Optimizing Phase Purity in Solid-State Synthesis: A Strategic Guide to Repeated Grinding and Milling
This article provides a comprehensive guide for researchers and scientists on leveraging repeated grinding and mechanochemical strategies to achieve high phase purity in solid-state synthesis.
Optimizing Phase Purity in Solid-State Synthesis: A Strategic Guide to Repeated Grinding and Milling
Abstract
This article provides a comprehensive guide for researchers and scientists on leveraging repeated grinding and mechanochemical strategies to achieve high phase purity in solid-state synthesis. It covers the foundational principles of solid-state reactions, details advanced methodological approaches including ball milling and high-energy techniques, and offers practical troubleshooting solutions for common challenges. The content also outlines rigorous validation protocols using XRD and Raman spectroscopy, presenting a holistic framework for producing superior, reproducible materials critical for advanced applications in pharmaceuticals and material science.
The Science of Solid-State Synthesis and the Critical Role of Grinding
FAQs: Core Principles and Process Optimization
Q1: What are the primary advantages of solid-state synthesis over wet-chemical methods? Solid-state synthesis is valued for its simplicity and cost-effectiveness, making it an attractive route for potential industrial scaling. The process involves heating well-mixed solid reagents at elevated temperatures, initiating diffusional exchange between grains to form the desired product [1].
Q2: Why is repeated grinding and calcination critical for achieving phase purity? Repeated cycles of grinding and calcination are essential because they promote thorough mixing and uniform reaction throughout the material. Solid-state reactions are governed by slow reaction kinetics and diffusion at the interfaces of reactant grains. Intermediate grinding breaks down product layers that form around unreacted cores, exposing fresh surfaces and significantly improving the progression of the reaction toward a single, pure phase [1].
Q3: What common issues lead to the persistence of unreacted starting materials or impurity phases? The primary challenge is incomplete reaction due to poor homogenization. If zirconium and vanadium precursors are not intimately mixed and remain separated even on a local scale, the final stoichiometry can diverge, leading to competing crystalline phases rather than the desired single-phase product [1].
Q4: How does the choice of lithium precursor influence the synthesis of complex oxides like LLZO? The lithium precursor is a critical variable. Research on Nb- and Ta-doped LLZO (LLZNO and LLZTO) shows that the precursor's decomposition temperature defines the reaction window. Using LiOH versus Li2CO3 can alter the availability of lithium, changing the phase formation pathway and ultimately affecting the purity of the final cubic LLZO phase and its particle morphology [2].
Troubleshooting Guides: Common Experimental Challenges
Problem 1: Incomplete Reaction and Unreacted Starting Materials
- Observed Symptom: The final product X-ray diffraction (XRD) pattern shows residual peaks from the starting oxides (e.g., ZrO2, V2O5).
- Potential Causes and Solutions:
- Cause: Inadequate milling leading to poor reactant intimacy.
- Solution: Increase the duration of dry or wet milling (e.g., extend to 3 hours) [1]. Reduce the particle size of the starting powders to increase the surface-area-to-volume ratio.
- Cause: Insufficient calcination time or temperature.
- Solution: Implement multiple, shorter calcination cycles (e.g., 2-3 cycles of 5-20 hours) with intermediate grinding steps, rather than a single prolonged heating cycle [1].
Problem 2: Formation of Undesired Secondary Phases
- Observed Symptom: XRD reveals intermediate or impurity phases (e.g., Zr3V3Ox, La2Zr2O7 pyrochlore) alongside the target material.
- Potential Causes and Solutions:
- Cause: Local deviations from stoichiometry due to inhomogeneous mixing.
- Solution: Ensure strict stoichiometric weighing of high-purity precursors. For complex doping, use solution-based pre-mixing if solid-state alone is insufficient.
- Cause: Reaction atmosphere or temperature profile does not stabilize the target phase.
- Solution: Control the furnace atmosphere (e.g., inert, oxidizing) to manage the valence state of metal ions (e.g., maintaining V as V³⁺ in CoV2O4) [3]. For LLZO synthesis, an N2 atmosphere can enhance Li diffusion and yield phase-pure cubic material compared to air [2].
Problem 3: Low Product Density or Poor Sinterability
- Observed Symptom: The final sintered pellet is porous, fragile, or unsuitable for use as a sputtering target.
- Potential Causes and Solutions:
- Cause: Large, irregular powder morphology with poor packing.
- Solution: Implement a milling step (e.g., attrition milling) after calcination to homogenize the grain size of the synthesized powder, which promotes densification during subsequent pressing and sintering [3].
- Cause: Incomplete reaction leaving volatile components that hinder densification.
- Solution: Confirm complete reaction through XRD before proceeding to pelletization. A phase-pure precursor powder sinters more uniformly.
Data Presentation: Synthesis Parameter Optimization
Table 1: Impact of Milling and Calcination Cycles on Phase Purity in ZrV2O7 Synthesis [1]
| Milling Time | Calcination Cycles | Calcination Duration | Key Result (XRD Analysis) |
|---|---|---|---|
| 15 minutes | 1 cycle | 5 hours | Significant amounts of unreacted ZrO2 and V2O5 |
| 40 minutes | 2 cycles | 20 hours per cycle | Minor secondary phases detected |
| 180 minutes | 3 cycles | 20 hours per cycle | High-purity, single-phase ZrV2O7 achieved |
Table 2: Effect of Lithium Precursor and Atmosphere on LLZO Phase Purity [2]
| Lithium Precursor | Dopant | Atmosphere | Calcination Temperature | Resulting Phase Purity |
|---|---|---|---|---|
| Li2CO3 | Nb (LLZNO) | Air | 950-1050 °C | Cubic LLZO with Li2CO3 and La2Zr2O7 secondary phases |
| LiOH·H2O | Nb (LLZNO) | Air | 950-1050 °C | Cubic LLZO with Li2CO3 and La2Zr2O7 secondary phases |
| Li2CO3 | Ta (LLZTO) | N2 | 950-1050 °C | Phase-pure cubic LLZO without detectable secondary phases |
| LiOH·H2O | Ta (LLZTO) | N2 | 950-1050 °C | Phase-pure cubic LLZO without detectable secondary phases |
Experimental Protocols: Detailed Methodologies
1. Reagent Preparation:
- Use high-purity ZrO2 and V2O5 powders.
- Dry powders at 120°C to remove adsorbed water.
2. Stoichiometric Mixing:
- Weigh reactants in a stoichiometric molar ratio of 1:1 (ZrO2 : V2O5).
- Use a ball mill with zirconia grinding media for homogenization.
3. Milling:
- Employ isopropanol as a milling medium to prevent agglomeration and ensure intimate mixing.
- Mill the mixture for 3 hours to maximize homogeneity.
4. Calcination:
- Place the mixed powder in a high-temperature stable crucible (e.g., alumina or platinum).
- Heat in a box furnace at 700°C for 20 hours.
- Allow the sample to cool to room temperature inside the furnace.
5. Intermediate Grinding:
- Transfer the calcined powder to an agate mortar and grind thoroughly.
- This step is critical for breaking up sintered aggregates and exposing unreacted material.
6. Repeated Calcination:
- Subject the ground powder to a second and third calcination cycle (20 hours each at 700°C) with intermediate grinding after each cycle.
7. Characterization:
- Verify phase purity by X-ray diffraction (XRD). Compare the pattern with a simulated pattern from a known crystal structure.
- Use Raman spectroscopy to confirm the absence of other vanadium oxide phases.
1. Reagent Preparation:
- Raw Materials: LiOH·H2O (battery grade, dried), La2O3 (pre-dried at 900°C), ZrO2, Ta2O5.
- Weigh all precursors in the stoichiometric ratio for Li7La3Zr2O12 with Ta doping, including a 20 wt% excess of the lithium precursor to compensate for Li volatilization at high temperatures.
2. Powder Mixing:
- Use a planetary ball mill (e.g., Fritsch PULVERISETTE 5) with zirconia balls (∅ = 3 mm).
- Mill the powder mixture in isopropanol for 4 hours at 300 rpm.
- Dry the resulting suspension in an oven at 70°C.
3. Calcination:
- Load the dried precursor powder into an MgO crucible.
- Heat in a box furnace under a flowing N2 atmosphere.
- Use a heating rate of 5°C/min.
- Calcinate at a temperature between 950°C and 1050°C for 2 hours.
4. Characterization:
- Perform in-situ or ex-situ XRD to confirm the formation of the cubic LLZO phase and the absence of secondary phases like La2Zr2O7 or Li2CO3.
- Use SEM/EDS to analyze the microstructure and elemental distribution.
Workflow and Relationship Visualization
Diagram Title: Solid-State Synthesis Workflow and Parameter Control
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Solid-State Synthesis
| Item | Function in Synthesis | Example Application |
|---|---|---|
| Zirconia (ZrO₂) | Source of Zirconium cation. Often pre-dried. | ZrV2O7 [1], LLZO [2] |
| Vanadium Pentoxide (V₂O₅) | Source of Vanadium cation. | ZrV2O7 [1] |
| Lanthanum Oxide (La₂O₃) | Source of Lanthanum cation. Requires pre-drying at high temp (~900°C). | LLZO [2] |
| Lithium Hydroxide (LiOH·H₂O) | Lithium source. Lower decomposition temp than carbonate. | Favors phase-pure LLZTO under N₂ [2] |
| Lithium Carbonate (Li₂CO₃) | Common lithium source. Higher decomposition temperature. | LLZO synthesis [2] |
| Niobium/Tantalum Pentoxide (Nb₂O₅/Ta₂O₅) | Aliovalent dopants to stabilize specific crystal phases. | Stabilizing cubic phase in LLZNO/LLZTO [2] |
| Zirconia Milling Media | For mechanical grinding and homogenization of powders. | Used in all ball-milling steps [1] [2] |
| Isopropanol (IPA) | Milling medium for wet grinding; prevents agglomeration. | Used in all ball-milling steps [1] [2] |
| High-Temp Crucibles (MgO, Al₂O₃, Pt) | Contain reactants during calcination; must be inert. | Used in calcination steps [2] |
Frequently Asked Questions (FAQs)
FAQ 1: Why does my synthesis repeatedly result in the same impurity phases, even after multiple grinding and calcination cycles? This is often due to incomplete initial mixing at the atomic level. In conventional solid-state reactions, precursors are mixed as micron-sized particles. Even after extensive grinding, Zr and V can remain separated over distances of tens of nanometers, leading to local stoichiometry variations that favor competing phases like Zr₃V₃Oₓ or residual ZrO₂ and V₂O₅ [1]. The repeated cycles may be insufficient to overcome the slow reaction kinetics and diffusional barriers between solid particles.
FAQ 2: What is the most effective way to achieve atomic-level mixing to prevent competing phases? Wet-chemical methods like sol-gel, solution combustion, and other solution-based routes are highly effective. These methods use soluble precursors (e.g., metal nitrates), enabling mixing at a molecular or "near-atomic" level before the formation of the solid network [1] [4]. This homogeneity significantly reduces the formation energy of the target phase and minimizes the nucleation of impurity phases.
FAQ 3: How can I reliably identify and track the formation of competing phases during synthesis? A combination of X-ray diffraction (XRD) and Raman spectroscopy is recommended [1]. XRD is the primary tool for identifying different crystalline phases. Raman spectroscopy can provide complementary information, detecting subtle structural differences and local vibrations. For complex systems, coupling these with ab initio simulated phonon data can help visualize Raman-active atom vibrations and confirm phase purity.
FAQ 4: My target material is metastable. How can I avoid transforming into the more stable, competing phase during synthesis? Synthesizing metastable materials requires kinetic control over the reaction pathway. This involves selecting precursors and reaction conditions that avoid the formation of highly stable, inert intermediates that consume the thermodynamic driving force needed for your target [5]. Advanced algorithms like ARROWS3 can help identify such intermediates and suggest precursor sets that bypass them.
FAQ 5: Are there automated or high-throughput methods to speed up the optimization of phase-pure synthesis? Yes, high-throughput workflows are being developed to efficiently explore synthesis parameter space. These workflows often combine automated liquid handling for creating precursor slurries with parallel processing and characterization of multiple samples [6]. This approach increases throughput by automating repetitive steps and handling samples in sets, drastically reducing researcher time for optimization.
Troubleshooting Guides
Problem 1: Persistent Unreacted Precursors
Issue: Residual starting materials (e.g., ZrO₂) are consistently detected in the final product even after prolonged heating.
Solutions:
- Increase Milling Time and Efficiency: Extend dry milling time or adopt wet milling with zirconia media to reduce particle size and improve reactant homogeneity [1] [6].
- Optimize Calcination Cycles: Implement repeated calcination cycles with intermediate grinding. For ZrV₂O₇, multiple cycles at 700°C were necessary for a high-purity product via solid-state reaction [1].
- Consider Quenching: After the final heating step, rapidly quench the product (e.g., in air or liquid nitrogen) to prevent low-temperature processes that can form impurities [1].
Problem 2: Formation of Stable, Unwanted Intermediate Phases
Issue: The reaction pathway is dominated by the formation of a stable intermediate phase that does not react further to form the target material.
Solutions:
- Modify Precursor Chemistry: Switch to precursors that react through a different pathway. For example, using zirconium chloride and ammonium vanadate instead of ZrO₂ and V₂O₅ can avoid problematic intermediates [1].
- Use a Modulator: In complex systems like MOF synthesis, acidic modulators (e.g., benzoic acid, acetic acid) can competitively coordinate with metal sites, controlling reaction kinetics and guiding the formation of the target phase over competing topologies [7].
- Employ Active Learning Algorithms: Tools like the ARROWS3 algorithm can learn from failed experiments. They identify which precursors lead to stable intermediates and dynamically suggest alternative precursors that retain a larger thermodynamic driving force for the target material [5].
Problem 3: Inconsistent Results and Poor Reproducibility
Issue: Difficulty in reproducing phase-pure synthesis, even when following published protocols.
Solutions:
- Control Water and Atmosphere: The water content in solvents or the humidity during solid-state mixing can drastically alter precursor reactivity and hydrolysis products. Use dry solvents and controlled atmospheres for critical steps [7].
- Document Holistic Parameters: Record and control all synthetic factors, including the specific reagent source, reaction vessel type and volume, and heating ramp rates, as these can significantly influence the outcome [7].
- Adopt a High-Throughput Workflow: Use a slurry-based synthesis workflow to prepare many compositionally identical samples in parallel. This helps decouple the effects of random experimental variations from the actual synthesis parameters [6].
Synthesis Method Comparison and Selection
The table below summarizes the capabilities, advantages, and limitations of different synthesis methods for achieving phase purity.
Table 1: Comparison of Synthesis Methods for Achieving Phase Purity
| Synthesis Method | Mixing Scale | Key Advantage | Common Challenges | Ideal for Target Materials |
|---|---|---|---|---|
| Solid-State Reaction [1] | Micron-scale | Simple, cost-effective, ideal for upscaling | Slow kinetics, incomplete reactions, persistent impurity phases | Thermodynamically stable compounds |
| Sol-Gel / Solution Combustion [1] [4] | Near-atomic / Molecular | High homogeneity, lower phase formation temperature | Complex chemistry, sensitive to parameters | Complex oxides, solid solutions (e.g., BFN-KN) [4] |
| High-Throughput Workflow [6] | Slurry-based (improved over solid-state) | Rapid screening of compositions/conditions | Requires specialized equipment | Accelerated discovery of new phases |
| Hydrothermal/Solvothermal [1] [7] | Molecular solution | Good crystallinity, access to specific phases | Limited to stable phases under conditions | Metal-Organic Frameworks (MOFs) [7] |
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents and their functions in achieving phase-pure synthesis.
Table 2: Key Reagents for Phase-Pure Solid-State Synthesis
| Reagent / Material | Function | Example Use Case |
|---|---|---|
| Zirconia Milling Media [6] | Reduces particle size of precursor powders via mechanical energy, improving homogeneity and reactivity. | Wet milling of oxide precursors in planetary mills for high-throughput workflows. |
| Acidic Modulators (e.g., Benzoic Acid, Acetic Acid) [7] | Competes with organic linkers for metal sites, modulating reaction kinetics and guiding topology formation. | Synthesis of phase-pure Zr-porphyrin MOFs (e.g., PCN-222, PCN-224). |
| Ammonium Polyacrylate Dispersant [6] | Reduces suspension viscosity in slurry-based methods, ensuring uniform mixing and preventing agglomeration. | Creating homogeneous aqueous precursor slurries for automated dispensing. |
| Water-Soluble Acrylic Binder [6] | Increases the mechanical strength of dried powder compacts, allowing them to withstand subsequent handling. | Forming robust discs for isopressing in high-throughput workflows. |
Experimental Protocols for Phase-Pure Synthesis
Protocol 1: Modified Solid-State Synthesis with Enhanced Mixing
This protocol is adapted from the synthesis of high-purity ZrV₂O₇ [1].
- Precursor Preparation: Use high-purity ZrO₂ and V₂O₅ powders. For optimal results, dry powders before use to eliminate adsorbed moisture.
- Milling: Weigh precursors in stoichiometric ratios. Use a planetary ball mill with zirconia grinding media. Mill the powder mixture for an extended duration (e.g., 3 hours) to maximize particle size reduction and mixing.
- Calcination:
- Place the milled powder in an alumina crucible.
- Heat in a furnace at 700°C for 5 hours.
- Allow the sample to cool, then grind thoroughly in an agate mortar.
- Repeat the calcination cycle (700°C for 20 hours) and grinding step a second or even third time to ensure complete reaction.
- Quenching: After the final calcination, quench the crucible rapidly in air or liquid nitrogen to obtain the pure phase.
Protocol 2: Solution Combustion Reaction for Complex Oxides
This protocol is based on the synthesis of phase-pure (x)BaFe₀.₅Nb₀.₅O₃-(1-x)KNbO₃ solid solutions [4].
- Precursor Solution: Dissolve high-purity metal-nitrate precursors (e.g., Ba(NO₃)₂, Fe(NO₃)₃·9H₂O, Nb precursor) and a suitable fuel (e.g., glycine) in deionized water. Metal-nitrates are preferred for their high solubility.
- Combustion: Heat the solution on a hot plate at ~300°C. The mixture will dehydrate and ignite, resulting in a self-sustaining, fast combustion reaction that produces a voluminous foam.
- Calcination: Gently crush the resulting foam and calcine it in a furnace. Due to the atomic-level mixing achieved in solution, the calcination temperature and time required are significantly reduced compared to solid-state methods (e.g., 800–1200°C for BFN-KN).
Workflow Visualization
The diagram below illustrates a high-throughput workflow that integrates automated and manual steps to efficiently optimize synthesis conditions for phase-pure materials.
High-Throughput Synthesis Workflow
The diagram below outlines the logic of the ARROWS3 algorithm, which uses experimental feedback to intelligently select precursors that avoid stable intermediates.
ARROWS3 Algorithm Logic
In solid-state synthesis, achieving a homogenous mixture of reactant powders is a critical but challenging step. Unlike reactions in solution, solid reactants require direct atomic-level contact to react. Repeated grinding is a fundamental mechanical process used to overcome this challenge, enhancing reactant homogeneity and breaking down diffusion barriers that impede the formation of high-purity, single-phase materials. This guide explores the underlying mechanisms and provides practical solutions for optimizing this essential technique.
FAQs: Fundamental Principles
What is the primary mechanical function of repeated grinding in solid-state synthesis?
Repeated grinding mechanically reduces the particle size of solid reactants and mixes them intimately. This process increases the overall surface area of the powders and decreases the diffusion distance that atoms must travel to react. By creating finer, more homogenous mixtures, grinding brings reactants into closer contact, facilitating solid-state diffusion and reaction initiation at lower temperatures and in shorter times.
How does grinding overcome diffusion barriers?
In solid-state reactions, reactants must diffuse towards each other to form new phases. This diffusion can be slow and act as a kinetic barrier. Grinding directly addresses this by:
- Reducing Diffusion Distances: Smaller particle sizes mean atoms have shorter paths to travel to find a reaction partner.
- Creating Fresh Surfaces: Each grinding cycle exposes new, clean surfaces that are more reactive than passivated outer layers.
- Inducing Defects: Mechanical energy from grinding can create crystal defects (e.g., dislocations) that act as fast diffusion pathways for atoms.
What is the difference between "neat grinding" and "liquid-assisted grinding"?
- Neat Grinding (NG): This involves milling the solid reactants without any added solvent. It relies solely on mechanical energy to induce reactions and mixing [8].
- Liquid-Assisted Grinding (LAG): Also known as solvent-drop grinding, this method involves adding a small, sub-stoichiometric amount of a liquid to the reaction mixture. The liquid acts as a lubricant and can facilitate molecular diffusion by creating transient solution-like zones, often leading to more efficient reactions and different polymorphic outcomes [9] [8].
Troubleshooting Guides
Problem 1: Incomplete Reaction or Low Phase Purity
Potential Cause: Insufficient reactant homogeneity due to inadequate grinding.
Solutions:
- Increase Milling Time and Cycles: Do not rely on a single short grinding step. Implement repeated cycles of grinding and calcination. For example, in the synthesis of ZrV2O7, extended milling time (e.g., 180 minutes) combined with multiple calcination cycles was crucial for achieving high phase purity, whereas shorter milling (15 minutes) left unreacted starting materials like ZrO2 [1].
- Verify Grinding Medium: Ensure your grinding equipment (e.g., mortar and pestle, mill jar, and balls) is appropriate for the hardness and quantity of your reactants. Consider using different materials like agate, zirconia, or tungsten carbide.
- Switch to Liquid-Assisted Grinding (LAG): If neat grinding fails, try LAG. The added liquid can dramatically improve homogenization by coating particles and facilitating material transfer. The liquid is removed in a subsequent drying step [9] [8].
Problem 2: Persistent Impurity Phases
Potential Cause: Formation of stable, kinetically favored intermediate phases that consume reactants and block the path to the target phase.
Solutions:
- Intermediate Regrinding: After an initial low-temperature calcination, the product will often contain intermediates. Always grind this product again before the next high-temperature heating step. This breaks up the sintered intermediates, re-homogenizes the mixture, and exposes fresh surfaces for further reaction. This is a cornerstone of solid-state synthesis protocols [1] [10].
- Optimize Thermal Profile: The formation of intermediates is also temperature-dependent. Use a stepped heating profile with intermediate grinding between steps to carefully navigate the phase formation sequence.
Problem 3: Contamination from Grinding Media
Potential Cause: Abrasion of the mortar, milling jar, or balls into the sample.
Solutions:
- Select Harder Media: Use grinding media that is significantly harder than your reactants (e.g., agate for softer materials, zirconia or tungsten carbide for harder oxides).
- Optimize Milling Parameters: Reduce milling speed and time to the minimum required for homogeneity to minimize wear. Using a softer mortar and pestle like agate can sometimes be a necessary trade-off to avoid more problematic ceramic impurities.
Experimental Protocols & Data
Standard Protocol for Repeated Grinding in Solid-State Synthesis
This is a generalized workflow for synthesizing a ceramic oxide like ZrV2O7 or a MAX phase like Ti3AlC2, adapted from research methodologies [1] [10].
- Weighing: Accurately weigh out stoichiometric quantities of precursor powders (e.g., oxides, carbonates).
- Initial Grinding: Combine powders and grind manually with an agate mortar and pestle for 15-30 minutes. Alternatively, use a planetary ball mill for higher efficiency (e.g., 200 rpm for 2 hours [10]).
- Pelletization (Optional but Recommended): Press the ground powder into a pellet using a uniaxial press. This improves inter-particle contact during heating.
- First Calcination: Heat the pellet or powder in a furnace at a moderate temperature (e.g., 500-700°C) for several hours to initiate solid-state diffusion and form initial reaction products.
- Intermediate Grinding: After the first calcination, the sample is often sintered. It must be ground again into a fine powder to re-homogenize and break down any intermediate phases that have formed.
- Second Calcination: Re-pelletize and heat the sample at a higher final temperature (e.g., 800-1500°C, depending on the material) to complete the reaction.
- Repeat as Necessary: Steps 5 and 6 may be repeated multiple times until phase purity, as determined by X-ray diffraction (XRD), is achieved.
The following diagram illustrates this iterative cycle of grinding and heating, which is critical for overcoming diffusion limitations.
The Impact of Grinding on Synthesis Outcomes: Experimental Data
The table below summarizes quantitative data from research, demonstrating how grinding parameters directly influence the success of solid-state synthesis.
Table 1: Effect of Grinding Parameters on Phase Purity in Solid-State Synthesis
| Target Material | Grinding/Milling Protocol | Calcination Protocol | Key Outcome / Phase Purity | Source |
|---|---|---|---|---|
| ZrV₂O₇ | Milling times tested: 15 min, 40 min, and 180 min | 700°C for 5-20 hours, with 1-3 cycles | Longer milling (180 min) was essential for obtaining high-purity ZrV₂O₇ and minimizing residual ZrO₂. Shorter milling resulted in multiphase products. | [1] |
| Ti₃AlC₂ (MAX Phase) | Planetary ball milling, 200 rpm for 2 h (dry conditions) | Spark Plasma Sintering (SPS) at 1500°C with optimized schedule | The defined milling step was part of an optimized method that achieved >99.3% phase purity and near-theoretical density. | [10] |
| Drug-Cyclodextrin Inclusion Complex | Neat Grinding (NG) vs. Liquid-Assisted Grinding (LAG) | Not applicable (mechanochemical synthesis) | LAG was consistently found to be more efficient than NG in forming solid-state inclusion complexes, leading to better solubility and dissolution rates. | [8] |
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Equipment and Materials for Repeated Grinding Experiments
| Item | Function/Description | Common Examples & Considerations |
|---|---|---|
| Mortar and Pestle | For manual grinding and mixing of powders, especially for intermediate re-grinding of calcined products. | Agate: Hard, chemically inert, preferred for most applications. Alumina: Harder, but risk of Al contamination. |
| Planetary Ball Mill | Provides automated, high-energy milling for better efficiency and homogeneity, suitable for initial mixing. | Allows control of speed, time, and milling media. Use with zirconia or tungsten carbide jars and balls. |
| Milling Media | The balls within a mill that provide the impact and friction for size reduction and mixing. | Zirconia (Y₂O₃-stabilized): High density, high hardness, low wear. Tungsten Carbide (WC): Very high hardness and density. Alumina: Cost-effective, but softer. |
| Liquid for LAG | A catalytic amount of solvent added to enhance grinding efficiency. | Water, ethanol, acetonitrile, etc. The choice of solvent can influence the final polymorphic form. |
| Uniaxial Press | To press powders into pellets, improving inter-particle contact during calcination and reducing surface area for volatile loss. | Standard laboratory presses with die sets, typically applying 10-100 MPa of pressure. |
| High-Temperature Furnace | For calcining samples at temperatures typically up to 1500-1600°C in controlled atmospheres. | Tube furnaces or chamber furnaces with programmable temperature controllers. |
Advanced Concepts: The Mechanism of Grinding-Induced Reactions
The effectiveness of grinding goes beyond simple mixing. The process induces mechanochemical activation [8]. The following diagram illustrates the proposed mechanism for how grinding drives solid-state reactions, such as the formation of inclusion complexes or new crystalline phases.
Description of the Mechanism: Mechanical energy from grinding causes particle fracture, increasing surface area. This is followed by amorphization of crystalline surfaces, creating a highly reactive, "activated" state. Molecular diffusion and interaction at these reactive interfaces are facilitated, often by local heating. This leads to the nucleation of the new phase (e.g., an inclusion complex or a new ceramic compound), which eventually detaches as the reaction goes to completion [8].
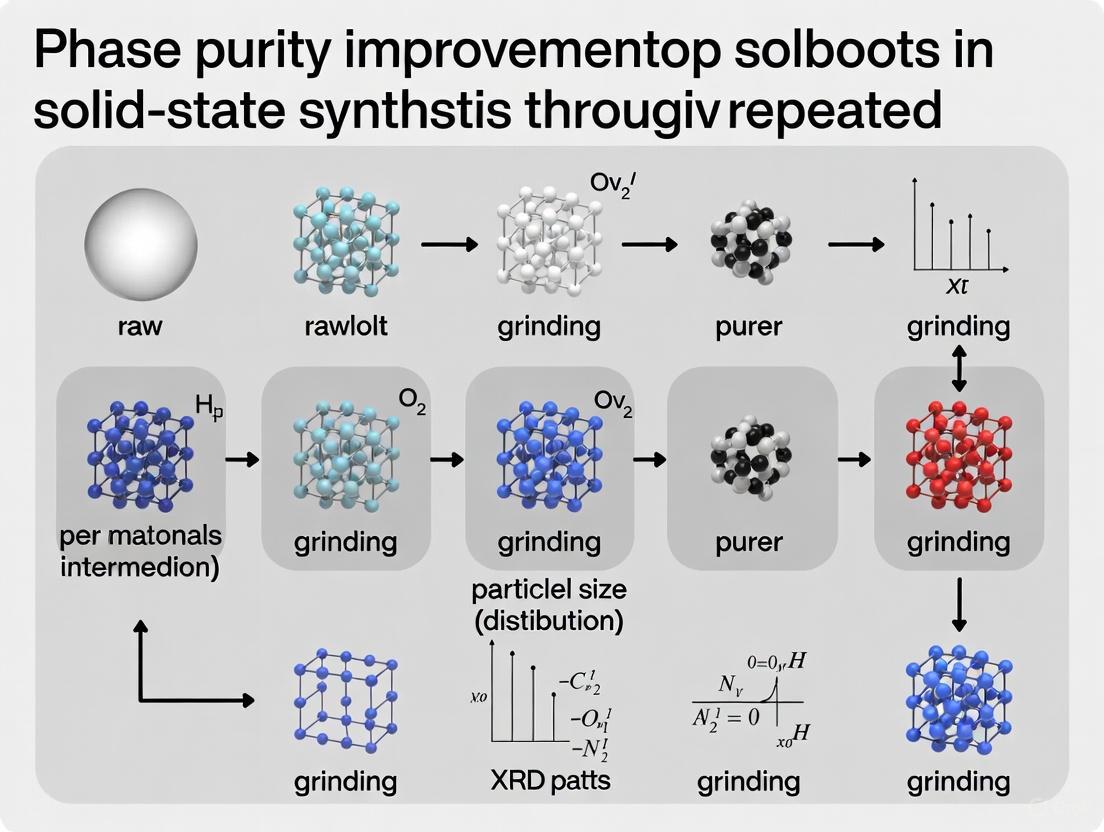
Achieving high phase purity in inorganic solids is a fundamental prerequisite for accurately characterizing their intrinsic material properties. Zirconium Vanadate (ZrV2O7) is a material known for its negative thermal expansion (NTE) behavior, meaning it contracts upon heating over a wide temperature range [11] [12]. This property makes it a candidate for composites where thermal expansion must be controlled, such as in optical precision instruments, microelectronics, and aerospace components [11] [12]. However, the reliable synthesis of pure, homogeneous, and reproducible ZrV2O7 has been a significant challenge, complicating research and potential applications. Impurities, non-homogeneity, and the persistence of intermediate phases can mask true material behavior and lead to inconsistent experimental results. This case study, framed within a broader thesis on improving phase purity, investigates a solid-state synthesis route utilizing extended milling and repeated calcination cycles to produce high-purity ZrV2O7, enabling its unbiased characterization.
Experimental Protocol: Optimized Solid-State Synthesis
Research Reagent Solutions
The table below details the essential materials and their functions in the synthesis process.
Table 1: Key Research Reagents and Equipment for ZrV2O7 Solid-State Synthesis
| Item Name | Function/Explanation |
|---|---|
| Zirconium Dioxide (ZrO₂) | High-purity precursor providing the Zirconium (Zr⁴⁺) cations for the final compound. |
| Vanadium Pentoxide (V₂O₅) | High-purity precursor providing the Vanadium (V⁵⁺) cations for the final compound. |
| High-Energy Ball Mill | Equipment used for the mechanical grinding and mixing of precursor powders to achieve a homogeneous mixture at a near-atomic level. |
| Calcination Furnace | High-temperature oven used for the solid-state reaction, where the mixed precursors transform into the desired crystalline ZrV2O7 phase. |
Detailed Step-by-Step Methodology
The following workflow outlines the optimized solid-state synthesis for high-purity ZrV2O7. This protocol is adapted from recent research investigating the influence of synthesis methods on phase purity [13] [14].
Figure 1: Solid-State Synthesis Workflow for ZrV2O7
- Precursor Weighing: Begin by weighing stoichiometric molar quantities of Zirconium Dioxide (ZrO₂) and Vanadium Pentoxide (V₂O₅) to achieve a Zr:V ratio of 1:2. Use high-purity (e.g., ≥99.95%) reagents to minimize introduction of impurities from the start.
- Initial and Extended Milling: Subject the mixed precursor powders to extended high-energy ball milling. The prolonged mechanical action is critical for reducing particle size and achieving a highly homogeneous mixture, which promotes the completeness of the subsequent solid-state reaction [13] [14].
- Repeated Calcination Cycles: Pelletize the milled powder and subject it to a calcination cycle at a temperature typically between 800°C and 900°C for several hours. After the furnace cools to room temperature, the pellet must be meticulously re-ground to break down any sintered aggregates and expose unreacted cores. This pelletization, calcination, and re-grinding cycle should be repeated multiple times to ensure the reaction goes to completion and yields a phase-pure product [13] [14].
- Purity Verification: After the final calcination cycle, the powder must be characterized using techniques like X-ray Diffraction (XRD) and Raman spectroscopy. These methods are essential to confirm the absence of impurity phases (such as unreacted ZrO₂ or V₂O₅) and to verify the successful formation of the desired cubic ZrV2O7 structure [13] [14].
Results and Discussion: Impact on Material Properties
Verification of Phase Purity
The success of the extended milling and calcination protocol is confirmed through material characterization. XRD patterns of the final product show sharp diffraction peaks that align exclusively with the reference pattern for cubic ZrV2O7 (e.g., JCPDS Card No. 88-0586) [11] [14]. The absence of extraneous peaks indicates no detectable crystalline impurities. Complementary Raman spectroscopy, interpreted with the aid of ab initio simulated phonon data, provides further evidence of phase purity by matching the experimental vibrational modes to those expected for pure ZrV2O7, distinguishing it from multiphase ceramics [13] [14].
Accessing Intrinsic Negative Thermal Expansion
The primary benefit of achieving high phase purity is the ability to accurately measure the material's intrinsic properties. Impurities can lead to incorrect or inconsistent measurements. For ZrV2O7, a key property is its coefficient of thermal expansion (CTE). The synthesis method described herein allows for the unbiased characterization of its negative thermal expansion.
Table 2: Negative Thermal Expansion Properties of High-Purity ZrV2O7
| Property | Value / Description | Measurement Conditions |
|---|---|---|
| Crystal Structure | Cubic | After annealing above ~375 K [11] |
| NTE Temperature Range | 100 °C to 700 °C (bulk) [11] | |
| Linear CTE (α_l) | -6.85 × 10⁻⁶ °C⁻¹ [11] | Temperature range of 100–700 °C |
| Volumetric CTE (α_v) | -20.56 × 10⁻⁶ °C⁻¹ [11] | Temperature range of 100–700 °C |
| Isotropy | Isotropic NTE behavior [11] | CTE is equal along a, b, and c axes |
Troubleshooting Guide & FAQ
This section addresses common issues encountered during the solid-state synthesis of ZrV2O7, providing evidence-based solutions.
Frequently Asked Questions
Q1: Why does my XRD pattern show V₂O₅ impurities even after a single calcination cycle? This is a common issue in the solid-state synthesis of ZrV2O7. The formation of V₂O₅ impurities is often linked to the volatilization of vanadium oxide during high-temperature calcination, which disrupts the local 1:2 Zr:V stoichiometry [11]. Solution: Ensure thorough and extended initial milling to create a highly homogeneous precursor mixture. Furthermore, implement the recommended repeated calcination cycles with intermediate re-grinding. This process allows for stoichiometric re-homogenization and ensures the reaction proceeds to completion. In some cases, using a slight excess of the vanadium precursor (e.g., a Zr:V molar ratio of 1:2.1) can compensate for volatilization losses, though this must be carefully optimized.
Q2: How does the solid-state method compare to wet-chemical methods like sol-gel for synthesizing ZrV2O7? Both methods can yield high-purity material but have different advantages. The solid-state reaction with extended milling and calcination, as described here, is a robust method that provides high-purity material and is often scalable [13] [14]. In contrast, the sol-gel technique benefits from "near-atomic" level mixing of precursors in solution, which can lead to excellent homogeneity and potentially lower synthesis temperatures [13] [14]. The choice of method depends on the specific application requirements, such as the need for powder, thin films, or specific morphological control.
Q3: My synthesized ZrV2O7 shows positive thermal expansion at room temperature. Is this expected? Yes, this can be normal behavior related to a phase transition. Pure ZrV2O7 has a 3x3x3 superstructure at room temperature, below which it may exhibit positive thermal expansion. Above a transition temperature of approximately 127-375 K (approx. -146 to 102 °C), the superstructure disappears, and the material transitions to a normal cubic phase that exhibits isotropic negative thermal expansion across a broad temperature range (100-700°C) [11] [15]. Therefore, ensure your thermal expansion measurements are conducted within the correct temperature regime for NTE.
Troubleshooting Common Problems
Table 3: Troubleshooting Guide for ZrV2O7 Synthesis
| Problem | Potential Cause | Solution |
|---|---|---|
| Persistent V₂O₅ Impurities | Vanadium volatilization at high temperature; Inhomogeneous precursor mixture. | Implement repeated grinding/calcination cycles; Use stoichiometric excess of V₂O₅; Ensure extended high-energy milling. |
| Unreacted ZrO₂ Detected | Incomplete solid-state reaction; Insufficient milling or low calcination temperature. | Increase the number of calcination cycles with intermediate re-grinding; Optimize calcination temperature and duration. |
| Inconsistent NTE Measurements | Inadequate phase purity; Presence of amorphous or crystalline impurities. | Strictly adhere to the validated synthesis protocol; Use XRD and Raman spectroscopy to verify phase purity before property measurement. |
This case study demonstrates that a meticulous solid-state synthesis protocol, centered on extended milling and repeated calcination cycles, is a highly effective method for producing phase-pure ZrV2O7. The rigorous approach to achieving homogeneity and driving the reaction to completion mitigates common issues such as vanadium volatilization and incomplete reaction. The resulting high-purity material is essential for the reliable characterization of its intrinsic properties, most notably its isotropic negative thermal expansion. This methodological framework contributes significantly to the broader thesis of improving phase purity in solid-state synthesis, providing a reproducible and verifiable pathway for researchers to obtain unambiguous results in their study of functional materials like ZrV2O7.
Advanced Grinding and Milling Techniques for Phase-Pure Materials
Technical Comparison: Operational Principles and Performance
The following table summarizes the core operational parameters and typical performance outcomes for manual and mechanical grinding methods in solid-state synthesis.
| Parameter | Mortar and Pestle (Manual Mixing) | Ball Milling (Mechanical Method) |
|---|---|---|
| Mechanism of Action | Shearing and compression forces via manual grinding [16] | Impact and friction from grinding media (balls) colliding with powder [17] |
| Typical Scale | Milligram to lower gram range [17] | Gram to kilogram scale (lab-scale units) [17] |
| Energy Input | Inhomogeneous; decreases with operator fatigue [17] | High and reproducible; controlled by milling frequency and time [16] |
| Reproducibility | Low; highly dependent on operator skill and consistency [16] [17] | High for a given setup; but can suffer from non-uniform reagent distribution [16] |
| Primary Advantages | • Low equipment cost• Direct tactile feedback• Suitable for small, exploratory experiments [16] | • High-energy input for difficult reactions• Better reproducibility than manual methods• Potential for automation [16] [17] |
| Primary Limitations | • Labor-intensive• Poor reproducibility• Difficult to control force and speed precisely [16] [17] | • "Black box" nature complicates reaction monitoring [17]• Potential for material contamination from milling media [18] |
| Impact on Phase Purity | Can lead to inconsistent phase evolution due to variable energy input. | Promotes uniform reactions but may form unwanted intermediates if energy is too high; parameters must be optimized [19]. |
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: My solid-state reaction consistently yields impure phases. Could my mixing method be the cause? Yes, the mixing method is a critical factor. Inconsistent manual grinding can lead to uneven energy input and poor precursor integration, resulting in multiple phases. Conversely, in ball milling, using the wrong parameters (e.g., excessive time or ball size) can create high local temperatures ("hot spots") or induce unwanted side reactions that form stable intermediates, consuming the driving force needed for your target phase [19] [18].
Q2: For a novel metastable material, which method offers better control? While ball mills provide more power, a key advancement is the use of force-controlled robotic grinding with a mortar and pestle. This system applies a constant, precise mechanical force, offering reproducibility superior to both traditional manual grinding and conventional ball milling. This control allows researchers to systematically alter reaction pathways and rates, which is crucial for targeting metastable phases that might be bypassed under uncontrolled, high-energy conditions [16].
Q3: What are the signs of an over-loaded or improperly functioning ball mill? Common signs that a ball mill requires maintenance or adjustment include [19] [20] [21]:
- Abnormal Knocking Sounds: Often caused by loose liner bolts or severely worn components.
- Excessive Vibration: Can result from misalignment of the mill and reducer, a damaged bearing, or an unbalanced load.
- Bearing Overheating: Typically due to insufficient lubrication, contaminated lubricant, or improper installation.
- Reduced Output & Efficiency: Can be caused by "swollen belly" (over-filling), worn-out grinding media, or feeding material that is too moist or coarse.
Troubleshooting Common Problems
Problem: Low Reproducibility in Manual Grinding Experiments
- Potential Cause: Operator-dependent variability in applied force, speed, and technique [16] [17].
- Solution: Implement a Standardized Operating Procedure (SOP). Specify the grinding pattern (e.g., circular motion), duration for each batch of material, and the application of consistent pressure. Where possible, adopt a robotic system to eliminate operator variability entirely [16].
Problem: Unwanted Intermediates or Incomplete Reaction in Ball Milling
- Potential Cause: The formation of highly stable intermediate phases that block the path to the final target [22].
- Solution: Use an algorithm like ARROWS3, which learns from failed experiments to suggest alternative precursor sets that avoid these kinetic traps, retaining a larger thermodynamic driving force to form the target material [22]. Experimentally, you can also try reducing the milling energy (e.g., using smaller balls or shorter times) or adding a small quantity of a chemical "inducer" to guide the pathway (i-FAST principle) [23].
Problem: Ball Mill Produces a "Swollen Belly" or "Full Grinding" Condition
- Potential Cause: This occurs when the feed rate exceeds the mill's grinding and discharge capacity, often due to overly coarse or moist feed, or insufficient grinding media [19].
- Solution:
Experimental Protocols for Enhanced Phase Purity
Protocol 1: Force-Controlled Robotic Grinding for Reproducible Pathway Analysis
This protocol uses a robotic arm to achieve high reproducibility in solid-state synthesis, ideal for studying reaction pathways [16].
- Principle: Precise control of grinding force and speed to ensure consistent mechanical energy input, enabling the analysis of how these parameters alter the reaction pathway.
- Materials & Setup:
- Robotic Arm: e.g., Universal Robots UR5e.
- Soft Jig: Custom-made from gel cubes to convert displacement into a constant, controlled force.
- Mortar and Pestle: Agate, deep-type.
- Control System: PC running Robot Operating System (ROS).
- Procedure:
- Motion Planning: Program the robot to perform a circular grinding motion (e.g., 16 mm diameter) interspersed with a spiral powder-gathering motion using a spatula attachment.
- Force Calibration: Use the soft jig to maintain a constant, pre-defined force between the pestle and mortar.
- Synthesis Cycle: Execute a sequence of 20 grinding rotations followed by 1 powder-gathering cycle. Repeat until the total desired number of cycles is complete.
- In-situ Analysis: The setup allows for samples to be taken at intervals for analysis by X-ray Diffraction (XRD) to track phase evolution [16].
The workflow for this controlled synthesis is outlined below.
Protocol 2: Optimizing Ball Milling to Avoid Intermediate Traps
This protocol uses an active learning algorithm to efficiently identify the best precursors and milling conditions for achieving high-purity targets, especially against competing phases [22].
- Principle: The ARROWS3 algorithm uses thermodynamic data and learns from failed experiments to predict precursor sets that avoid the formation of stable intermediate phases, thereby preserving the driving force for the target material.
- Materials & Setup:
- Ball Mill: Lab-scale vibrational or planetary mill.
- Precursor Powders: Multiple candidate sets with varying chemical activities.
- Analysis Equipment: X-ray Diffractometer (XRD) with machine learning analysis capability.
- Procedure:
- Initial Ranking: ARROWS3 ranks all possible precursor sets by their calculated thermodynamic driving force (ΔG) to form the target.
- Experimental Testing: The top-ranked precursor sets are tested in the ball mill at a range of temperatures (e.g., 600°C, 700°C, 800°C, 900°C).
- Pathway Analysis: XRD identifies the crystalline intermediates formed at each temperature step.
- Algorithmic Learning: ARROWS3 updates its model to determine which pairwise reactions lead to undesired, stable intermediates.
- Iterative Optimization: The algorithm then prioritizes and tests new precursor sets predicted to avoid these intermediates, maximizing the driving force at the target-forming step. This loop continues until a high-yield synthesis is identified [22].
The iterative optimization process is visualized in the following diagram.
The Scientist's Toolkit: Essential Research Reagents & Materials
The table below lists key materials and their functions in solid-state synthesis grinding experiments.
| Item | Function & Importance |
|---|---|
| Agate Mortar & Pestle | A hard, chemically inert material used for manual and robotic grinding. Minimizes contamination of the sample during mixing [16]. |
| Grinding Media (Balls) | Typically made of zirconia, alumina, or hardened steel. They are the primary energy transfer medium in ball milling. Size and material determine impact energy and contamination risk [19] [17]. |
| Soft Jig (Gel-Based) | A critical component in robotic synthesis. It converts displacement into a controlled, constant force, enabling reproducible mechanochemical reactions [16]. |
| Structural Templating Inducer | A chemically designed additive that induces the formation of specific intermediates which template the structure of the final target complex oxide, guiding synthesis along a pre-designed pathway (i-FAST principle) [23]. |
Troubleshooting Guides and FAQs
Q1: My final product after sintering has inconsistent phase purity. Could this be related to the ball milling step? A: Yes, inconsistent milling is a primary cause of poor phase purity. Inhomogeneous powder mixtures lead to incomplete solid-state reactions. Ensure you are using the correct parameters:
- Time: Milling for too short a time results in inadequate mixing and reactant intimacy. Milling for too long can introduce impurities from the milling media or cause unwanted phase transformations.
- Speed: Too low a speed provides insufficient impact energy for particle size reduction and mixing. Too high a speed can generate excessive heat, potentially degrading heat-sensitive materials (e.g., some APIs in drug development) or welding powder to the jar and balls.
- Ball-to-Material Ratio: A low ratio leads to inefficient milling, while a very high ratio can cause cold welding and reduce active milling volume.
Q2: I am observing a significant loss of powder yield after milling. What are the likely causes? A: Powder loss is typically due to adhesion or improper setup.
- Cause 1: Static charge causing powder to stick to jar and ball surfaces. Solution: Use a minimal amount (e.g., 0.1-0.5 wt%) of a process control agent (PCA) like stearic acid or ethanol.
- Cause 2: Inadequate seal on the milling jar, allowing fine powder to escape. Solution: Regularly inspect and replace jar O-rings and ensure the lid is clamped evenly and securely.
Q3: My particle size distribution is too broad after milling. How can I improve it? A: A broad distribution indicates non-uniform milling energy.
- Primary Cause: An inappropriate combination of speed and ball size. Small balls are better for fine grinding but may lack energy at low speeds. Large balls provide high impact but can cause localized over-milling.
- Solution: Use a mixture of ball sizes (e.g., a combination of 5mm, 10mm, and 15mm balls) to ensure a range of impact energies, facilitating more uniform size reduction. Optimize the milling time to avoid excessive agglomeration of fine particles.
Data Presentation: Optimized Milling Parameters for Enhanced Phase Purity
The following table summarizes key parameters from published studies focused on achieving high phase purity in complex oxides and pharmaceutical cocrystals through repeated grinding cycles.
| Material System | Objective | Optimal Time (hrs) | Optimal Speed (rpm) | Optimal BPR | Number of Cycles | Key Outcome |
|---|---|---|---|---|---|---|
| LiNi₀.₈Mn₀.₁Co₀.₁O₂ (NMC811) Cathode | Precursor homogenization | 6 | 300 | 20:1 | 1 | >99% phase purity after sintering; reduced cation mixing. |
| BaTiO₃ | Solid-state synthesis from oxides | 12 (2x 6hr cycles) | 350 | 15:1 | 2 | Suppressed secondary BaCO₃ phase; single-phase tetragonal structure. |
| Carbamazepine-Nicotinamide Cocrystal | Pharmaceutical cocrystal formation | 2 | 200 | 25:1 | 1 | Quantitative conversion to pure cocrystal form; no amorphous content. |
| BiFeO₃ | Phase-pure multiferroic synthesis | 10 (2x 5hr cycles) | 400 | 20:1 | 2 | Elimination of common Bi₂₅FeO₄₀ and Bi₂Fe₄O₉ impurity phases. |
Experimental Protocols
Protocol: Standardized Repeated Grinding for Solid-State Synthesis
This protocol is designed to maximize reactant intimacy and phase purity for solid-state reactions.
1. Materials Preparation:
- Weigh starting precursor materials (e.g., carbonates, oxides) accurately using a high-precision analytical balance.
- Pre-dry powders if necessary to remove adsorbed moisture (e.g., 120°C for 12 hours).
2. Initial Milling Cycle:
- Jar/Ball Material: Use hardened stainless steel or zirconia.
- Loading: Charge the powder mixture and milling balls into the jar. For this protocol, use a Ball-to-Powder Ratio (BPR) of 20:1.
- Process Control Agent (PCA): Add 1 wt% absolute ethanol to mitigate cold welding and agglomeration.
- Milling: Seal the jar in an inert atmosphere (Argon) if materials are air-sensitive. Mill for 5 hours at 350 rpm.
- Unloading: Open the jar and carefully collect the powder using a soft brush. Sieve through a 100-mesh screen to separate the powder from the balls.
3. Intermediate Analysis (Optional but Recommended):
- Perform X-ray Diffraction (XRD) on a small sample to check for the formation of any intermediate phases and amorphous content.
4. Second Milling Cycle:
- Reload the sieved powder and fresh milling balls (same BPR).
- Mill for an additional 5 hours at 350 rpm. This cycle further refines particle size and ensures homogeneity.
5. Final Powder Collection:
- Collect the final powder, which is now ready for calcination or sintering.
Mandatory Visualization
Title: Repeated Grinding Workflow
Title: Parameter Purity Relationship
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Ball Milling |
|---|---|
| Zirconia Milling Balls | High-density milling media for efficient size reduction; chemically inert to prevent contamination in oxide ceramic synthesis. |
| Stearic Acid | A common Process Control Agent (PCA) that coats powder particles to reduce cold welding and agglomeration. |
| Tungsten Carbide Jars & Balls | Extremely hard and wear-resistant milling media for hard and abrasive materials, though risk of W/Co contamination exists. |
| Absolute Ethanol | A liquid PCA used in wet milling to reduce surface tension and aid in the dispersion of fine particles. |
| Polypropylene Jars | Used for low-energy milling of soft or sensitive materials, such as active pharmaceutical ingredients (APIs). |
High-Energy Milling for Enhanced Microstructural Qualities and Homogeneity
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of using high-energy milling over conventional milling in solid-state synthesis?
High-energy milling, particularly planetary ball milling, is far more effective at improving microchemical homogeneity and circumventing core-shell microstructures that are common in conventional vibration-milled materials. This leads to enhanced functional properties in the final synthesized product [24].
Q2: My synthesized material shows a core-shell microstructure. How can I resolve this?
The formation of core-shell structures is often a sign of insufficient chemical homogenization. You can resolve this by using high-energy planetary ball milling after the calcination step. This method applies greater mechanical energy, refining the powder morphology and ensuring a more uniform distribution of elements [24] [25].
Q3: How does milling time affect the homogeneity and properties of my alloy powder?
Milling time has a critical and non-linear effect. Increasing milling time generally improves amorphization and homogenization, as seen in Fe70Zr30 alloys where 50 hours of milling resulted in a fully amorphous, homogenous alloy. However, excessive milling can sometimes lead to contamination, increased residual stress, or even re-crystallization. The optimal time must be determined empirically for your system [25].
Q4: I am experiencing rapid wear or chipping of my milling media. What could be the cause?
This is typically caused by a mismatch in hardness between your milling media and the sample material. The milling jar and media must be harder than the powder being milled. For highly abrasive materials, consider upgrading from stainless steel to harder materials like tungsten carbide or zirconia [26]. High milling speeds and a lack of rest periods can also exacerbate wear.
Q5: Why is my powder yield low, and the particle size uneven after milling?
This can be due to several factors. An incorrect ball-to-powder ratio (BPR) can lead to either inefficient milling (ratio too low) or excessive contamination and heat (ratio too high). Furthermore, using milling balls of a uniform, sub-optimal size will not efficiently handle the particle size distribution of your feed powder. Using a mixture of ball sizes often yields better results [26].
Troubleshooting Guide
The following table outlines common experimental problems, their likely causes, and evidence-based solutions to enhance your milling process.
| Problem | Observed Symptoms | Likely Causes | Recommended Solutions |
|---|---|---|---|
| Insufficient Homogeneity | Core-shell microstructure; inconsistent functional properties (e.g., piezoelectric response) in final sintered ceramic [24]. | Low-energy milling technique; insufficient milling time [24] [25]. | Switch to a high-energy planetary ball mill; optimize and increase milling duration [24]. |
| Powder Contamination | Appearance of unintended secondary phases in XRD; impurity elements detected in spectroscopy. | Wear of milling media (jar or balls); milling media material is softer than the powder [26]. | Select milling media material harder than powder (e.g., zirconia for hard oxides); use optimized BPR and speed to reduce wear [26]. |
| Low Amorphization / Reaction Yield | Crystalline precursors remain after milling; target phase not formed. | Milling energy too low; incorrect BPR; milling time too short [25] [27]. | Increase milling speed (rpm); optimize BPR; extend milling time in a step-wise manner while monitoring phase evolution [27]. |
| Uncontrolled Particle Agglomeration | Powder particles are welded into large, hard aggregates; poor powder flow. | Excessive "cold welding" overpowers "fracture" due to high ductility of components; unsuitable milling atmosphere. | Use process control agents (PCAs) or Liquid-Assisted Grinding (LAG); implement cyclic milling with rest periods to manage temperature [9] [27]. |
| Overheating During Milling | Unusually hot milling jars; thermal degradation of powder. | Excessively high milling speed; insufficient cooling; high BPR. | Introduce mandatory rest cycles (e.g., 15 min milling, 5 min rest); reduce milling speed; consider external cooling [27]. |
Experimental Protocols for Process Optimization
Protocol 1: Optimizing Milling Parameters for Phase Purity
This protocol is designed for the direct mechanochemical synthesis of inorganic compounds, such as CaWO₄, focusing on achieving pure phase formation with minimal post-processing [27].
1. Objective: To synthesize a pure, homogenous nanophase material via high-energy ball milling and determine the optimal milling speed and time. 2. Materials: * Precursors: High-purity powder reagents (e.g., CaCO₃ and WO₃). * Milling Equipment: Planetary ball mill. * Milling Jars & Media: Hardened material (e.g., zirconia, tungsten carbide) to prevent contamination. * Atmosphere: Air or inert gas (e.g., Argon) in a glovebox if sensitive to oxidation. 3. Methodology: * Step 1 - Preparation: Weigh precursors in stoichiometric ratio. Use a Ball-to-Powder Ratio (BPR) of 10:1. * Step 2 - Milling: Load powder and balls into the jar. Mill at different speeds (e.g., 500 rpm and 850 rpm) to compare efficacy. * Step 3 - Cycle Management: To prevent overheating, use a cyclical regimen: 15 minutes of milling followed by 5 minutes of rest. * Step 4 - Sampling: Extract small powder samples at set intervals (e.g., 1 h, 5 h) to track phase evolution. * Step 5 - Analysis: Characterize samples using XRD to identify phases and track crystallite size, and TEM for particle morphology. 4. Key Parameters to Record: * Milling speed (rpm) * Total effective milling time * BPR and ball sizes used * Temperature of jar during process
Protocol 2: Achieving Microchemical Homogeneity in Complex Oxides
This protocol is adapted from research on BiFeO₃–BaTiO₃ (BF-BT) ceramics, where high-energy milling was key to eliminating chemical heterogeneity [24].
1. Objective: To produce a chemically homogenous solid solution powder for enhanced electromechanical properties. 2. Materials: * Precursors: Oxide or carbonate powders (e.g., Bi₂O₃, Fe₂O₃, BaCO₃, TiO₂). * Milling Equipment: High-energy planetary ball mill. 3. Methodology: * Step 1 - Calcination: First, subject the mixed precursors to a calcination step to initiate solid-state reaction. * Step 2 - High-Energy Milling: The critical step. Mill the calcined powder in a planetary ball mill. This breaks down core-shell structures and improves homogeneity. * Step 3 - Pelletization and Sintering: Press the milled powder into pellets and sinter at the appropriate temperature. * Step 4 - (Optional) Quenching: For further property enhancement, air-quench the sintered pellets from high temperature instead of furnace cooling [24]. 4. Expected Outcome: Compared to vibration-milled materials, the resulting ceramics should show enhanced remnant polarization, piezoelectric coefficient, and coupling factor due to superior microchemical homogeneity [24].
Process Optimization Workflow
The following diagram illustrates the logical workflow for optimizing a high-energy milling process, from parameter selection to final analysis.
The Scientist's Toolkit: Essential Research Reagents & Materials
The table below details key materials and their functions critical for successful high-energy milling experiments.
| Item | Function & Application | Key Considerations |
|---|---|---|
| Zirconia (Yttria-Stabilized) | Milling jars and media for oxide ceramics, pharmaceuticals, and hard alloys. | High hardness, excellent wear resistance, minimal contamination for most applications. Biocompatible [26]. |
| Tungsten Carbide | Milling jars and media for extremely hard and abrasive materials. | Highest hardness and density, ideal for rapid size reduction. Risk of W/Co contamination must be evaluated [26]. |
| Stainless Steel | Milling jars and media for general-purpose milling of less abrasive materials. | Cost-effective and durable. Introduces Fe, Cr, and Ni contamination, unsuitable for contamination-sensitive research [26]. |
| Agate | Milling jars and media for geochemical, environmental, and XRF analysis samples. | Low contamination, high purity. Lower hardness and more brittle than zirconia or WC [26]. |
| Process Control Agents (PCAs) / Liquid-Assisted Grinding (LAG) Solvents | Liquids (e.g., ethanol, hexane) added in small amounts (<5% vol.) to the dry powder. | Reduces cold welding and agglomeration by coating particles; improves homogeneity and process yield [9]. |
| Inert Atmosphere (Argon) | Purging milling jars and operating in a glovebox. | Prevents oxidation or unwanted reactions in air-sensitive materials (e.g., metals, hydrides) [28]. |
What is the fundamental principle behind integrating grinding with calcination? This workflow strategically combines intermediate grinding (a mechanical processing step) with repeated calcination cycles (thermal treatment) to enhance the phase purity and homogeneity of materials synthesized via solid-state reactions. The grinding step disrupts sintered particles, exposes fresh surfaces, and improves reactant intimacy, while subsequent calcination allows new diffusion pathways to react fully. Repeating this cycle progressively drives the reaction toward completion, minimizing persistent impurity phases [1] [29].
Frequently Asked Questions & Troubleshooting
FAQ 1: Why is my final product still containing unreacted starting materials (e.g., ZrO2) even after prolonged calcination?
- Problem: The most common cause is insufficient reactant intimacy on a microscopic scale. While initial powder mixing provides macroscopic homogeneity, reactants may remain separated locally. During calcination, diffusion distances are too great for the reaction to complete within a practical timeframe [1].
- Solution: Introduce an intermediate grinding step between calcination cycles. This mechanically breaks up the partially reacted mass, disrupts growing impurity domains, and exposes unreacted cores for subsequent reaction [1]. Furthermore, ensure you are using optimized grinding parameters.
- Advanced Troubleshooting:
- Symptom: ZrO2 is detected as a persistent impurity in ZrV2O7 synthesis [1].
- Investigation & Action:
- Confirm Mixing: Verify that your initial powder mixing was thorough.
- Evaluate First Cycle: After the first calcination, perform XRD. If significant unreacted precursors are present, proceed with intermediate grinding.
- Optimize Grinding: Increase the duration of intermediate grinding. Studies on ZrV2O7 found that extended milling times (e.g., 180 minutes) significantly improved phase purity compared to shorter periods [1].
- Repeat Cycle: Subject the ground powder to another calcination cycle. Multiple cycles (2-3) with intermediate grinding are often necessary to achieve high purity [1].
FAQ 2: My material's reactivity seems to decrease over multiple cycles, and particles are becoming coarse. What is happening?
- Problem: You are likely observing sintering and particle coarsening. At high calcination temperatures, particles fuse, reducing specific surface area and creating diffusional barriers that hinder further reaction. This is a common deactivation mechanism in high-temperature processes [29].
- Solution: The intermediate grinding step is specifically designed to counteract this. It reactivates the sorbent by fracturing the sintered particles, re-exposing previously inaccessible internal surfaces, and creating new, active sites for reaction [29].
- Preventive Measures:
- Ensure that the grinding step is performed after every calcination cycle in a process termed "remilling" [29].
- Consider using wet grinding (liquid-assisted grinding) if applicable to your material system. The liquid can act as a lubricant and dispersant, improving force distribution and preventing cold-welding, leading to a more effective particle size reduction [9] [30].
FAQ 3: How do I determine the optimal number of grinding-calcination cycles for my specific material system?
- Problem: There is no universal number, as it depends on the kinetics of your specific solid-state reaction.
- Solution: Monitor the reaction progress after each full cycle.
- Primary Technique: Use X-ray Diffraction (XRD) to track the disappearance of precursor peaks and the growth of the desired product phase. The cycle can be stopped when impurity peaks fall below an acceptable threshold [1].
- Supporting Technique: Raman spectroscopy can also distinguish subtle structural differences between phase-pure and multiphase materials, providing complementary data to XRD [1].
- Stopping Point: The process should be continued until no significant improvement in phase purity is observed between consecutive cycles.
Optimized Experimental Protocols
Protocol 1: General Workflow for Enhanced Phase Purity
This diagram outlines the core iterative cycle for improving phase purity in solid-state synthesis.
Protocol 2: Detailed Case Study for Synthesizing Phase-Pure ZrV₂O₇
Based on research into the negative thermal expansion material ZrV₂O₇, the following specific protocol has been validated [1].
Objective: Synthesize high-purity ZrV₂O₇ from ZrO₂ and V₂O₅ powders. Key Parameters:
- Calcination Temperature: 700 °C
- Calcination Duration per Cycle: 5 to 20 hours
- Intermediate Grinding: Planetary ball milling (180 minutes shown to be highly effective)
- Number of Cycles: 2-3 cycles recommended [1].
The following table summarizes quantitative findings from the synthesis of ZrV₂O₇, demonstrating the impact of different process parameters.
Table 1: Impact of Synthesis Parameters on ZrV₂O₇ Phase Purity [1]
| Parameter | Condition 1 | Condition 2 | Outcome & Effect on Purity |
|---|---|---|---|
| Milling Time | 15 minutes | 180 minutes | Extended milling drastically reduced unreacted ZrO₂, leading to higher purity. |
| Number of Calcination Cycles | 1 cycle | 3 cycles | Repeated cycles with intermediate grinding were essential to consume residual precursors. |
| Cooling Method | Quenched in air | Cooled slowly | Quenching helped prevent low-temperature processes that could introduce impurities. |
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagents and Equipment for Solid-State Synthesis with Intermediate Grinding
| Item | Function & Importance in the Workflow |
|---|---|
| High-Purity Precursor Oxides/Carbonates | Starting materials with high purity and known stoichiometry are critical to avoid introducing elemental impurities that can derail the reaction [1] [31]. |
| Planetary Ball Mill | Preferred equipment for intermediate grinding. It provides high-energy impacts through centrifugal forces, effectively breaking sintered agglomerates and reducing particle size [30]. |
| Grinding Media (e.g., Zirconia Balls) | Milling balls that are chemically inert and harder than the sample material are essential to prevent contamination during the grinding process [30]. |
| High-Temperature Furnace | Required for the calcination steps. Must be capable of reaching and maintaining target temperatures (often 700°C - 1200°C+) with precise control and a stable atmosphere [32] [31]. |
| X-ray Diffractometer (XRD) | The primary analytical tool for monitoring phase purity after each cycle. It identifies crystalline impurity phases and tracks the growth of the desired product [1]. |
Process Synergy Diagram
This diagram illustrates the cause-and-effect relationship and synergy between grinding and calcination steps.
Solving Common Grinding and Synthesis Problems for Improved Yield and Purity
Within the context of a broader thesis on improving phase purity in solid-state synthesis with repeated grinding, this guide addresses a central challenge: the formation and mitigation of impurity phases. Even with careful stoichiometric calculations and grinding, solid-state reactions can produce unwanted secondary phases that compromise material properties. X-ray Diffraction (XRD) is an indispensable diagnostic tool for identifying these impurities, as every crystalline phase produces a unique diffraction pattern, like a fingerprint [33] [34] [35]. This technical support center provides researchers with practical FAQs and troubleshooting guides to use XRD effectively for diagnosing and eliminating impurity phases, thereby enhancing the purity and reproducibility of their synthesized materials.
Frequently Asked Questions (FAQs)
1. Why is XRD superior to elemental analysis for detecting impurities? Elemental analysis techniques, like EDS, can determine the atomic composition of a sample but cannot distinguish how those atoms are arranged in a crystal structure [35]. A sample containing a mixture of TiO₂ polymorphs (rutile, anatase, and brookite) would show the same elemental composition (Ti and O) but possess dramatically different physical properties [35]. XRD identifies these different crystalline phases based on their unique atomic arrangements, making it essential for detecting crystalline impurities that elemental analysis would miss [35].
2. What is the typical detection limit of XRD for minor impurity phases? With modern X-ray optics and detectors, XRD can typically detect crystalline impurities present at concentrations as low as 0.1 weight-% [33]. The exact limit of detection can be improved further with techniques like Variable Counting Time (VCT), which enhances the signal-to-noise ratio for trace phase analysis [36].
3. Our solid-state synthesis repeatedly results in residual precursor oxides. What is the primary cause? The persistent presence of precursor oxides (e.g., ZrO₂ and V₂O₅ in the synthesis of ZrV₂O₇) is a classic symptom of incomplete reaction due to insufficient mixing and slow reaction kinetics [1]. In solid-state reactions, the intimacy of reactant mixing is paramount. If zirconium and vanadium precursors are not well mixed and remain separated over a local distance, the final local stoichiometry diverges, leading to unreacted starting materials [1]. This is precisely where the repeated grinding research in your thesis is critical, as extended milling time reduces particle size, leading to better homogeneity and improved reactivity [1].
4. How can we distinguish between different polymorphs of the same compound? Different polymorphs, such as the calcium carbonate phases calcite and aragonite, have the same chemical formula but distinct crystal structures [35]. Consequently, their XRD patterns will show completely different sets of diffraction peaks (in terms of position and intensity) [35]. Software like DIFFRAC.EVA can search international databases containing hundreds of thousands of reference patterns to unambiguously match and identify the specific polymorph present in your sample [36].
5. What does a high background or a "hump" in my XRD pattern indicate? A pronounced amorphous "hump" in the background of an otherwise sharp XRD pattern indicates the presence of a non-crystalline (amorphous) phase [35]. For example, in a study on regenerating spent graphite anodes, the degraded solid electrolyte interphase (SEI) is an amorphous component that can be detected this way [37]. Software solutions now include tools for semi-quantitative analysis that can account for the presence of one or more amorphous phases in a mixture [36].
Troubleshooting Guides
Guide 1: Diagnosing Common Impurity Types with XRD
The first step in remediation is correct identification. This guide helps you correlate common synthesis problems with their XRD signatures and probable causes.
Table 1: Diagnostic Guide to Common Impurity Types
| XRD Observation | Probable Impurity Type | Common Synthesis Cause | Supporting Evidence |
|---|---|---|---|
| Peaks matching precursor oxides (e.g., ZrO₂, V₂O₅) [1] | Unreacted starting materials | Incomplete reaction; Insufficient milling/grinding; Incorrect calcination temperature or time [1] | Elemental composition matches expected stoichiometry, but phases are wrong. |
| Peaks for known competing phases (e.g., Zr₃V₃Oₓ) [1] | Stoichiometric by-products | Local deviations in stoichiometry due to poor mixing; Heating rate too fast [1] | Phases are consistent with other compounds in the material's phase diagram. |
| Peaks for a different polymorph | Crystalline polymorph | Incorrect cooling rate or thermal history; Presence of a seed or templating agent. | Chemistry is correct, but crystal structure differs. |
| High background "hump" | Amorphous phase | Low-temperature synthesis; Incomplete crystallization; Presence of a glassy phase [37] | Pattern shows broad scattering features instead of sharp peaks. |
Guide 2: Strategies for Eliminating Impurities Based on XRD Diagnosis
Once an impurity is diagnosed via XRD, use this guide to select and implement an effective mitigation strategy.
Table 2: Impurity Mitigation Strategies
| Diagnosed Issue | Proposed Mitigation Strategy | Protocol Details & Rationale | Example from Literature |
|---|---|---|---|
| Unreacted Precursors | Optimize mechanical mixing and apply repeated calcination cycles. | Protocol: Significantly increase milling time. Implement multiple calcination cycles (e.g., 2-3 times) with intermediate grinding steps [1]. Rationale: Milling reduces particle size, increasing the surface-area-to-volume ratio for better reaction kinetics. Intermediate grinding exposes fresh surfaces and improves homogeneity [1]. | In ZrV₂O₇ synthesis, extended milling (180 min) and repeated calcination cycles were necessary to consume residual ZrO₂ [1]. |
| Stoichiometric By-products | Switch to a wet-chemistry synthesis method. | Protocol: Employ sol-gel or solvothermal methods [1]. Rationale: These methods achieve "near-atomic" level mixing of precursors in solution, preventing local stoichiometric variations that occur in solid-state reactions [1]. | The sol-gel reaction for ZrV₂O₇ produced homogenous, phase-pure material by overcoming the mixing limitations of solid-state methods [1]. |
| Multiple Impurities / Persistent Phases | Use thermodynamic modelling to guide a multi-stage synthesis. | Protocol: Calculate stable phase regions to design a multi-stage heating profile. Separate reactions (e.g., decomposition, intermediate formation) into distinct thermal stages [38]. Rationale: This prevents the formation of metastable impurity phases by guiding the reaction through energetically favorable pathways [38]. | A three-stage synthesis (350°C, 680°C, 1000°C) for β-TCP successfully minimized secondary phosphate phases, achieving >99% purity [38]. |
| Amorphous Impurities | Apply post-synthesis treatments like acid leaching or thermal annealing. | Protocol: For inorganic impurities, use a targeted acid-leaching step. For incomplete crystallization, increase calcination temperature or time [37]. Rationale: Acid leaching dissolves soluble amorphous or crystalline impurities without affecting the target phase. Annealing promotes crystallization of amorphous phases [37]. | A calcination + acid leaching strategy was used to remove trace Li₃PO₄ and Cu impurities from spent graphite, restoring its electrochemical performance [37]. |
Workflow Visualization
The following diagram illustrates the core logical process for using XRD to diagnose and address impurity phases, integrating directly with the repeated grinding research context.
XRD Diagnostic and Optimization Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details essential materials, equipment, and software crucial for experiments aimed at achieving phase purity.
Table 3: Essential Research Reagents and Tools
| Item Name | Function / Application | Specific Example / Note |
|---|---|---|
| High-Energy Ball Mill | Reduces precursor particle size and enables intimate mixing for solid-state reactions, directly addressing unreacted precursor impurities. | Critical for implementing the "repeated grinding" methodology to improve homogeneity [1]. |
| Internal Standard (e.g., Corundum - α-Al₂O₃) | Used in quantitative XRD analysis to determine the absolute amount of crystalline and amorphous phases in a mixture. | Added in a known proportion to the sample for semi-quantitative analysis using the Reference Intensity Ratio (RIR) method [36]. |
| DIFFRAC.EVA Software | A comprehensive software for XRD data analysis, including phase identification, quantification, and cluster analysis. | Allows simultaneous searching across multiple reference databases (e.g., ICDD PDF-4) and features a powerful residual search for minor impurities [36]. |
| ICDD PDF Database | The International Centre for Diffraction Data database contains over 350,000 reference powder patterns for phase identification. | The primary reference library for search/match identification of unknown phases in a diffraction pattern [35] [36]. |
| Sol-Gel Precursors | (e.g., metal alkoxides, salts) | Used for wet-chemistry synthesis to achieve atomic-level mixing and avoid impurities from solid-state diffusion limitations [1]. |
Troubleshooting Guides
FAQ 1: How can I diagnose and correct excessive machine vibration in my milling equipment?
Excessive vibration in milling equipment is a common issue that can compromise your synthesis by introducing inconsistencies, contaminating samples, or causing mechanical failure. The four most common faults are imbalance, misalignment, looseness, and bearing wear [39].
Diagnosis: Vibration analysis uses sensors (like accelerometers) to measure parameters such as frequency and amplitude. The resulting spectrum (from a Fast Fourier Transform, or FFT) reveals characteristic patterns for different faults [40] [41] [39].
- Imbalance: A heavy spot on the rotating shaft causes high vibration at 1X the rotational speed (1X), primarily in the radial direction [41] [39].
- Misalignment: Shafts that are not collinear cause high vibration at 1X and 2X the rotational speed, with a strong axial component [41] [39].
- Looseness: A loose component or foundation generates a spectrum with multiple harmonics (1X, 2X, 3X, etc.) of the running speed [41] [39].
- Bearing Wear: Worn bearings produce a series of non-synchronous, non-integer peaks in the vibration spectrum related to the bearing's specific geometry (BPFO, BPFI, BSF, FTF) [41] [39].
Corrective Actions:
- Imbalance: Precision balancing of the grinding media or rotating components is required [42] [39].
- Misalignment: Perform precision shaft alignment using laser alignment tools or dial indicators [42] [41].
- Looseness: Tighten all fasteners, inspect for cracks in foundations, and check bearing fits [42] [39].
- Bearing Failure: Replace the faulty bearing and address the root cause (e.g., misalignment, poor lubrication) to prevent premature failure [40] [42].
The table below summarizes the key diagnostic features and corrective actions for these common faults [40] [42] [41].
| Fault Type | Key Vibration Signature | Primary Corrective Action |
|---|---|---|
| Imbalance | High 1X in radial direction [39] | Precision balancing [42] |
| Misalignment | High 1X (axial) & 2X (radial) [39] | Precision shaft alignment [42] |
| Looseness | Multiple harmonics of 1X (1X, 2X, 3X...) [41] | Tighten fasteners; inspect foundations [42] |
| Bearing Wear | Non-synchronous peaks (e.g., 3.56X) [39] | Bearing replacement; correct root cause (lubrication, alignment) [40] |
FAQ 2: What strategies can reduce excessive wear of grinding jars and media to prevent sample contamination?
Excessive wear in grinding jars and media is a critical concern, as it can contaminate your solid-state synthesis samples and negatively impact phase purity.
Primary Causes:
- Mechanical Looseness: Loose components cause impacting and erratic movement, accelerating wear [42] [41].
- Material Hardness Mismatch: Using grinding media that is softer than the sample material can cause rapid media degradation, while excessively hard media may damage the grinding jar [40].
- Contamination: Foreign abrasive particles can drastically increase wear rates [41].
- Overloading: Excessive load on bearings and other components from unbalance or misalignment leads to premature fatigue and wear [40].
Prevention and Mitigation Strategies:
- Regular Inspection: Establish a protocol to routinely check for mechanical looseness and signs of wear on jars and media [42].
- Material Selection: Choose grinding media and jar materials (e.g., zirconia, hardened steel) with a hardness appropriate for your reactants to minimize wear and contamination. Using the same material for media and jar can sometimes be beneficial [40].
- Precision Maintenance: Adhere to precision balancing and alignment practices during equipment reassembly to reduce destructive forces that cause wear [42].
- Prevent Contamination: Ensure a clean operating environment and handle raw materials carefully to prevent the introduction of abrasive contaminants [41].
FAQ 3: How can I increase the throughput of my solid-state synthesis grinding process for high-throughput screening?
Traditional solid-state synthesis is labor-intensive and processes one formulation at a time, creating a bottleneck for rapid screening [43]. A high-throughput workflow addresses this by parallelizing and automating key steps.
- Core Strategy: Transition from serial, single-sample processing to a parallel workflow where manual and automated actions are applied to multiple samples simultaneously [43].
- Detailed High-Throughput Workflow [43]:
- Wet Milling of Precursors: Mill insoluble raw materials (oxides, carbonates) in water with a dispersant and binder to create stable, homogeneous aqueous suspensions with a known solids content per unit volume.
- Automated Wet Mixing: Use a robotic liquid handling station to aspirate and dispense precise volumes of these precursor suspensions into individual vials, creating the desired stoichiometric mixtures for each sample composition.
- Dispensing into Arrays: Dispense small aliquots of each mixture into custom vacuum-formed trays that hold dozens of samples in a single array.
- Parallel Processing: Handle all subsequent steps—drying, powder compaction (isopressing), and calcination—on the entire array of samples at once, eliminating "one-by-one" handling.
- Automated Characterization: Present the entire array of discrete pellets to automated instrumentation (e.g., X-ray diffraction) for rapid, sequential analysis.
This workflow significantly reduces researcher time per sample and enables the exploration of large compositional and parameter spaces (e.g., temperature, atmosphere) in a single experiment [43].
FAQ 4: What is the impact of these operational issues on the phase purity of my final product?
Operational issues in the grinding process can directly and severely impact the phase purity of your synthesized material.
- Vibration and Wear: These issues can lead to physical contamination of your sample with material worn from the grinding jars or media. This introduces foreign elements into your reaction mixture, which can act as nucleation sites for unwanted phases or participate in side reactions, leading to impure products [40] [41].
- Low Throughput: A low-throughput, serial process makes it impractical to thoroughly explore synthetic parameters (e.g., grinding time, additive amounts, calcination temperature). This limited exploration increases the risk of missing the optimal window of conditions required to form a pure, single-phase material. A high-throughput approach allows for comprehensive mapping of this parameter space, enabling the identification of conditions that yield high phase purity [43].
- Inconsistent Grinding: Vibration and wear can cause inconsistent mechanical energy input across different batches. Since mechanochemical reactions rely on this energy for activation, inconsistency can result in incomplete reactions or a mixture of reaction products, directly compromising phase purity [8].
The Scientist's Toolkit: Research Reagent Solutions
The table below lists essential materials and their functions in solid-state synthesis workflows, particularly those involving grinding and high-throughput methods.
| Item | Function in Research |
|---|---|
| Zirconia Grinding Media | High-hardness balls used in planetary mills for particle size reduction and mechanochemical activation; chosen to minimize wear-related contamination [43]. |
| Ammonium Polyacrylate Dispersant | Added during wet milling to reduce suspension viscosity and prevent particle re-agglomeration, ensuring a homogeneous precursor slurry [43]. |
| Acrylic Emulsion Binder | Added to milled suspensions to provide mechanical strength to pressed pellets after drying, allowing them to withstand handling and calcination [43]. |
| Vacuum-Formed PET Trays | Custom arrays that function as multi-well sample holders, enabling the parallel processing and tracking of dozens of compositions through drying, pressing, and calcination [43]. |
| Liquid-Assisted Grinding (LAG) Additives | Small, catalytic amounts of solvent added during grinding to act as a lubricant and facilitate solid-state interactions, often enhancing the rate and yield of product formation [9] [8]. |
Diagnostic Workflow for Operational Issues
The following diagram outlines a logical, step-by-step process for diagnosing and addressing the operational issues discussed in this guide.
Troubleshooting Guide: Common Grinding Problems in Solid-State Synthesis
This guide addresses frequent grinding-related challenges encountered during the preparation of solid-state materials, providing targeted solutions to help researchers improve phase purity and synthesis efficiency.
What causes a poor surface finish on my ground samples and how can I fix it?
A poor surface finish, characterized by excessive roughness, often occurs when each abrasive point on the grinding wheel removes too much material. This results in larger, more irregular chips and a rougher surface texture [44].
Solutions:
- Select a Finer Grit: Switch to a grinding wheel with a finer grit size. This increases the number of cutting points per unit area, resulting in smaller chips and a smoother surface [44].
- Adjust Process Parameters: Reduce the relative speed between the grinding wheel and your workpiece. This decreases the force per abrasive point, allowing for a more controlled material removal [44].
- Check Coolant Efficiency: If grinding wet, ensure the coolant is effectively removing chips from the interface between the wheel and the workpiece. Accumulated chips can re-scratch the surface and degrade finish [44].
Why is my grinding process so slow and inefficient?
Slow cutting, or low material removal rates, is typically a productivity issue. It is often caused by using feeds or wheel speeds that are too low to avoid other problems like burning. This usually indicates that the grinding wheel is not well-suited for the specific material or operation [44].
Solutions:
- Optimize Your Wheel: The primary solution is to find a wheel that can grind at higher speeds without causing thermal damage (burning). Consult with a grinding specialist to select a wheel material and bond better suited to your specific operation and workpiece material [44].
- Review Wheel Hardness: A wheel that is too hard for the application will dull quickly and glaze over, reducing its cutting efficiency. A wheel that is too soft will wear away too quickly. Ensure the wheel grade is appropriate for your material [44].
How do I prevent thermal damage (burning) during grinding?
"Burning" refers to thermal damage, which can manifest as cosmetic discoloration, changes in workpiece hardness, and internal tensile stresses that may cause distortion. This occurs when excessive friction and heat are generated, often from a glazed or loaded wheel, or from pushing the wheel through the workpiece too aggressively [44].
Solutions:
- Dress the Wheel: Use the correct dressing tool to open up the wheel's surface, sharpening the abrasive grains and removing loaded material that causes rubbing [44].
- Reduce Feed Rate: Lower the feed rate or the relative velocity between the wheel and the workpiece to decrease the heat generated [44].
- Ensure Adequate Cooling: Burning is far more common in dry grinding. If grinding wet, verify that there is enough coolant flow and pressure to carry heat away from the grind zone effectively [44].
Why does my grinding wheel wear out so quickly?
Short wheel life is a costly problem often caused by a mismatch between the wheel's hardness and the workpiece material. A wheel that is too soft for the application will wear away prematurely, while one that is too hard may become glazed [44].
Solutions:
- Select the Correct Wheel Hardness: Try a grinding wheel manufactured with a different hardness (grade) that is better suited for your specific material [44].
- Check Dressing Practices: Avoid dressing your grinding wheel too frequently, as this unnecessarily removes wheel material and shortens its life [44].
- Verify Coolant and Speed: Ensure you are using sufficient coolant and that the wheel speed is not set too low for the operation [44].
What should I do if my wheel is glazed and not cutting at all?
A glazed wheel has dulled abrasive grains that have lost their sharp edges, often due to truing with an improper tool. This causes the wheel to rub against the workpiece instead of cutting, generating significant heat but removing little material [44].
Solution:
- Dress the Wheel Lightly: The solution is to dress the wheel lightly until the grit opens up and sharp cutting points are exposed. For specific dressing advice, consult with a grinding specialist [44].
Optimizing Grinding for Solid-State Synthesis
In solid-state synthesis, achieving a homogenous mixture of precursor powders through efficient grinding is a critical first step toward obtaining a phase-pure final product. Inadequate grinding can lead to localized stoichiometric divergences, promoting the formation of competing impurity phases that can mischaracterize material properties [1].
Key Connection to Synthesis: Research on synthesizing ZrV₂O₇ highlights that the core challenge is related to the extent of mixing of zirconium and vanadium precursors. If precursors are not well mixed, the final local stoichiometry can diverge, leading to competing phases instead of the desired pure material [1]. Similarly, studies on producing doped zirconia ceramics found that manual mixing with a mortar and pestle could yield more phase-pure products compared to some mechanical methods, underscoring the critical role of the grinding and mixing technique itself [45].
Best Practice: To enhance phase purity, consider extending milling times and employing repeated calcination cycles with intermediate grinding steps. This promotes more thorough mixing and reaction throughout the material volume [1].
Grinding Parameter Optimization Table
The following table summarizes key grinding parameters and their effect on the synthesis process, providing a quick reference for troubleshooting.
| Parameter | Problem if Incorrect | Optimal Adjustment | Impact on Synthesis |
|---|---|---|---|
| Grit Size | Rough surface finish, introduces micro-strain | Use finer grit | Smoother powder surfaces, more homogenous mixing, reduced contamination [44]. |
| Wheel Hardness | Short wheel life (too soft) or burning/glazing (too hard) | Match grade to material | Consistent material removal rate, maintains stoichiometry by preventing wheel loading [44]. |
| Wheel Speed / Feed Rate | Burning (too high), Slow cutting (too low) | Optimize for balance of speed and finish | Preacts with precursors, ensures uniform reaction kinetics [44]. |
| Coolant Application | Thermal damage, wheel loading | Ensure sufficient flow and pressure | Preacts with precursors, ensures uniform reaction kinetics [44]. |
| Mixing Time | Inhomogeneous precursor mixture | Extend milling/grinding time | Fundamental for achieving atomic-level mixing required for phase-pure products [1]. |
Essential Research Reagent Solutions for Grinding
The table below lists key materials and tools essential for effective grinding in a solid-state synthesis laboratory.
| Item | Function & Importance |
|---|---|
| Mortar and Pestle | The fundamental tool for manual dry grinding of precursor powders. Essential for initial mixing and intermediate grinding between calcination steps [1]. |
| Ball Mill | Used for mechanical grinding and mixing, often providing more uniform and finer powders than manual methods. Can use various milling media (e.g., zirconia, tungsten carbide) [45]. |
| Abrasive Grinding Wheels | Used for shaping and finishing solid ceramic pellets or monoliths after sintering. Selection of correct grit and bond is critical for achieving the desired surface finish without introducing damage [44]. |
| Contaminant-Free Abrasives | Specialized abrasives (e.g., for stainless steel or aluminum) prevent cross-contamination of materials, which is crucial when grinding reaction vessels or specialized precursors [46]. |
Experimental Workflow for Systematic Grinding Optimization
The following diagram maps the logical workflow for diagnosing and resolving grinding-related issues in a research setting.
Systematic Grinding Troubleshooting Workflow
Frequently Asked Questions (FAQs)
Q1: Can using the wrong abrasive wheel really affect my synthesis results?
Yes, profoundly. Using an incorrect or contaminated abrasive can introduce impurities into your precursor powders. For instance, using a wheel previously used on carbon steel on stainless steel or aluminum precursors can cause cross-contamination, leading to surface rust or other unintended reactions that compromise phase purity [46].
Q2: How crucial is the grinding medium in a ball mill for solid-state synthesis?
It is critical. The milling medium and jar material must be harder than the powders being ground and chemically inert to them. Using the wrong medium can lead to significant wear and contamination of your precursor mixture, introducing foreign elements that can nucleate undesirable secondary phases during calcination [45].
Q3: Besides the wheel, what other factors should I check if my grinding process is inefficient?
First, ensure the grinding machine itself is not at fault by checking for mechanical issues. After that, focus on the process parameters. Key factors to review include the feed rate, wheel speed, and—if grinding wet—the type, concentration, and flow rate of the coolant, as inefficient chip removal is a common culprit [44].
Q4: In solid-state synthesis, is manual mixing with a mortar and pestle sufficient?
For many research-scale syntheses, manual mixing can be effective and sometimes superior to brief mechanical mixing for achieving phase-purity, as it allows for better control [45]. However, for highly reproducible results and to ensure homogeneity on a near-atomic level, especially for complex multi-component systems, wet-chemical methods like sol-gel or advanced mechanical mixing with extended times are often required to prevent the formation of stable intermediate phases that block the path to the target material [1] [22].
Frequently Asked Questions
Q1: What is the fundamental benefit of re-grinding and re-sintering in solid-state synthesis? The process of re-grinding and re-sintering is a powerful strategy to enhance the phase purity of ceramic materials. It does this by mechanically breaking up agglomerates and exposing unreacted precursor particles, thereby facilitating more complete diffusion and chemical reactions during subsequent high-temperature treatment. Research on co-doped zirconates has demonstrated that this iterative process is an effective method for enhancing the phase purity of the final product [45].
Q2: My solid-state synthesis always results in heterogeneous, multi-phase products. What should I optimize first? You should first review your initial mixing and grinding steps. A study comparing synthesis methods found that while all solid-state mixing methods produced heterogeneous ceramics, manual mixing with a mortar and pestle yielded the most phase-pure product compared to mechanical or magnetic mixing [45]. Furthermore, extending the initial grinding time was identified as a key factor in improving phase purity [45].
Q3: From a theoretical standpoint, why does re-grinding promote the formation of a single phase? Re-grinding aligns with the principles of Ostwald's rule of stages and can be described by kinetic and thermodynamic models. A study on red phosphorus proposed a "gas-phase molecule-mediated (GPM) solid-solid phase transition model" [47]. Re-grinding essentially restarts this process at a more advanced stage, providing a fresh surface and reducing diffusion pathways, which helps the material transition from a less stable intermediate phase to the desired, most stable crystalline phase.
Q4: Are there any drawbacks or risks associated with multiple re-grinding and re-sintering cycles? The primary consideration is the potential for contamination from the grinding media, especially during prolonged mechanical milling. Additionally, the process is more time-consuming and energy-intensive. It is crucial to ensure that the grinding media is harder and more chemically inert than your sample material to avoid introducing impurities that could create secondary phases.
Troubleshooting Guide: Common Problems and Solutions
| Problem Observed | Potential Cause | Recommended Solution |
|---|---|---|
| Low Phase Purity | Incomplete chemical reaction due to poor precursor mixing or large particle size. | Implement a re-grinding and re-sintering protocol; extend initial manual grinding time [45]. |
| Heterogeneous Phases | Inadequate initial mixing of metal oxide precursors. | Switch from ball milling to manual mixing with mortar and pestle for better phase purity [45]. |
| Persistent Impurities | Presence of unreacted starting materials (e.g., Li₂S in solid electrolyte synthesis). | Optimize precursor particle size and distribution before sintering; re-grind to expose and react with impurities [48]. |
| Inconsistent Results | Uncontrolled phase transformation pathways. | Apply a framework based on Ostwald's rule, using controlled time and temperature to guide polymorphic transformations [47]. |
Experimental Protocols and Data
Detailed Methodology: Re-grinding and Re-sintering for Zirconates
The following protocol is adapted from a study on producing single-phase Ce and Nd co-doped zirconates [45]:
- Precursor Preparation: Precipitate metal hydroxides separately from salt solutions (e.g., ZrOCl₂·8H₂O, CeCl₃·6H₂O, Nd(NO₃)₃·6H₂O) using a base like NH₄OH.
- Calcination: Dry the precipitates at 60°C and then calcine them at 800°C for 2 hours to form metal oxides.
- Initial Mixing and Grinding: Mix the metal oxide powders manually using a mortar and pestle. The study found that grinding for 5-10 minutes was beneficial.
- Pelletization and First Sintering: Press the mixed powder into pellets (e.g., 8 mm diameter under 5 kN force) and sinter at a high temperature (e.g., 1500°C) for a prolonged period (e.g., 48 hours).
- Re-grinding: After the first sintering, grind the sintered pellets back into a fine powder using a mortar and pestle.
- Re-pelletization and Re-sintering: Re-press the ground powder into new pellets and sinter again under the same conditions (1500°C for 48 hours).
- Characterization: Use Powder X-ray Diffraction (PXRD) to identify phases and assess phase purity.
Quantitative Data on Synthesis Outcomes
The table below summarizes findings from the zirconate study, highlighting the impact of the synthesis route [45]:
| Synthesis Method | Mixing Technique | Resulting Phase Homogeneity | Key Finding |
|---|---|---|---|
| Coprecipitation | Liquid-phase mixing | Predominantly phase-pure products for all compositions. | Achieved monoclinic, cubic defect fluorite, and cubic pyrochlore structures by varying dopant concentration. |
| Solid-State Synthesis | Manual Mixing (Mortar & Pestle) | Produced the most phase-pure product among solid-state methods. | Extending grinding time and re-sintering enhanced phase purity. |
| Solid-State Synthesis | Mechanical Mixing (Ball Mill) | Multiple phases with impurities. | Identified impurity phases of ZrO₂, CeO₂, and Nd₂O³. Increasing milling time did not improve purity. |
| Solid-State Synthesis | Magnetic Mixing (in Slurry) | Multiple phases with impurities. | Method resulted in heterogeneous ceramics. |
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function in Synthesis |
|---|---|
| Zirconium Oxychloride (ZrOCl₂·8H₂O) | Primary source of zirconium for forming the host matrix [45]. |
| Cerium Chloride (CeCl₃·6H₂O) | Acts as a dopant and a surrogate for tetravalent actinides like plutonium [45]. |
| Neodymium Nitrate (Nd(NO₃)₃·6H₂O) | Co-dopant used to stabilize cubic defect fluorite and pyrochlore structures [45]. |
| Ammonium Hydroxide (NH₄OH) | Precipitating agent for converting metal salts to hydroxides in coprecipitation routes [45]. |
| High-Purity Alumina Crucibles | Sample container for high-temperature (e.g., 1500°C) calcination and sintering steps [45]. |
Visualizing the Process and Theory
The following diagrams illustrate the experimental workflow for enhancing phase purity and the theoretical framework that guides polymorphic control.
Diagram Title: Re-grinding and Re-sintering Workflow
Diagram Title: Phase Transformation via Ostwald's Rule
Analytical Techniques and Comparative Analysis for Quality Assurance
Troubleshooting Guides
Raman Spectroscopy Troubleshooting
This table outlines common problems encountered during Raman spectroscopy experiments, their potential causes, and recommended solutions.
| Problem | Spectrum/Error Message | Possible Explanation | Recommended Solution |
|---|---|---|---|
| No Communication | "Unable To Find Device With Serial:" or "Error Opening USB Device" [49] | Software cannot communicate with the spectrometer due to incorrect settings [49]. | Restart the software. If the problem persists, contact technical support [49]. |
| Flat Spectrum | Spectrum is absolutely flat, all Y-values are zero [49]. | Computer and spectrometer are not communicating; laser may be off [49]. | Check that the spectrometer is connected and powered on. Ensure the laser is turned on and all safety keys (e.g., interlock key) are engaged. Do not look directly into the laser beam. [49] |
| No Peaks, Only Noise | Spectrum shows no Raman peaks, only background noise is visible [49]. | Laser power is too low or the laser is off completely [49]. | Verify the laser is on. Use a power meter to check output at the probe tip (e.g., ~200 mW for 785 nm systems; 25-50 mW for 532 nm systems) [49]. |
| Incorrect Peak Locations | Peaks are present but their positions (Raman shifts) do not match known references [49]. | The spectrometer system has not been properly calibrated [49]. | Perform a wavelength calibration using a standard reference material (e.g., 4-acetamidophenol) [50]. For a 785 nm system, use the verification cap; for 532 nm, use isopropyl alcohol [49]. |
| Saturated Peaks | Peaks are cut off at the top [49]. | The detector (CCD) is saturated because the signal is too intense [49]. | Reduce the integration time. If saturation continues, slightly defocus the laser beam by moving the probe away from the sample [49]. |
| Broad Fluorescent Background | Raman peaks are superimposed on a very broad, intense background [49] [51]. | Sample fluorescence is overwhelming the weaker Raman signal [49] [51]. | Consider using a laser with a longer wavelength (e.g., 785 nm or 1064 nm) to minimize fluorescence excitation [51]. Employ background correction algorithms during data processing [50]. |
| Cosmic Spikes | Sharp, narrow spikes appear at random positions in a single scan [50]. | High-energy cosmic rays strike the detector during measurement [50]. | Most modern software includes algorithms for cosmic spike removal. Increase the number of accumulations to average out the sporadic spikes [50]. |
| Over-Optimized Preprocessing | Model performance is overestimated after baseline correction and normalization [50]. | Preprocessing parameters were optimized to maximize model performance instead of using spectral markers, leading to overfitting [50]. | Use a grid search for preprocessing parameters based on the merit of spectral markers rather than the final model's performance [50]. |
X-ray Diffraction (XRD) Troubleshooting
This table outlines common problems encountered during XRD experiments, their potential causes, and recommended solutions.
| Problem | Possible Explanation | Recommended Solution |
|---|---|---|
| No Peaks or Very Weak Peaks | Sample is non-crystalline (amorphous), amount of material is insufficient, or sample preparation is poor [52]. | Ensure the sample is crystalline. Use a sufficient amount of finely ground powder (<10 µm) and pack it evenly into the sample holder to create a flat surface [52]. |
| Broad and Diffuse Peaks | Crystallites are too small (nanocrystalline), there is significant microstrain in the material, or the instrument is poorly aligned [53]. | If nanoscale crystallites are expected, use Scherrer's equation for analysis. For instrumental issues, check alignment and use a standard sample to verify resolution [53]. |
| Peak Shifts from Reference | Residual stress/strain in the sample, or incorporation of dopants causing lattice expansion/contraction [53]. | The shift is often the key information. Use the peak positions to calculate precise lattice parameters. A standard (e.g., Si powder) can be added to correct for systematic errors [52]. |
| High Background Noise | Sample fluoresces under X-ray radiation (common with Fe-containing samples), or sample holder contributes to scattering [53]. | Use a monochromator to filter fluorescent radiation. For powder samples, ensure the sample is not too thin, and use a zero-background holder if necessary [52]. |
| Preferential Orientation (Texture) | Plate- or needle-like crystallites align non-randomly during sample preparation, changing relative peak intensities [52] [53]. | Improve sample preparation to ensure random orientation. Use a back-loading sample holder. For smears, use a random rather than oriented smear [52]. |
| Unidentifiable Peaks | Presence of an unknown crystalline impurity or phase [52]. | Compare the entire pattern to databases like the Powder Diffraction File (PDF) for phase identification. The detection limit for minor phases is typically ~2% [52]. |
Frequently Asked Questions (FAQs)
General Principles
Q1: What is the fundamental physical principle behind XRD? A1: XRD is based on the constructive interference of monochromatic X-rays scattered by the periodic arrangement of atoms in a crystal lattice. When the path difference between X-rays scattered from parallel crystal planes is an integer multiple of the wavelength, the beams reinforce each other, producing a detectable signal. This condition is defined by Bragg's Law: nλ = 2d sinθ, where n is an integer, λ is the X-ray wavelength, d is the interplanar spacing, and θ is the angle of incidence [54] [53].
Q2: How does Raman spectroscopy provide a "molecular fingerprint"? A2: The Raman effect is an inelastic scattering process. When light interacts with a molecule, a tiny fraction of photons (∼1 in 10⁶) exchange energy with the molecular vibrations. This energy shift, measured as a wavenumber (cm⁻¹) in the spectrum, is unique to specific chemical bonds and symmetries. The resulting pattern of peaks provides a characteristic fingerprint that can be used to identify and characterize the material [55] [51].
Q3: What are the key differences between single-crystal and powder XRD? A3:
- Single-Crystal XRD: Requires a single, well-ordered crystal. It provides a full set of 3D structural data, allowing for the determination of the complete molecular and crystal structure, including atomic coordinates and bond lengths/angles [54] [53].
- Powder XRD: Uses a polycrystalline powder sample with randomly oriented microcrystals. It produces a one-dimensional pattern of concentric rings (Debye rings) and is primarily used for phase identification, quantification, and determining lattice parameters, but provides less structural detail than single-crystal XRD [52] [53].
Application in Solid-State and Grinding Synthesis
Q4: How can XRD be used to confirm the success of a solid-state synthesis via grinding? A4: Powder XRD is the primary tool for this purpose. After grinding, the XRD pattern of the product is compared to the patterns of the starting materials.
- Formation of a New Phase: The appearance of new, distinct peaks not present in the reactants indicates the formation of a new crystalline phase (e.g., a cocrystal or coordination polymer) [8].
- Consumption of Reactants: The disappearance or reduction of peaks corresponding to the starting materials confirms their consumption [8].
- Phase Purity: A clean pattern matching a known reference without extra peaks indicates a pure phase [53].
Q5: Why is Raman spectroscopy particularly useful for analyzing products from liquid-assisted grinding (LAG)? A5: Raman spectroscopy is highly sensitive to molecular-level interactions, such as hydrogen bonding and π-π stacking, which are crucial in supramolecular chemistry. It can detect:
- Conformational Changes: Shifts in peak positions can indicate changes in molecular conformation upon inclusion or complex formation [8] [56].
- Molecular Environment: Changes in the spectrum can reveal how a molecule's environment has altered due to the formation of a new phase during LAG [9] [8].
- Complementarity with XRD: While XRD reveals long-range order and crystal structure, Raman probes short-range interactions and bonding, providing complementary information [56].
Q6: What is a common data analysis mistake in Raman spectroscopy for diagnostic models? A6: A critical mistake is information leakage during model validation. If data splits (training/validation/test sets) are created randomly from all measured spectra without considering that multiple spectra come from the same biological replicate or patient, the model's performance can be drastically overestimated. Spectra from the same source must be kept entirely within one data subset to ensure a reliable performance estimate [50].
Data Analysis and Interpretation
Q7: What is the correct order for preprocessing Raman spectra? A7: The standard pipeline is:
- Cosmic Spike Removal: Remove sharp spikes from cosmic rays [50].
- Wavenumber & Intensity Calibration: Correct for instrumental drifts to generate stable, setup-independent spectra [50].
- Baseline Correction: Remove the broad fluorescent background before normalization [50].
- Normalization: Standardize spectral intensity to make different measurements comparable. Normalizing before baseline correction will bias the data [50].
- Denoising & Feature Extraction: Final steps to clean and reduce data dimensionality for analysis [50].
Q8: How do I identify an unknown mineral or compound from its XRD pattern? A8: The primary method is to measure the d-spacing (from the peak position via Bragg's Law) and relative intensity of each diffraction peak. This set of d-I data is then compared to a standard reference database, such as the International Centre for Diffraction Data (ICDD) Powder Diffraction File (PDF), for a match [52].
Experimental Protocols
Protocol for Sample Preparation and Analysis via Powder XRD
This protocol ensures high-quality data for phase identification in solid-state synthesis products.
1. Sample Preparation (Crucial Step)
- Grinding: Grind the solid product (e.g., from a mechanochemical synthesis) into a fine powder using a mortar and pestle or a mill. The ideal particle size is <10 micrometers to minimize systematic errors and ensure a sufficient number of crystallites are in the beam path [52].
- Loading: Pack the fine powder into a sample holder. The goal is to create a flat, level surface with a random orientation of crystallites.
- Method: Place the holder on a flat surface, fill it with powder, and use a glass slide or blade to press and smooth the top surface flat. Avoid stroking in a single direction, which can induce preferred orientation [52].
- Note on Clays: For clay minerals that require analysis of preferred orientation, specialized oriented smear techniques are used [52].
2. Instrumental Setup and Data Collection
- Standard Scan Parameters:
- 2θ Range: Typically from ~5° to 70° [52].
- Step Size: 0.01° - 0.02° for high-resolution scans.
- Time per Step: 0.5 - 2 seconds.
- The goniometer rotates the sample by θ and the detector by 2θ, recording the intensity of diffracted X-rays at each angle [52] [53].
3. Data Analysis Workflow
- Peak Finding: Identify the center of each diffraction peak, typically measured at 80% of the peak height [52].
- Conversion to d-spacing: Apply Bragg's Law to convert each peak's 2θ position into a d-spacing value [52] [53].
- Phase Identification: Input the list of d-spacings and their relative intensities into a search/match algorithm against the PDF database to identify the crystalline phases present [52].
Protocol for Routine Raman Spectroscopy of Solid Materials
This protocol guides the measurement of a solid sample to obtain a high-quality Raman spectrum.
1. Initial Instrument Check
- Laser Safety: Ensure the interlock key is engaged and all safety protocols are followed. Never look directly into the laser beam or its reflections. [49]
- Verification/Calibration: Before measuring samples, perform a wavelength verification using a standard (e.g., a verification cap for 785 nm systems or isopropyl alcohol for 532 nm systems) to ensure peak positions are accurate [49].
2. Sample Mounting
- For powders, gently press the material onto a glass slide or into a sample vial. Ensure the laser is focused on the sample material.
3. Data Acquisition Optimization
- Laser Power: Start with a low power (e.g., 25% of maximum) to avoid damaging the sample. Increase gradually if the signal is weak. All samples have a power density threshold beyond which they can be damaged [51].
- Integration Time: Start with 1-10 seconds. If peaks are saturated (cut off at the top), decrease the time. If the signal is noisy, increase it [49].
- Number of Accumulations: Accumulating and averaging multiple scans significantly improves the signal-to-noise ratio.
4. Data Preprocessing Pipeline Follow this sequence to correctly prepare raw spectra for analysis [50]:
- Remove Cosmic Spikes: Apply a cosmic spike removal algorithm.
- Calibrate Wavenumber Axis: Use a daily measurement of a wavenumber standard (e.g., 4-acetamidophenol) to correct for any instrumental drift [50].
- Correct Baseline: Subtract the fluorescent background using an appropriate algorithm (e.g., polynomial fitting). This must be done before normalization. [50]
- Normalize Spectra: Scale the spectra to a standard intensity (e.g., to the unit vector or a prominent peak) to compare samples from different days [50].
Research Reagent Solutions
This table lists key materials and reagents essential for experiments involving XRD and Raman spectroscopy, particularly in the context of solid-state grinding synthesis.
| Reagent/Material | Function/Explanation | Example Use Case |
|---|---|---|
| Liquid-Assisted Grinding (LAG) Solvents | Small amounts of solvent act as a lubricant and facilitate molecular diffusion, often controlling the polymorphic outcome of a reaction [9] [8]. | Used in mechanochemical synthesis to selectively form specific cocrystals or polymorphs that are not accessible via neat grinding [9]. |
| Cyclodextrins (e.g., β-CD, HP-β-CD) | Multifunctional excipients that form inclusion complexes, improving drug solubility, dissolution rate, and bioavailability [8]. | Preparation of drug-cyclodextrin inclusion complexes via solid-state grinding to enhance the physicochemical properties of a poorly water-soluble API [8]. |
| Wavenumber Standard (e.g., 4-Acetamidophenol) | A reference material with a high number of well-defined Raman peaks across a broad wavenumber range [50]. | Daily calibration of the Raman spectrometer's wavenumber axis to correct for instrumental drift and ensure peak position accuracy [50]. |
| Intensity Standard | A material with a known and stable Raman cross-section used to correct for the spectral transfer function of the instrument [50]. | Correction of the Raman spectrum's intensity response to generate setup-independent spectra, crucial for quantitative comparisons [50]. |
| Powder XRD Standards (e.g., Si, Al₂O₃) | Highly crystalline materials with precisely known lattice parameters and diffraction patterns [52]. | Mixed with an unknown sample to correct for systematic errors in peak position. Used to verify instrument alignment and resolution [52]. |
| Zero-Background Sample Holders | Sample holders made from a single crystal of silicon cut along a non-diffracting plane or from amorphous material [52]. | Mounting powder samples for XRD to minimize background scattering from the holder itself, thereby improving the signal-to-noise ratio [52]. |
## FAQs and Troubleshooting Guides
Frequently Asked Questions
Q1: What is the fundamental difference in the XRD pattern of a phase-pure ceramic compared to a multi-phase ceramic? A phase-pure ceramic will exhibit a diffraction pattern where every peak can be indexed to a single crystal structure and its space group [57]. In contrast, a multi-phase ceramic will show a pattern that is a superposition of multiple sets of peaks, with each set corresponding to a different crystalline phase present in the sample [58]. You must identify all peaks and match them to the reference patterns for all suspected phases.
Q2: My XRD pattern shows broad, low-intensity peaks. What could be the cause and how can I confirm? Broad and low-intensity peaks often suggest the presence of very small crystallites or amorphous content [57]. This is a common issue if the solid-state reaction is incomplete, potentially due to insufficient grinding, calcination time, or temperature. To confirm, you can:
- Perform Rietveld refinement to quantify the amorphous fraction.
- Increase the calcination temperature or time and re-analyze to see if sharper, more intense peaks develop, indicating further crystallization.
- Use microscopy techniques (SEM/TEM) to observe crystallite size and morphology.
Q3: After repeated grinding and calcination, a minor impurity phase persists. How should I proceed? A persistent minor phase indicates a thermodynamic or kinetic barrier. Troubleshooting steps include:
- Verify precursor purity and stoichiometry: Use highly pure precursors and confirm your weighing calculations. Characterize precursors with XRF to check for stoichiometric deviations [57].
- Adjust synthesis parameters: Modify the calcination atmosphere (e.g., oxidizing vs. inert) or use a different heating and cooling profile.
- Consider alternative synthesis routes: Methods like Spark Plasma Sintering (SPS) can achieve high density and phase purity with shorter processing times, limiting unwanted grain growth and phase interactions [58] [59].
Q4: In a multi-phase ceramic, how can I determine which phase is hosting a specific element? XRD identifies crystal structures but not elemental partitioning. To link elements to phases:
- Use µXRF (Micro X-ray Fluorescence): This technique creates high-resolution elemental maps, showing the spatial distribution of elements across your sample's surface [57]. By correlating these maps with microscopy images, you can see which elements are concentrated in which grains.
- Perform SEM/EDS (Energy Dispersive X-ray Spectroscopy): EDS can provide elemental analysis from specific, microscopic points on your sample corresponding to different phases seen in the image [60].
Troubleshooting Common Experimental Issues
Problem: Inconsistent XRD Results Between Synthesis Batches
- Potential Cause: Inhomogeneous precursor mixing due to manual grinding.
- Solution: Implement a more consistent and controlled powder processing method. High-throughput workflows that use automated slurry mixing and milling can significantly improve homogeneity and reproducibility [6]. Ensure grinding time and pressure are standardized.
Problem: Difficulty Detecting Minor or Amorphous Impurity Phases
- Potential Cause: The crystalline main phase dominates the XRD pattern, obscuring weak signals from minor phases.
- Solution: Use more sensitive data collection strategies, such as longer scan times or synchrotron XRD for higher resolution and intensity. Pair XRD with Raman spectroscopy, which can be more sensitive to local structural disorder and specific amorphous phases.
Problem: Apparent Phase Purity by XRD, but Poor Sinterability or Unexpected Properties
- Potential Cause: Residual chemical heterogeneity at the sub-micron level that is not detected by conventional XRD.
- Solution: Employ local structural probes like TEM for nanoscale phase identification. Thermal analysis (DSC/TGA) can also reveal phase transitions or reactions that XRD might miss.
## Essential Analytical Techniques for Phase Identification
The following table summarizes the key techniques used to distinguish between phase-pure and multi-phase ceramics.
Table 1: Key Analytical Techniques for Phase Identification in Ceramics.
| Technique | Primary Function | Key Output for Phase Purity Assessment | Sample Requirements |
|---|---|---|---|
| X-ray Diffraction (XRD) [57] | Identifies and quantifies crystalline phases. | A pattern where all peaks match a single crystal structure. Extra peaks indicate secondary phases. | Powder or solid pellet. |
| Micro X-ray Fluorescence (µXRF) [57] | Maps elemental composition and distribution. | Homogeneous distribution of all elements suggests a single phase. Clustering of elements reveals secondary phases. | Solid surface, minimal preparation. |
| X-ray Microscopy (XRM) [57] | Non-destructive 3D imaging of internal structure. | Reveals internal defects, pores, and cracks that can influence phase stability and interpretation. | Small solid sample. |
| Scanning Electron Microscopy with Energy Dispersive X-ray Spectroscopy (SEM/EDS) [60] | Visualizes microstructure and provides localized elemental analysis. | Reveals grain morphology and allows point-and-shoot chemical analysis of different grains to confirm phase composition. | Solid sample, often requires conductive coating. |
## Experimental Protocols for Phase Characterization
Objective: To identify the crystalline phases present in a synthesized ceramic powder and assess phase purity.
Materials & Equipment:
- X-ray diffractometer with Cu Kα radiation source
- Sample holder
- Flat plate or glass slide
- Mortar and pestle or ball mill
- Ceramic powder sample
Methodology:
- Sample Preparation: If the sample is not already a fine powder, grind it gently in a mortar and pestle to a consistent fine powder (typically < 10 µm) to minimize preferred orientation.
- Loading: Evenly pack the powder into the cavity of a sample holder or onto a low-background silicon wafer. Use a glass slide to create a flat, smooth surface.
- Instrument Setup: Load the sample into the diffractometer. Set the scan parameters (e.g., 2θ range from 10° to 80°, step size of 0.02°, and counting time of 1-2 seconds per step).
- Data Collection: Initiate the scan. The instrument will rotate the sample and detector to collect the diffraction pattern.
- Data Analysis:
- Import the data into analysis software.
- Perform background subtraction and smoothing if necessary.
- Identify the peaks in the pattern.
- Use a database such as the ICDD PDF-4+ to match the observed peaks to reference patterns for known phases.
- For a phase-pure material, all peaks should correspond to a single phase. Any unindexed peaks indicate the presence of impurity phases.
Objective: To automate and parallelize the synthesis of multiple ceramic compositions for rapid screening of phase formation and stability.
Materials & Equipment:
- Precursor oxides and carbonates
- Planetary mill with zirconia media
- Automated liquid handling station
- Vacuum-formed PET trays
- Freeze drier
- Isostatic press
- High-temperature furnace
Methodology:
- Wet Milling: Mill insoluble raw materials in deionized water with a dispersant and binder to create homogeneous aqueous suspensions of each precursor.
- Automated Wet Mixing: Use a robotic liquid handler to aspirate and mix precise volumes of the precursor suspensions in glass vials to achieve the target compositions.
- Dispensing: Dispense small aliquots of each mixture into custom vacuum-formed trays.
- Freeze Drying: Manually transfer the trays to a freezer, then to a freeze drier to remove the solvent, forming porous powder discs.
- Isopressing: Place the dried discs in custom silicone holders, vacuum seal in bags, and isopress at high pressure (105–210 MPa) to increase disc density and strength.
- Calcination: Transfer the trays to a furnace for high-temperature calcination to react the precursors and form the desired crystalline phases.
- Characterization: The resulting discs are free-standing and can be directly presented to automated XRD for high-throughput phase identification.
## Workflow and Data Interpretation Diagrams
Diagram 1: Phase Analysis Workflow
Diagram 2: Data Interpretation Guide
## The Scientist's Toolkit: Key Reagents and Materials
Table 2: Essential Research Reagents and Materials for Solid-State Synthesis.
| Item | Function in Synthesis | Example in Context |
|---|---|---|
| High-Purity Oxide/Carbonate Precursors | Provide the cationic species for the final ceramic phase. Impurities can lead to secondary phases. | BaCO₃, Cr₂O₃, and TiO₂ for hollandite synthesis [58]. |
| Zirconia Milling Media | Used in ball milling to reduce particle size and homogenize precursor mixtures, increasing reactivity [6]. | Critical for achieving a complete solid-state reaction in Mg₂Si synthesis [61]. |
| Polyacrylate Dispersant | Aids in de-agglomerating powder particles in suspension during wet milling, promoting a more uniform mixture [6]. | Used in high-throughput workflows to create stable precursor slurries. |
| Binder (e.g., Acrylic Emulsion) | Provides mechanical strength to pressed powder compacts (green bodies) before calcination, preventing handling damage [6]. | Added to suspensions before freeze-drying to form robust discs for isopressing. |
| Graphite Paper/ Foil | Used as a sacrificial layer between the sample and the die during Spark Plasma Sintering (SPS) to prevent reaction and aid release [58] [59]. | Essential for fabricating high-density "designer waste forms" and SiC-composites via SPS. |
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of wet-chemistry methods like coprecipitation and sol-gel over solid-state synthesis? The primary advantage is the achievement of atomic-level mixing of precursors. In solid-state reactions, precursors are mixed as solid powders with micron-sized particles, making atomic-scale homogeneity difficult to achieve. Wet-chemistry methods use soluble precursors (e.g., metal nitrates) that are mixed in solution, enabling a much more homogeneous mixture at the molecular level before any heat treatment is applied. This intimate mixing significantly reduces the diffusion distances required for reaction, leading to higher phase purity, lower synthesis temperatures, and shorter reaction times [4].
Q2: Can solid-state synthesis ever produce phase-pure materials? Yes, solid-state synthesis can produce phase-pure materials, but it often requires significant optimization. Key parameters include extended milling times to reduce particle size and improve reactant homogeneity, and repeated calcination cycles with intermediate grinding steps to promote a more thorough reaction. For example, high-purity ZrV2O7 was achieved via solid-state reaction through extended milling and repeated calcination [1]. However, achieving sub-nanometric level purity can be challenging, and the process is often more time- and energy-intensive compared to wet-chemical routes [4].
Q3: How does the choice of synthesis method impact the electrochemical properties of battery cathode materials? The synthesis method directly influences the microstructure, which in turn affects electrochemical performance. Materials produced via wet-chemical routes often demonstrate improved initial discharge capacity and better cycling stability. For instance, LiNiO2 (LNO) synthesized via coprecipitation (C-LNO) showed a higher initial discharge capacity (221 mA h g⁻¹) compared to solid-state synthesized LNO (199 mA h g⁻¹) when cycled between 2.7–4.3 V. C-LNO also exhibited better capacity retention after 100 cycles [62] [63]. The enhanced homogeneity achieved through coprecipitation helps minimize defects that can lead to capacity fade.
Q4: What are the main drawbacks of solid-state synthesis? The main drawbacks are its inability to achieve atomic-level mixing and its slow reaction kinetics. This often necessitates high temperatures and prolonged heating durations to facilitate solid-state diffusion, consuming more time and energy. Furthermore, incomplete reactions can lead to the persistence of impurity phases or unreacted starting materials, which can be difficult to detect and may hinder the target material's functional properties [1] [4].
Q5: Are there modern approaches to optimizing solid-state synthesis? Yes, research is focused on integrating computational and automated methods. Algorithms like ARROWS3 (Autonomous Reaction Route Optimization with Solid-State Synthesis) have been developed to guide precursor selection. These algorithms use thermodynamic data and actively learn from experimental outcomes (e.g., X-ray diffraction patterns) to identify and avoid reaction pathways that lead to stable intermediate byproducts, thereby increasing the success rate of synthesizing the desired phase with high purity [5].
Troubleshooting Guides
Issue 1: Persistent Impurity Phases in Solid-State Synthesis
Problem: Despite repeated grinding and calcination, X-ray diffraction (XRD) analysis continues to show traces of unreacted starting materials or secondary phases.
Solutions:
- Increase Milling Time and Efficiency: The initial mixing step is critical. Extend the milling duration significantly. For example, in the synthesis of ZrV2O7, milling times of 180 minutes were investigated to improve homogeneity, as opposed to shorter durations of 15 or 40 minutes [1].
- Implement Multiple Calcination-Grinding Cycles: Do not rely on a single, long heating step. Instead, perform several shorter calcination cycles (e.g., 5-20 hours at the target temperature), each followed by a thorough grinding step to expose fresh surfaces and improve diffusion. Studies on ZrV2O7 used up to three repeated calcination cycles at 700°C to achieve high purity [1].
- Optimize Precursor Chemistry: Consider using alternative precursor compounds that are more reactive. For example, some synthesis routes for ZrV2O7 use zirconium chloride and ammonium vanadate instead of the standard oxides (ZrO2 and V2O5) to improve reaction kinetics [1].
- Verify Phase Purity with Multiple Techniques: Use Raman spectroscopy in conjunction with XRD to confirm phase purity, as it can reveal subtle structural differences and the presence of amorphous or low-concentration impurities that XRD might miss [1].
Issue 2: Inconsistent Results in Coprecipitation Synthesis
Problem: Reproducibility is low between batches; the final product's composition, particle size, or morphology varies.
Solutions:
- Strictly Control Solution Conditions: For coprecipitation, parameters such as pH, temperature, stirring rate, and concentration of the precursor solutions must be meticulously controlled and kept consistent across all batches. Any fluctuation can lead to variations in the precipitated precursor, affecting the final product.
- Employ a Fuel in Combustion Methods: For methods like Solution Combustion Reaction (SCR), ensure the correct stoichiometric ratio between metal nitrates (oxidizers) and the organic fuel (e.g., glycine, urea). This ensures a self-sustaining exothermic reaction that produces a voluminous, fluffy foam with high purity, as demonstrated in the synthesis of BFN-KN solid solutions [4].
- Characterize the Precipitated Intermediate: Before the final calcination, analyze the precipitated powder (often a hydroxide or carbonate) for its chemical composition, phase, and particle size distribution. This intermediate's quality directly dictates the quality of the final oxide material.
Issue 3: Low Product Yield or Poor Sinterability
Problem: The final product has low yield, or the powders do not densify well during sintering, leading to weak mechanical properties in ceramics.
Solutions:
- Switch to a Wet-Chemistry Route: Wet-chemical methods like sol-gel or coprecipitation typically produce nano-sized, highly reactive powders with a large surface area. These powders have higher sinterability, meaning they can be densified at lower temperatures and in less time. The SCR method is noted for reducing the phase formation temperature [4].
- Optimize Calcination Temperature: Avoid excessively high calcination temperatures that can cause premature particle coarsening (Ostwald ripening) and reduce sinterability. Use the minimum temperature required to form the desired crystalline phase, as determined by in-situ XRD or thermal analysis.
Detailed Methodology: Solution Combustion Reaction (SCR) for Ceramic Oxides
The following protocol is adapted from the synthesis of (x)BaFe0.5Nb0.5O3-(1-x)KNbO3 (BFN-KN) solid solutions [4].
1. Reagent Preparation:
- Precursors: Use high-purity metal nitrates (e.g., Ba(NO3)2, Fe(NO3)3·9H2O, Nb precursor, KNO3). If oxides or carbonates are the only available starting materials, they must first be dissolved in nitric acid to form nitrate solutions.
- Fuel: Urea (CO(NH2)2) or glycine (C2H5NO2) is commonly used.
- Stoichiometry: Calculate the required amounts of metal nitrates to achieve the target cationic stoichiometry. The fuel quantity should be calculated based on the total oxidizing valence of the metal nitrates to ensure a balanced, stoichiometric redox reaction.
2. Procedure:
- Dissolution: Dissolve all the metal nitrate precursors and the fuel in a minimum quantity of deionized water in a large beaker (use a beaker with a capacity much larger than the solution volume to accommodate the foam produced during combustion).
- Heating/Combustion: Place the beaker on a hot plate inside a fume hood. Heat the solution gradually to around 300-500°C. The solution will undergo dehydration, followed by frothing, and finally ignite spontaneously, resulting in a rapid, self-propagating combustion reaction that yields a voluminous solid foam.
- Calcination: Gently grind the resulting foam into a powder. Calcine (heat) this powder in a furnace at a temperature determined to be optimal for phase formation (e.g., 800-1200°C for several hours) to obtain the final crystalline oxide phase.
Quantitative Data Comparison
The tables below summarize key performance metrics from recent studies comparing synthesis methods.
Table 1: Synthesis Parameters and Phase Purity
| Material System | Synthesis Method | Key Synthesis Parameters | Phase Purity Outcome | Citation |
|---|---|---|---|---|
| ZrV₂O₇ | Solid-State | Milling: 15-180 min; Calcination: 700°C, 1-3 cycles (5-20 h each) | High purity achieved with extended milling (180 min) and repeated calcination. | [1] |
| ZrV₂O₇ | Sol-Gel | "Near-atomic" level mixing of precursors. | Homogeneous, phase-pure material achieved. | [1] |
| BFN-KN Solid Solutions | Solid-State (SSR) | Calcination: 800-1200°C | Phase-pure samples difficult to achieve; impurity phases often detected. | [4] |
| BFN-KN Solid Solutions | Solution Combustion (SCR) | Calcination: 800-1200°C | Phase-pure samples achieved due to molecular-level mixing of precursors. | [4] |
| LiNiO₂ (LNO) | Solid-State (SS) | Sintering: 800°C in O₂ | Phase pure, but with higher cation mixing. | [62] [63] |
| LiNiO₂ (LNO) | Coprecipitation (C) | Sintering: 1 h at 800°C in O₂ | Pristine, phase-pure material achieved in a short time. | [62] [63] |
Table 2: Electrochemical Performance of LiNiO₂ (LNO) Cathodes
| Material | Synthesis Method | Voltage Window (V vs. Li⁺/Li) | Initial Discharge Capacity (mA h g⁻¹) | Capacity Retention (after 100 cycles) | Citation |
|---|---|---|---|---|---|
| SS-LNO | Solid-State | 2.7 - 4.3 | 199 | 41% | [62] [63] |
| C-LNO | Coprecipitation | 2.7 - 4.3 | 221 | 47% | [62] [63] |
| SS-LNO | Solid-State | 2.7 - 4.1 | 144 | 84% | [62] [63] |
| C-LNO | Coprecipitation | 2.7 - 4.1 | 168 | 76% | [62] [63] |
Synthesis Method Selection Workflow
The following diagram outlines a logical decision process for selecting a synthesis method, based on target material priorities and available resources.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Their Functions in Materials Synthesis
| Item | Function in Synthesis | Common Examples |
|---|---|---|
| Oxide Precursors | Source of metal cations in solid-state reactions; often the most stable and readily available form. | ZrO₂, V₂O₅, α-Fe₂O₃, Nb₂O₅ [1] [4]. |
| Carbonate Precursors | Source of alkali or alkaline earth metal cations; decompose upon heating to release the metal oxide and CO₂. | BaCO₃, K₂CO₃, Li₂CO₃ [4] [62]. |
| Nitrate Precursors | Soluble sources of metal cations for wet-chemical methods; also act as oxidizers in combustion synthesis. | Ba(NO₃)₂, Fe(NO₃)₃·9H₂O, Metal nitrates [4]. |
| Organic Fuel | Serves as a reductant in solution combustion synthesis; reacts exothermically with metal nitrates to produce the desired oxide. | Urea (CO(NH₂)₂), Glycine (C₂H₅NO₂) [4]. |
| Grinding Media | Used to reduce particle size and mix precursors homogeneously in solid-state and some wet-chemical methods. | Agate mortar and pestle, Zirconia milling balls in a ball mill [1] [4]. |
Frequently Asked Questions
Q1: My solid-state synthesis consistently results in unwanted secondary phases. How can I improve phase purity?
This is a common challenge often rooted in insufficient reactant mixing or suboptimal thermal profiles. To achieve high phase purity, you must ensure "near-atomic" level mixing of precursors and optimize your calcination parameters.
- Extended Milling: For solid-state reactions, extend dry milling times significantly. One study achieved high-purity ZrV2O7 only after 180 minutes of milling, compared to shorter durations of 15 or 40 minutes [1].
- Repeated Calcination: Implement multiple calcination cycles with intermediate grinding steps. Research shows that two or three repeated calcination cycles at 700°C were necessary to consume remnant starting materials like ZrO2 and achieve high purity [1].
- Lithium Precursor Control (for Oxide Ceramics): If synthesizing lithium-containing ceramics like LLZO, the choice of lithium precursor (e.g., Li2CO3 vs. LiOH) dictates the reaction pathway. LiOH, with its lower decomposition temperature, can provide faster lithium availability and diffusion, leading to a more direct route to the phase-pure product without persistent secondary phases like La2Zr2O7 [2].
Q2: How can I quantitatively track and confirm improvements in phase purity?
Rely on a combination of quantitative characterization techniques rather than a single method.
- X-ray Diffraction (XRD): Use Rietveld refinement on your XRD patterns to quantify the weight fractions of crystalline phases present. Compare your experimental pattern against a simulated pattern from a known crystal structure to identify subtle differences [1].
- Raman Spectroscopy: This technique is highly sensitive to local structural changes and bonding. You can confirm phase purity by matching your spectrum to ab initio simulated phonon data, which allows for the interpretation of Raman-active atom vibrations [1].
Q3: Why do my synthesis results lack reproducibility, even when following published procedures?
Reproducibility is a major hurdle in materials synthesis, often caused by uncontrolled variables in processing. A recent interlaboratory study on battery materials highlighted that even with the same starting materials and recipe, results can vary widely [64].
- Pressure and Duration Control: Document and control the precise pressures and durations used during powder compaction and pelletization. Studies show that applied pressures can vary by hundreds of MPa and pressing times can differ by orders of magnitude between labs, drastically affecting the final microstructure and performance [64].
- Report in Triplicate: Always perform and report key experiments in triplicate. This provides a statistical basis for your results and demonstrates the robustness of your protocol [64].
- Detailed Documentation: Maintain exhaustive records of all procedural details, including powder handling, mixing, and environmental conditions (e.g., humidity in gloveboxes), as these are common failure points [64].
Troubleshooting Guides
Issue: Inconsistent Batch Quality and Performance
Problem: Batch-to-batch inconsistencies are observed, affecting material properties and experimental outcomes.
| Troubleshooting Step | Action | Key Metric for Success |
|---|---|---|
| 1. Raw Material Audit | Qualify and audit suppliers. Implement strict incoming material testing for purity and potency [65]. | Certificate of Analysis (CoA) confirming purity and stoichiometry. |
| 2. Process Validation | Identify Critical Process Parameters (CPPs) like heating rates, hold temperatures, and atmosphere. Validate the ranges for these parameters [65]. | Successful reproduction of phase-pure material across 3+ separate batches, confirmed by XRD. |
| 3. In-Process Control | Implement real-time quality checks during synthesis. For powder processing, use automated tools for tapped density and angle of repose to reduce operator dependency [66]. | Hausner Ratio repeatability of < 2% standard deviation between measurements [66]. |
Issue: Formation of Impurity Phases During Calcination
Problem: Unwanted secondary phases (e.g., Zr3V3Ox, La2Zr2O7) persist even after high-temperature treatment.
| Troubleshooting Step | Action | Key Metric for Success |
|---|---|---|
| 1. Pathway Analysis | Use In-situ HT-XRD to map the phase evolution from precursors to final product. Identify temperature windows where intermediates form and decompose [2]. | Identification of the precise temperature for the disappearance of key intermediate phase diffraction peaks. |
| 2. Atmosphere Optimization | Experiment with different calcination atmospheres (e.g., air vs. N2). An inert atmosphere like N2 can sometimes suppress secondary phase formation and alter Li diffusion kinetics [2]. | Achievement of a phase-pure XRD pattern with no detectable secondary phases. |
| 3. Quenching | After the final calcination step, rapidly quench the sample (e.g., in air or liquid nitrogen) to prevent low-temperature phase decomposition or transformation [1]. | Retention of high-temperature phase at room temperature, confirmed by XRD. |
Quantitative Data Tables
Table 1: Impact of Synthesis Parameters on Phase Purity
This table summarizes quantitative findings on how specific parameters influence the success of solid-state synthesis, as evidenced by recent studies.
| Material System | Critical Parameter | Optimized Value | Impact on Phase Purity & Performance |
|---|---|---|---|
| ZrV2O7 [1] | Milling Time | 180 minutes | Achieved high purity; shorter times (15', 40') left remnant reactants. |
| ZrV2O7 [1] | Calcination Cycles | 2-3 cycles at 700°C | Required to consume remnant ZrO2; a single cycle was insufficient. |
| LaNi5 [67] | Synthesis Temperature & Time | ≥ 1000°C for ≥ 4 hours | Necessary for forming single-phase LaNi5; lower T/t led to impurity phases. |
| LLZO (Nb/Ta-doped) [2] | Lithium Precursor | LiOH under N2 | Enabled a direct reaction path to cubic LLZO without detectable secondary phases. |
| LLZO (Nb/Ta-doped) [2] | Final Calcination Temperature | 950–1050°C (in air) | Required to achieve phase-pure cubic LLZO when using Li2CO3 in air. |
Table 2: Metrics for Homogeneity and Reproducibility
This table outlines key metrics to track for ensuring material homogeneity and procedural reproducibility.
| Metric Category | Specific Metric | Measurement Technique | Target/Desired Outcome |
|---|---|---|---|
| Powder Homogeneity | Hausner Ratio | Tapped Density Analyzer (e.g., GranuPack) | Lower ratio (<1.25 indicates good flowability); high measurement repeatability [66]. |
| Powder Homogeneity | Angle of Repose (AOR) | Automated Heap Shape Analysis (e.g., GranuHeap) | Consistent, operator-independent AOR values between batches [66]. |
| Structural Reproducibility | Lattice Parameter Variation | XRD with Rietveld Refinement | Standard deviation of < 0.001 Å across multiple synthesis batches. |
| Performance Reproducibility | Initial Discharge Capacity | Electrochemical Cycling (e.g., for batteries) | Low coefficient of variation (< 5%) in initial capacity across cells [64]. |
| Protocol Reproducibility | Success Rate of Cell Assembly | Standardized Operating Procedure (SOP) | >80% of assembled cells (e.g., batteries) are operational without premature failure [64]. |
Experimental Protocols
Detailed Methodology: Solid-State Synthesis with High Phase Purity
This protocol is adapted from procedures used to synthesize high-purity ZrV2O7 and LaNi5 [1] [67].
1. Precursor Preparation
- Stoichiometric Weighing: Accurately weigh out precursor oxides (e.g., ZrO2 and V2O5 for ZrV2O7) in the required stoichiometric ratio.
- Lithium Compensation: For lithium-containing materials, add an excess of lithium precursor (e.g., 10-20 wt%) to compensate for volatile losses during high-temperature calcination [2].
- Initial Mixing: Use a mortar and pestle for an initial dry mix to homogenize the powders.
2. High-Energy Ball Milling
- Equipment: Place the mixed powders in a zirconia milling jar with zirconia grinding media.
- Milling Medium: Add isopropanol as a wetting agent to prevent caking and improve mixing efficiency.
- Milling Parameters: Mill at 300 rpm for 4 hours. For some systems, extended milling times (e.g., 3 hours) are critical for achieving the necessary homogeneity [1] [2].
- Drying: Pour the resulting suspension into a glass dish and dry in an oven at 70°C overnight. Gently break up the dried cake with a mortar and pestle.
3. Calcination and Sintering
- Crucible Selection: Choose an appropriate crucible (e.g., MgO, Al2O3) that is chemically inert with the sample.
- Thermal Profile:
- Intermediate Grinding: After the first calcination cycle, remove the powder and grind it thoroughly again to expose fresh surfaces and enhance solid-state diffusion. Repeat the calcination cycle 1-2 more times for complete reaction [1].
4. Post-Synthesis Treatment (if applicable)
- Hydrometallurgical Treatment: For some materials (e.g., calciothermic synthesis of LaNi5), a slaking (controlled reaction with water) and leaching process is used to remove by-products and reduce oxygen content. The duration of slaking must be optimized, as it decisively affects final impurity levels [67].
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Synthesis | Example from Literature |
|---|---|---|
| Zirconia Milling Media | Used in planetary ball mills to reduce particle size and achieve homogeneous mixing of precursor materials, crucial for reaction kinetics [1] [2]. | 3 mm zirconia balls used for 4-hour milling of LLZO precursors [2]. |
| Lithium Hydroxide Monohydrate (LiOH·H2O) | A lithium precursor with a lower decomposition temperature than Li2CO3, enabling faster Li availability and a cleaner reaction pathway to phase-pure products [2]. | Used in the synthesis of Nb/Ta-doped LLZO under N2 atmosphere to avoid secondary phases [2]. |
| Lithium Carbonate (Li2CO3) | A common, stable lithium precursor. Requires higher temperatures to decompose and can lead to different reaction intermediates compared to LiOH [2]. | Used in solid-state synthesis of LLZO; requires temperatures of 950-1050°C in air to achieve phase purity [2]. |
| Niobium Pentoxide (Nb2O5) | An aliovalent dopant precursor used to stabilize the high-conductivity cubic phase in LLZO solid electrolytes [2]. | Dopant for LLZNO (Li7La3Zr2O12 doped with Nb) [2]. |
| Indium Foil | Serves as a reference or counter electrode material in all-solid-state battery research for benchmarking performance [64]. | Provided as a standard material to 21 research groups for assembling and benchmarking solid-state battery cells [64]. |
Workflow and Relationship Diagrams
Synthesis Optimization Workflow
Parameter and Metric Relationships
Conclusion
The strategic implementation of repeated grinding is a powerful, scalable, and cost-effective method for achieving exceptional phase purity in solid-state synthesis. By understanding the fundamental principles, meticulously applying optimized milling protocols, proactively troubleshooting common issues, and rigorously validating outcomes with advanced characterization, researchers can reliably produce high-quality materials. This holistic approach directly addresses key challenges in pharmaceutical solid forms and advanced material development, paving the way for more reproducible drugs and tailored material properties. Future directions should focus on integrating smart, data-driven monitoring of milling parameters and exploring novel mechanochemical pathways to further push the boundaries of synthesis control.
References
- 1. Synthesis and phase purity of the negative thermal expansion ... [https://pubs.rsc.org/en/content/articlehtml/2025/tc/d4tc04095c]
- 2. Phase Formation Study of Solid-State LLZNO and LLZTO ... [https://www.mdpi.com/2571-6131/8/4/132]
- 3. Solid-state synthesis of stoichiometric and dense CoV2O4 ... [https://www.sciencedirect.com/science/article/pii/S0955221925001062]
- 4. (1-x)KNbO3 solid solutions on synthesis method and their ... [https://www.sciencedirect.com/science/article/abs/pii/S0022369725007693]
- 5. Autonomous and dynamic precursor selection for solid- ... [https://www.nature.com/articles/s41467-023-42329-9]
- 6. A high throughput synthetic workflow for solid state synthesis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10866347/]
- 7. Strategies to achieve reproducible synthesis of phase-pure ... [https://www.nature.com/articles/s43246-024-00690-2]
- 8. Grinding as Solvent-Free Green Chemistry Approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6321573/]
- 9. Recent advances in liquid assisted grinding chemistry [https://www.sciencedirect.com/science/article/pii/S2211715625006587]
- 10. An optimized method for synthesizing phase-pure Ti3AlC2 ... [https://www.sciencedirect.com/science/article/abs/pii/S0955221921007603]
- 11. Synthesis, thermal expansion and optical properties of ZrV ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884224001913]
- 12. Two Decades of Negative Thermal Expansion Research [https://pmc.ncbi.nlm.nih.gov/articles/PMC5448970/]
- 13. Synthesis and phase purity of the negative thermal ... [https://chemrxiv.org/engage/chemrxiv/article-details/66967ed35101a2ffa88a4481]
- 14. Synthesis and phase purity of the negative thermal ... [https://pubs.rsc.org/zh-hans/content/articlelanding/2025/tc/d4tc04095c]
- 15. Influence of W doped ZrV2O7 on structure, negative ... [https://www.sciencedirect.com/science/article/abs/pii/S0169433214011489]
- 16. Force-controlled robotic mechanochemical synthesis [https://pubs.rsc.org/en/content/articlehtml/2024/dd/d4dd00189c]
- 17. The mechanochemical synthesis of polymers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8978534/]
- 18. Synthesis of SnTe by repeated cold-pressing [https://www.sciencedirect.com/science/article/abs/pii/S092150939900043X]
- 19. 17 Signs of Problems with Ball Mills: Quickly Remove ... [https://www.ftmmachinery.com/blog/17-signs-of-problems-with-ball-mills-quickly-remove-hidden-troubles.html]
- 20. Common problems and troubleshooting of ball mill [https://www.mycrusherpart.com/news/common-problems-and-troubleshooting-of-ball-mill/]
- 21. Common Faults of Ball Mill and Their Solutions [https://www.baichychina.com/Products_Knowledge/Common-Faults-of-Ball-Mill-and-Their-Solutions.html]
- 22. Autonomous and dynamic precursor selection for solid- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10618174/]
- 23. Unveiling phase evolution of complex oxides toward ... [https://pubmed.ncbi.nlm.nih.gov/40712022/]
- 24. Microchemical homogeneity and quenching-induced ... [https://www.sciencedirect.com/science/article/pii/S2666539522001055]
- 25. Influence of Milling Time on the Homogeneity and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7014023/]
- 26. High-Energy Ball Mills: Parameters for Optimizing ... [https://www.msesupplies.com/blogs/news/high-energy-ball-mills-parameters-for-optimizing-milling-efficiency?srsltid=AfmBOopoluNBdiWI_ObYQjd2yPuM3O2lXDkVmlSPHeBj36xsFD-XxnoI]
- 27. The Influence of High-Energy Milling on the Phase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11313661/]
- 28. Phase Evolution During High-Energy Ball Milling and ... [https://www.mdpi.com/1996-1944/18/11/2494]
- 29. Process Enhancement of Calcium Looping through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12487757/]
- 30. Mechanochemical Synthesis of Advanced Materials for All ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12431395/]
- 31. Low-temperature solid-state reaction synthesis of SrY2O4 ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884224061121]
- 32. Investigating the Calcination Temperature and Grinding ... [https://www.mdpi.com/2412-3811/8/10/139]
- 33. XRD Phase Identification [https://www.malvernpanalytical.com/en/products/measurement-type/phase-identification]
- 34. Identification of crystalline materials with X-Ray Diffraction [https://forcetechnology.com/en/articles/identification-crystalline-materials-x-ray-diffraction]
- 35. X-ray Diffraction–Solving Problems with Phase Analysis [https://www.mccrone.com/mm/xray-diffraction-phase-analysis-solutions/]
- 36. DIFFRAC.EVA [https://www.bruker.com/en/products-and-solutions/diffractometers-and-x-ray-microscopes/x-ray-diffractometers/diffrac-suite-software/diffrac-eva.html]
- 37. An efficient recycling strategy to eliminate the residual “ ... [https://www.sciencedirect.com/science/article/pii/S2468025722001741]
- 38. Thermodynamic modelling assisted three-stage solid state ... [https://www.sciencedirect.com/science/article/pii/S0264127524000510]
- 39. Demystifying Vibration Monitoring Part 4 [https://www.pruftechnik.com/understanding-vibration-monitoring-common-faults-part-4/]
- 40. Troubleshooting with Vibration Monitoring [https://www.pumpsandsystems.com/troubleshooting-vibration-monitoring]
- 41. Top 10 Machine Failures – The ultimate guide in vibration ... [https://www.erbessd-instruments.com/articles/top-10-machine-failures/?srsltid=AfmBOood7C4QUwDGxraE1N1W7ZahDfmhqTV0wuUsZDXYWuanZSKHgAu4]
- 42. 7 Useful Tips For Treating Machine Vibration Problems [https://rms-reliability.com/vibration/treatment-for-machine-vibration/]
- 43. A high throughput synthetic workflow for solid state synthesis ... [https://pubs.rsc.org/en/content/articlehtml/2024/sc/d3sc05688k]
- 44. 6 Common Grinding Problems and How to Solve Them [https://www.cdtusa.net/blog/6-common-grinding-problems]
- 45. Optimizing synthesis strategies for the production of single- ... [https://link.springer.com/article/10.1557/s43580-024-00789-1]
- 46. Tips for Overcoming 6 Common Surface Grinding and ... [https://mexico.weilerabrasives.com/en/mx-articles/tips-for-overcoming-6-common-surface-grinding-and-cutting-problems]
- 47. Phase-selective synthesis and polymorphic transformation ... [https://www.nature.com/articles/s41467-025-63668-9]
- 48. High-purity Li₆PS₅Cl sulfide solid electrolytes via liquid ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725077903]
- 49. Welcome to Ramansystems - Troubleshooting [http://ramansystems.com/english/troubleshooting.htm]
- 50. Errors and Mistakes to Avoid when Analyzing Raman Spectra [https://www.spectroscopyonline.com/view/errors-and-mistakes-to-avoid-when-analyzing-raman-spectra]
- 51. Artifacts and Anomalies in Raman Spectroscopy: A Review on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11478279/]
- 52. X-ray Powder Diffraction (XRD) [https://serc.carleton.edu/msu_nanotech/methods/XRD.html]
- 53. XRD Explained: Principles, Analysis & Applications [https://www.technologynetworks.com/analysis/articles/x-ray-diffraction-xrd-xrd-principle-xrd-analysis-and-applications-404260]
- 54. X-Ray Diffraction Basics | Chemical Instrumentation Facility [https://www.cif.iastate.edu/services/acide/xrd-tutorial/xrd]
- 55. Raman Techniques: Fundamentals and Frontiers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6626094/]
- 56. Raman Spectroscopy - an overview [https://www.sciencedirect.com/topics/materials-science/raman-spectroscopy]
- 57. Analytical X-ray techniques for chemical and structural ... [https://bulletin.ceramics.org/article/analytical-x-ray-techniques-for-chemical-and-structural-characterization-of-ceramics/]
- 58. Preparation and characterization of multiphase ceramic ... [https://www.nature.com/articles/s41598-021-84014-1]
- 59. Ceramics, Volume 8, Issue 4 (December 2025) – 14 articles [https://www.mdpi.com/2571-6131/8/4]
- 60. Comparison of analytical methods in materials science [https://www.cleancontrolling.com/en/news/newsdetails/comparison-of-analytical-methods-in-materials-science]
- 61. Mechanically induced reaction for solid-state synthesis of ... [https://www.sciencedirect.com/science/article/abs/pii/S0966979505002116]
- 62. Optimising the synthesis of LiNiO 2 : coprecipitation versus solid-state, and the effect of molybdenum doping - Energy Advances (RSC Publishing) DOI:10.1039/D3YA00046J [https://pubs.rsc.org/en/content/articlehtml/2023/ya/d3ya00046j]
- 63. Optimising the synthesis of LiNiO2: coprecipitation versus solid-state, and the effect of molybdenum doping - Energy Advances (RSC Publishing) [https://pubs.rsc.org/en/content/articlelanding/2023/ya/d3ya00046j]
- 64. Benchmarking the reproducibility of all-solid-state battery ... [https://www.nature.com/articles/s41560-024-01634-3]
- 65. Troubleshooting Common Pharmaceutical Manufacturing ... [https://globalpharmacenter.com/manufacturing/troubleshooting-common-pharmaceutical-manufacturing-challenges/]
- 66. POWDER CHARACTERISATION FOR PHARMACEUTICALS [https://www.ondrugdelivery.com/powder-characterisation-for-pharmaceuticals-beyond-standards/]
- 67. Effect of synthesis parameters on phase composition, purity ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838824047583]