular structures is fundamentally constrained by the initial, critical step of crystal nucleation. This process is often characterized by its slowness and irreproducibility, presenting a significant bottleneck in fields ranging from drug discovery to structural biology. The inherent challenges of biomolecular crystallization—including the complex energy landscapes, sensitivity to environmental conditions, and the fragile nature of large molecules—have spurred extensive research into both understanding and controlling nucleation mechanisms. This technical guide examines contemporary strategies to accelerate biomolecular crystal nucleation, framed within the ongoing scientific dialogue between Classical Nucleation Theory (CNT) and emerging nonclassical perspectives. By integrating recent advances in theoretical modeling, interface engineering, and solution chemistry, researchers can now deploy more rational and effective approaches to overcome the kinetic barriers that have traditionally limited progress in this field.
Theoretical Framework: Classical vs. Nonclassical Nucleation
Classical Nucleation Theory (CNT) and Its Limitations
Classical Nucleation Theory has served as the predominant framework for understanding crystallization processes for decades. According to CNT, nucleation occurs when solute molecules form ordered clusters that, upon reaching a critical size, become stable enough to initiate spontaneous crystal growth. This critical nucleus represents a balance between the bulk free energy gain and the surface free energy cost of forming a new phase interface [47]. The nucleation rate (J) in CNT is formally expressed as:
J = knexp(-ΔG/RT) [33]
where kn is the nucleation rate kinetic constant, ΔG is the Gibbs free energy of nucleation, R is the gas constant, and T is temperature.
While CNT provides a valuable foundational model, its application to biomolecules reveals significant limitations. Experimental observations often deviate from CNT predictions, particularly when nucleation rates differ substantially from theoretical values or when critical nucleus sizes don't align with calculations [47]. Biomolecules like proteins frequently require extremely high supersaturation levels to overcome the nucleation energy barrier, conditions that can promote undesirable outcomes such as aggregation or amorphous precipitation rather than ordered crystallization.
Nonclassical Nucleation Pathways
In contrast to the single-step mechanism of CNT, nonclassical nucleation theories propose multistep processes involving metastable intermediate states. For biomolecules, evidence suggests that nucleation often proceeds through the initial formation of liquid-like or disordered protein-rich clusters that subsequently reorganize into crystalline structures [47]. This two-step nucleation mechanism, first proposed for lysozyme by Galkin and Vekilov in 2000, can significantly reduce the kinetic barriers to nucleation by temporarily bypassing the high surface energy costs associated with direct crystal nucleus formation [47].
Studies on insulin crystallization have provided compelling evidence for nonclassical pathways. At intermediate precipitant concentrations (2.3 mM ZnCl2) and elevated temperatures (20-40°C), as well as at low precipitant concentrations (1.6 mM) and low temperature (5°C), insulin nucleation deviates from CNT predictions [47]. These nonclassical pathways often involve the formation of metastable intermediate states before crystal formation, characterized by rheological transitions from Newtonian to shear-thinning behavior [47].
Diagram 1: Comparison of classical and nonclassical nucleation pathways. CNT follows a direct path from solution to critical nucleus, while nonclassical mechanisms involve intermediate metastable clusters [47].
Quantitative Modeling of Nucleation Kinetics
Metastable Zone Width (MSZW) and Nucleation Rate Prediction
Recent advances in nucleation modeling have enabled more precise quantification of biomolecular crystallization kinetics. A 2025 study by Vashishtha and Kumar introduced a mathematical model based on CNT that predicts nucleation rates and Gibbs free energy of nucleation using metastable zone width (MSZW) data as a function of solubility temperature [33]. The MSZW defines the range of supersaturation where no spontaneous nucleation occurs but crystal growth is possible, making it a critical parameter for crystallization process design.
The model establishes a relationship between experimentally measurable parameters and nucleation kinetics:
ln(ΔCmax/ΔTmax) = ln(kn) - ΔG/RTnuc [33]
where ΔCmax is the maximum supersaturation at nucleation, ΔTmax is the MSZW, and Tnuc is the nucleation temperature. This approach has been successfully validated across 22 solute-solvent systems, including 10 active pharmaceutical ingredients (APIs), lysozyme, glycine, and various inorganic compounds [33].
Thermodynamic and Kinetic Parameters for Biomolecules
Application of this model to experimental data reveals key nucleation parameters for biomolecules:
Table 1: Experimentally Determined Nucleation Parameters for Various Biomolecules and Compounds [33]
| Compound | Solvent | Nucleation Rate Constant, kn | Gibbs Free Energy of Nucleation, ΔG (kJ mol-1) | Nucleation Rate, J (molecules m-3 s-1) |
|---|---|---|---|---|
| Lysozyme | NaCl | 1.08 × 1024 | 87.0 | 1020 - 1034 |
| Glycine | Water | 2.61 × 109 | 26.7 | - |
| APIs (various) | Various | - | 4.0 - 49.0 | 1020 - 1024 |
The significantly higher Gibbs free energy barrier for lysozyme (87 kJ mol-1) compared to most APIs (4-49 kJ mol-1) and smaller molecules like glycine (26.7 kJ mol-1) illustrates the substantial kinetic challenges in nucleating large biomolecules [33]. The extremely high nucleation rates observed for lysozyme (up to 1034 molecules m-3 s-1) under certain conditions further highlight the complex nucleation behavior of protein systems.
From these fundamental parameters, additional critical nucleation characteristics can be derived:
Table 2: Derived Nucleation Parameters for System Characterization [33]
| Parameter | Calculation Method | Physical Significance |
|---|---|---|
| Surface Free Energy (γ) | γ = [ΔG/(3πrc2)]1/3 | Energy cost for creating new surface area |
| Critical Nucleus Radius (rc) | rc = 2γ/ΔG | Minimum stable nucleus size |
| Number of Unit Cells in Critical Nucleus | N = (4/3)πrc3/Vcell | Molecular content of critical nucleus |
These quantitative relationships enable researchers to move beyond empirical optimization toward predictive control of biomolecular crystallization.
Experimental Strategies and Methodologies
Solid/Liquid Interface-Induced Crystallization
The use of solid/liquid interfaces has emerged as a powerful strategy to promote and modulate nucleation events in biomolecular systems. By reducing the energy barrier for nucleus formation, engineered surfaces can significantly accelerate the nucleation process and improve crystal quality [77]. The effectiveness of different interface types depends on their specific interactions with target biomolecules:
- Porous substrates provide confined environments that concentrate biomolecules and lower nucleation barriers
- Hydrophobic and charged surfaces interact with complementary regions on protein surfaces to promote molecular alignment
- Functionalized substrates with specific chemical groups (e.g., APTES, nitrilotriacetic acid) can form directed interactions with target molecules
- Rough surfaces increase the available area for nucleation and may provide preferential nucleation sites [77]
The mechanism of surface-induced nucleation typically involves either electrostatic complementarity (e.g., matching surface charge with protein pI) or non-electrostatic interactions such as hydrophobic matching, chemical recognition, and geometric confinement [77].
Additive-Assisted Nucleation Enhancement
The strategic introduction of additives represents another effective approach to accelerate biomolecular nucleation:
- Micro- and macroparticles (e.g., porous polystyrene-divinylbenzene) provide heterogeneous nucleation surfaces
- Nanoparticles (gold, platinum, nanodiamonds) offer high surface area-to-volume ratios and tunable surface chemistry
- Carbon nanomaterials (graphene, graphene quantum dots) present ordered surfaces that can template molecular assembly
- Molecularly imprinted polymers (MIPs) create biomimetic recognition sites for specific target molecules [77]
These additives function through various mechanisms including molecular confinement, surface patterning, electrostatic steering, and reduction of nucleation energy barriers.
Deep Eutectic Solvent-Mediated Crystallization
Deep eutectic solvents (DESs) have recently emerged as sustainable media for modulating nucleation and crystal growth of biomolecules and pharmaceuticals. These systems can regulate polymorphism, crystal habit, and cocrystal formation through their unique solvent properties and ability to form specific interactions with solute molecules [78]. The tunable nature of DES composition allows researchers to systematically optimize crystallization conditions for specific biomolecular targets.
Detailed Experimental Protocol: Insulin Crystallization
The following protocol for insulin crystallization illustrates key principles in controlling nucleation kinetics, incorporating elements for both classical and nonclassical pathway analysis:
Table 3: Research Reagent Solutions for Insulin Crystallization Studies [47]
| Reagent | Composition | Function | Critical Parameters |
|---|---|---|---|
| Insulin Solution | 0.25-2.5 mg/mL recombinant human insulin in HCl (pH 1.6) | Target protein for crystallization | Concentration, pH, storage conditions |
| Precipitant Solution | 1.6-4.7 mM ZnCl2, 62.5 mM trisodium citrate, 12.5% (v/v) acetone (pH 6.2) | Induces supersaturation and provides essential cofactors | Zinc concentration, pH, acetone percentage |
| Buffer Components | Trisodium citrate, HCl | pH control and maintenance of protein stability | Buffer capacity, ionic strength |
| Cosolvents | Acetone, potential alternatives: PEG, phenol | Modifies solvent properties and promotes molecular assembly | Polarity, hydrogen bonding capacity |
Procedure:
Solution Preparation: Prepare insulin stock solution at 2.5 mg/mL in HCl (pH 1.6). Prepare precipitant solution with varying ZnCl2 concentrations (1.6-4.7 mM) to probe different nucleation pathways [47].
Sample Mixing: Combine insulin and precipitant solutions in appropriate ratios to achieve final target concentrations. Mix gently to avoid introducing air bubbles or generating excessive shear forces during initial mixing.
Nucleation Monitoring: Utilize a combination of techniques to detect nucleation events and characterize pathways:
- Rotational rheometry: Measure shear viscosity curves using cone-and-plate geometry with controlled temperature (5-40°C). Newtonian behavior suggests CNT pathway, while Newtonian to shear-thinning transitions indicate nonclassical mechanisms [47].
- Dynamic Light Scattering (DLS): Monitor scattering intensity fluctuations to detect cluster formation and size evolution during early nucleation stages [47].
- Optical microscopy: Periodically examine samples for crystal appearance, noting induction times and crystal morphology.
Pathway Analysis: Correlate rheological measurements with structural characterization:
- Constant viscosity maintenance suggests direct crystal formation via CNT
- Shear-thinning behavior indicates aggregation/agglomeration events
- Viscosity transitions suggest multistep nucleation mechanisms [47]
Characterization: For formed crystals, perform additional analysis using Energy-Dispersive Spectroscopy (EDS), Scanning Electron Microscopy (SEM), and Differential Scanning Calorimetry (DSC) to confirm crystal identity, morphology, and stability [47].
Diagram 2: Experimental workflow for analyzing insulin nucleation pathways. Multiple parallel monitoring techniques enable discrimination between classical and nonclassical mechanisms [47].
Integrated Approaches and Future Directions
The most effective strategies for overcoming biomolecular nucleation slowness increasingly combine multiple complementary approaches. Integrating surface engineering with additive assistance, for example, can create synergistic effects that substantially reduce nucleation barriers. Similarly, combining traditional crystallization screens with real-time monitoring techniques (rheology, DLS, in situ imaging) enables researchers to not only identify successful conditions but also understand the mechanistic pathways involved.
Future advances in this field will likely focus on several key areas:
- Predictive modeling that incorporates molecular-level interactions to forecast nucleation behavior
- High-throughput automated systems for rapid screening of crystallization conditions and additives [61]
- Advanced in situ characterization techniques that provide atomic-level insights into early nucleation events
- Bio-inspired approaches that mimic natural mineralization and crystallization control mechanisms
- Machine learning applications to identify patterns in crystallization outcomes and optimize conditions [77]
The continuing dialogue between classical and nonclassical nucleation perspectives remains essential for developing a comprehensive understanding of biomolecular crystallization. By acknowledging the limitations of CNT while leveraging its predictive capabilities where appropriate, and by recognizing the prevalence of nonclassical pathways in biomolecular systems, researchers can develop more effective, rational strategies to overcome the persistent challenge of slow biomolecular crystal nucleation.
As experimental and computational capabilities continue to advance, the integration of multiscale modeling with high-precision experimentation will likely yield new generations of nucleation control strategies, ultimately transforming crystal nucleation from a persistent bottleneck into a precisely controllable process in biomolecular research and pharmaceutical development.
Validating Theories: A Critical Comparison of CNT and Nonclassical Models
Nucleation, the initial step in the formation of a new phase from a parent phase, is a fundamental process governing phenomena ranging from cloud formation to pharmaceutical crystallization. For decades, Classical Nucleation Theory (CNT) has served as the primary framework for describing this process. However, a growing body of experimental and computational evidence reveals significant discrepancies between CNT predictions and observed behavior across diverse systems, prompting the development of nonclassical nucleation theories [12] [4].
This whitepaper provides an in-depth technical comparison of classical and nonclassical nucleation theories, focusing on their predictions and validation through experimental data. Within the context of organic crystals research—particularly relevant for drug development—we examine critical deviations from CNT, explore alternative mechanistic pathways, and present quantitative data from state-of-the-art experimental techniques.
Theoretical Frameworks: Core Principles and Predictions
Classical Nucleation Theory (CNT)
CNT describes nucleation as a process where atomic or molecular clusters form spontaneously within a metastable parent phase. If these clusters exceed a critical size, they become stable and grow into the new phase. The theory is built on the capillarity approximation, which assumes that the properties of these nascent nuclei (e.g., surface tension) are identical to those of the bulk macroscopic phase [4].
The free energy cost for forming a spherical nucleus of radius r is given by:
ΔG = 4πr²γ - (4/3)πr³|ΔGv|
where γ is the interfacial free energy and ΔGv is the bulk free energy gain per unit volume. This results in an energy barrier, ΔG/, that must be overcome for nucleation to proceed. The nucleation rate *J is exponentially dependent on this barrier: J = K exp(-ΔG//kBT), where *K is a kinetic prefactor [4].
Nonclassical Nucleation Theories
Nonclassical nucleation theories challenge the CNT framework by proposing alternative pathways that do not rely on the capillarity approximation or the formation of a critical-sized nucleus in a single step. Key mechanisms include:
- Pre-Nucleation Cluster (PNC) Pathways: Stable clusters exist in solution before the appearance of a nascent crystalline phase [12].
- Two-Step Nucleation: A dense liquid phase (DLP) or amorphous intermediate forms first, which subsequently solidifies into a crystalline entity [12].
- Stepwise Nucleation and Coalescence: Systems circumvent the high energy barrier of homogeneous nucleation via the coalescence of subcritical clusters or other multi-step processes [7].
These pathways are not mutually exclusive, and evidence suggests that nucleation may often proceed via an amalgamation of multiple nonclassical mechanisms [12].
Quantitative Data: Theory vs. Experiment
The table below summarizes key quantitative comparisons between CNT predictions, nonclassical theory predictions, and experimental observations across various material systems.
Table 1: Quantitative Comparison of CNT Predictions, Nonclassical Theory, and Experimental Data
| Material System | CNT Prediction | Nonclassical Prediction | Experimental/Observed Data | Discrepancy/Magnitude | Source |
|---|---|---|---|---|---|
| Hard Sphere Colloids | Nucleation Rate Density (NRD) at Φ ≈ 0.52 | Not Applicable (CNT used as baseline) | NRD measured via confocal microscopy | Disagreement by 22 orders of magnitude in NRD [79] | |
| Iron (BCC nucleation in FCC) | High energy barrier for homogeneous nucleation | Low-energy pathway via coalescence of subcritical clusters & stepwise nucleation | Molecular Dynamics (MD) simulations show nonclassical pathways | Atomic system circumvents the high CNT energy barrier [7] | |
| Flufenamic Acid (Pharmaceutical) | Single-step nucleation via critical nucleus | Two-step nucleation via PNCs and DLP | Liquid Phase Electron Microscopy (LPEM) shows PNC pathway | Observation of intermediate pre-crystalline stages [12] | |
| Nanoscale Gaseous Nuclei (Cavitation) | Blake threshold pressure | Lower cavitation pressures | MD simulations and extended CNT (Tolman/VdW corrections) | New CNT formulation predicts lower pressures, matching MD data [80] | |
| Binary Patchy Particle Mixture | Identical nucleation for all polymorphs | Variable nucleation rates based on melt structure | MD simulations show radically different nucleation properties | Nucleation properties depend on polymorph unit cell size [4] |
Experimental Protocols and Methodologies
Direct observation of nucleation events, especially for small organic molecules, presents significant challenges due to their small size and the transient nature of intermediate stages. The following advanced techniques have been crucial in gathering evidence for nonclassical pathways.
Table 2: Key Experimental and Computational Methodologies in Nucleation Research
| Technique | Key Application | Technical Details | System Studied |
|---|---|---|---|
| Laser-Scanning Confocal Microscopy (LSCM) | Direct-space observation of crystallization at particle level | Fluorescent PMMA particles in gravity-matched solvent; particle coordinate tracking with local bond order analysis [79] | Colloidal Hard Spheres |
| Liquid Phase Electron Microscopy (LPEM) | Direct visualization of nanoscale early-stage nucleation | High electron flux to induce nucleation in native solvent environment; imaging of intermediate stages [12] | Flufenamic Acid (FFA) |
| Molecular Dynamics (MD) Simulations | Atomic-level analysis of nucleation mechanisms | Simulation of homogeneous nucleation processes; analysis of atomic trajectories and cluster formation [7] [4] | Iron, Binary Patchy Particles |
| Successive Umbrella Sampling | Computing free energy landscapes | Used to calculate free energy cost of interface formation between polymorphs and liquid [4] | Binary Patchy Particles |
Detailed Protocol: Direct Observation of Hard Sphere Nucleation
A comprehensive investigation of nucleation in colloidal hard spheres using LSCM provides a benchmark for quantitative comparison with theory [79].
- Sample Preparation: Sterically stabilized, fluorescent Poly(methyl methacrylate) (PMMA) particles (diameter = 1.388 ± 0.002 μm) are dispersed in a mixture of cis-decalin (CDL) and tetrachloroethylene (TCE) to match both mass density and refractive index.
- Sample Cell Preparation: Custom cells with screw caps are used, allowing for highly accurate volume fraction adjustments (~1 mL suspension). All container walls are coated with larger PMMA particles to eliminate heterogeneous nucleation.
- Shear Melting: Before kinetics studies, samples are shear-molten by tumbling for several hours at ~1 Hz.
- Data Acquisition: A white light LSCM (e.g., Leica TCS-SP8) scans 25 volumes of 82 × 82 × 60 μm³ (~3 × 10⁶ particles) in the lateral center of the cell. Voxel size is ~80 × 80 × 130 nm³, with a scan time of ~50 s per volume.
- Data Analysis: Particle coordinates are determined using custom algorithms. Crystalline clusters are identified using local bond order parameters (q₄, q₆). A particle is crystalline if it has ≥8 nearest neighbors within 1.4× the particle diameter and a scalar product >0.5. Clusters of ≥4 connected crystalline particles are identified as a crystalline cluster.
Detailed Protocol: Observing Nonclassical Pathways in Pharmaceuticals
LPEM has been used to capture the nonclassical crystallization pathway of the pharmaceutical Flufenamic Acid (FFA) [12].
- Solution Preparation: A 50 mM solution of FFA in ethanol is prepared.
- LPEM Experiment: The solution is loaded into a liquid cell holder with silicon nitride windows. A high electron dose (>150 e⁻/Ų/s) is used to induce nucleation via radiolysis of the solvent, which alters the local chemical environment.
- Imaging: The process is monitored in real-time, capturing the formation of pre-nucleation clusters and their evolution into a dense liquid phase before solidification into crystalline FFA.
- Data Interpretation: The temporal evolution of clusters is tracked, providing direct evidence for a PNC pathway followed by two-step nucleation.
Visualization of Nucleation Pathways
The following diagrams illustrate the key differences between the classical and nonclassical nucleation pathways.
Classical Nucleation Theory (CNT) Pathway
Classical Nucleation Theory Pathway: This pathway involves a single, reversible step where a cluster must overcome a well-defined energy barrier to become a critical nucleus.
Nonclassical Nucleation Pathway
Nonclassical Nucleation Pathway: This multi-step pathway involves the formation of several intermediate states, such as pre-nucleation clusters and a dense liquid phase, before crystallization.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Nucleation Studies
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| Fluorescent PMMA Particles | Model colloidal system for direct observation | Serves as a hard sphere model system for confocal microscopy studies [79] |
| cis-Decalin (CDL) / Tetrachloroethylene (TCE) Mixture | Dispersion medium for colloids | Matches refractive index and mass density of PMMA particles for optimal imaging [79] |
| Flufenamic Acid (FFA) | Model pharmaceutical compound | Study of nonclassical nucleation pathways in small organic molecules [12] |
| DNA Origami/Patches | Engineered specific interactions | Used to create patchy particles with programmable bonding for falsifiability tests of CNT [4] |
| Particle Coatings (e.g., poly-hydroxy-stearic-acid) | Steric stabilization | Prevents aggregation in colloidal suspensions, maintaining system stability [79] |
| Catalyst Nanoparticles (e.g., Fe, Co) | Catalyze CNT growth | Serves as a substrate for carbon nanotube synthesis in CVD processes [9] |
The accumulation of high-quality experimental and computational data across diverse systems—from colloidal hard spheres to pharmaceuticals and metals—consistently reveals profound quantitative and mechanistic failures of Classical Nucleation Theory. Discrepancies of up to 22 orders of magnitude in nucleation rates and the direct observation of multi-step, nonclassical pathways necessitate a paradigm shift in how nucleation is conceptualized and modeled [79] [12] [4].
For researchers and drug development professionals, this has critical implications. The reliance on CNT for predicting crystallization behavior, polymorph selection, and product stability in pharmaceuticals is inherently risky. Embracing nonclassical frameworks that account for system-specific intermediate states and pathways is essential for advancing predictive control in materials synthesis and pharmaceutical manufacturing. Future research must integrate advanced in situ characterization techniques with multi-scale computational models to fully elucidate the complex mechanisms governing the birth of crystals.
The transition from a disordered liquid or solution to an ordered crystalline solid represents one of the most fundamental phase transformations in materials science, with profound implications for drug development, pharmaceutical formulation, and advanced materials design. For decades, Classical Nucleation Theory (CNT) has served as the predominant theoretical framework for describing this process, modeling nucleation as a single-step activation barrier crossing where nascent crystal clusters compete between favorable bulk free energy and unfavorable surface energy [81]. However, the persistent inability of CNT to consistently predict experimental nucleation rates, even in simple glass-forming systems, has revealed significant limitations in this idealized model [81] [82]. This has sparked a vigorous scientific debate centered on the energy landscapes of crystallization pathways and prompted the development of non-classical theories that account for more complex, multi-step nucleation mechanisms involving metastable intermediate phases [82].
The core of this debate hinges on a thermodynamic and kinetic dichotomy: CNT describes a smooth, single-barrier landscape, while non-classical models propose more complex landscapes with multiple minima and barriers corresponding to transient intermediate states. For researchers developing organic crystalline drugs, this distinction is not merely academic—it directly impacts crystallization outcomes, polymorph selection, crystal habit, and ultimately, drug bioavailability and stability. This review synthesizes current understanding of these competing models, their experimental validation, and practical methodologies for probing these energy landscapes directly.
Theoretical Frameworks: Classical vs. Non-Classical Perspectives
Classical Nucleation Theory (CNT) and Its Extensions
Classical Nucleation Theory provides a thermodynamic description of crystal formation using a continuum approximation. The core model posits that the free energy cost to form a spherical nucleus of radius r is given by:
ΔG = - (4/3)πr³|Δμ| + 4πr²γₗₛ
where |Δμ| is the thermodynamic driving force (typically proportional to supersaturation or supercooling), and γₗₛ is the liquid-solid interfacial tension [58]. This relationship produces a well-defined energy barrier, ΔG, at a critical nucleus size *r, beyond which growth becomes thermodynamically favorable. The nucleation rate *I follows an Arrhenius dependence on this barrier:
I = A exp(-ΔG/kT)
where A is a kinetic prefactor [58]. Despite its simplicity, CNT has demonstrated remarkable robustness, even in heterogeneous nucleation scenarios involving chemically heterogeneous surfaces where its underlying assumptions of fixed contact angle might be expected to fail [58].
Recent work has extended CNT to address specific limitations. For nanoscale nuclei where curvature effects become significant, the Tolman correction introduces curvature-dependent surface tension, while the Van der Waals correction accounts for real-gas behavior in systems like nanoscale gaseous nuclei [80]. These refinements are crucial for accurate prediction in systems like cavitation inception but remain within the conceptual framework of single-step nucleation.
Non-Classical Nucleation and the Two-Step Model
Non-classical nucleation challenges CNT's single-step premise by proposing that crystal formation frequently proceeds through metastable intermediate phases. The two-step nucleation (2S) model suggests that a dense, disordered liquid phase or a stable intermediate phase initially forms, subsequently serving as a template for the final crystalline phase [82]. This mechanism is particularly relevant to perovskite solar cells and organic crystal formation, where solvent-induced intermediates and low-dimensional phases are commonly observed [82].
The thermodynamic advantage of this pathway lies in the significantly lower nucleation energy barrier (ΔG₂) required to form the intermediate phase compared to the direct barrier (ΔG₁) to form the stable crystal [82]. The intermediate, while not the global free energy minimum, acts as a kinetic stepping stone. The resulting energy landscape features multiple minima and barriers, explaining phenomena like pre-nucleation clusters and polymorph selection that are difficult to reconcile with CNT. This framework has proven essential for understanding and controlling crystallization in complex organic and hybrid organic-inorganic systems where direct nucleation is kinetically hindered.
Table 1: Comparison of Classical and Non-Classical Nucleation Theories
| Feature | Classical Nucleation Theory (CNT) | Two-Step (2S) Non-Classical Theory | ||
|---|---|---|---|---|
| Fundamental Pathway | Single-step, direct formation of crystalline phase from disordered phase | Multi-step, involves metastable intermediate phases (e.g., dense liquid, pre-nucleation clusters) | ||
| Energy Landscape | Single activation barrier (ΔGₕₒₘ*) | Multiple minima and barriers; lower initial barrier (ΔG₂ < ΔG₁) [82] | ||
| Nucleus Structure | Sharp interface, spherical cap (heterogeneous) or spherical (homogeneous) | Diffuse interface; intermediate phase may lack long-range order | ||
| Key Parameters | Interfacial tension (γₗₛ), thermodynamic driving force ( | Δμ | ) | Properties and stability of the intermediate phase |
| Strengths | Simple, intuitive, robust for many heterogeneous systems [58] | Explains polymorph control, pre-nucleation, crystallization in complex systems like perovskites [82] | ||
| Limitations | Often underestimates/overestimates rates; fails in non-ideal, complex systems | More complex, requires characterization of transient intermediates |
Computational and Experimental Methodologies
Computational Modeling of Nucleation Pathways
Computational approaches are indispensable for probing nucleation mechanisms at the atomic and molecular level, providing direct insight into energy landscapes.
Energy Landscape Modeling: This method involves mapping the potential energy surface U(x₁, y₁, z₁, ... xₙ, yₙ, zₙ) of the system to identify local minima (inherent structures) and the first-order saddle points connecting them [81]. For barium disilicate glass, this approach successfully predicted nucleation rates and liquidus temperature by independently calculating CNT parameters (interfacial energy, kinetic barrier, free energy difference) from the landscape, validating CNT's underlying physics in this specific system [81].
Molecular Dynamics (MD) Simulations: MD tracks the temporal evolution of atom positions by integrating Newton's equations of motion. Standard MD is often insufficient for observing spontaneous nucleation due to timescale limitations. Studies on model atomic liquids using the Lennard-Jones potential simulate systems with thousands of atoms (e.g., ~10,752 A-type particles) in periodic boxes with dimensions on the scale of tens of particle diameters (e.g., lₓ ≈ 34σₐₐ) [58]. Simulations are run at reduced temperatures (kT/εₐₐ) below the melting point to observe supercooled liquid behavior.
Enhanced Sampling Techniques: Methods like Jumpy Forward Flux Sampling (jFFS) overcome MD's timescale limitation by efficiently sampling the sequence of configurations that lead from the metastable liquid to the stable crystal [58]. This technique is particularly valuable for quantifying nucleation rates and examining the structure of critical nuclei on patterned or chemically heterogeneous surfaces, providing direct tests of CNT's geometric assumptions [58].
Experimental Characterization Techniques
Experimental validation of nucleation theories requires techniques capable of detecting nascent nuclei and transient intermediates.
Differential Scanning Calorimetry (DSC): A mainstay for studying crystallization kinetics in glasses and organic systems, DSC measures heat flow associated with phase transitions as a function of temperature. It can determine nucleation rates indirectly by analyzing the crystallization exotherms of pre-nucleated samples, providing data on the temperature dependence of nucleation [81].
Electron Microscopy: Transmission and scanning electron microscopy (TEM/SEM) offer direct visualization of crystal nuclei, their spatial distribution, and morphology. Advanced techniques like cryo-TEM are particularly powerful for capturing the structure of transient intermediate phases in solution before they transform into stable crystals, providing direct evidence for non-classical pathways [82].
X-ray Scattering Methods: Both wide-angle (WAXS) and small-angle (SAXS) X-ray scattering are used to probe structural evolution during crystallization. WAXS identifies the emergence of long-range crystalline order, while SAXS is sensitive to the formation of nanoscale density fluctuations, pre-nucleation clusters, and intermediate phases, even when they lack long-range order [82].
Table 2: Key Experimental Parameters in Nucleation Studies
| Parameter | Description | Measurement Techniques | Impact on Nucleation | ||||
|---|---|---|---|---|---|---|---|
| Interfacial Tension (γₗₛ) | Free energy per unit area of the crystal-liquid interface | Energy landscape modeling [81], Contact angle measurements (heterogeneous) | Directly determines CNT energy barrier; ΔG* ∝ γₗₛ³ [58] | ||||
| Thermodynamic Driving Force ( | Δμ | ) | Free energy difference between liquid and crystal per molecule | DSC, Calculation from undercooling/supersaturation | Drives nucleation; ΔG* ∝ 1/ | Δμ | ² [58] |
| Contact Angle (θc) | Angle between nucleus and substrate in heterogeneous nucleation | MD simulations, jFFS analysis on model surfaces [58] | Determines potency factor fc(θc); lowers effective barrier [58] | ||||
| Zeldovich Factor (Ze) | Probability a critical nucleus grows vs. dissolves | ~0.1 used as an approximation in computational studies [81] | Kinetic factor in nucleation rate | ||||
| Diffusivity (D) | Molecular mobility in the supercooled liquid/solution | MD simulations, analysis of relaxation time <*τ*(*T*)> [81] | Kinetic prefactor in nucleation rate |
Case Studies and Applications
Inorganic Glass-Ceramics: Barium Disilicate
The barium disilicate (BaO·2SiO₂) system serves as a model for testing nucleation theories in inorganic glasses. Energy landscape modeling of this system, using 768-atom simulation cells, successfully calculated the temperature dependence of the nucleation rate by independently determining the interfacial energy, kinetic prefactor, and thermodynamic driving force [81]. The results showed fair agreement with experimental data, providing a parameter-free validation of CNT for this specific system [81]. This study demonstrated that all CNT parameters could be computed ab initio from the landscape, reinforcing CNT's utility while also highlighting the computational intensity required.
Organic-In Hybrid Perovskites for Photovoltaics
The crystallization of perovskite films for solar cells is a prime example where CNT fails and non-classical models excel. The rapid, conventional solution crystallization often leads to defective films. Intermediate phase engineering leverages the two-step nucleation model by deliberately forming and controlling metastable intermediates (e.g., solvent-induced or additive-induced complexes) [82]. These intermediates act as thermodynamic templates, regulating subsequent crystal growth, reducing defect densities, and enhancing film uniformity [82]. This control over the energy landscape has been pivotal in achieving high-performance, large-area perovskite solar cells with power conversion efficiencies exceeding 26% [82].
Heterogeneous Nucleation on Engineered Surfaces
Molecular dynamics and jFFS studies of heterogeneous nucleation on chemically patterned "checkerboard" surfaces (with alternating liquiphilic and liquiphobic patches) reveal the surprising robustness of CNT [58]. Despite surface heterogeneity, the nucleation rate retained its canonical temperature dependence, and critical nuclei maintained an almost fixed contact angle through a pinning mechanism at patch boundaries [58]. This explains CNT's unexpected success in many experimental contexts involving impure or imperfect surfaces and provides design principles for engineered nucleants in pharmaceutical crystallization.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for Nucleation Studies
| Reagent / Tool | Type/Class | Function in Nucleation Research |
|---|---|---|
| Lennard-Jones Potential | Interatomic Potential | A computationally efficient model potential (e.g., U(r) = 4ε[(σ/r)¹² - (σ/r)⁶]) for MD simulations of nucleation in model atomic liquids [58]. |
| LAMMPS | Software Package | A widely used open-source molecular dynamics simulator for performing nucleation simulations with various force fields and boundary conditions [58]. |
| Jumpy Forward Flux Sampling (jFFS) | Sampling Algorithm | An enhanced sampling technique to calculate nucleation rates and mechanisms in MD simulations by efficiently traversing the free energy barrier [58]. |
| Molecular Additives (e.g., MACl, DMSO) | Chemical Modifier | Used in perovskite and organic crystal formation to form stable intermediate phases (e.g., low-dimensional perovskites), controlling crystallization kinetics and final film quality [82]. |
| Harmonic Spring Potentials | Computational Tool | Used in MD to tether atoms in substrate surfaces (spring constant ~500 ε/σ²), modeling rigid but atomistically detailed nucleating substrates [58]. |
The debate between classical and non-classical nucleation theories is not a winner-take-all contest but a refinement of our understanding of energy landscapes across different material systems. CNT, often extended with corrections for curvature and non-ideal behavior, remains a powerful and surprisingly robust framework, especially for heterogeneous nucleation on complex surfaces [58] [80]. However, the two-step and other non-classical models have proven indispensable for explaining and engineering crystallization in complex, functional materials like organic pharmaceuticals and hybrid perovskites, where metastable intermediates dictate the final outcome [82].
The future of this field lies in the integration of high-fidelity computational modeling, advanced experimental characterization, and data science. Energy landscape modeling offers a pathway to parameter-free prediction of nucleation phenomena [81]. Furthermore, artificial intelligence and machine learning are poised to accelerate the discovery of molecular additives and predict the structure of low-dimensional intermediate phases, thereby enabling the rational design of crystallization pathways for next-generation drugs and materials [82]. This multidisciplinary approach will ultimately provide researchers with a unified and predictive framework for controlling one of nature's most fundamental self-assembly processes.
Nucleation kinetics fundamentally control material synthesis, geochemical processes, and pharmaceutical development, yet significant discrepancies between predicted and observed nucleation rates persist across scientific disciplines. Traditional interpretations within Classical Nucleion Theory (CNT) heavily emphasize the thermodynamic energy barrier (ΔG*). However, emerging quantitative evidence from diverse systems—including small organic molecules, proteins, and calcium carbonate—increasingly implicates the pre-exponential kinetic factor (A) and its associated apparent activation energy (Ea) as primary sources of these discrepancies. This whitepaper synthesizes current experimental data and theoretical analyses, demonstrating that heterogeneous surfaces and molecular-specific interactions can alter pre-exponential factors by orders of magnitude without significantly changing interfacial energies. The findings necessitate a critical re-evaluation of CNT and broader adoption of non-classical perspectives that explicitly account for kinetic parameters in organic crystal research and drug development.
Classical Nucleation Theory (CNT) has long provided the foundational framework for describing the initial step of phase formation from a supersaturated solution. The theory expresses the steady-state nucleation rate (J) as the product of a kinetic pre-factor and an exponential thermodynamic term:
[J = A \exp\left(-\frac{E_a}{RT}\right) \exp\left(-\frac{\Delta G^*}{RT}\right)]
where (A) is the pre-exponential kinetic factor, (E_a) is the apparent activation energy, (R) is the ideal gas constant, (T) is temperature, and (\Delta G^*) is the thermodynamic free energy barrier associated with forming a critical nucleus [83] [81].
Historically, research emphasis has centered on the exponential term, particularly how (\Delta G^) relates to interfacial energies and supersaturation. This focus stems from the profound influence small changes in interfacial energy exert on the nucleation rate due to its cubic relationship with (\Delta G^). Consequently, the pre-exponential factor (A) was often assumed constant or its variability considered negligible [83]. However, persistent, orders-of-magnitude discrepancies between theoretical predictions and experimental measurements of nucleation rates, especially in protein crystallization and complex organic systems, have challenged this simplification [84] [81].
A paradigm shift is underway, driven by evidence that the kinetic factors (A) and (E_a) are not mere constants but are sensitive to molecular composition, solvent interactions, and the nature of heterogeneous surfaces. This whitepaper collates quantitative evidence establishing the pre-exponential factor's critical role in nucleation rate discrepancies, framing these insights within the broader debate between classical and non-classical nucleation theories.
Quantitative Data: Compiling the Evidence
Direct experimental determinations of (A) and (E_a) reveal that kinetic parameters vary significantly across different systems and are critically dependent on specific molecular interactions.
Table 1: Experimentally Determined Pre-exponential Factors and Activation Energies
| System | Nucleation Type | Pre-exponential Factor (A) | Apparent Activation Energy (Ea) | Key Finding | Reference |
|---|---|---|---|---|---|
| Calcium Carbonate on Quartz | Heterogeneous | 10¹²·⁰ ± ¹·¹ nuclei μm⁻² min⁻¹ | 45 ± 7 kJ mol⁻¹ | Direct measurement of (A) and (E_a); temperature shortens induction time without changing nucleus size. | [83] |
| Glycine/Diglycine in Aqueous Solution | Heterogeneous (Templated) | Increase of ≥2-fold vs. homogeneous | Unchanged | Heterogeneous surface enhances nucleation via increased (A), not by reducing interfacial energy. | [85] |
| Lysozyme Crystallization | Homogeneous (Protein) | Multiple estimates vary widely | N/A | Large discrepancies in (A) and (J) between different experimental methods. | [84] |
| Barium Disilicate Glass | Homogeneous | Computed from energy landscape | Computed from energy landscape | Energy landscape modeling validates CNT form but requires accurate (A) and surface energy. | [81] |
The data in Table 1 underscores several key points. A landmark study on calcium carbonate nucleation on quartz provided direct, experimental quantification of these previously elusive parameters [83]. Meanwhile, research on glycine and diglycine demonstrated that a heterogeneous template could double the pre-exponential factor without altering the system's interfacial energy, a mechanism attributed to hydrogen bond formation between the solute and template surface [85]. Perhaps most strikingly, a comparison of three independent experimental estimates of lysozyme crystal nucleation rates revealed "large discrepancies," with models that successfully describe small-molecule systems grossly overpredicting protein nucleation rates—in some cases by tens of orders of magnitude [84]. This failure points to fundamental gaps in our understanding of the kinetic prefactor for complex molecules.
Experimental Protocols for Kinetic Parameter Determination
Accurately determining the parameters (A) and (E_a) requires sophisticated techniques capable of probing early-stage nucleation and quantifying rates.
Induction Time Measurements and Statistical Analysis
This common methodology involves conducting numerous identical crystallization experiments under constant supersaturation and measuring the time elapsed before nucleation is detected (the "induction time"). The statistical distribution of these induction times directly relates to the nucleation rate. As established by Skripov and refined by others, the probability distribution of nucleation events can be analyzed to extract both the pre-exponential factor (K_v) and the activation energy (\Delta G^*) [86]. Monte Carlo simulations have verified that for a data set of 100 experimental runs, the uncertainty in the exponential factor and the log of the pre-exponential factor is approximately 10%, assuming precise temperature control [86].
In Situ Grazing Incidence Small-Angle X-Ray Scattering (GISAXS)
For heterogeneous nucleation, GISAXS is a powerful tool for direct, real-time observation. This protocol involves:
- Sample Preparation: A substrate of interest (e.g., a quartz crystal) is immersed in a metastable supersaturated solution (e.g., with IAP/Ksp = 10¹·⁶⁵ for CaCO₃).
- In Situ Measurement: The solution-substrate interface is probed with a focused X-ray beam at a grazing incidence angle. Scattering patterns are collected at regular time intervals.
- Data Analysis: The scattering intensities are analyzed using model fitting to determine the number density and size of nuclei forming on the substrate over time. The slope of the nucleus number versus time plot gives the nucleation rate ((J)) [83].
- Variable Temperature Studies: Repeating the experiment at different temperatures (e.g., 12, 25, and 31°C) allows for the construction of an Arrhenius plot ((\ln(J)) vs. (1/T)). The slope of this plot yields the combined energy barrier ((\Delta G^* + Ea)), from which (Ea) can be derived after independently calculating (\Delta G^*) [83].
Complementary Ex Situ Atomic Force Microscopy (AFM)
GISAXS data can be validated and supplemented with ex situ AFM measurements. After nucleation experiments, the substrate is carefully removed from solution, dried, and imaged with AFM. This provides direct, real-space visualization of nucleus density and morphology, offering a cross-check for the quantitative data extracted from GISAXS [83].
The following workflow diagram illustrates the integration of these techniques for determining kinetic parameters in a heterogeneous nucleation study:
Diagram 1: Experimental workflow for determining kinetic parameters in heterogeneous nucleation.
The Scientist's Toolkit: Key Research Reagents and Materials
The following table details essential materials and their functions in nucleation kinetics research, as derived from the cited studies.
Table 2: Essential Research Reagents and Materials
| Material/Reagent | Function in Nucleation Research | Specific Example |
|---|---|---|
| Quartz Substrate | A well-defined, chemically homogeneous surface for studying heterogeneous nucleation kinetics. | Used as a model silicate substrate in CaCO₃ nucleation studies [83]. |
| Amino Acids (Glycine, Diglycine) | Model organic compounds for studying molecular-specific interactions in templated nucleation. | Used to demonstrate the 2-fold increase in pre-exponential factor due to hydrogen bonding with a heterosurface [85]. |
| Lysozyme | A model protein for investigating the unique challenges of macromolecular crystallization. | Used to highlight large discrepancies in nucleation rate estimates between different experimental methods [84]. |
| Ultrafine Carbon Particles | Nucleating agents for studying heterogeneous nucleation of organic vapors. | Used as heterogeneous nuclei for compounds like dibutyl phthalate and octadecanol [87]. |
| Pharmaceutical Compounds (ABT-072, ABT-333) | Complex organic molecules for studying the impact of crystal packing and polymorphism on nucleation and solubility. | Used in crystal structure prediction (CSP) and molecular dynamics (MD) studies to link molecular structure to nucleation behavior [88]. |
Implications for Organic Crystals and Pharmaceutical Research
The recognition that kinetic factors drive nucleation discrepancies has profound implications for organic crystal research, particularly in pharmaceuticals.
- Rational Co-Crystal Design: Crystal engineering, which employs pharmaceutical co-crystals to improve drug solubility and stability, relies on understanding intermolecular interactions. The finding that hydrogen bonding can directly increase the pre-exponential factor provides a mechanistic rationale for selecting coformers that not only alter thermodynamics but also enhance nucleation kinetics [85] [89].
- Predictive Modeling Challenges: Early-stage drug development often uses machine learning (ML) to profile compounds. However, ML models that lack explicit 3-D structural and dynamic information struggle to differentiate structural analogs like ABT-072 and ABT-333. Physics-based simulations, such as Crystal Structure Prediction (CSP) and Molecular Dynamics (MD), are required to capture conformational flexibility and kinetic accessibility that govern polymorphism and nucleation [88].
- Addressing Solubility Challenges: The aqueous solubility of a drug is drastically reduced by crystallization. Formulation efforts must account for the fact that small structural changes can significantly alter the kinetic factor (A), thereby affecting the nucleation rate and the resulting crystal form, with direct consequences for bioavailability [88].
The following diagram conceptualizes how molecular and environmental factors influence the kinetic and thermodynamic parameters of nucleation, ultimately determining the observed nucleation rate.
Diagram 2: Factors influencing nucleation rate through kinetic and thermodynamic parameters.
The collective body of quantitative evidence firmly establishes that the pre-exponential factor (A) and the apparent activation energy (E_a) are critical, variable components in the nucleation rate equation. Discrepancies between theory and experiment cannot be resolved by focusing solely on thermodynamic barriers. Instead, a comprehensive understanding must incorporate the kinetic complexities of nucleation, including the profound influence of heterogeneous surfaces via hydrogen bonding and molecular complementarity, as well as the unique challenges posed by macromolecular and flexible organic systems.
For researchers and drug development professionals, this underscores the necessity of moving beyond purely classical interpretations. Embracing non-classical perspectives and advanced computational tools that explicitly account for kinetic parameters and molecular-level interactions is essential for accurately predicting and controlling crystallization across the materials and pharmaceutical sciences. The future of rational crystal design hinges on integrating these nuanced understandings of both kinetics and thermodynamics.
Crystallization, a fundamental process in the production of materials ranging from pharmaceuticals to semiconductors, was traditionally understood through Classical Nucleation Theory (CNT). This model posits that crystals form via the step-by-step addition of monomers, with a critical-sized nucleus emerging directly from solution through density fluctuations [12] [90]. However, a growing body of experimental evidence now challenges this oversimplified view. Non-classical crystallisation (NCC) pathways involve intermediate stages that are more complex than the one-step process described by CNT, often featuring precursor particles such as pre-nucleation clusters, dense liquid phases, or amorphous aggregates [12]. For organic crystals and active pharmaceutical ingredients (APIs), understanding these pathways is not merely academic; it is critical for controlling polymorph selection, crystal habit, and ultimately, drug efficacy and bioavailability [12]. This guide synthesizes current techniques and evidence for the direct observation of these intermediate stages, providing a technical framework for researchers aiming to validate non-classical pathways in their own systems.
The limitations of CNT have become increasingly apparent, particularly for complex molecular systems. CNT fails to explain phenomena such as polymorphic prevalence and the appearance of transient metastable forms that often precede the final, most stable crystal [12]. Non-classical theories, in contrast, provide a more nuanced framework. These include the Pre-Nucleation Cluster (PNC) pathway, where stable clusters exist in solution before any solid phase appears, and the two-step nucleation model, where a dense, liquid-like intermediate serves as a precursor to the crystalline state [12] [90]. Directly observing these low-electron-density, often transient intermediates in small organic molecules presents significant signal-to-noise and contrast challenges [12]. This guide details the advanced methodologies overcoming these hurdles, enabling the direct visualization that is paramount for validating these pathways and harnessing them for rational crystal design.
Key Experimental Techniques for Direct Observation
Validating non-classical pathways requires observational techniques that can probe nanoscale events in real-time, often within a native liquid environment. The following table summarizes the primary methods, their operating principles, and their applications in characterizing intermediate stages.
Table 1: Techniques for Direct Observation of Crystallization Intermediates
| Technique | Fundamental Principle | Key Application in NCC | Spatial/Temporal Resolution | Notable System(s) |
|---|---|---|---|---|
| Liquid Phase Electron Microscopy (LPEM) | High-energy electron beam traverses a thin liquid cell containing the sample. | Direct, in situ visualization of pre-nucleation clusters and intermediate phases in native solvent [12]. | Nanoscale spatial resolution; temporal resolution limited by beam effects and detector speed. | Flufenamic Acid (FFA) in ethanol [12]. |
| Fluorescence Spectroscopy/Microscopy | Tracks changes in emission properties (color, intensity) during molecular assembly. | Probing molecular states (monomer, amorphous aggregate, crystal) via environment-sensitive fluorophores [90]. | Micron-scale spatial; millisecond to second temporal resolution. | Dibenzoylmethane boron complex (BF2DBMb) [90]. |
| Tuned Ionic Colloidal Assembly | Uses microscopic charged particles as "model atoms" to observe assembly pathways. | Direct observation of two-step process: gas → amorphous blob → crystal [16]. | Resolution sufficient to track individual "model atom" particles. | Binary mixtures of oppositely charged colloids [16]. |
| Differential Phase Contrast STEM (DPC-STEM) | Aberration-corrected STEM with segmented detector to map magnetic induction. | Visualizing nucleation intermediate states in magnetic skyrmion systems [91]. | Atomic-scale spatial resolution; lower temporal resolution. | FeGe1-xSix thin plates [91]. |
Core Methodologies and Workflows
The techniques outlined in Table 1 require specialized protocols to successfully capture transient nucleation events.
Liquid Phase Electron Microscopy (LPEM) Protocol: The LPEM experiment for observing flufenamic acid (FFA) crystallization involves several critical steps [12]:
- Sample Preparation: A 50 mM solution of FFA in ethanol is prepared. The solution is loaded into a microfluidic cell with electron-transparent silicon nitride windows.
- Beam-Induced Nucleation: The electron beam is condensed using a monochromator to achieve a high electron flux (>150 e⁻/Ų/s). This high dose is used deliberately to exploit radiolysis of the solvent, which generates reactive species that alter the local chemical environment and induce nucleation in an otherwise undersaturated solution [12].
- In Situ Imaging: The beam interaction region is monitored continuously. High temporospatial imaging captures the evolution of features from low-contrast, low-electron-dense entities into defined crystals.
- Data Interpretation: The observed phenomena, such as the appearance and evolution of pre-crystalline phases, are directly linked to existing non-classical theories like the PNC pathway and two-step nucleation.
Fluorescence-Based Observation Protocol: The workflow for visualizing two-step nucleation using the mechanofluorochromic molecule BF2DBMb is as follows [90]:
- System Characterization: First, the fluorescence profiles of the distinct molecular states are established: purple for the monomer in dilute solution, greenish-orange for the amorphous state, and blue for the crystal [90].
- Evaporative Crystallization: A droplet of BF2DBMb in 1,2-dichloroethane (3.1 × 10⁻² mol·dm⁻³) is deposited and allowed to evaporate under a fluorescence microscope.
- Spectral Acquisition: Fluorescence images and spectra are captured in real-time as the solvent evaporates and the solution becomes supersaturated.
- Quantitative Deconvolution: The time-dependent fluorescence spectra are deconvoluted using non-linear least-squares fitting with Gaussian peaks corresponding to the monomer, amorphous, and crystal species. The relative abundance of each state is plotted over time to reveal the consecutive reaction pathway: Monomer → Amorphous → Crystal [90].
Quantitative Data and Observed Intermediates
Direct observation yields quantitative data on the kinetics and thermodynamics of intermediate phases, providing concrete validation for NCC pathways.
Table 2: Characterized Intermediate States and Their Properties
| System | Observed Intermediate | Key Identifying Feature | Quantitative Metrics | Final Crystal Structure |
|---|---|---|---|---|
| Flufenamic Acid (FFA) | Pre-nucleation clusters (PNCs) and dense liquid phase [12]. | Low-electron-density entities preceding crystal formation via LPEM. | Crystallization induced by electron dose >150 e⁻/Ų/s [12]. | Hexagonal crystals (Form I) [12]. |
| BF2DBMb | Amorphous cluster state [90]. | Greenish-orange fluorescence (λ~550 nm) distinct from monomer and crystal. | Amorphous fraction peaks at ~0.6 relative abundance before decaying as crystal fraction rises [90]. | Blue-fluorescing crystals (λ~445, 470 nm) [90]. |
| Ionic Colloids | Metastable amorphous blobs [16]. | Condensed, liquid-like phase containing both positive and negative particles. | Formation depends on Debye length (λD); occurs in a specific window of interaction strength [16]. | Faceted binary crystals (e.g., CsCl, NaCl structures) [16]. |
| Magnetic Skyrmion (FeGeSi) | Antiskyrmion-like spin texture [91]. | Theoretical predicted twist in a helical stripe, pinned by artificial surface pits. | Observed at specific temperature (95 K) and magnetic field (160 mT) [91]. | Magnetic skyrmion lattice [91]. |
The data in Table 2 demonstrates that intermediate states are a universal phenomenon across diverse systems. For instance, in the BF2DBMb system, quantitative analysis of fluorescence spectra showed the amorphous fraction reaching approximately 60% of the total population before rapidly decaying as the crystalline fraction emerged, providing unambiguous kinetic evidence for the amorphous state acting as a direct precursor to the crystal [90]. Similarly, in ionic colloidal systems, the existence of the amorphous blob phase was shown to be highly dependent on the interaction potential, which can be finely tuned by the solution's Debye length [16]. This tunability allows researchers to map phase diagrams and identify the specific conditions that favor non-classical over classical pathways.
The Scientist's Toolkit: Essential Research Reagents and Materials
Success in these experiments relies on a carefully selected set of reagents and materials.
Table 3: Key Research Reagent Solutions and Materials
| Item | Function in Experiment | Example from Literature |
|---|---|---|
| Model Active Pharmaceutical Ingredient (API) | A well-characterized, often polymorphic, small organic molecule for studying nucleation pathways. | Flufenamic Acid (FFA), a non-steroidal anti-inflammatory drug [12]. |
| Mechanofluorochromic Fluorophore | A molecule whose fluorescence properties change between amorphous and crystalline states, acting as a built-in probe. | Dibenzoylmethane boron complex (BF2DBMb) [90]. |
| Tunable Ionic Colloids | Monodisperse, oppositely charged particles that serve as "model atoms" for direct optical observation of assembly. | Polystyrene particles with polymer brush coating, suspended in solvents with controlled salt concentration [16]. |
| Liquid Cell with Electron-Transparent Windows | Encapsulates the liquid sample for in situ observation within the high-vacuum environment of an electron microscope. | Microfluidic cell with silicon nitride (SiN) windows [12]. |
| Continuous Dialysis Setup | Enables fine spatiotemporal control over interaction strength by dynamically changing solution conditions. | Observation cell connected to a deionized water reservoir to gradually reduce salt concentration [16]. |
Experimental Workflow and Pathway Visualization
The following diagram illustrates the general experimental workflow for validating non-classical pathways using direct observation techniques, integrating multiple approaches from the cited studies:
The mechanistic relationship between observed intermediates and the final crystalline product is complex. The following diagram synthesizes the key non-classical pathways identified in the search results, highlighting the multi-step nature of crystallization:
The direct observation of intermediate stages, as detailed in this guide, has fundamentally shifted the understanding of crystallization from a simple one-step process to a complex, multi-pathway phenomenon. Techniques like LPEM, advanced fluorescence microscopy, and tuned colloidal assembly have provided incontrovertible evidence for non-classical pathways involving pre-nucleation clusters, dense liquid phases, and amorphous intermediates across diverse systems, from small-molecule pharmaceuticals to functional materials [12] [90] [16]. For researchers in drug development, this paradigm offers both a challenge and an opportunity. The challenge lies in the increased complexity of controlling crystallization processes. The opportunity, however, is the potential to direct these pathways toward previously inaccessible polymorphs with more desirable properties, ultimately enabling the rational design of advanced crystalline materials.
The initial step in crystallization, nucleation, fundamentally determines critical material properties such as polymorphism, crystal size distribution, and purity. For decades, Classical Nucleation Theory (CNT) has served as the primary theoretical framework for quantifying this process, modeling it as a direct, single-step assembly of individual atoms or molecules into a stable crystal nucleus [1] [72]. However, a growing body of experimental and computational evidence across diverse materials—from metallic iron to active pharmaceutical ingredients (APIs)—reveals nucleation pathways that are more complex and nuanced than CNT suggests [7] [33] [72]. These observations have given rise to nonclassical nucleation theories, which include mechanisms like the two-step pathway and pre-nucleation clusters.
This whitepaper contends that a dogmatic adherence to either purely classical or nonclassical views is insufficient for a complete understanding of nucleation, particularly in industrially relevant inorganic and pharmaceutical crystals. Instead, an integrative approach is necessary—one that recognizes the coexistence of these mechanisms and provides a framework for identifying the dominant pathway under specific thermodynamic and kinetic conditions. By synthesizing the latest research, this guide aims to equip researchers and drug development professionals with the models and methodologies to navigate this complex landscape.
Theoretical Foundations: Classical vs. Nonclassical Nucleation
Core Principles of Classical Nucleation Theory (CNT)
CNT provides a quantitative, thermodynamic description of nucleation as an activated process where a stable crystal nucleus forms via the sequential addition of single molecules or atoms [1] [72]. The central concept is the free energy barrier (ΔG*), which arises from the competition between the bulk free energy gain and the surface free energy cost of forming a new phase.
For a spherical nucleus, the work of formation is given by:
ΔG = (4/3)πr³Δg_v + 4πr²σ [1]
Where:
Δg_vis the Gibbs free energy change per unit volume (the thermodynamic driving force, negative for a stable nucleus).σis the interfacial tension (surface free energy per unit area).ris the radius of the nucleus.
This relationship yields a critical nucleus radius (r_c) and the critical free energy barrier (ΔG*):
r_c = 2σ / |Δg_v| and ΔG* = 16πσ³ / (3|Δg_v|²) [1] [92]
The kinetic expression for the steady-state nucleation rate (J), which gives the number of nuclei formed per unit volume per unit time, is a cornerstone of CNT:
J = J_0 exp(-ΔG* / k_B T) [1] [33] [72]
Here, the pre-exponential factor J_0 encompasses kinetic factors like molecular attachment frequency and the concentration of nucleation sites, while the exponential term embodies the thermodynamic probability of overcoming the energy barrier [1].
Limitations of the Classical Framework and the Emergence of Nonclassical Concepts
Despite its widespread utility, CNT possesses well-documented limitations. It often predicts nucleation rates that are many orders of magnitude lower than those observed experimentally, particularly in solutions [72]. Furthermore, its assumption that the internal structure and surface energy of a microscopic nucleus are identical to those of the bulk macroscopic phase is a significant simplification [92].
These limitations have spurred the development of nonclassical theories, which propose alternative pathways to circumvent the high energy barrier of classical nucleation. Key nonclassical mechanisms include:
- The Two-Step Nucleation Mechanism: In this pathway, the formation of a crystalline nucleus is preceded by the creation of a dense, metastable liquid cluster. The crystal nucleus then forms within this pre-structured environment, which can significantly lower the interfacial energy barrier compared to forming directly from the dilute solution [72].
- Coalescence and Stepwise Nucleation: Molecular dynamics simulations of phase transformations in solids, such as the nucleation of BCC phase in FCC iron, show that systems can avoid the high CNT energy barrier through the coalescence of subcritical clusters or a stepwise nucleation process, rather than single-atom additions [7].
These mechanisms are not merely theoretical curiosities; they have practical implications for controlling crystal polymorphism and size distribution [72].
Quantitative Data: Comparing Energetics and Kinetics
The following tables synthesize quantitative data from recent studies, highlighting the energetic and kinetic parameters across different materials and nucleation pathways. This data is crucial for comparing classical predictions with experimental observations that often reflect nonclassical behaviors.
Table 1: Gibbs Free Energy of Nucleation (ΔG) and Kinetic Constants for Various Materials in Solution [33]
| Material Category | Specific System | Gibbs Free Energy, ΔG (kJ mol⁻¹) | Nucleation Rate Kinetic Constant, k_n |
|---|---|---|---|
| APIs | Various (10 systems) | 4 – 49 | > 0.97 (r² for model fit) |
| Large Biomolecule | Lysozyme/NaCl | 87 | > 0.99 (r² for model fit) |
| Amino Acid | Glycine | Fits model well | > 0.97 (r² for model fit) |
| Inorganic Compounds | 8 different systems | Fits model well | ~0.89 – >0.97 (r² for model fit) |
Table 2: Key Nucleation Parameters and their Significance in Classical and Nonclassical Frameworks
| Parameter | Classical Nucleation Theory Interpretation | Nonclassical Considerations | ||
|---|---|---|---|---|
| Critical Radius (r_c) | `r_c = 2σ / | Δg_v | `; determined by surface energy and volume driving force [1] [92]. | May represent the size of a dense liquid droplet or a pre-nucleation cluster rather than a perfect crystal [72]. |
| Work of Formation (W_c/ΔG*) | ΔG* = 16πσ³ / (3(Δg_v)²); the primary barrier for homogeneous nucleation [1] [92]. |
Can be significantly lowered by a two-step mechanism or cluster coalescence, explaining faster-than-predicted rates [7] [72]. | ||
| Nucleation Rate (J) | J = J₀ exp(-ΔG*/k_B T); highly sensitive to the energy barrier [1] [33]. |
Pre-exponential factor J₀ and pathway can be altered by diffusion through intermediate metastable phases [72]. |
||
| Surface Energy (σ) | Treated as a constant, equivalent to the macroscopic planar interface (capillarity approximation) [92]. | Can be variable and dependent on cluster size and structure, potentially leading to a "solution-crystal spinodal" at high supersaturation [72]. |
Experimental Protocols for Disentangling Mechanisms
Distinguishing between classical and nonclassical nucleation requires carefully designed experiments. Below are detailed methodologies for key experimental approaches cited in contemporary research.
Determining Metastable Zone Width (MSZW) and Nucleation Rate
The MSZW is the region between the solubility curve and the supersolubility curve where crystal growth is possible without spontaneous nucleation. Measuring it via the polythermal method is a foundational technique for studying nucleation kinetics [33].
Detailed Protocol:
- Solution Preparation: Prepare a saturated solution of the solute (e.g., an API or inorganic salt) at a known reference temperature, T* (e.g., 5 °C above the saturation temperature). Ensure complete dissolution and equilibration [33].
- Cooling Crystallization: Cool the solution from T* at a constant, predefined cooling rate (dT*/dt). This can be performed in an automated reactor system (e.g., Technobis Crystalline) for improved reproducibility [33] [61].
- Nucleation Detection: Monitor the solution in situ for the first appearance of crystals. This can be achieved using:
- Focused Beam Reflectance Measurement (FBRM): Detects a sudden increase in particle count.
- Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) Spectroscopy: Detects a change in solute concentration.
- In-situ Imaging: Directly observes crystal formation [61].
- Data Recording: Record the temperature at which nucleation is detected as Tnuc. The MSZW is ΔTmax = T* - Tnuc. The corresponding maximum supersaturation is Δcmax [33].
- Modeling and Analysis: The nucleation rate (J) can be related to the cooling rate and MSZW. A model based on CNT allows for the extraction of the Gibbs free energy of nucleation (ΔG) and the nucleation rate kinetic constant (k_n) by linearizing the following relation [33]:
ln(Δc_max / ΔT_max) = ln(k_n) - (ΔG / R) * (1 / T_nuc)A linear plot ofln(Δc_max / ΔT_max)vs.1 / T_nucyields a slope of-ΔG/Rand an intercept ofln(k_n).
Molecular Dynamics (MD) Simulations for Atomic-Scale Mechanism Observation
MD simulations provide atomic-level insight into nucleation pathways that are extremely difficult to capture experimentally, making them a powerful tool for identifying nonclassical events [7].
Detailed Protocol:
- System Setup: Create a simulation box containing a large number of atoms (e.g., >100,000) arranged in the initial metastable phase (e.g., FCC iron for a solid-state transformation) [7].
- Potential Definition: Employ an accurate interatomic potential (or force field) that correctly describes the energies and forces between atoms for both the parent and product phases.
- Equilibration: Run the simulation at the target temperature and pressure to equilibrate the initial structure.
- Production Run and Quenching: Perform multiple, long-timescale MD runs. To simulate a phase transformation, the system may be quenched to a temperature below the phase transition point.
- Cluster Analysis: Analyze the trajectory to identify and track the formation and evolution of atomic clusters of the new phase. Key metrics include:
- Size of clusters (number of atoms).
- Structural signature (e.g., Common Neighbor Analysis) to identify crystal structure.
- Lifetime and trajectory of clusters.
- Mechanism Identification: The analysis can reveal nonclassical behavior, such as the continuous coalescence and dissociation of subcritical clusters, or the stepwise formation of a stable nucleus via intermediate structural states, as demonstrated in FCC to BCC iron transformations [7].
Discriminating Surface vs. Bulk Nucleation in Membrane Crystallization
This protocol is designed to differentiate between heterogeneous nucleation (on a surface like a membrane) and homogeneous nucleation (in the bulk solution), which is critical for understanding and controlling scaling [93].
Detailed Protocol:
- Experimental Setup: Use a membrane distillation crystallizer. The membrane surface provides a primary site for heterogeneous nucleation, while the bulk solution can support homogeneous nucleation.
- Induction Time Measurement: Non-invasively measure the induction time for nucleation in two discrete domains simultaneously:
- At the Membrane Surface: Use visual observation or microscopy.
- In the Bulk Solution: Use particle size analysis (e.g., using a particle analyzer) or turbidity probes.
- Parameter Control: Systematically adjust the temperature (T) and the temperature difference across the membrane (ΔT). ΔT is a key parameter that controls the supersaturation level in the concentration boundary layer adjacent to the membrane surface [93].
- Data Correlation: Correlate the measured induction times with the calculated supersaturation in the boundary layer and the bulk. A log-linear relation between nucleation rate and boundary layer supersaturation is characteristic of CNT-controlled behavior.
- Morphology Analysis: Characterize the crystals formed on the membrane (scaling) and in the bulk separately using techniques like scanning electron microscopy (SEM). Different crystal habits or polymorphs indicate distinct nucleation mechanisms and pathways [93].
Visualization of Integrative Nucleation Pathways
The following diagram synthesizes the concepts of classical and nonclassical nucleation into a unified pathway, illustrating the potential coexistence and competition of these mechanisms.
This diagram shows that from a common metastable solution, the system can traverse multiple pathways. The classical route proceeds via single-molecule additions, while nonclassical routes may involve the formation of a dense liquid phase or the coalescence of smaller, sub-critical clusters. These pathways can converge at the critical nucleus stage, beyond which growth proceeds irreversibly.
The Scientist's Toolkit: Essential Reagents and Materials
This section details key reagents, materials, and equipment essential for conducting the experiments described in this whitepaper.
Table 3: Research Reagent Solutions and Key Experimental Materials
| Item Name | Function/Application | Specific Examples/Notes |
|---|---|---|
| Automated Crystallization Platform | Enables precise control and in-situ monitoring of temperature, antisolvent addition, and nucleation events. Essential for reproducible MSZW and kinetic studies. | Technobis Crystalline [61]. |
| In-situ Analytical Probes | For real-time detection and monitoring of nucleation and growth. | FBRM (for particle count/size), ATR-FTIR (for concentration), PVM (for direct imaging) [61]. |
| Molecular Dynamics (MD) Software | Simulates nucleation mechanisms at the atomic scale, allowing observation of nonclassical pathways. | LAMMPS, GROMACS; requires accurate interatomic potentials [7]. |
| Model Solute-Solvent Systems | Well-characterized systems for method validation and fundamental studies. | APIs (e.g., various from [33]), inorganic salts (KCl, K₂SO₄ [61]), amino acids (Glycine [33]), biomolecules (Lysozyme [33] [72]). |
| Antisolvents / Mixed Solvents | Used to modulate supersaturation by changing solubility, particularly in studies of strong electrolytes. | Ethanol-water mixtures for potassium salts [61]. |
| Activity Coefficient Models | Critical for accurate supersaturation calculation in electrolyte solutions (e.g., inorganic salts), where ideal solution assumptions fail. | Implemented in process modeling software for kinetics estimation [61]. |
The evidence is clear: classical and nonclassical nucleation mechanisms are not mutually exclusive but are often parallel pathways whose dominance is dictated by system-specific conditions such as supersaturation, temperature, and the presence of interfaces or impurities. The integrative model presented here provides a more holistic and accurate framework for interpreting nucleation phenomena in inorganic and pharmaceutical crystals.
For researchers and drug development professionals, adopting this integrative perspective is more than an academic exercise; it is a practical necessity. The ability to identify and influence the dominant nucleation pathway offers powerful levers for controlling critical quality attributes of crystalline products. By leveraging the quantitative data, experimental protocols, and conceptual visualizations outlined in this guide, scientists can design smarter crystallization processes that reliably target desired polymorphs and particle size distributions, ultimately accelerating development and enhancing product performance.
Conclusion
The paradigm of crystal nucleation has fundamentally shifted from the simplistic view of CNT to a more complex, yet accurate, picture involving multiple nonclassical pathways. The evidence is clear that mechanisms such as two-step nucleation via dense liquid phases or pre-nucleation clusters are not mere exceptions but are prevalent in systems ranging from inorganic metals to pharmaceutical crystals. This understanding provides powerful levers for control: by manipulating solvent environments, additives, and supersaturation conditions, scientists can steer nucleation toward desired polymorphs, suppress pathological crystallization, and optimize material properties. Future directions in biomedical research will heavily rely on this knowledge, enabling the design of novel crystallization inhibitors for diseases like malaria and gout, and the robust manufacturing of complex pharmaceutical polymorphs with tailored bioavailability and efficacy. The challenge and opportunity lie in quantitatively applying these principles to predict and design crystalline outcomes from the molecular level up.
References
- 1. Classical nucleation theory [https://en.wikipedia.org/wiki/Classical_nucleation_theory]
- 2. Classical Nucleation Theory - an overview [https://www.sciencedirect.com/topics/engineering/classical-nucleation-theory]
- 3. A simulation of oral absorption using classical nucleation ... [https://www.sciencedirect.com/science/article/abs/pii/S037851730900355X]
- 4. A Falsifiability Test for Classical Nucleation Theory [https://arxiv.org/html/2503.23124v1]
- 5. Toward a Deterministic Nucleation Theory for Chirality ... [https://arxiv.org/html/2510.25425v3]
- 6. First principles study on the early nucleation process of ... [https://www.sciencedirect.com/science/article/abs/pii/S0039602825000810]
- 7. Fundamental study of nonclassical nucleation mechanisms ... [https://www.sciencedirect.com/science/article/pii/S1359645422000404]
- 8. Advancing Carbon Nanotube Fibers: Addressing Challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12203435/]
- 9. Progress in computational methods and mechanistic insights ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d4nr05487c]
- 10. Therapeutic approach of carbon nanotube - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11698598/]
- 11. Role of clusters in nonclassical nucleation and growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3918807/]
- 12. Non-classical crystallisation pathway directly observed for ... [https://www.nature.com/articles/s41598-020-75937-2]
- 13. ' Prenucleation ' published in 'Encyclopedia of Nanotechnology' Clusters [https://link.springer.com/rwe/10.1007/978-94-007-6178-0_380-2]
- 14. Recent advances in the understanding of two-step ... [https://pubmed.ncbi.nlm.nih.gov/25859918/]
- 15. Classical nucleation theory approach to two-step ... [https://www.sciencedirect.com/science/article/abs/pii/S0022024819305159]
- 16. Direct observation and control of non-classical crystallization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12003862/]
- 17. of Formation and transformation to ZnSe... prenucleation clusters [https://pubmed.ncbi.nlm.nih.gov/40040559/]
- 18. Mesoscale clusters of organic solutes in solution and their role ... [https://pubs.rsc.org/en/content/articlehtml/2022/ce/d2ce00718e]
- 19. Investigation of molecular and mesoscale clusters in ... [https://www.sciencedirect.com/science/article/abs/pii/S0927775719302742]
- 20. Nucleation of protein mesocrystals via oriented attachment [https://pmc.ncbi.nlm.nih.gov/articles/PMC8222410/]
- 21. Investigation of the role of filtration in suppressing laser ... [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00516g]
- 22. Formation and stabilization mechanism of mesoscale ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8895010/]
- 23. Mesoscale properties of protein clusters determine the size ... [https://www.nature.com/articles/s42005-025-02134-w]
- 24. On the role of crystal-liquid interfacial energy in ... [https://www.sciencedirect.com/science/article/pii/S0376738825002911]
- 25. Facile supersaturation control strategies for regulating ... [https://www.sciencedirect.com/science/article/abs/pii/S1383586625028497]
- 26. Molecular nucleation mechanisms and control strategies ... [https://pubmed.ncbi.nlm.nih.gov/29620730/]
- 27. Competing nucleation pathways in nanocrystal formation [https://www.nature.com/articles/s41524-024-01371-x]
- 28. Dynamic light scattering: a practical guide and applications in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5425802/]
- 29. Dynamic Light Scattering Particle Size Distribution Analysis [https://www.horiba.com/int/technology/particle-analysis/dynamic-light-scattering-dls-particle-size-distribution-analysis/]
- 30. A basic introduction to Dynamic Light Scattering (DLS) for ... [https://www.malvernpanalytical.com/en/learn/knowledge-center/insights/a-basic-introduction-to-dynamic-light-scattering-dls-for-particle-size-analysis-qa]
- 31. Dynamic light scattering and transmission electron microscopy ... [https://pubs.rsc.org/en/content/articlehtml/2023/mh/d3mh00717k]
- 32. Recent developments in molecular modeling tools and ... [https://www.sciencedirect.com/science/article/pii/S0731708523006052]
- 33. Nucleation rate and Gibbs free energy of nucleation of APIs ... [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00467e]
- 34. A classical view on nonclassical nucleation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5617248/]
- 35. Two-step nucleation of the Earth's inner core - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8764699/]
- 36. Molecular dynamics simulation of heterogeneous ... [https://www.sciencedirect.com/science/article/pii/S1359646225004816]
- 37. Hcp/fcc nucleation in bcc iron under different anisotropic ... [https://www.nature.com/articles/s41598-018-25758-1]
- 38. Deciphering the complexities of crystalline state(s) with ... [https://www.nature.com/articles/s42004-025-01667-z]
- 39. A Colloidal Crystal Model for Controlled Polymorph Selection [https://www.tohoku.ac.jp/en/press/colloidal_crystal_model_for_controlled_polymorph_selection.html]
- 40. Selection mechanism of crystal polymorphs revealed by ... [https://www.kanazawa-u.ac.jp/en/miraichi/172385/]
- 41. Polymorphic transitions during nonclassical nucleation and ... [https://www.nature.com/articles/s42005-025-02062-9]
- 42. A colloidal crystal model for controlled polymorph selection [https://www.sciencedaily.com/releases/2025/04/250422131951.htm]
- 43. Change in the pathways of polymorphic transitions during ... [https://pubmed.ncbi.nlm.nih.gov/41295956/]
- 44. Mechanisms of hematin crystallization and inhibition by the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4413305/]
- 45. A nonclassical pathway of β-hematin crystal nucleation ... [https://www.nature.com/articles/s42004-025-01612-0]
- 46. Nonclassical mechanisms to irreversibly suppress β- ... [https://www.nature.com/articles/s42003-023-05046-z]
- 47. Classical and Nonclassical Nucleation Mechanisms of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11154923/]
- 48. Synthesis, characterization, inhibition of β-hematin ... [https://www.sciencedirect.com/science/article/abs/pii/S0162013425002211]
- 49. The effect of salt additives on the glycine crystallization ... [https://pubmed.ncbi.nlm.nih.gov/40035758/]
- 50. Crystal measurement technologies for ... [https://www.sciencedirect.com/science/article/abs/pii/S0263224124005578]
- 51. Statistical Analysis of Crystallization Database Links ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4079662/]
- 52. Quantitative analysis of molecular-level DNA crystal growth ... [https://www.nature.com/articles/srep02115]
- 53. Classical Nucleation Theory [https://encyclopedia.pub/entry/25427]
- 54. Non-Classical Crystallization in Soft and Organic Materials [https://www.pnnl.gov/publications/non-classical-crystallization-soft-and-organic-materials]
- 55. Modeling the surface stress and solid–liquid interface ... [https://www.sciencedirect.com/science/article/abs/pii/S0022369722000804]
- 56. Review Control of crystal nucleation, size and morphology ... [https://www.sciencedirect.com/science/article/abs/pii/S1383586623001405]
- 57. Prospects of Nanoscience with Nanocrystals: 2025 Edition [https://pmc.ncbi.nlm.nih.gov/articles/PMC12445017/]
- 58. Robustness of classical nucleation theory to chemical ... [https://arxiv.org/html/2510.01420v1]
- 59. Crystal Nucleation Kinetics and Mechanism: Influence of ... [https://pubmed.ncbi.nlm.nih.gov/40904127/]
- 60. The effect of solvent on crystal growth and morphology [https://www.sciencedirect.com/science/article/abs/pii/S0009250900004590]
- 61. Extension to Inorganic Salts [https://www.sciencedirect.com/science/article/pii/S0263876225005064]
- 62. Role of Solvents in Improvement of Dissolution Rate of Drugs [https://pmc.ncbi.nlm.nih.gov/articles/PMC4352216/]
- 63. Solvent Effects on Crystallization Kinetics: Investigating ... [https://www.sciencedirect.com/org/science/article/pii/S1083616025002361]
- 64. Studying Solvent Effect on Crystal Growth and Morphology [https://blog.3ds.com/brands/biovia/crystal-growth-and-morphology/]
- 65. - Wikipedia Nucleation [https://en.wikipedia.org/wiki/Nucleation]
- 66. Gypsum heterogenous nucleation pathways regulated by ... [https://www.nature.com/articles/s41467-025-55993-w]
- 67. Nucleation rate and Gibbs free energy ... [https://pubs.rsc.org/en/content/articlelanding/2025/ce/d5ce00467e]
- 68. The mechanisms of crystal growth inhibition by organic and ... [https://www.nature.com/articles/s41467-018-04022-0]
- 69. Classification of additives and their influence mechanisms ... [https://www.sciencedirect.com/science/article/abs/pii/S0013935125006279]
- 70. Comprehensive Investigation of Polymorphic Stability and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12300558/]
- 71. Continuum Crystallization Model Derived from Pharmaceutical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8161475/]
- 72. Nucleation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2995260/]
- 73. Cooling crystallization monitoring and control in API ... [https://www.vaisala.com/en/blog/2025-09/cooling-crystallization-monitoring-and-control-api-production-processes-ri-measurements]
- 74. Energy-Efficient Crystallization: Pioneering Sustainable ... [https://pharmafeatures.com/energy-efficient-crystallization-pioneering-sustainable-methods-for-active-pharmaceutical-ingredients/]
- 75. Crystallization Strategies for API Development and Scale ... [https://finance.yahoo.com/news/crystallization-strategies-api-development-scale-133000701.html]
- 76. Crystallization Strategies for API Development and Scale-Up [https://xtalks.com/webinars/crystallization-strategies-for-api-development-and-scale-up/]
- 77. Solid/liquid interface induced protein crystallization [https://www.sciencedirect.com/science/article/abs/pii/S096089742500018X]
- 78. Advances in deep eutectic solvent-mediated crystallization [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00434a]
- 79. The missing billions in hard sphere nucleation - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/sm/d5sm00776c]
- 80. Extending classical nucleation theory to consider curvature ... [https://www.sciencedirect.com/science/article/pii/S1350417725004286]
- 81. Energy landscape modeling of crystal nucleation [https://www.sciencedirect.com/science/article/abs/pii/S1359645421005437]
- 82. Unraveling the thermodynamic origins and kinetic ... [https://www.sciencedirect.com/science/article/abs/pii/S2590238525001870]
- 83. The apparent activation energy and pre-exponential kinetic ... [https://www.nature.com/articles/s42004-018-0056-5]
- 84. Comparison of experimental estimates and model ... [https://www.sciencedirect.com/science/article/abs/pii/S0927775701006641]
- 85. Studying the Impact of Pre-exponential Factor on ... [https://pubs.rsc.org/en/content/articlelanding/2021/fd/d1fd00101a]
- 86. Monte Carlo testing of the statistical analysis of nucleation ... [https://www.sciencedirect.com/science/article/abs/pii/S1359645497004394]
- 87. Comparison of Experimental and Theoretical ... [https://pubmed.ncbi.nlm.nih.gov/26317330/]
- 88. 3D conformation and crystal interaction insights into drug ... [https://www.nature.com/articles/s42004-025-01618-8]
- 89. Crystal engineering considerations for pharmaceutical co- ... [https://pubs.rsc.org/en/content/articlelanding/2025/ce/d5ce00820d]
- 90. Direct Visualization of the Two-step Nucleation Model by ... [https://www.nature.com/articles/srep22918]
- 91. Direct visualization of nucleation intermediate state ... [https://www.sciencedirect.com/science/article/abs/pii/S0304885321002523]
- 92. Application of the Nucleation Theorem to Crystallization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7514492/]
- 93. Unifying nucleation and crystal growth mechanisms in ... [https://www.sciencedirect.com/science/article/pii/S0376738825003345]