Beyond Nucleation: Controlling Ion Migration in Advanced Materials Through Grain Boundary Engineering and Additive Design
This article synthesizes the latest research on the intricate relationship between nucleation processes, grain boundary formation, and ion migration in advanced materials like halide perovskites and solid-state electrolytes.
Beyond Nucleation: Controlling Ion Migration in Advanced Materials Through Grain Boundary Engineering and Additive Design
Abstract
This article synthesizes the latest research on the intricate relationship between nucleation processes, grain boundary formation, and ion migration in advanced materials like halide perovskites and solid-state electrolytes. It explores the paradigm shift from traditional nucleation-centric models to mechanisms where additives primarily govern post-nucleation ion mobility. For researchers and scientists, we provide a foundational understanding of ion migration mechanisms, detail methodological advances in nucleation control and interface engineering, address critical troubleshooting and optimization strategies for device stability, and present validation techniques and comparative analyses of material systems. This comprehensive overview aims to equip professionals with the knowledge to design next-generation, stable electronic and energy storage devices by strategically managing ion migration from the bottom up.
The Fundamentals of Ion Migration and Its Link to Material Nucleation
Ion migration refers to the movement of ions within a solid material or across interfaces when subjected to driving forces such as electric fields, concentration gradients, or mechanical stress. This phenomenon is particularly prevalent in materials with mixed ionic-electronic conductivity, including metal halide perovskites, solid-state electrolytes, and various electrochemical systems. While ion migration can be exploited for beneficial applications like memristors and artificial synapses, it more commonly presents significant challenges by causing device degradation, operational instability, and performance hysteresis in electronic and energy devices [1].
Understanding ion migration is fundamental to advancing nucleation control research, as the initial formation and growth of crystals directly influence defect populations and ion migration pathways. By managing nucleation processes, researchers can create microstructures that inherently resist detrimental ion movement, thereby enhancing device longevity and reliability [2].
Fundamental Mechanisms of Ion Migration
Primary Driving Forces
Ion migration occurs through several distinct mechanisms, often acting in concert:
- Electric Field-Driven Drift: Charged ions experience a force in the presence of an electric field, causing them to move toward oppositely charged electrodes. This is a primary driver in operational electronic devices under bias [3] [1].
- Concentration Gradient Diffusion: Ions naturally move from regions of high concentration to low concentration, even without an applied field, leading to gradual compositional changes over time [3].
- Defect-Assisted Hopping: In crystalline materials, ions typically move by hopping between vacancy sites or through interstitial positions. This process requires overcoming an activation energy barrier and is thermally activated [4] [5].
- Mechanically-Induced Transport: Mechanical stresses and membrane deformations can activate mechanosensitive ion channels in biological systems and soft materials, converting physical force into ionic signals [6].
Key Mobile Species in Different Systems
Table 1: Mobile Ionic Species in Various Material Systems
| Material System | Primary Mobile Ions | Impact on Device Performance |
|---|---|---|
| Halide Perovskites (e.g., MAPbI₃, FAPbI₃) | Iodide vacancies (Iâ»), Cations (MAâº, FAâº) | Hysteresis in J-V curves, Phase segregation, Enhanced non-radiative recombination [1] [7] |
| Solid-State Electrolytes (e.g., Li₆PS₅Cl) | Li⺠ions | Determines ionic conductivity, Influences interfacial stability with electrodes [4] |
| Multivalent-Ion Battery Electrodes | Zn²âº, Mg²âº, Al³âº, Ca²⺠| Sluggish diffusion kinetics, High diffusion barriers, Structural degradation [5] |
| Biological Cell Membranes | Ca²⺠(via MS ion channels) | Regulation of cell migration, Mechanotransduction, Signaling pathways [6] |
Experimental Characterization and Quantification
Electrical Testing Methods
Current-Voltage (J-V) Hysteresis Analysis Hysteresis in current-voltage measurements is a primary indicator of ion migration in perovskite and other electronic devices. The hysteresis index (HI) can quantitatively characterize ion migration effects, though it must be interpreted carefully with appropriate scan rates [8].
Experimental Protocol:
- Use a solar simulator with appropriate light intensity calibration
- Perform J-V scans at multiple rates (e.g., 0.1 V/s to 1.0 V/s)
- Calculate HI using the formula: HI = (PCEreverse - PCEforward) / PCE_reverse
- Correlate HI values with ion migration intensity and device stability [8]
Impedance Spectroscopy Electrochemical impedance spectroscopy (EIS) can distinguish between electronic and ionic transport processes through analysis of characteristic time constants and equivalent circuit modeling. Features such as low-frequency arcs and negative capacitance often indicate ionic movement [1].
Direct Compositional Analysis
Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS) TOF-SIMS provides depth profiling of elemental distributions with high sensitivity, enabling direct tracking of ion migration across layers and interfaces [3].
Experimental Protocol:
- Prepare device cross-sections or layer-by-step etching
- Use primary ion beam (e.g., Cs⺠or Bi³âº) for sputtering
- Detect secondary ions with mass spectrometry
- Create 3D reconstruction of elemental distributions, particularly tracking iodide penetration into charge transport layers [3]
X-ray Photoelectron Spectroscopy (XPS) Under Bias XPS can monitor chemical composition changes at interfaces under electrical bias, providing direct evidence of ion accumulation and chemical state alterations [3].
Advanced Simulation Techniques
Classical Molecular Dynamics (CMD) with Constant Potential Method CMD simulations under constant potential can reveal atomistic mechanisms of ion migration in solid electrolytes and electrode materials under realistic operating conditions [4].
Simulation Protocol:
- Construct atomistic model of the material system with appropriate force fields
- Apply constant potential boundary conditions using methods like the Constant Potential Method (CPM)
- Simulate at relevant temperatures (300K-800K) for sufficient time scales (nanoseconds to microseconds)
- Calculate diffusion coefficients from mean square displacement and ionic conductivity from the Nernst-Einstein relation [4]
Multi-Physics Coupled Simulations Comprehensive models integrating carrier transport, ion migration, thermodynamics, and mechanical stress provide holistic understanding of device behavior, particularly at grain boundaries and interfaces [9].
Troubleshooting Common Ion Migration Issues
Problem 1: Efficiency Hysteresis in Perovskite Solar Cells
Symptoms: Power conversion efficiency (PCE) varies significantly with voltage scan direction; unstable power output under maximum power point tracking.
Root Causes:
- Migration of halide vacancies (particularly iodide) under electric field
- Ion accumulation at interfaces with charge transport layers
- Non-radiative recombination at ion-induced defects
Solutions:
- Implement interface blocking layers (e.g., atomic-layer-deposited HfOâ‚‚) to create scattering barriers [3]
- Incorporate large cations (guanidinium, phenylethylammonium) to reduce vacancy concentration and increase migration activation energy [1]
- Optimize perovskite composition to reduce intrinsic defect density (e.g., mixed cation formulations) [1]
Problem 2: Rapid Performance Degradation Under Operation
Symptoms: Significant efficiency drop within initial operational hours; darkening of device areas; electrode corrosion.
Root Causes:
- Iodide ions migrating into charge transport layers and electrodes
- Electrochemical reactions at interfaces triggered by migrating ions
- Phase segregation and compositional inhomogeneity
Solutions:
- Quantify required barrier energy (typically 0.6-0.9 eV for iodide confinement) and design composite blocking layers to meet this threshold [3]
- Utilize dipole monolayers to create drift electric fields that counteract ion diffusion
- Implement defect passivation strategies at grain boundaries and interfaces [9] [7]
Problem 3: Inconsistent Device-to-Device Performance
Symptoms: Large variations in performance metrics across batches; poor reproducibility despite similar processing conditions.
Root Causes:
- Variations in nucleation and crystal growth affecting defect density and distribution
- Inhomogeneous grain boundary properties
- Fluctuations in interfacial composition
Solutions:
- Implement controlled nucleation via substrate temperature management, antisolvent treatment, and solvent engineering [2]
- Standardize crystallization protocols to ensure consistent grain size and boundary characteristics
- Utilize in-situ monitoring during film formation to detect and correct process deviations [2]
Quantitative Data and Performance Metrics
Table 2: Ion Migration Parameters and Mitigation Effectiveness in Various Systems
| Material/Strategy | Ion Diffusion Coefficient (cm²/s) | Activation Energy (eV) | Stability Improvement | Key Limitation |
|---|---|---|---|---|
| MAPbI₃ (3D Perovskite) | ~10â»â¸ | 0.20-0.30 | T80: 5-12 hours | High ion mobility leading to rapid degradation [1] |
| FA-based Mixed Perovskites | 10â»â¸-10â»Â¹Â¹ | 0.30-0.50 | T80: 10-100 hours | Phase instability issues [1] |
| 2D/Quasi-2D Perovskites | 10â»Â¹Â²-10â»Â¹âµ | 0.50-0.80 | T80: >750 hours | Reduced charge carrier mobility [1] |
| HfOâ‚‚ Scattering Barrier | N/A | N/A | 30-50% reduction in iodide migration | Insufficient as standalone solution [3] |
| Composite HfO₂+Dipole Layer | N/A | >0.6 (barrier) | >95% efficiency retention after 1500h at 85°C | Complex fabrication process [3] |
| Li₆PSâ‚…Cl Solid Electrolyte | ~10â»â¸ (Liâº) | 0.20-0.32 | High cyclic stability in solid-state batteries | Interface resistance challenges [4] |
The Researcher's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagents for Ion Migration Studies
| Reagent/Material | Function | Application Context |
|---|---|---|
| HfOâ‚‚ (Hafnium Oxide) | Atomic-layer-deposited scattering barrier | Blocks iodide migration in perovskite solar cells [3] |
| CF₃-PBAPy Molecule | Self-assembled dipole monolayer | Creates drift electric field to suppress ion migration [3] |
| Poly(N-vinylcarbazole) (PVK) | High work function hole transport material | Addresses band shift from interfacial electric fields [3] |
| Guanidinium (GAâº) | Large cation additive | Reduces dimensionality and ion migration in perovskites [1] |
| Phenylethylammonium (PEAâº) | Spacer cation for 2D perovskites | Suppresses ion migration through structural confinement [1] |
| Li₆PS₅Cl (LPSC) | Solid electrolyte material | Study of Li⺠migration mechanisms in solid-state batteries [4] |
| Sophorose | 2-O-beta-D-Glucopyranosyl-D-glucose|High-Purity | This high-purity 2-O-beta-D-Glucopyranosyl-D-glucose (Kojibiose) is For Research Use Only (RUO). Not for human, veterinary, or household use. |
| QS 7 | QS 7, MF:C83H130O46, MW:1863.9 g/mol | Chemical Reagent |
Mechanisms and Workflows Visualization
Ion Migration Mechanism in Perovskite Solar Cells
Defect Engineering for Ion Migration Control
Frequently Asked Questions
Q1: Why does ion migration cause efficiency hysteresis in perovskite solar cells? Ion migration creates a dynamic redistribution of charges within the device under operation. As ions move and accumulate at interfaces, they modify the local electric field and band alignment, leading to different current-voltage characteristics depending on the scan direction and history. This manifests as hysteresis in J-V measurements and unstable power output [8] [1].
Q2: What is the relationship between nucleation control and ion migration? Nucleation control directly influences the crystal quality, grain size, and defect density in polycrystalline materials. By optimizing nucleation through techniques like substrate temperature control, antisolvent treatment, and additive engineering, researchers can create films with larger grains, fewer grain boundaries, and lower defect concentrations. This reduces the pathways and driving forces for ion migration, significantly enhancing device stability [2] [9].
Q3: How can I quantitatively measure ion migration in my devices? Several complementary techniques are available:
- J-V Hysteresis Analysis: Calculate hysteresis index at different scan rates [8]
- Impedance Spectroscopy: Identify low-frequency features associated with ionic transport [1]
- TOF-SIMS: Directly profile elemental distribution changes after operation [3]
- XPS Under Bias: Monitor chemical state changes at interfaces in operando [3]
- CMD Simulations: Compute diffusion coefficients and activation energies from atomistic models [4]
Q4: What barrier energy is needed to effectively suppress iodide migration? Research indicates that a barrier energy of approximately 0.6-0.9 eV is required to effectively confine iodide ions within the perovskite layer and prevent their migration into charge transport layers. This threshold was determined by measuring the potential drop needed to establish dynamic equilibrium between diffusion and drift motions at the perovskite/HTL interface [3].
Q5: Can ion migration ever be beneficial for device functionality? Yes, in certain applications, ion migration is exploited for novel functionalities. In memristors, the dynamics of ion migration are used to tune resistance states for information storage and neuromorphic computing. Ion migration also enables adaptive interfaces and self-healing properties in some systems. The key is controlling rather than completely eliminating ion movement for specific applications [1].
Ion migration represents a critical challenge in advancing modern electronic and energy devices, particularly in perovskite photovoltaics, solid-state batteries, and related technologies. Through comprehensive characterization techniques, multi-physics modeling, and targeted mitigation strategies—especially those rooted in nucleation control—researchers can significantly suppress detrimental ion movement while maintaining high device performance. The quantitative frameworks and troubleshooting guidelines presented here provide a foundation for developing more stable and reliable devices across multiple technology platforms.
Frequently Asked Questions (FAQs)
Q1: The nucleation-centric theory has long guided our additive selection. Why is it failing to predict grain morphology in our titanium alloy AM processes? The nucleation-centric theory often relies solely on the growth restriction factor (Q) to predict equiaxed microstructures. However, recent research demonstrates that this mechanism is less reliable than previously thought. In studies involving Ti alloys, compositions with high Q values (e.g., Ti-12Mo with Q=54 K) still resulted in coarse columnar grains, contradicting the theory. The evidence suggests that a mechanism based on increasing the alloy's freezing range (ΔT) provides a more accurate prediction. A sufficiently large ΔT, when coupled with the rapid cooling of AM, causes significant undercooling, which drives a high nucleation rate and leads to the formation of equiaxed grains [10].
Q2: In our lab, we are working on Sn-based perovskite solar cells. How do additives influence grain morphology and ion migration in these materials? In Sn-based perovskites, additives play a crucial role in managing crystallization and suppressing ion migration, which is a major degradation factor. The extremely fast crystallization of Sn-perovskites leads to defective films. Additives like SnF2 are used to control crystallization kinetics, leading to larger, more uniform grains with fewer defects. This improved grain morphology reduces the pathways and vacancies that facilitate the migration of Sn²⺠ions and halide vacancies, thereby enhancing both device performance and operational stability [11].
Q3: Our team is struggling with inconsistent grain structures in Laser Directed Energy Deposition (L-DED). What process control strategy can we implement to improve homogeneity? Inconsistent thermal conditions during printing are a primary cause of variable grain structure. Implementing real-time process control that uses a thermal signature (e.g., melt pool area or thermal intensity) as a feedback signal can dramatically improve homogeneity. Research shows that using a coaxial camera to monitor thermal intensity and dynamically adjusting the laser power or velocity to maintain a constant target value can reduce the standard error of the thermal signature by up to 45%. This controlled thermal environment leads to a more uniform solidification front, which in turn reduces grain area variability by up to 94% [12].
Q4: What are the best practices for accurate and reproducible grain size analysis in our quality control lab? Modern digital image analysis is key to overcoming the inaccuracies of manual chart comparison methods. For reliable results, you should:
- Use a 10X metallurgical objective lens on an inverted microscope.
- Employ image analysis software (e.g., MIPAR, ZEISS ZEN core) that complies with standards like ASTM E112.
- Utilize automated methods like the intercept or planimetric method, which remove operator subjectivity.
- Ensure your digital camera has sufficient resolution (typically ≥3 megapixels) to meet the Nyquist sampling criteria for the features you are analyzing [13] [14]. This approach ensures accurate, repeatable, and well-documented results that are reproducible across different operators [13] [15] [14].
Key Experimental Data & Protocols
Quantitative Comparison of Additive Mechanisms in Titanium Alloys
The following table summarizes critical experimental data from additive manufacturing of binary Ti alloys, comparing the predictive power of the nucleation-centric theory (Growth Restriction Factor, Q) versus the freezing range (ΔT) mechanism [10].
Table 1: Predictive Performance of Different Mechanisms for Grain Morphology in Ti Alloys
| Alloy Composition | Growth Restriction Factor, Q (K) | Freezing Range, ΔT (K) | Predicted Morphology (via Q) | Predicted Morphology (via ΔT) | Actual Observed Morphology | Average Grain Aspect Ratio |
|---|---|---|---|---|---|---|
| Ti-20V | ~0 | ~10 | Columnar | Columnar | Coarse Columnar | 3.1 |
| Ti-12Mo | 54 | ~110 | Equiaxed | Columnar | Coarse Columnar | 4.4 |
| Ti-2.5Cu | ~18 | ~672 | Columnar | Equiaxed | Refined Equiaxed | 1.6 |
| Ti-6.8Cu | ~52 | ~623 | Columnar | Equiaxed | Refined Equiaxed | 1.8 |
| Ti-8.5Cu | 62 | ~603 | Equiaxed | Equiaxed | Equiaxed | N/A |
Quantifying Ion Migration in Perovskite Materials
For research on ion migration, key parameters can be quantified to compare different material compositions. The following table contrasts metal halide perovskites (MHPs) with other technologies [16].
Table 2: Quantitative Metrics for Ion Migration in Different Material Systems
| Material / Device | Mobile Ion Concentration, No (cmâ»Â³) | Ionic Mobility, μ (cm²/Vs) | Electronic Mobility (cm²/Vs) |
|---|---|---|---|
| Solid State Electrolyte (LLZO) | 5 x 10¹⸠to 5 x 10²Ⱐ| 10â»Â¹â° to 10â»Â¹â´ | 0.06 |
| MAPbI₃ Perovskite | 2 x 10¹ⷠ| 8 x 10â»â¶ | 20 to 71 |
| Triple-Halide Perovskite | 5 x 10¹ⵠ| 3 x 10â»â´ | 11 to 40 |
| Silicon Solar Cell | 0 | 0 | ~160 |
Experimental Protocol: Determining the Dominant Mechanism in AM
This protocol is designed to test whether grain refinement is governed by the growth restriction factor (Q) or the freezing range (ΔT) [10].
Step 1: Alloy Selection & Calculation
- Select a series of binary alloys (e.g., Ti-Cu, Ti-V, Ti-Mo).
- Use computational thermodynamics software (e.g., Pandat, Thermocalc) with accurate databases to calculate the values of Q and ΔT for each alloy composition.
Step 2: Sample Fabrication
- Process the selected alloy powders using a fusion-based AM technique (e.g., Directed Energy Deposition - DED or Laser Powder Bed Fusion).
- Maintain consistent processing parameters (e.g., laser power, scan speed, hatch spacing) across all compositions to isolate the effect of the alloy chemistry.
Step 3: Microstructural Characterization
- Prepare metallographic samples from the as-built components using standard grinding and polishing techniques.
- Analyze the grain structure using Electron Backscatter Diffraction (EBSD).
- Obtain Inverse Pole Figure (IPF) maps to visualize grain morphology and orientation.
Step 4: Quantitative Image Analysis
- Import EBSD maps or optical micrographs into image analysis software (e.g., MIPAR, ZEISS ZEN core).
- Use the software to determine the average grain aspect ratio. An aspect ratio >3.0 typically indicates a columnar morphology, while a ratio closer to 1.0 indicates an equiaxed morphology [10].
- Analyze the grain size distribution and texture strength.
Step 5: Data Correlation and Mechanism Validation
- Correlate the measured grain morphologies with the calculated Q and ΔT values.
- A mechanism is validated if it consistently predicts the correct morphology (equiaxed vs. columnar) across all tested alloys. Research indicates that a high ΔT (>~110 K) is a more reliable predictor than a high Q value [10].
Experimental Protocol: Process Control for Grain Homogeneity in L-DED
This protocol outlines the setup for a real-time control system to achieve consistent grain structures [12].
Step 1: System Setup and Calibration
- Integrate a coaxial, high-resolution CMOS or CCD camera into your L-DED system.
- Calibrate the camera and software to ensure accurate measurement. The pixel size should be small enough to resolve the melt pool features (meeting Nyquist criteria).
Step 2: Define Control Variable and Target
- Define the control variable. A pixel-wise sum of the coaxial image (termed "thermal intensity") has been shown to be effective and well-correlated with melt pool area [12].
- Set a target value for this thermal intensity through initial test runs that produce a satisfactory deposit.
Step 3: Implement the Control Loop
- Use in-house or commercial control software (e.g., a Proportional-Integral-Derivative controller) to adjust the laser power or velocity.
- The control loop should run at a high frequency (e.g., corrections every 0.15 seconds). The software adjusts the parameter (power/velocity) to minimize the difference between the measured thermal intensity and the target value.
Step 4: Validation and Analysis
- Build components with and without the control system active.
- Compare the standard error of the thermal intensity signal between controlled and uncontrolled builds.
- Perform microstructural analysis (as in Protocol 2.3) on cross-sections of both samples to quantify the reduction in grain area variability.
Signaling Pathways and Workflows
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Materials for Grain Morphology Research
| Item / Solution | Function / Application |
|---|---|
| Computational Thermodynamics Software (e.g., Pandat, Thermocalc) | Used to calculate critical parameters for predicting grain morphology, including growth restriction factors (Q) and freezing ranges (ΔT), prior to costly experimental work [10]. |
| Image Analysis Software (e.g., MIPAR, ZEISS ZEN core) | Provides automated, accurate, and reproducible grain size and aspect ratio measurements from micrographs, complying with international standards like ASTM E112 and eliminating operator subjectivity [13] [15] [14]. |
| Inverted Metallurgical Microscope | The preferred microscope configuration for examining prepared metallographic samples, allowing flat, polished samples to lay flat on the stage for consistent focus and observation [13]. |
| 10X Metallurgical Objective Lens | The standard magnification required for performing grain size analysis according to ASTM E112 and other international standards [13]. |
| Electron Backscatter Diffraction (EBSD) System | A crucial tool for detailed microstructural characterization, providing data on grain orientation, morphology, and phase distribution, which is used to create Inverse Pole Figure (IPF) maps [10]. |
| SnFâ‚‚ (Tin Fluoride) Additive | A common additive used in Sn-based perovskite research to control crystallization kinetics, suppress the oxidation of Sn²⺠to Snâ´âº, and reduce defect density, thereby improving film morphology and device stability [11]. |
| Coaxial CMOS/CCD Camera & Control Software | Enables real-time monitoring of the melt pool (thermal intensity/area) during additive manufacturing processes like L-DED. This data is used for feedback control of laser power/velocity to achieve consistent thermal conditions and homogeneous grain structures [12]. |
| CCR1 antagonist 9 | CCR1 antagonist 9, MF:C20H16FN5O3S, MW:425.4 g/mol |
| TRPV3 antagonist 74a | TRPV3 antagonist 74a, CAS:1432051-63-2, MF:C17H17F3N2O2, MW:338.32 g/mol |
Frequently Asked Questions
Q1: What are the key parameters for quantifying ion migration in metal halide perovskites (MHPs)? The two most critical parameters for quantifying ion migration are Mobile Ion Concentration (Nₒ) and Ionic Mobility (μ) [16].
- Nâ‚’ is the number of mobile ions present in the material.
- μ measures how easily these ions move under an electric field. Research indicates that Nₒ has a more significant impact on device stability than ionic mobility. Reducing Nₒ is therefore a primary target for enhancing the operational stability of perovskite solar cells (PSCs) [16].
Q2: How does the choice of electrode material affect mobile ion concentration? Experimental evidence suggests that the selection of the top electrode in a device structure has a more substantial impact on the measured mobile ion concentration (Nâ‚’) than other factors, such as the introduction of small alkali metal cation additives (e.g., Na+, K+, Rb+) or exposure to moisture [16]. The wrong electrode material can interact with migrating ions, leading to irreversible degradation.
Q3: What are the practical design principles for blocking ion migration pathways? The primary design principle is to focus on defect passivation to reduce the source of mobile ions (Nₒ) [16]. Furthermore, understanding the impact of operational conditions is crucial, as ionic mobility (μ) increases with temperature due to its low activation energy. Therefore, effective strategies must include both material-level passivation and device-level engineering for stable operation under thermal stress [16].
Q4: What is a common experimental method to measure mobile ion concentration (Nₒ)? A developed method to measure Nₒ involves transient current measurements in the dark [16]. This technique, along with others like electrochemical impedance spectroscopy to measure ionic conductivity, allows researchers to decouple and quantify the individual contributions of Nₒ and μ to overall ion migration.
Q5: How do intrinsic defects lead to material degradation? Intrinsic vacancies in MHPs, particularly halide vacancies, act as mobile ions. These ions can migrate and participate in detrimental chemical reactions. For example, in MAPbI₃, mobile I⻠ions can oxidize to form I₂, which then triggers a series of cyclic reactions that decompose the perovskite material and produce volatile byproducts, leading to irreversible device degradation [16].
Troubleshooting Guides
Problem: Rapid Performance Degradation Under Bias and Light
- Symptoms: Significant drop in power conversion efficiency, increased hysteresis, and non-recoverable performance loss after operation.
- Investigation & Resolution:
- Quantify Ion Migration: Perform transient current measurements in the dark to determine the mobile ion concentration (Nâ‚’) of your film or device [16].
- Compare to Benchmarks: Compare your Nâ‚’ value to known stable compositions. For instance, a Triple Halide composition has been shown to have a lower Nâ‚€ (~5 × 10¹ⵠcmâ»Â³) compared to standard MAPbI₃ (Nâ‚€ ~2 × 10¹ⷠcmâ»Â³) [16].
- Action: If Nâ‚’ is high, implement a more robust defect passivation strategy during film fabrication. This could involve adding specific cation or anion additives that suppress vacancy formation.
Problem: High Ionic Mobility Leading to Instability at Elevated Temperatures
- Symptoms: Device performance is acceptable at room temperature but degrades rapidly when temperature increases (e.g., >85°C).
- Investigation & Resolution:
- Check Activation Energy: Determine the activation energy (Eâ‚) for ion migration in your material. MHP halide vacancies have low Eâ‚ (e.g., 0.58 eV for VIâº), meaning mobility (μ) increases significantly with temperature [16].
- Verify Electrode Stability: Ensure your top electrode is not reacting with migrating ions, which accelerates at higher temperatures [16].
- Action: Explore different A-site cation additives or alloy compositions to increase the activation energy for ion migration. Also, review and change the top electrode material to one that is more inert.
Quantitative Data on Ion Migration
Table 1: Mobile Ion Concentration (Nₒ) and Ionic Mobility (μ) Across Different Systems [16]
| Device / Material | Nâ‚’ (cmâ»Â³) | μ (cm²/Vs) | Electronic Mobility (cm²/Vs) |
|---|---|---|---|
| Solid State Electrolyte (LLZO) | ~5 × 10¹⸠to 5 × 10²Ⱐ| ~10â»Â¹â° to 10â»Â¹â´ | 0.06 |
| MAPbI₃ | ~2 × 10¹ⷠ| ~8 × 10â»â¶ | 20 to 71 |
| Triple Halide | ~5 × 10¹ⵠ| ~3 × 10â»â´ | 11 to 40 |
| Silicon | 0 | 0 | ~160 |
Table 2: Activation Energies (Eâ‚) for Vacancy Formation in MAPbI₃ [16]
| Vacancy Type | Activation Energy (Eâ‚) |
|---|---|
| VI⺠(Iodide Vacancy) | 0.58 eV |
| VMAâ» (Methylammonium Vacancy) | 0.84 eV |
| VPb²⻠(Lead Vacancy) | 2.31 eV |
Experimental Protocols
Protocol 1: Quantifying Mobile Ion Concentration (Nâ‚’) via Transient Current Measurement Objective: To decouple and measure the mobile ion concentration (Nâ‚’) in a metal halide perovskite film or device. Methodology: [16]
- Setup: Place the sample (film or full device) in the dark to eliminate photogenerated carriers.
- Polarization: Apply a constant DC bias voltage to polarize the sample, prompting mobile ions to drift and accumulate at interfaces.
- Transient Measurement: Switch the circuit to short-circuit or reverse bias condition while measuring the transient current over time.
- Analysis: The total extracted charge (Q), calculated by integrating the transient current, is proportional to the number of mobile ions. The mobile ion concentration Nâ‚’ can be derived using the equation involving Q, the sample volume, and the elementary charge.
Protocol 2: Investigating Defect Passivation Efficacy Objective: To evaluate the effectiveness of different passivation agents (e.g., alkali metal cations) in reducing ion migration. Methodology: [16]
- Sample Preparation: Fabricate a series of MHP films or devices with identical base compositions but incorporating different passivating additives (e.g., control, Naâº, Kâº, Rbâº).
- Characterization: Subject all samples to Protocol 1 (Transient Current Measurement) to determine and compare their Nâ‚’ values.
- Stability Testing: Perform accelerated aging tests on the devices under light, heat, and bias.
- Analysis: Correlate the reduction in initial Nâ‚’ with the improvement in long-term operational stability to rank the efficacy of the passivation agents.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Ion Migration Research
| Item | Function / Relevance |
|---|---|
| Alkali Metal Salts (e.g., NaI, KBr, RbCl) | Used as A-site cation additives to modify the perovskite crystal lattice, passivate defects, and influence vacancy formation energies [16]. |
| Lead Precursors (e.g., PbIâ‚‚, PbBrâ‚‚) | Source of the B-site cation (Pb²âº) and halides. Stoichiometry and purity are critical for controlling intrinsic defect (vacancy) concentrations [16]. |
| Organic Salts (e.g., MAI, FAI) | Source of the A-site organic cation (e.g., Methylammonium, Formamidinium). Their decomposition products can be involved in ion migration pathways [16]. |
| Metal Electrode Materials (e.g., Au, Ag, Al) | Used as top contacts. The choice of metal is critical as it can react irreversibly with migrating halide ions, leading to electrode corrosion and device failure [16]. |
| N-Acetylcytisine | N-Acetylcytisine, CAS:6018-52-6, MF:C13H16N2O2, MW:232.28 g/mol |
| Cytochalasin F | Cytochalasin F, CAS:36084-18-1, MF:C29H37NO5, MW:479.6 g/mol |
Signaling Pathways & Experimental Workflows
Ion Migration Degradation Pathways
Nucleation Control Research Workflow
FAQs: Analytical Techniques for Precursor Inks
1. How can NMR spectroscopy characterize precursor inks and what specific information does it provide? Nuclear Magnetic Resonance (NMR) spectroscopy is used to determine the chemical structure and composition of precursor inks. It exploits the magnetic properties of atomic nuclei with non-zero spin quantum numbers (like ( ^1H ) and ( ^{13}C )). When placed in a magnetic field, these nuclei align with or against the field, creating energy levels. Radiofrequency radiation induces transitions between these levels, generating a signal [17]. For polymers and complex formulations, NMR can quantify monomer ratios in copolymers, measure the degree of branching and crosslinking, investigate polymer dynamics, determine tacticity, and analyze end-groups in polymerization reactions [17]. It provides detailed structural information and quantitative analysis, making it indispensable for understanding ink composition.
2. What role does UV-Vis spectroscopy play in analyzing the electronic properties of precursor inks? Ultraviolet-Visible (UV-Vis) spectroscopy measures the absorption of UV and visible light by a sample, which corresponds to electronic transitions between molecular orbitals [17]. This technique is crucial for identifying chromophores and conjugated systems within the ink, such as aromatic rings or unsaturated bonds (e.g., C=C, C=O) [17]. It helps analyze the wavelength and intensity of absorption bands, determine the band gap of conjugated polymers, quantify the concentration of chromophores and dyes, and monitor polymer degradation under UV exposure [17]. This is particularly valuable for assessing the optical properties and electronic structure of inks intended for optoelectronic applications.
3. Why is electrical conductance measurement critical in the development of conductive inks? Electrical conductance measurement directly assesses the effectiveness of an ink formulation in forming conductive pathways, such as copper interconnections in printed electronics [18]. The transition from a non-conductive precursor ink (e.g., copper formate) to a conductive metal layer (e.g., copper metal) is confirmed through these measurements [18]. Furthermore, optimizing process parameters, like intense pulsed light (IPL) curing, relies on conductance data to find the correct energy settings that yield low-resistance patterns without damaging the substrate [18].
4. How are these analytical techniques complementary in the study of precursor inks? NMR, UV-Vis, and electrical conductance provide a comprehensive view of ink properties. NMR offers detailed structural and quantitative data, UV-Vis probes electronic transitions and optical characteristics, and conductance gives a direct functional performance metric [17]. Combining these methods overcomes the limitations of any single technique, allowing researchers to correlate the ink's chemical makeup (from NMR) with its light-absorption behavior (from UV-Vis) and its final conductive performance, leading to more robust and reliable ink development [17].
Troubleshooting Guides
Issue 1: Inconsistent Electrical Conductance in Cured Conductive Inks
Problem: Cured ink patterns show high or inconsistent electrical resistance. Solution:
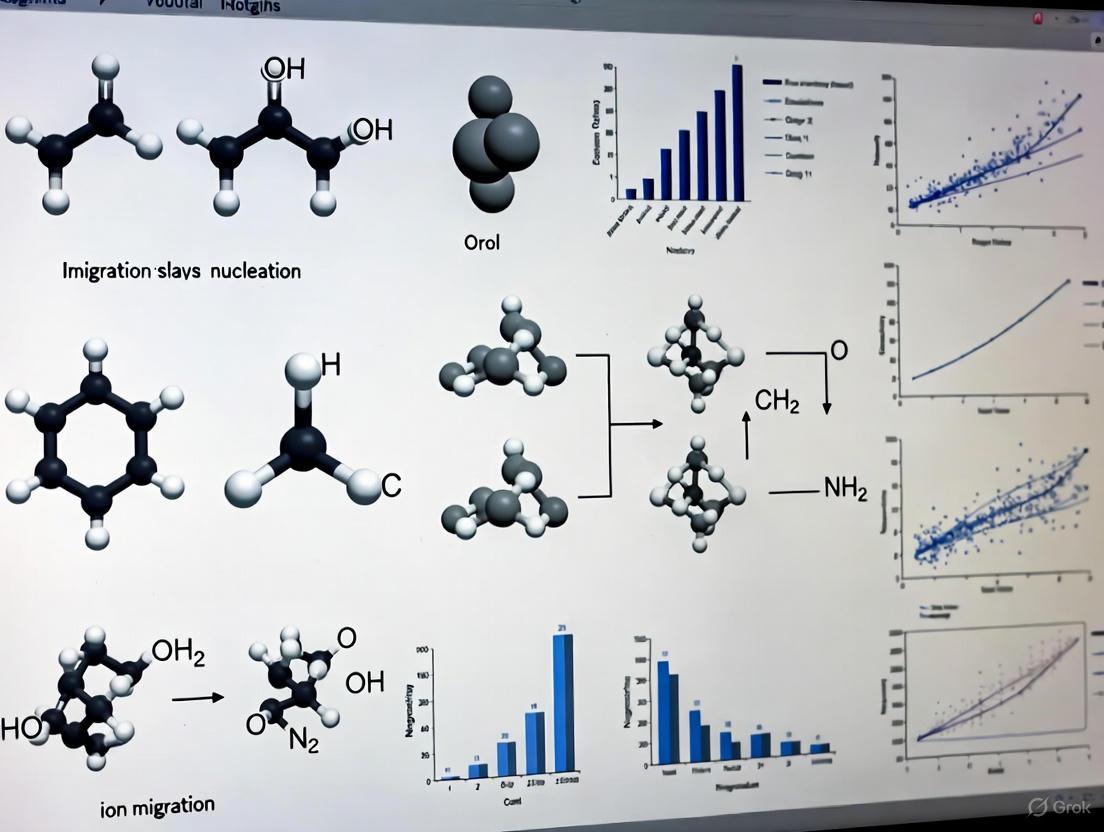
- Verify Curing Parameters: For photonic curing processes like Intense Pulsed Light (IPL), ensure a strict set of pulse parameters (duration, intensity, and number of repetitions) is used. Inconsistent conductance often arises from stochastic nucleation and uneven energy delivery during curing [18] [19].
- Optimize Light Absorption: Enhance the ink's absorptance by incorporating efficient light absorbers like carbon nanotubes (CNTs). Adding as little as 0.5 wt % single-wall CNTs can increase absorptance by about 50% and decrease the threshold energy required for obtaining a conductive pattern by approximately 25% [18].
- Check Layer Thickness: Ensure the printed layer is uniform. Partial decomposition can occur if the pulse energy is sufficient to decompose only the top layer but cannot penetrate to the lower part of the film, resulting in a non-conductive bottom layer [18].
Issue 2: Poor Nucleation and Crystal Growth in Perovskite Inks
Problem: Formation of perovskite films with poor morphology, such as small grains, pinholes, or incomplete coverage, which adversely affects device performance and increases ion migration [20]. Solution:
- Control Supersaturation: The supersaturation stage, prerequisite for nucleation, can be induced faster by modifying the chemical potential of the system. Techniques include:
- Substrate Temperature Treatment: Pre-conditioning the coating substrate with thermal energy facilitates faster nucleation, leading to more uniform thin films [20].
- Antisolvent Treatment: During spin-coating, applying an antisolvent promotes rapid supersaturation, leading to a denser and more uniform nucleation layer [20].
- Solvent Engineering: Modifying the precursor solvent composition can control the rate of solvent extraction and the formation of intermediate perovskite precursor solvates, guiding subsequent crystal growth [20].
- Target Heterogeneous Nucleation: Aim for nucleation to occur directly on the substrate rather than spontaneously within the solution (homogeneous nucleation). Nucleation on the substrate is the ideal approach for achieving high-quality, compact films with fewer defects [20].
Issue 3: Interpreting Complex Spectroscopic Data
Problem: Difficulty in assigning signals in NMR or IR spectra to specific structural features in complex ink formulations. Solution:
- Use Combined Theoretical and Experimental Methods: Employ Density Functional Theory (DFT) calculations to predict vibrational wavenumbers, NMR chemical shifts, and UV-Vis absorption wavelengths. Compare these computed values directly with experimental data (FT-IR, FT-Raman, NMR, UV-Vis) for validation and accurate assignment [21].
- Consult Reference Databases: Compare obtained IR spectra with reference databases to identify functional groups and structural features. Characteristic absorption bands can signal specific groups, such as carbonyl (C=O) stretches in polyesters or C-H stretches in polyethylene [17].
- Analyze HOMO-LUMO Energies: For UV-Vis, theoretical calculations can provide HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) energies, helping to interpret electronic transitions and the nature of chromophores [21].
Experimental Protocols & Data Presentation
Protocol 1: Photonic Curing of Copper Precursor Inks with IPL
This protocol is adapted from methods used to decompose copper formate-based inks into conductive copper patterns [18].
Objective: To obtain conductive copper patterns from a self-reducing copper formate particle ink using Intense Pulsed Light (IPL).
Materials & Reagents:
Procedure:
- Ink Preparation: Disperse submicron copper formate particles in a suitable solvent. For enhanced light absorption, prepare a hybrid ink by adding 0.5 wt % SWCNTs to the copper formate ink and mix thoroughly [18].
- Printing: Deposit the ink onto the plastic substrate via screen printing to form the desired pattern. Allow the printed layer to dry [18].
- IPL Curing Setup: Place the printed substrate in the IPL system. The process is conducted in an air environment [18].
- Parameter Screening: Systematically vary the IPL parameters:
- Pulse duration: Test from 2 to 10 ms in intervals.
- Pulse intensity: Controlled by the applied voltage to the xenon lamp (e.g., from 180 to 450 V).
- Number of pulses: Apply 1 to 4 pulses at a frequency of 1 Hz [18].
- Curing: Execute the pulse sequence. Successful decomposition is indicated by a visual color change from blue (copper formate) to a shiny reddish color (copper metal) [18].
- Analysis: Measure the electrical resistance of the resulting patterns. Characterize morphology using techniques like scanning electron microscopy (SEM) [18].
Expected Outcomes: A specific combination of pulse parameters will yield conductive patterns. The addition of CNTs should lower the energy threshold required for achieving conductivity.
Protocol 2: Spectroscopic Characterization of a Model Compound
This protocol outlines a general approach for the multi-technique characterization of a chemical compound, as demonstrated in studies of molecules like 3-fluorophenylboronic acid [21].
Objective: To comprehensively characterize a compound's structure using FT-IR, NMR, and UV-Vis spectroscopy, supported by quantum chemical calculations.
Materials & Reagents:
- Sample of the compound to be analyzed.
- Deuterated solvents for NMR (e.g., DMSO-d6).
- Spectroscopic-grade solvents for UV-Vis (e.g., ethanol, water).
Procedure:
- FT-IR Spectroscopy:
- Record the infrared spectrum in the region of 4000-400 cmâ»Â¹ [21].
- Analyze the absorption bands to identify functional groups and types of bonds.
- NMR Spectroscopy:
- Dissolve the sample in a deuterated solvent.
- Record ( ^1H ) and ( ^{13}C ) NMR spectra [21].
- Analyze chemical shifts, integration, and coupling constants to determine structure.
- UV-Vis Spectroscopy:
- Prepare solutions of the compound in different solvents (e.g., ethanol and water).
- Record the absorption spectrum in the range of 200-400 nm [21].
- Identify absorption maxima (λ_max) corresponding to electronic transitions.
- Theoretical Calculations:
- Perform quantum chemical calculations (e.g., DFT at the B3LYP/6-311++G(d,p) level) to optimize the compound's geometry [21].
- Calculate the theoretical vibrational wavenumbers, NMR chemical shifts, and UV-Vis absorption wavelengths.
- Compare theoretical and experimental results to validate the findings.
- FT-IR Spectroscopy:
Expected Outcomes: Successful correlation between experimental spectroscopic data and theoretical predictions, leading to a confirmed molecular structure and understanding of its electronic properties.
Quantitative Data from Search Results
Table 1: Effect of CNT Additives on IPL Curing of Copper Formate Ink [18]
| Parameter | Without CNTs | With 0.5 wt % SWCNTs | Change |
|---|---|---|---|
| Light Absorptance | Baseline | Increased by ~50% | +50% |
| Threshold Energy for Conduction | Baseline | Decreased by ~25% | -25% |
Table 2: Key Spectroscopic Techniques for Ink Analysis
| Technique | Principle | Key Information Obtained | Example Applications in Ink Analysis |
|---|---|---|---|
| NMR | Magnetic nuclei in RF field [17] | Chemical structure, composition, dynamics, tacticity [17] | Quantify monomer ratios, analyze end-groups [17] |
| UV-Vis | Electronic transitions [17] | Chromophores, band gap, degradation, optical properties [17] | Study conjugated polymers, monitor UV stability [17] |
| FT-IR | Molecular vibrations [21] [17] | Functional groups, chemical bonds, reaction monitoring [17] | Identify carbonyl groups, monitor polymerization [17] |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Precursor Ink Development and Analysis
| Item | Function / Application |
|---|---|
| Copper Formate | A self-reducing precursor for conductive copper inks; decomposes to copper metal, COâ‚‚, and Hâ‚‚ upon energy application [18]. |
| Carbon Nanotubes (CNTs) | Added to inks to enhance light absorption during photonic curing, improving energy efficiency and reducing the threshold for obtaining conductive patterns [18]. |
| Antisolvents | Used in perovskite and crystal growth processes to rapidly induce supersaturation during spin-coating, leading to controlled nucleation and dense film formation [20]. |
| Deuterated Solvents (e.g., DMSO-d6) | Required for NMR spectroscopy to provide a signal-free environment for analyzing the structure of compounds in solution [21]. |
| Intense Pulsed Light (IPL) System | A photonic curing tool that uses short, powerful light pulses to sinter metal inks or decompose precursors on heat-sensitive substrates rapidly [18]. |
| Neomenthoglycol | p-Menthane-3,8-diol (PMD) |
| Quininib | Quininib, CAS:4838-66-8, MF:C17H13NO, MW:247.29 g/mol |
Workflow Visualization
Troubleshooting Workflow for Precursor Ink Development
IPL Curing Mechanism for Conductive Inks
Strategic Control of Crystallization and Interface Engineering to Suppress Ion Migration
FAQs: Understanding Additive Mechanisms
Q1: I thought additives primarily work by slowing down nucleation to grow larger crystals. Is this correct? Recent interdisciplinary studies challenge this established view. Evidence now indicates that many popular Lewis-base additives do not predominantly impact the nucleation phase. Instead, they facilitate coarsening grain growth by boosting ion mobility across grain boundaries during the annealing step, after solvent removal and initial crystallization have already occurred [22].
Q2: What is the direct link between increased ion mobility at grain boundaries and final solar cell performance? Enhanced ion mobility allows for grain coarsening, which leads to:
- Larger perovskite grains, reducing the density of grain boundaries in the final layer [22].
- A reduction of deep trap states and non-radiative recombination at these boundaries [22].
- This directly translates to higher open-circuit voltage (Voc) and overall improved power conversion efficiency (PCE) [23].
Q3: Can the effect of additives be linked to other post-processing techniques? Yes. The mechanism of additive-mediated grain growth is directly applicable to post-processing methods like thermal hot-pressing. In both cases, the underlying principle is the same: increasing the mobility of the ions within the perovskite structure to enable grain boundary movement and grain coarsening [22].
Troubleshooting Guides
Issue: Inconsistent Grain Size Despite Additive Use
| Possible Cause | Diagnostic Check | Solution |
|---|---|---|
| Additive concentration is sub- or supra-optimal | Perform a concentration series (e.g., 0.5, 1.0, 1.5 mol%) and analyze films with SEM. | Systematically optimize the additive amount to find the concentration that maximizes grain size without forming secondary phases. |
| Annealing conditions are inadequate | Ensure the annealing temperature and time are sufficient to activate ion migration. | Extend annealing time or moderately increase temperature to provide the thermal energy needed for additive-mediated coarsening. |
| Additive is degrading or reacting prematurely | Check if the additive is stable at the processing temperatures used. | Consider additives with higher thermal stability or adjust the thermal profile to avoid decomposition. |
Issue: Poor Device Stability or Hysteresis
| Possible Cause | Diagnostic Check | Solution |
|---|---|---|
| Unpassivated grain boundaries | Fabricate a thin-film transistor (TFT); low hole mobility indicates poor grain boundary quality [23]. | Select additives with multi-functional groups (e.g., pyridine N and -NHâ‚‚) that can passivate both cationic and anionic defects [23]. |
| Residual additive at grain boundaries | Use FTIR spectroscopy to detect characteristic additive peaks in the final film. | Optimize the annealing protocol to ensure complete removal of the volatile additive components, leaving only the passivating moieties. |
| Oxidation of Sn²⺠(in Sn-Pb perovskites) | Use X-ray diffraction (XRD) to detect SnOâ‚‚ phases. | Incorporate reducing agents or antioxidant additives like DBPDA to suppress the oxidation of Sn²⺠to Snâ´âº, which causes p-type self-doping and instability [23]. |
Experimental Protocols & Data Analysis
Validating the Ion Mobility Mechanism via Phase-Field Simulations
- Objective: To model grain growth as a coarsening process limited by ion mobility across grain boundaries, mediated by the presence of an additive.
- Methodology:
- Setup: Develop a phase-field model where the grain boundary energy and mobility are key parameters.
- Parameterization: Treat the additive's effect as a localized reduction of the energy barrier for ion migration at the grain boundaries. This effectively increases the grain boundary mobility in the model.
- Simulation: Simulate the grain growth process with and without the enhanced mobility parameter.
- Validation: Compare the simulated grain size and morphology with experimental results from SEM of films processed with and without the additive [22].
- Expected Outcome: The simulation with increased ion mobility at boundaries should qualitatively and quantitatively reproduce the larger grain sizes observed experimentally when the additive is used.
Protocol: Incorporating the Additive DBPDA in Mixed Sn-Pb Perovskites
This protocol is adapted from a study that achieved a PCE of 21.24% using the additive 2,5-Dibromo-3,4-pyridinediamine (DBPDA) [23].
Precursor Solution Preparation:
- Prepare your standard mixed Sn-Pb perovskite precursor solution (e.g., in DMF:DMSO solvent).
- Add DBPDA directly to the precursor solution. The study found an optimal concentration relative to the lead content.
- Stir the solution thoroughly to ensure complete dissolution and homogenization.
Film Deposition and Annealing:
- Deposit the precursor solution onto your substrate using your preferred method (e.g., spin-coating).
- During the spin-coating process, initiate crystallization with an anti-solvent quench.
- Transfer the film onto a hotplate and anneal at the required temperature (e.g., 65°C) for 10-15 minutes. The annealing step is critical for activating the additive-mediated grain coarsening.
Characterization and Verification:
- X-ray Diffraction (XRD): Analyze the films to assess crystallinity and check for the suppression of Snâ´âº-related phases.
- Scanning Electron Microscopy (SEM): Image the surface morphology to confirm the increase in grain size and improved film coverage.
- FTIR Spectroscopy: Use to verify the interaction between DBPDA's functional groups (pyridine N, -NHâ‚‚) and the perovskite constituents, confirming defect passivation [23].
Quantitative Data from DBPDA Study
The following table summarizes key performance metrics achieved by incorporating the DBPDA additive in mixed Sn-Pb perovskite solar cells and thin-film transistors [23].
| Performance Parameter | Control Device (No DBPDA) | Target Device (With DBPDA) | Improvement |
|---|---|---|---|
| Power Conversion Efficiency (PCE) | 17.86% | 21.24% | ~19% relative increase |
| Open-Circuit Voltage (Voc) | Data in source | Enhanced | Suppressed Snâ´âº formation and defect passivation |
| Hole Mobility (TFT) | 0.18 cm²/V·s | 1.43 cm²/V·s | ~8x increase |
| Thermal Stability (65°C, unencapsulated) | - | Retained 72% of initial PCE after 240 hours | Significant improvement |
| Long-Term Stability (Nâ‚‚ glovebox) | - | Retained 80% of initial PCE after 1,008 hours | Significant improvement |
Solvent and Additive Properties
This table provides key properties of common solvents and an example additive to inform selection.
| Material | Chemical Formula / Structure | Function / Role | Key Property (Donor Number) |
|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | (CH₃)â‚‚SO | Lewis-base solvent | High DN (~29.8 kcal molâ»Â¹), forms strong complexes with Pb²⺠[22] |
| Dimethylformamide (DMF) | HCON(CH₃)â‚‚ | Lewis-base solvent | Moderate DN (~26.6 kcal molâ»Â¹) [22] |
| N-Methyl-2-pyrrolidone (NMP) | Câ‚…H₉NO | Lewis-base solvent | Moderate DN (~27.3 kcal molâ»Â¹) [22] |
| DBPDA Additive | Câ‚…Hâ‚…Brâ‚‚N₃ | Multi-functional Lewis-base additive | Pyridine N and amine groups passivate under-coordinated Sn²âº/Pb²⺠and alleviate micro-strain [23] |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Research |
|---|---|
| Lewis-base Solvents (DMSO, DMF, NMP) | Dissolve perovskite precursors and form intermediate complexes via coordinate bonds with the Lewis-acidic Pb²⺠center [22]. |
| Multi-functional Additives (e.g., DBPDA) | Passivate defects at grain boundaries, suppress cation oxidation (Sn²âº), and modulate crystallization kinetics to reduce micro-strain and enhance ion mobility [23]. |
| Anti-solvents (e.g., Chloroform, Toluene) | Rapidly extract the processing solvent during spin-coating, triggering supersaturation and initiating the crystallization process. |
| Reducing Agents (e.g., SnFâ‚‚) | Specifically added to tin-containing perovskite inks to mitigate the oxidation of Sn²⺠to Snâ´âº, reducing unwanted p-type doping [23]. |
| 6-Hydroxytropinone | (1R,5S)-8-methyl-8-azabicyclo[3.2.1]octan-3-one|Tropane Alkaloid Scaffold |
| 1,2-Epoxydecane | 1,2-Epoxydecane, CAS:68413-40-1, MF:C10H20O, MW:156.26 g/mol |
Mechanism Visualization
Ion migration is a critical degradation mechanism in metal halide perovskites, leading to current-voltage hysteresis, phase segregation, and rapid performance decline in solar cells and other optoelectronic devices. This phenomenon is particularly pronounced in mixed halide and all-inorganic perovskites, where halide ions and vacancies can move readily through the crystal lattice under operational stressors like electric fields, light, and heat. The intrinsic softness of the perovskite lattice and the relatively low formation energy of point defects, such as vacancies, create pathways for ion diffusion. Research has demonstrated that uncontrolled ion migration is a primary factor undermining the long-term operational stability of perovskite devices, making its suppression a central challenge for the field [24] [25].
Compositional engineering, specifically the alloying of tin (Sn) and lead (Pb), has emerged as a powerful strategy to mitigate ion migration. This approach functions by tailoring the fundamental structural and chemical properties of the perovskite material. Tin-lead (Sn-Pb) alloying directly addresses the root causes of ion mobility by tightening the lattice structure, enhancing the strength of ionic bonds within the inorganic framework, and reducing the concentration of deep-level defects that act as migration pathways [24]. These combined effects work to immobilize ionic species, particularly halide ions, thereby enhancing the intrinsic stability of the material. This technical support article, framed within the broader thesis of managing ion migration through nucleation control research, provides a detailed guide for researchers and scientists seeking to implement Sn-Pb alloying strategies in their experimental work. The following sections offer troubleshooting advice, methodological protocols, and resource toolkits to facilitate the successful application of these stabilization techniques.
Troubleshooting Guides & FAQs
Frequently Asked Questions (FAQs)
Q1: What is the fundamental mechanism by which Sn-Pb alloying suppresses ion migration? A1: Sn-Pb alloying operates through two synergistic mechanisms. First, the introduction of smaller-sized Sn²⺠cations reduces the unit cell volume, resulting in a tighter lattice structure. This physical constriction directly impedes the movement of ions through the lattice [24]. Second, Sn substitution has been shown to significantly reduce the density of anti-site defects (e.g., ICs and IPb), which are considered primary pathways for ion migration. By suppressing these defects, the alloying process effectively blocks common migration channels [24].
Q2: My Sn-Pb alloyed perovskite films oxidize and degrade rapidly. How can I improve their stability? A2: The oxidation of Sn²⺠to Snâ´âº is a common challenge. To mitigate this:
- Control the A-site composition: Research on Csâ‚â‚‹â‚“FAâ‚“PbI₃ shows that the A-site cation can influence vacancy formation and ion migration. Mixed A-site cations can enhance stability compared to pure FAPbI₃ [26].
- Employ additive engineering: Incorporate reducing agents or antioxidant additives into the precursor solution to protect Sn²⺠from oxidation.
- Optimize fabrication atmosphere: Process films in an inert environment (e.g., inside a nitrogen glovebox) to minimize exposure to oxygen and moisture, which are primary drivers of Sn²⺠oxidation [27].
Q3: For all-inorganic mixed halide perovskites, what is the optimal Sn:Pb ratio? A3: While the optimal ratio can depend on the specific application (e.g., single-junction vs. tandem solar cells), studies on all-inorganic perovskites have shown that Sn substitution effectively suppresses ion migration across a range of compositions [24]. For CsPbI₃, a composition such as CsPb₀.₇Sn₀.₃I₃ has demonstrated a record efficiency of 17.55%, indicating a well-balanced trade-off between bandgap tuning, efficiency, and stability [28]. We recommend a systematic investigation around this ratio as a starting point.
Q4: How can I experimentally verify that my alloying strategy has successfully suppressed ion migration? A4: You can use several characterization techniques:
- Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS): To track and visualize halide ion distribution and diffusion under stress.
- Galvanostatic Measurements: To quantify ion migration activity by measuring current response under a constant bias.
- Optical Microscopy: To observe morphological changes, such as halide segregation, under operational conditions [24].
- Ultralow-dose Transmission Electron Microscopy (TEM): To directly observe vacancy ordering and ion migration at the atomic scale [26].
Troubleshooting Common Experimental Issues
Problem: Poor Film Morphology with Pinholes and Incomplete Coverage
- Potential Cause: Inadequate control over the nucleation and crystal growth kinetics during the film formation process.
- Solution:
- Implement anti-solvent engineering. The precise timing and choice of anti-solvent (e.g., toluene, chlorobenzene) during spin-coating can rapidly induce supersaturation, leading to a uniform and dense nucleation layer [2].
- Apply substrate temperature treatment. Pre-heating the substrate before film deposition can tailor the Gibbs free energy and chemical potential of the system, promoting faster and more uniform nucleation [2].
Problem: Significant Hysteresis in J-V Curves
- Potential Cause: Residual ion migration within the perovskite bulk or at the interfaces.
- Solution:
- Verify alloy homogeneity: Ensure Sn and Pb are uniformly distributed to achieve consistent lattice tightening. Techniques like EDX mapping can confirm this.
- Implement interface passivation: Passivate the top surface and grain boundaries of the perovskite layer with halide salts (e.g., KI, PbIâ‚‚) or organic molecules to immobilize ions at the interfaces [28].
- Re-optimize the precursor stoichiometry: A slight excess of PbIâ‚‚ or SnIâ‚‚ can help reduce the concentration of halide vacancies, which are primary charge carriers in ion migration.
Problem: Phase Instability (Transition from Black to Yellow Phase)
- Potential Cause: The perovskite phase is metastable at room temperature due to intrinsic thermodynamic factors and lattice strain.
- Solution:
- Utilize strain engineering: Alloying with Sn can introduce compressive strain that stabilizes the black perovskite phase (α-phase) [28].
- Dope with smaller B-site cations: Doping with elements like Mn²⺠or Zn²⺠can further enhance the octahedral bonding and increase the energy barrier for phase transition [27].
- Control the crystallization pathway: Use vapor-assisted annealing or solvent annealing to guide the growth of the thermodynamically stable black phase [2].
Quantitative Data on Sn-Pb Alloying Effects
Table 1: Comparative Effects of Sn-Pb Alloying on Perovskite Properties
| Property | Pure Pb-based Perovskite | Sn-Pb Alloyed Perovskite | Measurement Technique | Reference |
|---|---|---|---|---|
| Ion Migration Activity | High | Greatly Suppressed | TOF-SIMS, Galvanostatic measurement | [24] |
| Vâ‚€ Formation Energy | Lower | Increased | Computational (DFT) | - |
| Anti-site Defect Density | High (e.g., ICs, IPb) | Significantly Reduced | Deep-level transient spectroscopy | [24] |
| Lattice Parameter | Larger | Reduced (Tightened) | X-ray Diffraction (XRD) | [24] |
| Hysteresis Index | Significant | Reduced | J-V Scan | [24] |
| Operational Stability | Fast decay | Improved | Maximum Power Point Tracking | [24] [28] |
Table 2: Impact of Boron Vacancies on Ionic Conductivity in LiBOâ‚‚ (A Comparative Model System)
| Material System | Defect Type | Effect on Li⺠Migration Energy Barrier (Eₘ) | Resulting Ionic Conductivity | Reference |
|---|---|---|---|---|
| m-LBO (Monoclinic LiBO₂) | Oxygen Vacancy | Lowered Eₘ | Enhanced | [29] |
| t-LBO (Tetragonal LiBO₂) | Oxygen Vacancy | Increased Eₘ | Reduced | [29] |
| m-LBO & t-LBO | Boron Vacancy | Significantly Reduced Eₘ | Remarkably Enhanced | [29] |
Detailed Experimental Protocols
Protocol 1: Suppressing Ion Migration in All-Inorganic Mixed Halide Perovskites via Sn-Pb Alloying
This protocol is adapted from research demonstrating that Sn-Pb alloying effectively inhibits ion migration in all-inorganic mixed halide perovskites [24].
Objective: To synthesize a CsPb₀.₇Sn₀.₃I₂Br thin film with suppressed ion migration and improved operational stability.
Materials: See Section 5 "The Scientist's Toolkit" for a detailed list.
Methodology:
- Precursor Solution Preparation:
- Prepare a 1.0 M precursor solution in a mixed solvent of DMF:DMSO (4:1 v/v).
- Cation Source: Dissolve CsI.
- B-site Source: Co-dissolve PbI₂ and SnI₂ in the molar ratio of 0.7:0.3. To prevent Sn²⺠oxidation, add 5-10 mol% of SnF₂ (relative to SnI₂) as a stabilizer.
- X-site Source: Include Br precursors (e.g., CsBr, PbBrâ‚‚) to achieve the target I:Br ratio for bandgap tuning.
- Stir the solution at 60°C for 2-4 hours until fully dissolved, then filter through a 0.45 μm PTFE filter.
Thin Film Fabrication (Spin-coating):
- Pre-clean the substrate (e.g., FTO/c-TiOâ‚‚/m-TiOâ‚‚) with UV-ozone for 15 minutes.
- Load the substrate and dynamically dispense the precursor solution.
- Spin-coat using a two-step program: 1000 rpm for 10 s (spread), followed by 4000 rpm for 30 s (thin).
- At the 5-second mark of the second step, rapidly inject 200 μL of chlorobenzene (anti-solvent) to instantaneously trigger uniform nucleation.
- Immediately after spin-coating, transfer the film to a hotplate and anneal at 100°C for 10 minutes to facilitate crystal growth and solvent removal.
Interface Passivation:
- After annealing and cooling, deposit a passivation layer by spin-coating a solution of, for example, phenethylammonium iodide (PEAI) in isopropanol. This step helps to passivate surface defects and further suppress ion migration at the interface.
Validation Measurements:
- Use TOF-SIMS to profile ion distribution before and after applying an external bias to visualize halide immobilization.
- Perform galvanostatic measurements to quantify the reduction in ionic current.
- Characterize the film with XRD to confirm phase purity and calculate lattice contraction.
- Conduct J-V scans in both reverse and forward directions to demonstrate reduced hysteresis.
Protocol 2: Active Nucleation Control for High-Quality Single Crystals (NanoAC Method)
This protocol, based on the NanoAC (Nanoscale Active Controls) method, provides deterministic control over nucleation, which is critical for growing high-quality crystals for fundamental studies [30].
Objective: To actively control the nucleation and growth of a single crystal using a nanopipette setup.
Materials: Nanopipette (40-150 nm radius), Ag/AgCl wires, potentiostat, optical microscope, sample and precipitant solutions.
Methodology:
- Setup Configuration:
- Backload the nanopipette with the precipitant solution (e.g., containing PEG and NaCl).
- Deposit a 20 μL droplet of the sample solution (e.g., 25 mg/mL HEWL in acetate buffer) into a custom cell.
- Insert the nanopipette into the sample droplet and place Ag/AgCl electrodes in both the sample (working electrode) and the nanopipette (reference/counter electrode).
Pre-conditioning:
- Apply a small negative bias (e.g., -0.1 V) upon initial contact. This counters the natural diffusion of the analyte and precipitants, preventing uncontrolled nucleation during setup and ensuring a consistent initial state.
Nucleation and Growth:
- At time zero, switch the applied potential to a positive value. This drives electrokinetic transport of the sample and precipitant molecules to the nanopipette tip, locally increasing the supersaturation.
- Monitor the ionic current in real-time. Nucleation and crystal growth will cause a detectable disruption or decrease in the ionic current limited by the nanotip.
- Use this current signature as feedback to fine-tune the applied potential and control the growth flux, thereby tuning the crystal habit and ensuring the growth of a single, high-quality crystal.
The Scientist's Toolkit
Table 3: Essential Research Reagents for Sn-Pb Perovskite and Nucleation Studies
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| SnIâ‚‚ & SnFâ‚‚ | B-site precursor and antioxidant stabilizer for Sn²âº. | Forming the Sn component in Sn-Pb alloyed perovskites; SnFâ‚‚ suppresses Sn²⺠oxidation [24] [28]. |
| CsI & FAI | Inorganic and organic A-site cations. | Tuning A-site composition (e.g., Csâ‚â‚‹â‚“FAâ‚“PbI₃) to influence vacancy formation and enhance stability [26]. |
| DMF & DMSO | Polar aprotic solvents for precursor dissolution. | Common solvent system for perovskite precursor inks; DMSO helps form intermediate phases for better film morphology. |
| Chlorobenzene | Anti-solvent for crystallization control. | Rapidly induces supersaturation during spin-coating to prompt uniform nucleation [2]. |
| Phenethylammonium Iodide (PEAI) | Surface passivation agent. | Passivates surface defects and grain boundaries, reducing non-radiative recombination and ion migration at interfaces. |
| COOH-PEG-COOH | Precipitant in controlled crystallization. | Used in nanopipette-based crystallization (NanoAC) to decrease solute solubility and drive nucleation [30]. |
| Solid-state Nanopipettes | Nano-fluidic device for localized transport control. | Serves as a nanoscale interface to control mass transport and supersaturation for single-entity nucleation studies [30]. |
| Niaprazine | Niaprazine, CAS:119328-74-4, MF:C20H25FN4O, MW:356.4 g/mol | Chemical Reagent |
| Arachidonoyl Thio-PC | Arachidonoyl Thio-PC, MF:C44H82NO6PS, MW:784.2 g/mol | Chemical Reagent |
Mechanisms and Workflow Visualizations
Diagram 1: Experimental workflow for creating stable perovskites via nucleation control and Sn-Pb alloying.
Diagram 2: Atomic-scale mechanisms of ion migration suppression via Sn-Pb alloying.
Troubleshooting Guides
Troubleshooting SnOâ‚‚/HfOâ‚‚ Barrier Layer Performance
Problem 1: Inconsistent Electrochemical Performance in Li-ion Battery Anodes
- Observation: Significant capacity fading is observed after relatively few charge-drain cycles.
- Potential Cause 1: Incomplete or non-conformal HfOâ‚‚ coating on the SnOâ‚‚ anode, leading to unprotected surfaces where irreversible reactions with the electrolyte can occur [31].
- Solution:
- Verify the conformality of your Atomic Layer Deposition (ALD) process. Ensure precursor pulse and purge times are optimized for your specific reactor and substrate geometry [31].
- Use characterization techniques like high-resolution transmission electron microscopy (HR-TEM) to inspect the coating uniformity and interface quality [31] [32].
- Potential Cause 2: Crystallization of the HfOâ‚‚ barrier layer. Crystalline HfOâ‚‚ may have different Li-ion diffusion pathways and reduced effectiveness in buffering volume changes compared to the amorphous phase [31].
- Solution:
- Optimize ALD deposition parameters (e.g., temperature, precursor chemistry) to maintain an amorphous HfOâ‚‚ structure. Amorphous HfOâ‚‚ has been shown to allow sufficient Li-ion diffusion for efficient anode operation while still providing excellent passivation [31].
Problem 2: High Leakage Current or Electrical Shorting in Thin-Film Devices
- Observation: Device exhibits high off-state current or fails due to short circuits.
- Potential Cause: Formation of conductive filaments through the HfOâ‚‚ layer, often linked to oxygen vacancies and surface roughness [32].
- Solution:
- Implement nanolaminated structures. Research shows that SnOâ‚‚ | HfOâ‚‚ | SnOâ‚‚ | HfOâ‚‚ laminated structures can exhibit stable resistive switching without destructive breakdown, as the interfaces help manage the formation and rupture of conductive paths [32].
- Ensure the use of smooth, clean electrode surfaces (e.g., TiN) before depositing the barrier layers to minimize defect-prone regions [32].
Problem 3: Poor Interfacial Adhesion or Stress-Induced Cracking
- Observation: The barrier layer delaminates or shows cracks after deposition or cycling.
- Potential Cause: High internal stress due to lattice mismatch or the large volume expansion (~200-300%) of the SnOâ‚‚ anode during lithiation [31] [33].
- Solution:
- Utilize the HfOâ‚‚ layer's ability to chemically interact with SnOâ‚‚ and act as a buffer against volume change [31].
- Consider using ultrathin layers or exploring the mechanical properties of the composite via Representative Volume Element (RVE) modeling to understand stress distribution, as demonstrated in Ag/SnOâ‚‚ composite studies [34].
Troubleshooting Material Synthesis and Deposition
Problem 1: Uncontrolled Crystallinity or Phase in SnOâ‚‚ Layers
- Observation: The SnOâ‚‚ layer does not exhibit the desired polymorph or crystallinity, affecting its electronic and electrochemical properties.
- Potential Cause: The synthesis method and parameters strongly influence the resulting SnOâ‚‚ phase. High-pressure polymorphs can be stabilized via substrate-induced strain or doping in thin films [33].
- Solution:
- For thin films, use techniques like sputtering with specific dopants (e.g., nitrogen, antimony) to stabilize cubic phases, or leverage epitaxial strain to stabilize orthorhombic phases [33].
- For nanoparticles, sol-gel methods typically yield the stable rutile phase. Control the precursor, solvent, temperature, and duration to tailor the morphology (nanorods, nanowires) for a higher surface-to-volume ratio, which can alleviate strain during cycling [33].
Problem 2: High-Temperature Annealing Requirement for Solution-Processed SnOâ‚‚
- Observation: Solution-processed SnO₂ films require annealing temperatures >400°C to achieve good performance, which is incompatible with flexible plastic substrates [35].
- Potential Cause: Conventional solution precursors require high thermal energy to remove organic impurities and form stable metal-oxygen-metal networks [35].
- Solution:
- Adopt a combustion-assisted solution process. This method uses exothermic reactions (e.g., with additives like ammonium nitrate and urea) to generate internal chemical energy, enabling the formation of high-quality SnO₂ films at temperatures below 300°C [35].
Frequently Asked Questions (FAQs)
Q1: Why are SnOâ‚‚ and HfOâ‚‚ an effective combination for blocking ion diffusion in electrodes?
A1: This combination creates a synergistic barrier system. SnOâ‚‚ is an attractive anode material with high theoretical capacity but suffers from large volume expansion and irreversible side reactions with the electrolyte. A nanoscale HfOâ‚‚ layer deposited conformally over SnOâ‚‚ acts as a physical barrier that protects the anode from the electrolyte, minimizes irreversible reactions, and buffers the mechanical stress from volume changes during ion insertion/extraction. Notably, the amorphous HfOâ‚‚ layer does not block Li-ion diffusion, allowing for efficient battery operation [31]. Furthermore, in laminated structures, these materials can work together to enable stable resistive switching for memory applications [32].
Q2: What is the best method to deposit uniform SnOâ‚‚/HfOâ‚‚ barrier layers?
A2: Atomic Layer Deposition (ALD) is highly recommended for creating high-performance barrier layers. ALD excels at depositing ultra-thin, pinhole-free, and perfectly conformal films over complex nanostructures. This is critical for ensuring complete coverage and uniform protection. ALD has been successfully used to deposit both HfOâ‚‚ passivation layers on SnOâ‚‚ anodes [31] and complex SnOâ‚‚-HfOâ‚‚ nanolaminates for electronic devices [32].
Q3: How does the HfOâ‚‚ barrier layer improve the performance of SnOâ‚‚ anodes in Li-ion batteries?
A3: The improvement is multi-faceted, as demonstrated by quantitative data [31]:
| Performance Metric | Uncoated SnOâ‚‚ Anode | HfOâ‚‚-Coated SnOâ‚‚ Anode | Improvement |
|---|---|---|---|
| Capacity after 100 cycles (at 150 mAgâ»Â¹) | 548 mAhgâ»Â¹ | 853 mAhgâ»Â¹ | +56% (305 mAhgâ»Â¹ increase) |
| Key Function | N/A | Protects from electrolyte, buffers volume change, allows Li-ion diffusion | Reversible capacity is significantly enhanced |
Q4: Can these layered structures be used for applications other than batteries?
A4: Yes. The SnOâ‚‚/HfOâ‚‚ material system is versatile. Research has shown its promise in:
- Memory Devices: Nanolaminated SnOâ‚‚-HfOâ‚‚ thin films exhibit stable bipolar resistive switching with a resistance state ratio of up to three orders of magnitude, making them suitable for non-volatile memory [32].
- Optical Coatings: HfOâ‚‚/VOâ‚‚/HfOâ‚‚ sandwich structures are used to make thermochromic films with low phase transition temperatures and excellent durability, where HfOâ‚‚ acts as a protective and antireflection layer [36].
- Transistors: SnOâ‚‚ is a promising channel material for thin-film transistors (TFTs), and HfOâ‚‚ can serve as a high-k gate dielectric [35] [37].
Q5: What are the critical characteristics of SnOâ‚‚ particles that affect composite material performance?
A5: In composite materials like Ag/SnOâ‚‚ electrical contacts, the particulate characteristics of SnOâ‚‚ are crucial for mechanical properties. Numerical simulations using Representative Volume Element (RVE) models reveal that both particle shape and mass fraction are critical [34].
- Shape: Optimizing shape is an effective strategy for both strengthening and toughening. Hollow spherical and regular polyhedral particles generally lead to better comprehensive mechanical properties compared to long prismatic particles [34].
- Mass Fraction: While a higher mass fraction strengthens the material, it typically reduces its toughness. An optimal balance must be found based on application requirements [34].
Experimental Protocols
Protocol: Atomic Layer Deposition of HfOâ‚‚ on SnOâ‚‚ for Li-ion Battery Anodes
This protocol is adapted from methods that have demonstrated improved battery performance through HfOâ‚‚ surface passivation [31].
1. Objective: To deposit a conformal, amorphous HfOâ‚‚ thin film on a SnOâ‚‚-based anode to act as a barrier against electrolyte decomposition and buffer volume expansion.
2. Materials and Equipment:
- Substrate: Prepared SnOâ‚‚ anode (e.g., nanoparticle film, thin film).
- ALD Reactor: Flow-type hot-wall ALD reactor.
- Precursors: Hafnium precursor (e.g., HfCl₄) and ozone (O₃, 220-250 g/m³).
- Carrier/Purge Gas: High-purity Nitrogen (Nâ‚‚, 99.999%).
- Heating System: Precursor evaporation ovens and heated reactor chamber.
3. Step-by-Step Procedure: 1. Loading: Place the SnO₂ anode substrate into the ALD reactor chamber. 2. Stabilization: Pump down the reactor and stabilize the temperature to 300°C. Maintain a pressure of approximately 2.2 mbar. 3. HfO₂ ALD Cycle: Execute the following cycle sequence repeatedly to achieve the desired film thickness (e.g., ~100 cycles for ~10 nm): * HfCl₄ Pulse: Introduce the HfCl₄ precursor vapor for 4 seconds. (HfCl₄ evaporation source temperature: ~162°C). * N₂ Purge: Purge the reactor with N₂ for 3 seconds to remove excess precursor and reaction by-products. * O₃ Pulse: Introduce the O₃ oxidizer for 2 seconds. * N₂ Purge: Purge the reactor again with N₂ for 7 seconds. 4. Cooling and Unloading: After the deposition is complete, allow the reactor to cool under N₂ flow before removing the coated sample.
4. Characterization:
- Electrochemical Testing: Assemble into coin cells with Li metal counter/reference electrodes. Perform galvanostatic cycling at various current densities to measure capacity retention and cycle life [31].
- Material Analysis: Use HR-TEM to confirm film conformality and amorphous nature. X-ray photoelectron spectroscopy (XPS) can probe chemical interactions at the SnOâ‚‚/HfOâ‚‚ interface [31] [32].
Protocol: Fabrication of SnOâ‚‚-HfOâ‚‚ Nanolaminates for Resistive Switching Studies
This protocol outlines the growth of laminated structures for potential memory applications [32].
1. Objective: To fabricate polycrystalline SnOâ‚‚-HfOâ‚‚ nanolaminated thin films on conductive TiN substrates to study their bipolar resistive switching behavior.
2. Materials and Equipment:
- Substrates: SiOâ‚‚/Si(100) wafers for characterization, and TiN-coated Si wafers for electrical measurements.
- ALD Reactor: As above.
- Precursors: SnI₄ (Tin(IV) iodide), HfCl₄, and O₃.
- Carrier/Purge Gas: Nâ‚‚.
3. Step-by-Step Procedure: 1. Substrate Preparation: Clean and etch the Si substrates. For TiN substrates, ensure a clean, conductive surface. 2. Film Deposition (Example Stack: HfO₂ | SnO₂ | HfO₂): * First HfO₂ Layer: Deposit ~10 nm HfO₂ using the HfO₂ ALD cycle described in Protocol 3.1. * SnO₂ Layer: Deposit ~10 nm SnO₂ using the following ALD cycle at 300°C: * SnI₄ Pulse: 5 seconds (SnI₄ source at ~83°C). * N₂ Purge: 2 seconds. * O₃ Pulse: 5 seconds. * N₂ Purge: 5 seconds. * Second HfO₂ Layer: Deposit another ~10 nm HfO₂ layer. 3. Top Electrorode Deposition: Use electron-beam evaporation through a shadow mask to deposit Ti/Au top electrodes (e.g., 0.204 mm² area) to complete the metal-insulator-metal (MIM) structure.
4. Characterization:
- Electrical Measurement: Use a semiconductor parameter analyzer (e.g., Keithley 2636A) to perform current-voltage (I-V) sweeps at room temperature. Look for bipolar resistive switching with a clear distinction between high and low resistance states [32].
- Structural Analysis: Grazing incidence X-ray diffraction (GIXRD) to determine crystallinity and phase. Energy-dispersive X-ray spectroscopy (EDX) for elemental composition profiling [32].
Visualization of Processes and Workflows
HfO2 Coating Improves SnO2 Anode Performance
ALD Process for SnO2/HfO2 Nanolaminates
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Materials for SnOâ‚‚/HfOâ‚‚ Barrier Layer Research
| Item | Function / Application | Critical Notes |
|---|---|---|
| HfCl₄ (Hafnium(IV) chloride) | Metal precursor for HfO₂ ALD. | Evaporates at ~162°C. Common, efficient precursor [32]. |
| SnI₄ (Tin(IV) iodide) | Metal precursor for SnO₂ ALD. | Evaporates at ~83°C. High purity (99.999%) is recommended [32]. |
| O₃ (Ozone) | Oxidizing agent for ALD processes. | Typical concentration: 220-250 g/m³. Crucial for obtaining pure oxide films [32]. |
| TiN-coated Substrate | Conductive bottom electrode for electrical tests. | Provides a smooth, stable surface for film growth and electrical contact [32]. |
| Tin(II) chloride dihydrate (SnCl₂·2H₂O) | Precursor for solution-processed SnO₂. | Used in combustion synthesis with NH₄NO₃ and urea for low-temperature processing [35]. |
| Ammonium Nitrate (NH₄NO₃) & Urea (CO(NH₂)₂) | Combustion agents in solution processes. | Generate exothermic reaction, enabling oxide formation at <300°C [35]. |
| 3-Hydroxytyramine hydrochloride | Bidirectional passivation agent for interfaces. | Can passivate oxygen vacancy defects on SnOâ‚‚ and improve contact with overlayers like perovskite [38]. |
| (Z,Z)-4,7-Decadienol | (Z,Z)-4,7-Decadienol, MF:C10H18O, MW:154.25 g/mol | Chemical Reagent |
This technical support center provides troubleshooting guides and FAQs for researchers working on managing ion migration through nucleation control, specifically for halide perovskites in applications like large-area solar modules.
Frequently Asked Questions (FAQs)
FAQ 1: What are the key quantitative thresholds in heterogeneous nucleation energy? The heterogeneous nucleation energy barrier ((\Delta G^{}_{\text{hetero}})) is a critical threshold determined by interface energy ((\sigma)), chemical potential ((\Delta \mu)), and the contact angle ((\theta)) between the solution and substrate [39]. The equation is: [ \Delta G^{}_{\text{hetero}} = \frac{16\pi}{3} \frac{\sigma^3 v^2}{\Delta \mu^2} \frac{2 - 3 \cos \theta + \cos^3 \theta}{4} ] A lower energy barrier promotes faster nucleation and denser film formation [39].
FAQ 2: How can I increase the nucleation rate and nucleus density in my perovskite films? The nucleation rate ((dN^{}_{\text{hetero}}/dt)) can be increased by manipulating specific experimental parameters [39]: [ \frac{dN^{}{\text{hetero}}}{dt} = \Gamma \exp\left[\frac{-\Delta G^{*}{\text{hetero}}}{k_B T}\right] ] To increase the rate, you can:
- Increase the coating temperature ((T)) to provide more thermal energy for nucleus formation [39].
- Increase the precursor concentration ((\Delta \mu)) to raise the supersaturation, which lowers the nucleation barrier [39].
- Increase the interface energy ((\sigma)) by using substrates with higher surface energy or through interface engineering to reduce the contact angle ((\theta)) [39].
FAQ 3: Why is it crucial to delay crystal growth after achieving fast nucleation, and how can it be done? Fast nucleation improves film compactness and uniformity, but rapid crystal growth can lead to poor crystal quality and more defects [39]. Delaying growth allows for larger, more ordered crystals, which enhances electronic properties like carrier diffusion length. The crystal growth rate ((R)) is directly linked to the rate of solute precipitation [39]: [ R = -\frac{d \Delta C}{dt} ] To slow growth, use additive engineering (e.g., with methylammonium chloride or 1,3-bis(cyanomethyl) imidazolium chloride) to manipulate the solute precipitation rate ((\Delta C)) [39].
FAQ 4: My large-area films are non-uniform. What experimental parameters should I adjust? Non-uniformity often stems from an imbalance between nucleation and growth [39].
- Problem: Insufficient nucleus density. Solution: Increase substrate temperature or precursor concentration during the coating process [39].
- Problem: Excessively rapid crystal growth. Solution: Incorporate crystallization-retarding additives into your precursor ink [39].
- Problem: Poor substrate wetting or interaction. Solution: Employ interface engineering (e.g., self-assembled monolayers) to modify the substrate surface energy and improve nucleation [39].
Quantitative Data on Nucleation and Growth
Table 1: Key Parameters Governing the Crystallization Kinetics of Halide Perovskites
| Parameter | Symbol | Role in Crystallization | How to Manipulate Experimentally |
|---|---|---|---|
| Nucleation Energy Barrier | (\Delta G^{*}_{\text{hetero}}) | Determines the number of initial nuclei; a lower barrier increases nucleation sites [39]. | Modify substrate surface energy; use additives that reduce contact angle ((\theta)) [39]. |
| Nucleation Rate | (dN^{*}_{\text{hetero}}/dt) | Controls the compactness and uniformity of the initial film [39]. | Adjust substrate temperature ((T)) and precursor concentration ((\Delta \mu)) [39]. |
| Interfacial Energy | (\sigma) | A higher value decreases the nucleation barrier, promoting nucleation [39]. | Interface engineering with different functional groups [39]. |
| Chemical Potential Difference | (\Delta \mu) | A higher supersaturation level lowers the nucleation barrier and accelerates nucleation [39]. | Increase solute concentration in the precursor ink [39]. |
| Crystal Growth Rate | (R) | A slower rate generally improves crystal quality and reduces defects [39]. | Use additives (e.g., MACl, BCMImCl) to slow solute precipitation [39]. |
Experimental Protocols
Protocol 1: Blade/Slot-Die Coating for Large-Area Perovskite Films
This protocol is for fabricating uniform large-area perovskite films via blade/slot-die coating, based on successful demonstrations for mini-modules [39].
- Substrate Preparation: Clean your substrate (e.g., ITO/glass). Apply an interface layer (e.g., PTAA, SnOâ‚‚) via spin-coating and anneal.
- Precursor Ink Formulation: Prepare your perovskite precursor solution (e.g., PbIâ‚‚, FAI, MABr in DMF/DMSO). For controlled crystallization, add nucleation promoters (e.g., specific polymers) or growth inhibitors (e.g., MACl, BCMImCl) [39].
- Coating Setup:
- Set the coating temperature (typically 50-100°C).
- Set the gap between the blade/slot-die head and the substrate (e.g., 100-500 µm).
- Set the coating speed (e.g., 1-10 mm/s).
- Film Deposition & Crystallization:
- Dispense the precursor ink in front of the coating head.
- Initiate the coating process. The heated substrate initiates solvent evaporation and nucleation.
- Immediately after coating, transfer the film to an annealing oven (e.g., 100°C for 10-20 minutes) to complete crystallization and solvent removal.
Protocol 2: Manipulating Crystallization via Additive Engineering
This protocol details the use of additives to achieve fast nucleation and slow growth [39].
- Additive Selection: Choose additives based on the desired function.
- Nucleation Promoters: Additives that increase interface energy ((\sigma)).
- Growth Retardants: Additives like MACl or BCMImCl that bind to precursors or crystals to slow down solute precipitation ((d\Delta C/dt)) [39].
- Ink Preparation: Co-dissolve your selected additive(s) with the perovskite precursors in the solvent. Typical additive concentrations range from 0.1 to 5 mol%.
- Film Fabrication: Follow the coating and annealing steps from Protocol 1.
- Validation: Use microscopy (SEM) to inspect crystal size and uniformity. Use X-ray Diffraction (XRD) to assess crystal quality and phase purity.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Halide Perovskite Crystallization
| Item | Function in Experiment |
|---|---|
| Lead Iodide (PbI₂) | Standard precursor providing the Pb²⺠cation and I⻠anion in the perovskite lattice. |
| Formamidinium Iodide (FAI) / Methylammonium Iodide (MAI) | Organic cations (FAâº, MAâº) that form the APbI₃ perovskite crystal structure. |
| Dimethylformamide (DMF) / Dimethyl Sulfoxide (DMSO) | Common solvent systems for preparing perovskite precursor inks. |
| Methylammonium Chloride (MACl) | A common volatile additive that retards crystal growth, leading to larger grains and higher quality films [39]. |
| 1,3-bis(cyanomethyl) imidazolium chloride (BCMImCl) | A non-volatile additive used to simultaneously promote fast nucleation and slow growth, enabling high-efficiency modules [39]. |
| PTAA / PEDOT:PSS | Common hole-transport layer (HTL) materials whose surface properties can influence heterogeneous nucleation. |
| SnOâ‚‚ / TiOâ‚‚ | Common electron-transport layer (ETL) materials that serve as the substrate for perovskite nucleation and growth. |
Supporting Diagrams
Experimental Workflow for Controlled Crystallization
Parameter Impact on Nucleation and Growth
Diagnosing and Solving Ion Migration in Real-World Device Architectures
Troubleshooting Guides
FAQ: Common Experimental Issues and Solutions
Q1: Why is my quantification of inactive lithium species yielding inconsistent results between different techniques?
A: Inconsistent quantification, particularly between Nuclear Magnetic Resonance (NMR) and titration methods, is often due to the presence of lithium hydride (LiH), which different techniques identify differently.
- Root Cause: Titration methods like Titration Gas Chromatography (TGC) quantify dead Li metal (Liâ°) by measuring hydrogen gas (Hâ‚‚) evolution upon reaction with water. LiH also produces Hâ‚‚ upon hydrolysis, leading to an overestimation of Liâ° if not accounted for. In contrast, operando NMR can differentiate metallic Li from Liâ° but may not always distinguish LiH [40].
- Solution: Employ a combined methodology. Use operando NMR to track the dynamic formation of dead Li metal and SEI during cycling. Validate these findings with ex situ TGC or Mass Spectrometry Titration (MST), explicitly accounting for LiH contribution in the TGC analysis. This multi-technique approach provides a more reliable quantification [40].
Q2: What could cause rapid capacity fade linked to transition metal dissolution in my cell tests?
A: Rapid fade often stems from dissolved transition metals (e.g., Mn²âº, Ni²âº) from the cathode migrating and depositing on the anode, disrupting the Solid Electrolyte Interphase (SEI).
- Root Cause: The dissolution is facilitated by electrolyte decomposition products at the cathode. Crucially, in degraded electrolytes, these transition metals coordinate strongly with fluorophosphate species (e.g., POâ‚‚Fâ‚‚â»), which alters their solvation environment and impacts their deposition mechanism at the anode [41].
- Solution:
- Introduce Additives: Use additives like LiPOâ‚‚Fâ‚‚, which modifies the SEI on both electrodes, making it more stable and reducing transition metal deposition [41].
- Employ Chelating Agents: Consider chelating agents (e.g., acetylacetonate) in the electrolyte or separator to sequester dissolved metals, preventing their migration to the anode [41].
- Material Robustness: Improve the robustness of cathode materials with coatings to suppress metal dissolution at the source [42].
Q3: How can I improve the sensitivity and accuracy of my metabolite analysis when studying ionic species in battery electrolytes or biological samples?
A: Poor sensitivity for polar or ionic analytes is frequently caused by their non-specific adsorption to metal surfaces in the chromatographic system.
- Root Cause: Analytes like phosphates can interact with Fe³⺠ions on stainless steel parts of LC systems, leading to poor peak shape and reduced detection sensitivity [43].
- Solution: Utilize a bioinert chromatographic system. These systems use hardware (e.g., titanium, PEEK) that minimizes metal-analyte interactions. For HILIC-MS analysis of metabolites, a bridged ethyl hybrid (BEH) amide column has been shown to provide excellent results. This eliminates the need for passivation with chelating additives and significantly improves data quality [43].
Table 1: Quantification of Inactive Lithium Species in an Anode-Free Cell (Cu||LiFePO₄) During the First Cycle (Baseline Electrolyte: 1M LiPF₆ in EC/EMC) [40]
| Measurement Method | Total Irreversible Capacity | Capacity Loss from Dead Li Metal (C~dead~) | Capacity Loss from SEI Formation (C~SEI~) |
|---|---|---|---|
| Electrochemistry & NMR | 27 μA·hour (9.4%) | 15 μA·hour (5.2%) | 12 μA·hour (4.2%) |
| Normalized values are relative to the first charge capacity (286 μA·hour). |
Table 2: Failure Mode Summary and Mitigation Strategies in Lithium-ion Batteries
| Failure Mode | Impact on Mobile Ion Concentration (Nâ‚€) | Primary Characterization Techniques | Proposed Mitigation Strategy |
|---|---|---|---|
| Dead Li Metal Formation | Permanent loss of active Liâ°, direct reduction of Nâ‚€ [40]. | Operando NMR, TGC, MST [40] | Optimize electrolyte formulation; modulate current density [40]. |
| Transition Metal Dissolution | Contributes to SEI growth, consuming Liâº; indirect reduction of usable Nâ‚€ [41] [44]. | NMR/EPR spectroscopy, Advanced Electron Microscopy [41] [44] | Use cathode coatings; electrolyte additives (e.g., LiPOâ‚‚Fâ‚‚); chelating agents [42] [41]. |
| Unstable SEI Growth | Continuous consumption of Li⺠ions from the electrolyte and anode, reducing N₀ [40]. | Titration methods (TGC), Advanced Electron Microscopy [44] [40] | Form stable SEI via electrolyte engineering (e.g., additives, high concentration salts) [40]. |
Experimental Protocols
Protocol 1: Quantifying Inactive Lithium Species via Operando NMR and Titration
Objective: To separately quantify the evolution of dead lithium metal (Liâ°) and solid electrolyte interphase (SEI) during battery cycling [40].
Materials:
- Customized operando NMR cell (e.g., Cu||LiFePOâ‚„ configuration).
- NMR spectrometer (e.g., 9.7 T magnet).
- Electrolyte of interest (e.g., 1 M LiPF₆ in EC/EMC).
- Titration Gas Chromatography (TGC) or Mass Spectrometry Titration (MST) setup.
- Glove box with inert atmosphere.
Methodology:
- Cell Assembly: Assemble the anode-free cell (Cu||LiFePOâ‚„) inside a glove box.
- Operando NMR Setup: Place the cell inside the NMR spectrometer. The â·Li NMR chemical shift differentiates metallic Li (~270 ppm) from diamagnetic Li⺠in the SEI (~0 ppm) [40].
- Electrochemical Cycling: Cycle the cell under the desired conditions (e.g., 0.5 mA cmâ»Â² between 2.8 V and 3.8 V).
- Data Acquisition:
- Continuously acquire â·Li NMR spectra during cycling.
- The integral of the signal at ~270 ppm is proportional to the amount of metallic Li.
- The signal remaining at the end of discharge (2.8 V) is quantified as dead Li metal (C~dead~) [40].
- Data Calculation:
- Total irreversible capacity (C~Ir~) is obtained from electrochemical data: C~Ir~ = Charge Capacity - Discharge Capacity.
- SEI capacity loss (C~SEI~) is calculated: C~SEI~ = C~Ir~ - C~dead~ (NMR measured) [40].
- Validation: After cycling, disassemble the cell and subject the electrodes to ex situ TGC or MST analysis to cross-validate the NMR findings and account for the presence of LiH [40].
Protocol 2: Probing Transition Metal Coordination in Electrolytes
Objective: To characterize the solvation sphere of dissolved transition metals (Mn²âº, Ni²âº) in pristine and degraded battery electrolytes [41].
Materials:
- Electrolyte samples (pristine and heat-degraded).
- Transition metal salts (e.g., Mn(TFSI)â‚‚).
- NMR spectrometer for ¹H and ¹â¹F relaxometry.
- Pulsed EPR spectrometer for HYSCORE and ENDOR experiments.
Methodology:
- Sample Preparation: Introduce Mn²⺠or Ni²⺠salts into both pristine and thermally degraded LiPF₆-based carbonate electrolytes.
- NMR Relaxometry: Perform ¹H and ¹â¹F NMR relaxometry at ambient temperatures. The relaxation rates are sensitive to the coordination environment of the paramagnetic metal ions, revealing interactions with solvent molecules and anions [41].
- Pulsed EPR Spectroscopy: Analyze the frozen solutions using double resonance techniques like HYSCORE and ENDOR. These methods can detect hyperfine interactions, directly revealing coordination to nuclei such as ³¹P in fluorophosphate degradation products (e.g., POâ‚‚Fâ‚‚â») [41].
- Control Experiments: Add potential coordinating agents (water, ethylene glycol, acetylacetonate, LiPOâ‚‚Fâ‚‚) to observe how they displace native solvent molecules from the metal's inner and outer coordination shells [41].
The Scientist's Toolkit: Key Reagents & Materials
Table 3: Essential Research Reagents for Ion Migration and Failure Mode Studies
| Reagent / Material | Function / Application | Key Consideration |
|---|---|---|
| LiPOâ‚‚Fâ‚‚ (Lithium Difluorophosphate) | Electrolyte additive that modifies CEI/SEI, reducing transition metal deposition and improving cycle life [41]. | Shown to coordinate directly with dissolved Mn²âº/Ni²âº, altering their deposition mechanism [41]. |
| Acetylacetonate (acac) | Chelating agent used to sequester dissolved transition metals in the electrolyte, preventing their migration to the anode [41]. | Can enhance or suppress metal dissolution depending on context (e.g., cathode material, state of charge) [41]. |
| Bioinert HILIC Column (e.g., BEH Amide) | For sensitive LC-MS analysis of polar metabolites and ionic degradation products in complex matrices [43]. | Minimizes non-specific adsorption of analytes, improving peak shape and sensitivity compared to standard steel columns [43]. |
| Ionic Liquids (e.g., imidazolium-based) | Tunable solvents or additives for synthesizing materials or modifying electrolyte properties; can act as catalysts or carriers [45]. | Structural tunability allows for customization of properties like hydrophilicity, viscosity, and toxicity [45]. |
Optimizing Additive Concentration and Processing for Enhanced Grain Coarsening
Welcome to the Technical Support Center for Grain Coarsening and Nucleation Control Research. This resource addresses the critical challenge of managing ion migration through precise nucleation control, a fundamental aspect of developing advanced materials and pharmaceuticals. The following guides and FAQs provide targeted solutions for common experimental issues, supported by detailed protocols and data analysis to enhance your research outcomes.
Troubleshooting Guides
Problem 1: Failure to Achieve Target Grain Size
Q: Despite following established protocols, the final grain size in my metal alloy/perovskite film is consistently too small or too large. What are the key factors I should adjust?
A: Achieving target grain size requires balancing multiple processing parameters. The problem often lies in an improper balance between cold work (deformation) and annealing conditions, or between precursor chemistry and thermal treatment.
Key Parameters to Investigate:
- Amount of Cold Work: Higher severity of cold work creates more nucleation sites, generally leading to finer grains after recrystallization. In tube fabrication, a higher amount of cold work makes it easier to achieve finer grains, all else being equal [46].
- Annealing Temperature and Time: Higher annealing temperatures and longer dwell times provide greater thermal energy for grain boundary movement, promoting grain growth. If grains are too small, you may need to increase the temperature or time; if they are too large, decrease them [46].
- Additive Concentration: In perovskite systems, additives like Lewis bases (e.g., DMSO) do not primarily retard nucleation but instead facilitate coarsening grain growth by increasing ion mobility across grain boundaries. Optimizing their concentration is crucial for controlling final grain size [47].
- Cooling Rate: A faster cooling rate after annealing completes the recrystallization process more quickly, reducing the time available for grains to grow [46].
Recommended Workflow:
- Characterize the initial grain size after cold work.
- Systematically vary the annealing temperature while holding time constant.
- If target size is not met, vary the annealing time at the optimal temperature.
- For perovskite films, perform a dose-response test with your additive to find the concentration that maximizes ion mobility and coarsening without introducing defects.
Problem 2: Uncontrolled or Heterogeneous Nucleation
Q: My experiments result in a non-uniform grain size distribution, with some very large and some very small grains. How can I achieve a more consistent microstructure?
A: A mixed-grain structure often arises from uncontrolled, stochastic nucleation events. The goal is to create a high density of uniform nucleation sites or to precisely control the nucleation process itself.
Key Parameters to Investigate:
- Solvent Removal Rate: In solution-processed materials like perovskites, the rate of solvent evaporation during film deposition can affect nucleation density. However, evidence suggests that the final grain size may be less sensitive to the drying technique (e.g., one-step, gas-quenching, anti-solvent-quenching) than previously thought, pointing to the annealing step and additive effects as more critical control points [47].
- Spatial Control of Supersaturation: Uncontrolled diffusion leads to localized variations in supersaturation, triggering multiple, asynchronous nucleation events. This is a fundamental limitation of ensemble-based crystallization methods [30].
- Substrate Properties: The wettability and surface energy of the substrate can influence initial nucleation. Testing both hydrophilic and hydrophobic substrates can determine if substrate interaction is a contributing factor [47].
Recommended Workflow:
- Ensure a homogeneous precursor ink by verifying the complete dissolution of components and the use of complexing solvents.
- Standardize the film deposition protocol (e.g., spin-coat speed, acceleration, environment) to minimize variability in initial film formation.
- Consider advanced nucleation control techniques, such as the NanoAC (Nanoscale Active Controls) method, which uses a nanopipette to localize and control supersaturation via electrokinetic transport, enabling deterministic nucleation and growth of single crystals [30].
Problem 3: Excessive Ion Migration During Processing
Q: Ion migration during thermal annealing or device operation leads to performance degradation and instability. How can this be suppressed?
A: Ion migration is driven by diffusion and electric field drift, and is accelerated by defects and low activation energies in the crystal structure. The strategy is to create energy barriers that confine ion movement.
Key Parameters to Investigate:
- Grain Boundary Engineering: Since grain boundaries can be fast pathways for ion migration, promoting grain coarsening itself reduces the number of boundaries. Additives that facilitate coarsening by increasing ion mobility at boundaries can paradoxically lead to a more stable final structure [47].
- Barrier Layers: Quantitatively designed blocking layers on the material's surface can effectively suppress ion migration. Research on perovskites has quantified that a barrier energy of approximately 0.911 eV is needed to prevent iodide ion loss. This can be achieved using composite layers, such as a thin HfO2 scattering layer combined with an ordered dipole monolayer that creates a drift electric-field [3].
- Nanochannel Functionalization: In battery systems, designing nanochannels with superhydrophobic interiors (e.g., using perfluorinated chains) can reduce interaction with ion solvation shells, promoting dehydration and lowering migration resistance [48].
Recommended Workflow:
- Identify the primary migrating ion (e.g., I⻠in perovskites, Zn²⺠in batteries).
- Apply a tailored blocking layer atop the active material and quantify its effectiveness using techniques like TOF-SIMS or XPS.
- For internal grain boundary modification, explore additives that passivate defects and increase the activation energy for ion hopping.
Frequently Asked Questions (FAQs)
Q1: What is the fundamental driving force behind grain coarsening? The primary driving force is the reduction of the total free energy of the system, particularly the grain boundary energy. The system lowers its energy by reducing the total area of grain boundaries, leading to boundary movement and grain growth [49].
Q2: How is grain size measured and reported? Grain size is typically measured from polished and etched cross-sections under magnification. The common standard is ASTM E112, which assigns a Grain Size Number (G). It is important to note that the grain size number is inversely related to the actual physical grain size. A higher number indicates finer grains [46].
Q3: For thin-walled components, what is a typical target grain size? As wall thickness decreases, required grain size becomes finer. For example:
- Wall ~0.050 inch: Grain Size 5 is often acceptable.
- Wall ~0.015 inch: Grain Size 6 or finer.
- Wall ~0.005 inch: Grain Size 7 or finer is required [46].
Q4: Do additives impact the nucleation stage or the growth stage of crystallization? For many systems, recent evidence challenges the common assumption that additives primarily retard nucleation. Instead, for several popular perovskite additives, the key impact occurs during the annealing step after nucleation, where they facilitate coarsening grain growth by increasing ion mobility across grain boundaries [47].
Q5: What are some advanced techniques for studying nucleation in real-time? A single-entity method called NanoAC uses a nanopipette to spatially confine the interface between a sample and a precipitant solution. An external potential controls ion transport, allowing real-time detection of nucleation and growth events through disruptions in the ionic current, providing direct electroanalytical feedback [30].
Quantitative Data Tables
Table 1: Grain Size Number vs. Actual Grain Size and Applications
Data derived from industrial standards and practices for metallic alloys [46].
| ASTM Grain Size Number (G) | Typical Application Context | Key Consideration |
|---|---|---|
| 3.5 to 6 | As-received material for further processing | Varies by alloy |
| 5 | Walls ≥ 0.050 inch | Acceptable strength |
| 6 | Walls ≈ 0.015 inch | Prevents splitting during fabrication |
| 7 or 8 | Walls ≈ 0.005 inch or Electropolishing | Ensures sufficient grains across thin walls; improves surface finish |
Data synthesized from research on halide perovskites and battery systems [3] [48].
| Strategy | Mechanism | Quantified Efficacy / Requirement | Key Trade-off |
|---|---|---|---|
| Composite Blocking Layer (HfO2 + Dipole) | Scattering + Drift Electric Field | 0.911 eV barrier energy to confine Iâ» migration; >99.9% reduction [3] | Layer thickness vs. carrier transport efficiency |
| Superhydrophobic Nanochannels (SPCOFs) | Reduced electrolyte-wall interaction; promoted ion dehydration | Enables 5000 hours of stable battery operation at 10 mA cmâ»Â² [48] | Complex synthesis of functionalized frameworks |
Detailed Experimental Protocols
Protocol 1: Nanopipette-Controlled Nucleation for Single Crystal Growth
This protocol is adapted from single-entity crystallization studies of biomacromolecules [30].
Objective: To achieve active control over nucleation and growth of a single crystal, bypassing stochastic ensemble behavior.
Materials:
- Nanopipette: Pulled to a tip radius of ~40-150 nm.
- Sample Solution: Analyte of interest (e.g., Lysozyme at 25 mg/mL) in a suitable buffer.
- Precipitant Solution: Contains precipitating agents (e.g., 10% wt COOH-PEG-COOH, 2 M NaCl) in the same buffer.
- Electroanalytical Setup: Amplifier with pre-amp, Ag/AgCl electrodes, and LabVIEW control software.
- Microscope: Optical microscope with camera for real-time monitoring.
Procedure:
- Loading: Backload the nanopipette with the precipitant solution.
- Setup: Deposit a 20 µL droplet of the sample solution into a custom cell. Insert the nanopipette into the droplet. Place Ag/AgCl electrodes in the sample droplet and inside the pipette.
- Pre-conditioning: Apply a small negative bias (e.g., -0.1 V) for 1-2 minutes. This counters diffusive transport and ensures a consistent initial state, suppressing uncontrolled nucleation.
- Nucleation Induction: At time zero, switch to a positive bias voltage. This drives the precipitant and analyte together via electrokinetic transport, locally increasing supersaturation at the nanopipette tip.
- Monitoring: Monitor the ionic current for a sudden disruption, which signals a nucleation event. Simultaneously observe the tip optically for crystal formation.
- Crystal Growth: Once a nucleus is detected, fine-tune the applied potential to control the flux of molecules, maintaining optimal supersaturation in the metastable zone for high-quality growth.
- Harvesting: Once the crystal reaches the desired size, carefully retract the nanopipette and retrieve the crystal for analysis (e.g., X-ray diffraction).
Protocol 2: In-situ Characterization of Additive-Mediated Grain Coarsening
This protocol is based on interdisciplinary studies of grain growth in perovskite films [47].
Objective: To correlate the presence of additives in a precursor ink with the dynamics of grain coarsening during the annealing stage.
Materials:
- Precursor Inks: Standard precursor solutions (e.g., for MAPbI₃) with and without the target additive (e.g., DMSO, other Lewis bases).
- Characterization Tools:
- Grazing Incidence Wide-Angle X-ray Scattering (GIWAXS): For in-situ monitoring of crystal structure formation during annealing.
- Ultraviolet Photoelectron Spectroscopy (UPS): To analyze the electronic structure and defect states at grain boundaries.
- Fourier Transform Infrared Spectroscopy (FTIR): To confirm coordination of the additive to precursor components (e.g., Pb²⺠sites).
- Scanning Electron Microscope (SEM): For ex-situ analysis of final grain morphology.
Procedure:
- Ink Characterization: First, probe the precursor inks using techniques like 207Pb NMR and electrical conductance to understand the complex formation between the additive and metal precursors in the liquid state.
- Film Deposition & In-situ Measurement: Deposit the precursor ink onto a substrate to form a thin film. Immediately transfer the sample to the GIWAXS setup equipped with a heating stage.
- Data Collection: Begin the annealing protocol (e.g., 100°C for 10 minutes) while collecting GIWAXS patterns at a high frame rate. This allows you to observe the transition from the initial amorphous or intermediate phase to the crystalline perovskite and to track the evolution of grain size in real-time.
- Post-Annealing Analysis: After annealing, characterize the final film with SEM to confirm the grain size and with UPS/FTIR to detect the presence and role of the additive at grain boundaries.
- Data Correlation: Correlate the GIWAXS data (grain growth kinetics) with the ex-situ analyses. A strong observation supporting the coarsening-facilitation theory is if the primary grain growth occurs after the initial perovskite phase has formed, coinciding with the presence of the additive.
Research Workflow and Signaling Pathways
Grain Coarsening Optimization Workflow
Ion Migration Suppression Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Grain Coarsening and Nucleation Control Experiments
| Reagent / Material | Function in Research | Example Context |
|---|---|---|
| Lewis Base Additives (e.g., DMSO, DMF) | Facilitate grain coarsening by increasing ion mobility across grain boundaries during annealing [47]. | Halide Perovskite Film Fabrication |
| Grain Refining Elements (e.g., Ti, Nb) | Inhibit grain growth by forming stable precipitates that pin grain boundaries [46]. | Aluminum Alloys, Stainless Steels |
| Carboxylic PEG (e.g., COOH-PEG-COOH) | Acts as a precipitant in controlled crystallization, forming concentration gradients that drive supersaturation [30]. | Biomacromolecule Crystallization |
| Perfluorohexyloxy Chains | Creates superhydrophobic nanochannels in frameworks (SPCOFs), reducing ion migration resistance by minimizing electrolyte-wall interactions [48]. | Zinc-ion Battery Anode Coating |
| Dipole Molecules (e.g., CF3-PBAPy) | Forms an ordered monolayer to create a uniform interfacial electric-field, providing a drift barrier to suppress ion migration [3]. | Perovskite Solar Cell Interface Engineering |
| Atomic-Layer-Deposited Metal Oxides (e.g., HfOâ‚‚) | Provides an ultra-thin, conformal scattering barrier to physically block ion migration [3]. | Protective Layer on Perovskite Films |
Frequently Asked Questions (FAQs)
Q1: What is the fundamental trade-off between ion-blocking and charge transport? Many surface passivation agents used to suppress ion migration (blocking) have poor electrical conductivity. This creates a conflict: while they effectively reduce defect states and non-radiative recombination, they can simultaneously impede the extraction and transfer of charge carriers (electrons or holes), often leading to a lower fill factor (FF) in devices like solar cells [50].
Q2: What are common experimental symptoms of this trade-off? You may observe the following issues in your device characterization:
- High Open-Circuit Voltage (VOC) but Low Fill Factor (FF): Effective defect passivation increases VOC, but poor charge transport through the passivation layer reduces FF [50].
- Slow Device Response: The transient response speed of a device can be limited by slow ion dynamics within the material, which hinders rapid switching or sensing capabilities [51].
- Inconsistent Performance: Material-drug or material-ion interactions can lead to batch variability and failure to meet dosing or electrical specifications over time [52].
Q3: What strategies can circumvent this ion-blocking vs. transport trade-off? Advanced strategies focus on creating synergies rather than compromises:
- Binary Synergistic Post-Treatment (BSPT): Using a blend of two or more passivation agents can simultaneously enhance defect passivation and improve the crystallinity and molecular packing of the passivation layer itself, facilitating better charge carrier transport [50].
- Use of Semiconducting Passivators: Replace conventional insulator-based passivation materials with semiconducting polymers. These materials provide defect passivation while maintaining good charge transport pathways, enabling high FF [50].
- Nucleation and Crystallization Control: Fabricating high-quality perovskite thin films with large, oriented grains and controlled morphology is a primary method to reduce intrinsic defect density and ion migration pathways, thereby lessening the reliance on thick or resistive blocking layers [20].
Q4: How does nucleation control help manage ion migration? Controlling the nucleation and crystal growth process is fundamental to managing ion migration. By inducing a high density of nucleation sites on the substrate and promoting the growth of large, uniform grains, you can minimize the formation of defects and grain boundaries that act as fast pathways for ion migration. This reduces the overall defect density, which is a primary driver of ion migration and subsequent device degradation [20].
Q5: How can I quickly diagnose if a performance issue is due to poor charge transport? A combination of characterization techniques is effective:
- Electrical Measurements: Use current-voltage (I-V) measurements on the passivation layer or complete device to directly assess conductivity and charge transport properties [50].
- Transient Response Analysis: For electrochemical devices like OECTs, analyzing the transient response to a gate voltage pulse can reveal whether the response speed is limited by electronic charge transport or slower ionic dynamics [51].
- Structural Analysis: Techniques like Grazing-Incidence Wide-Angle X-Ray Scattering (GIWAXS) can reveal the molecular orientation and packing of the passivation layer. More ordered packing is typically beneficial for charge transport [50].
Troubleshooting Guides
Problem: Inconsistent Elution or Dosing Rates in Drug-Delivery Devices
Potential Cause: Material-drug compatibility failures, where interactions between the polymer matrix and the active pharmaceutical ingredient (API) lead to unstable release profiles [52].
Solution:
- Early Compatibility Screening: Incorporate solvent resistance and chemical compatibility testing early in the material selection process. Screen for API absorption, polymer plasticization, and hydrolytic degradation [52].
- Process Control: For fabrication methods like hot-melt extrusion, use cryogenic milling to produce fine polymer powders. Ensure strict humidity control during milling and handling to prevent molecular degradation that affects performance [52].
- Material Tuning: Select or engineer polymers (e.g., thermoplastic polyurethanes - TPUs) where architecture (polyol type, segment distribution, crystallinity) can be tuned to precisely control elution rate, loading capacity, and stability [52].
Problem: Slow Transient Response in Organic Electrochemical Transistors (OECTs)
Potential Cause: The device's response speed is limited by slow ion transport within the organic mixed ionic–electronic conductor (OMIEC) channel, which is often slower than electronic charge transport [51].
Solution:
- Material and Morphology Optimization:
- Side-Chain Engineering: In aqueous electrolytes, ions move faster within OMIECs featuring specific side chains (e.g., alkoxy chains) [51].
- Crystallinity Control: Ions tend to transport more easily through the amorphous phases of a material than through crystalline regions due to lower energy barriers. Tuning the amorphous-to-crystalline ratio can optimize ion dynamics [51].
- Operational Conditions: Be aware that applied gate voltage and subsequent ion doping can cause channel swelling and microstructural changes (e.g., π–π stacking distances), which in turn affect ion transport. Characterize device performance under operational conditions [51].
Problem: Poor Fill Factor (FF) in Perovskite Solar Cells Despite Good Passivation
Potential Cause: The surface passivation layer, while reducing non-radiative recombination, is impeding hole or electron extraction due to its poor conductivity or misaligned energy levels [50].
Solution:
- Implement Binary Synergistical Post-Treatment (BSPT):
- Protocol: Blend two organic halide salts (e.g., 4-tert-butyl-benzylammonium iodide (tBBAI) and phenylpropylammonium iodide (PPAI)) in isopropanol (IPA). Spin-coat the blended solution onto the perovskite surface without further annealing [50].
- Expected Outcome: This method can enhance the crystallinity and molecular packing of the passivation layer, improve energy band alignment with the charge transport layer, and provide superior defect passivation, collectively boosting charge extraction and FF [50].
- Use Semiconducting Passivation Agents: Replace insulating passivation materials with semiconducting polymers that have been shown to enable high FF by providing better charge transport pathways [50].
Experimental Protocols & Data
Detailed Protocol: Binary Synergistical Post-Treatment (BSPT) for Perovskite Films
This protocol is adapted from methods used to achieve high-efficiency perovskite solar cells [50].
Objective: To apply a passivation layer that simultaneously suppresses surface defects and enhances charge carrier transport.
Materials:
- Substrate with deposited perovskite film (e.g., RbCl-doped FAPbI3).
- Anhydrous Isopropanol (IPA).
- 4-tert-butyl-benzylammonium iodide (tBBAI).
- Phenylpropylammonium iodide (PPAI).
- Spin coater.
- Glove box (Nâ‚‚ atmosphere).
Procedure:
- Solution Preparation: Prepare a blend solution by dissolving tBBAI and PPAI in a specific weight ratio (e.g., 1:1 by weight) in anhydrous IPA. The total concentration should typically be in the range of 0.5-1.5 mg/mL. Dissolve the salts completely using gentle stirring or shaking.
- Film Deposition: Place the perovskite substrate on the spin coater inside the glove box. Dynamicly spin-coat the BSPT solution onto the perovskite film at 4000-6000 rpm for 20-30 seconds.
- Drying: After spin-coating, do not perform a further annealing step. The film is ready for the subsequent deposition of the hole transport layer.
Characterization: Use GIXRD and GIWAXS to confirm the formation of a new, highly crystalline phase with ordered molecular packing. I-V measurements can verify improved conductivity of the passivation layer [50].
Detailed Protocol: Substrate Temperature Treatment for Nucleation Control
Controlling the substrate temperature is a direct method to tailor nucleation and crystal growth by modulating the system's chemical potential and Gibbs free energy [20].
Objective: To fabricate high-quality, uniform perovskite thin films with large grains and reduced defect density.
Materials:
- Coating substrate (e.g., ITO, FTO).
- Perovskite precursor solution.
- Hotplate or temperature-controlled spin coater.
Procedure:
- Pre-conditioning: Prior to spin-coating, pre-heat the substrate on a hotplate to a specific temperature. The optimal temperature (T_sub) must be determined empirically but often ranges from 30°C to 70°C [20].
- Nucleation: Immediately spin-coat the perovskite precursor solution onto the pre-heated substrate. The input of thermal energy facilitates a faster transition to a supersaturated state, leading to a higher density of nucleation sites on the substrate itself [20].
- Growth & Annealing: After spin-coating, transfer the film to a hotplate for annealing at a higher temperature (e.g., 100°C) to complete crystal growth and solvent removal.
Mechanism: Increasing the substrate temperature increases the chemical potential (µ) of the system, which lowers the energy barrier for nucleation (ΔG*), promoting heterogeneous nucleation on the substrate over homogeneous nucleation in the solution. This results in denser, more uniform films [20].
Table 1: Performance Outcomes of Different Passivation Strategies in Perovskite Solar Cells
| Passivation Strategy | Power Conversion Efficiency (PCE) | Fill Factor (FF) | Key Improvement |
|---|---|---|---|
| Unary Passivation (e.g., PPAI) [50] | Reported in literature | Increases at a slow rate | Defect passivation |
| Semiconducting Polymer Passivation [50] | High | Up to 83% | Enhanced charge transport |
| Binary Synergistical Post-Treatment (BSPT) [50] | Certified 26.0% | High | Combined defect passivation & enhanced charge transport |
| Theoretical S-Q Limit [50] | ~ | >83% | Ideal charge collection |
Table 2: Key Research Reagent Solutions for Managing Ion Migration & Charge Transport
| Reagent / Material | Function / Explanation | Example Application |
|---|---|---|
| Organic Halide Salts (e.g., PPAI, tBBAI) [50] | Surface passivators that bond with under-coordinated ions (e.g., Pb²âº) on perovskite surfaces, reducing defect-mediated ion migration. | Perovskite solar cells, light-emitting diodes. |
| Thermoplastic Polyurethanes (TPUs) [52] | Tunable polymer matrices for drug delivery; their chemistry can be engineered to control API elution rate and stability, preventing material-drug mismatches. | Long-acting injectables, implantable drug-delivery devices. |
| Organic Mixed Ionic-Electronic Conductors (OMIECs) [51] | Semiconducting polymers that allow simultaneous transport of ions and electronic charges. Their side chains and morphology can be optimized to control ion dynamics. | Organic electrochemical transistors (OECTs), biosensors, neuromorphic devices. |
| Antisolvents [20] | Used during spin-coating to rapidly induce supersaturation and control the nucleation density of perovskite crystals, leading to high-quality films. | Fabrication of pinhole-free perovskite thin films. |
Process Visualization
Fundamental Corrosion Mechanisms in Electrochemical Systems
Electrode corrosion is a pervasive challenge that compromises the efficiency, stability, and longevity of electrochemical devices. Understanding the underlying mechanisms is crucial for developing effective mitigation strategies.
Galvanic Corrosion
Galvanic corrosion occurs when two electrochemically dissimilar metals contact each other in the presence of an electrolyte, forming a bimetallic couple [53]. This creates an electrical potential difference where the more "active" metal becomes the anode and undergoes oxidation, while the more "noble" metal becomes the cathode and remains protected [53]. The driving force is the difference in electrochemical potential between the metals, which can be predicted using the galvanic series [53].
Table 1: Galvanic Series of Selected Metals in Seawater
| Metal | Galvanic Series Position | Potential (V) |
|---|---|---|
| Magnesium | Most Anodic (Most Active) | -1.75 |
| Zinc | -1.10 | |
| Aluminum Alloys | -0.90 to -1.00 | |
| Mild Steel | -0.68 | |
| Cast Iron | -0.60 | |
| Lead | -0.55 | |
| Brass | -0.40 | |
| Copper | -0.34 | |
| Chromium Steel (Passive) | -0.20 | |
| Nickel (Passive) | -0.15 | |
| Silver | -0.15 | |
| Titanium | -0.10 | |
| Gold | -0.05 | |
| Platinum | Most Cathodic (Least Active) | 0.00 |
The surface area ratio between the cathode and anode critically influences corrosion severity. An unfavorable ratio where the cathode area is much larger than the anode area dramatically accelerates anodic corrosion [53]. This is particularly problematic in coated systems where small defects (pinholes) expose a small area of the underlying, more active metal, creating a highly unfavorable cathode-to-anode ratio that concentrates corrosive attack [53].
Passivation and Cathodic Corrosion
Passivation describes the gradual formation of an insulating film (typically metal oxides/hydroxides) on the electrode surface, which hinders electron transfer and increases energy consumption [54]. In electrocoagulation, for instance, aluminum or iron anode dissolution generates metal hydroxyl complexes that can form an adherent passivation layer, blocking active sites and reducing process efficiency [54].
Conversely, cathodic corrosion is a less intuitive phenomenon where cathode materials degrade under strongly negative polarization [55]. This can involve:
- Alloying with alkali metals (e.g., from electrolyte)
- Cathodic etching in aqueous and aprotic media
- Formation of metal hydrides and organometallics [55]
This is a significant concern in electroorganic synthesis using heavy metal cathodes (e.g., Pb, Sn, Hg) prized for their high overpotential for the hydrogen evolution reaction (HER). Their deterioration releases toxic species and complicates product purification [55].
Figure 1: Galvanic corrosion mechanism between dissimilar metals in electrolyte.
Troubleshooting Common Electrode Failure Modes
FAQ: Performance Degradation and Failure Analysis
Q1: Why has my electrode's performance suddenly dropped despite using compatible materials? A: Sudden performance loss often indicates passivation layer formation or the initiation of cathodic corrosion [54] [55]. For anodes, analyze the surface for non-conductive oxide/hydroxide films using techniques like SEM-EDS or XPS [54]. For cathodes operating at high negative potentials, inspect for morphological changes, etching, or hydride formation [55]. Check for recent changes in operating parameters (e.g., increased current density, temperature) that may have accelerated these processes.
Q2: My electrode material is chemically stable, but I'm observing unexpected corrosion at connector points. What's causing this? A: This is classic galvanic corrosion at junctions [53]. Verify the materials' positions on the galvanic series—a potential difference exceeding 0.25V indicates high corrosion risk [53]. The problem exacerbates with unfavorable cathode-to-anode surface area ratios. Implement dielectric insulation between dissimilar metals or apply protective coatings to the more anodic material to break the electrical pathway [53].
Q3: How can I extend the lifespan of my Pb/Sn cathodes in reductive electrosynthesis? A: Cathodic corrosion of heavy metal cathodes can be mitigated by several approaches [55]:
- Use alloys instead of neat soft metals
- Apply protective cationic additives to the electrolyte
- Operate at the minimum necessary current density
- For Pb cathodes, consider introducing small amounts of chloride ions (if compatible with your chemistry) to promote protective layer formation [55]
Q4: What strategies can prevent passivation in electrocoagulation systems? A: Multiple effective strategies exist [54]:
- Implement periodic current reversal (PCR) or pulse electrolysis to dissolve forming passivation layers
- Optimize electrode design (e.g., serpentine flow, mesh electrodes) to improve hydrodynamics and reduce deposition
- Introduce chloride ions to form soluble complexes rather than precipitates
- Apply mechanical cleaning (ultrasound, scraping) or chemical cleaning with dilute acids in maintenance cycles [54]
Advanced Diagnostic Techniques
Table 2: Electrode Corrosion Detection and Analysis Methods
| Technique | Application | Information Obtained | References |
|---|---|---|---|
| In-situ XRD | Chemical stability at interface | Phase changes, impurity formation during heating | [56] |
| SEM-EDS | Surface morphology & composition | Passivation layer thickness, elemental distribution | [54] [56] |
| XPS | Surface chemistry | Oxidation states, chemical environment of elements | [56] [55] |
| Electrochemical Impedance Spectroscopy (EIS) | Interface resistance | Charge transfer resistance, interface degradation | [56] |
| Differential Scanning Calorimetry (DSC) | Thermal stability of interfaces | Reaction temperatures between electrode/electrolyte | [56] |
Experimental Protocols for Electrode Stabilization
Protocol: Surface Engineering via Protective Coating Application
Objective: Apply a defect-free barrier coating to prevent galvanic corrosion and surface degradation.
Materials:
- Armoloy Thin Dense Chrome (TDC) or equivalent protective coating
- Substrate electrode material
- Ultrasonic cleaning bath with isopropanol
- Surface profilometer for roughness measurement
Procedure:
- Surface Preparation: Polish electrode to mirror finish (Ra < 0.1 µm) and clean ultrasonically in isopropanol for 15 minutes to remove all contaminants.
- Coating Application: Apply TDC coating using industrial deposition process, ensuring:
- Uniform thickness between 2-5 µm
- Complete coverage without pinholes or defects
- Quality Control: Verify coating integrity using:
- Microscopic inspection at 100× magnification for defects
- Electrochemical testing in benign electrolyte to check for base metal exposure
- Implementation: Install coated electrode, ensuring proper insulation from dissimilar metals in the assembly.
Key Consideration: Even microscopic pinholes create extremely unfavorable cathode (coating)-to-anode (exposed base metal) area ratios, accelerating localized corrosion [53]. Multiple thin layers often provide better defect coverage than a single thick layer.
Protocol: Interface Stabilization via Entropy Stabilization
Objective: Create thermally stable electrode-electrolyte interfaces for high-temperature operation.
Materials:
- High-entropy disordered rock salt (HE-DRX) electrode materials
- Garnet-type solid-state electrolyte (e.g., LLZTO: Li₆.â‚„La₃Zrâ‚.â‚„Taâ‚€.₆Oâ‚â‚‚)
- Ultrafast high-temperature sintering (UHS) system
- X-ray diffraction (XRD) equipment for phase analysis
Procedure:
- Material Synthesis: Prepare HE-DRX precursor with multiple cations (e.g., Liâ‚.₃Mnâ‚‚âºâ‚€.â‚Coâ‚‚âºâ‚€.â‚Mn³âºâ‚€.â‚Cr³âºâ‚€.â‚Tiâ‚€.â‚Nbâ‚€.â‚‚Oâ‚.₇Fâ‚€.₃) to maximize configurational entropy [56].
- Interface Fabrication: Place HE-DRX precursor powder on dense LLZTO pellet.
- Rapid Sintering: Process using UHS technology at 1100°C for 3 seconds [56].
- Characterization: Validate interface quality using:
- XRD to confirm phase purity and absence of reaction products
- SEM to verify conformal contact and measure interface resistance
- Electrochemical impedance spectroscopy to measure interface resistance (target: <32 Ω·cm²) [56]
Key Findings: The entropy stabilization effect combined with rapid kinetics increases the chemically stable temperature (Tₛₜâ‚bâ‚—â‚‘) to 1100°C while achieving excellent wettability, reducing interface resistance by 700× compared to conventional LiCoOâ‚‚|LLZTO interfaces [56].
Figure 2: Interface stabilization via ultrafast high-temperature sintering.
Protocol: Passivation Mitigation via Polarity Reversal
Objective: Prevent anode passivation in electrocoagulation systems through operational modifications.
Materials:
- Bipolar power supply capable of polarity reversal
- Aluminum or iron electrodes
- pH meter and conductivity meter
Procedure:
- Baseline Establishment: Operate at constant current density (e.g., 10-50 A/m²) for 30 minutes to establish baseline voltage.
- Polarity Reversal Implementation: Program power supply to automatically reverse polarity at set intervals (typically 5-30 seconds) [54].
- Monitoring: Track system voltage and current efficiency over operation time.
- Optimization: Adjust reversal frequency based on:
- Rate of voltage increase (indicator of passivation)
- Treatment efficiency requirements
- Electrode material characteristics
Key Findings: Polarity reversal prevents continuous buildup of passivating layers by periodically dissolving forming deposits, maintaining lower operating voltages and higher Faradaic efficiency over extended operation [54].
Research Reagent Solutions for Corrosion Mitigation
Table 3: Essential Materials for Electrode Corrosion Prevention
| Reagent/Material | Function | Application Context | Key Considerations | |
|---|---|---|---|---|
| Armoloy TDC Coating | Barrier coating preventing electrolyte contact with base metal | Marine, plumbing, and transportation applications | Coating must be defect-free to prevent pinhole corrosion | [53] |
| High-Entropy DRX Materials | Multi-cation electrodes with enhanced thermal stability | All-solid-state batteries and high-temperature applications | Configurational entropy stabilizes interface with electrolytes | [56] |
| Chloride Salts (NaCl) | Forms soluble complexes preventing hydroxide precipitation | Electrocoagulation wastewater treatment | Concentration must be optimized to balance corrosion protection and process requirements | [54] |
| Cationic Additives | Protective layer formation on cathode surface | Electroorganic synthesis with heavy metal cathodes | Must not interfere with target electrochemical reaction | [55] |
| Dielectric Unions/Insulators | Physical separation of dissimilar metals | Plumbing systems, structural connections | Must withstand operational temperatures and environmental conditions | [53] |
Validating Ion Migration Suppression: Techniques and Comparative Material Analysis
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of using in-situ bias during TOF-SIMS analysis for ion migration studies? Applying an in-situ electrical bias during TOF-SIMS analysis allows for the direct observation of reversible ion migration, such as halide and lithium ions in perovskite devices, on the timescale of minutes. This setup creates a framework for actively investigating ion movement under operating conditions [57].
Q2: How can I ensure my TOF-SIMS data is collected without causing excessive surface damage? To stay within the "static limit" for organic materials and ensure your data is representative of the original surface chemistry, keep the primary ion dose at or below 1 × 10¹² ions/cm². The conventional static limit is typically set at 1 × 10¹³ ions/cm² [58].
Q3: What vacuum level is required for TOF-SIMS analysis? TOF-SIMS is an ultra-high vacuum (UHV) method. Typical operating pressures for the analysis chamber are in the range of 10â»â¸ to 10â»â¹ mbar [58].
Q4: My perovskite sample shows spectral instability. How can characterization techniques help? Techniques like in-situ GIWAXS can track structural dynamics in real-time, while TOF-SIMS can identify migrating ion species and their distribution. Combining these methods helps correlate structural changes with ion movement, which is a root cause of phase segregation and instability [2].
Q5: What is the purpose of using in-situ RBS/c for monitoring ion track formation? In-situ Rutherford Backscattering Spectrometry in channeling mode (RBS/c) is used to observe damage build-up in real-time during MeV heavy ion irradiation, for example, in materials like quartz (SiOâ‚‚) [59].
Troubleshooting Guides
Table 1: Troubleshooting Common Issues in Ion Tracking Experiments
| Problem | Possible Cause | Solution |
|---|---|---|
| Low mass resolution in TOF-SIMS | Energy spread of secondary ions; improper instrument tuning. | Use a reflectron to correct for energy dispersion; ensure proper calibration and alignment of the primary ion source and flight tube [58]. |
| Unclear in-situ GIWAXS data | Inadequate temporal resolution; sample degradation under beam. | Optimize beam intensity and exposure time; use a fast detector to capture rapid structural kinetics. |
| No ion migration signal in TOF-SIMS with bias | Poor electrical contact; insufficient bias voltage. | Verify ohmic contacts to the device; ensure the bias voltage is high enough to induce ion movement without damaging the sample [57]. |
| Surface contamination in TOF-SIMS spectra | Sample handling in ambient conditions; hydrocarbon contamination in vacuum. | Implement strict glove-box or clean-room protocols for sample preparation and transfer; use a load-lock system to maintain UHV [58]. |
| Charging effects in TOF-SIMS/XPS | Sample is electrically insulating. | Use a low-energy electron flood gun for charge compensation; consider using thinner samples or a metallic coating grid [58]. |
Table 2: Key Parameters for Tracking Ion Movement with TOF-SIMS
| Parameter | Typical Setting/Value | Technical Impact |
|---|---|---|
| Primary Ion Dose | ≤ 1 x 10¹² ions/cm² | Prevents surface damage, ensures data is within the "static limit" [58]. |
| In-situ Bias Voltage | Device-dependent (e.g., for perovskites) | Drives ion migration, allowing observation of reversible movement [57]. |
| Primary Ion Species | Biâ‚™âº, C₆₀âº, Arₙ⺠(Gas Cluster Ion Beams) | Bi/C₆₀ enhance molecular signal; Ar-GCIB enables efficient depth profiling with minimal damage [58]. |
| Analysis Depth | 1 - 3 nm | Provides true surface sensitivity for detecting initial ion movement and surface composition [58]. |
| Vacuum Pressure | 10â»â¸ - 10â»â¹ mbar | Reduces surface contamination, allows secondary ions to travel to the detector without collision [58]. |
Detailed Experimental Protocols
Protocol 1: In-Situ TOF-SIMS with Electrical Bias for Ion Migration
Objective: To actively track ion migration (e.g., halides, lithium) in a perovskite device under an applied electric field.
- Sample Preparation: Fabricate the perovskite device on a substrate suitable for electrical probing. Ensure electrical contacts are accessible.
- Electrical Connection: Mount the sample in the TOF-SIMS holder and use a custom-designed, UHV-compatible setup to make electrical connections to the device's anode and cathode [57].
- Instrument Setup:
- Select a bismuth (Biâ‚™âº) liquid metal ion gun as the primary source for high spatial resolution and good secondary ion yield.
- Set the primary ion dose to < 1 x 10¹² ions/cm² to remain in the static SIMS regime [58].
- In-Situ Biasing:
- With the sample under UHV, apply a DC bias voltage (e.g., +/- 2-5 V) across the device thickness using a feedthrough. The exact voltage should be below the device's breakdown voltage [57].
- The bias can be applied in cycles (on/off) to test for reversible migration.
- Data Acquisition:
- Collect mass spectra and/or chemical images (2D) at the surface facing the ion gun.
- Acquire data both with and without the applied bias to establish a baseline.
- Monitor specific negative ion masses (e.g., Iâ», Brâ») for halide migration and positive ion masses (e.g., Liâº) for cation migration [57].
- Data Interpretation: An increase in the intensity of specific ion signals (e.g., Iâ») at the biased electrode interface confirms migration of those species towards the applied field.
Protocol 2: Combining RBS/c and ToF-ERDA for In-Situ Ion Track Analysis
Objective: To monitor elemental composition changes and damage build-up during swift heavy ion irradiation.
- Sample Preparation: Prepare thin films of the target material (e.g., SrTiO₃, SiO₂) on a suitable substrate [59].
- Irradiation & Simultaneous Analysis:
- Place the sample in a chamber equipped for in-situ Rutherford Backscattering Spectrometry in channeling mode (RBS/c) and Time-of-Flight Elastic Recoil Detection Analysis (ToF-ERDA).
- Irradiate the sample with swift heavy ions (MeV energy range).
- Simultaneously, collect RBS/c spectra to monitor crystal damage and disorder.
- Simultaneously, use grazing incidence ToF-ERDA to monitor changes in the surface elemental composition and stoichiometry in real-time [59].
- Ex-Situ Validation: After irradiation, use Atomic Force Microscopy (AFM) to physically confirm the presence of ion tracks on the surface [59].
Research Reagent Solutions & Essential Materials
Table 3: Essential Materials for Ion Migration and Nucleation Control Studies
| Material/Reagent | Function in Experiment |
|---|---|
| Halide Perovskite Precursors (e.g., PbI₂, CH₃NH₃I) | The active material layer in which ion migration is studied and controlled via nucleation [2]. |
| Polyethylene Glycol (PEG) | A common precipitant used in controlled crystallization experiments to modulate chemical potential and induce supersaturation [30]. |
| Antisolvents (e.g., Toluene, Chlorobenzene) | Used in solvent engineering during spin-coating to rapidly induce supersaturation, triggering nucleation and controlling crystal growth [2]. |
| Silver/Silver Chloride (Ag/AgCl) Wire | Serves as a stable reference/counter electrode in electrochemical setups, such as those using nanopipettes for nucleation control [30]. |
| Single Asymmetric Nanopipettes | Provides spatially confined control over reagent delivery via electrokinetic transport, enabling deterministic nucleation of single crystals [30]. |
Experimental Workflow Diagrams
Diagram 1: General workflow for in-situ ion tracking.
Diagram 2: Logical flow from nucleation control to ion migration management.
Frequently Asked Questions (FAQs)
Q1: What are the primary computational methods for predicting ion migration barriers, and when should I use each?
A1: The primary methods are First-Principles (or ab initio) simulations and Molecular Dynamics (MD). The choice depends on your research goals, system size, and required accuracy.
First-Principles Methods (e.g., DFT-NEB): These methods, such as Density Functional Theory (DFT) combined with the Nudged Elastic Band (NEB) technique, calculate migration barriers from quantum mechanics. They are highly accurate but computationally expensive.
- Use for: Studying migration mechanisms in small unit cells or bulk crystals with high precision; identifying transition states; calculating activation energies without empirical parameters [60] [61].
- Example: Calculating the barrier for a single Li-ion hop between two specific sites in an anti-perovskite solid electrolyte [60].
Molecular Dynamics (MD) Simulations: These methods simulate the motion of atoms over time by solving classical equations of motion. They can be ab initio (AIMD), classical (CMD), or use machine-learned force fields (MLFF).
- Use for: Simulating ion transport over longer time and length scales; observing complex, multi-ion diffusion mechanisms (e.g., concerted motion); studying systems at finite temperatures [4] [61] [62].
- Example: Simulating Li-ion diffusion in a solid electrolyte under an applied electric field to compute its macroscopic conductivity [4].
Q2: My NEB calculation is failing to converge or is giving an unrealistically high energy barrier. What could be wrong?
A2: This is a common issue often related to the initial pathway guess.
- Cause 1: Poor Initial Pathway. A linear interpolation between initial and final sites can cause atoms to overlap, leading to high energies and slow convergence [60].
- Solution: Pre-optimize the migration path using faster, topological methods. The Topological Analysis of Procrystal Electron Density (TAPED) approach can efficiently find a pathway close to the true Minimum Energy Path (MEP), providing an excellent starting point for NEB [60].
- Cause 2: Finding a Local MEP. The NEB method converges to a local minimum on the Potential Energy Surface (PES). Your initial guess might be separated from the global MEP by a large energy hill [60].
- Solution: Use simple force fields or the TAPED method to scan the PES and identify the global MEP before refining with DFT-NEB [60].
Q3: How can I accurately simulate ion migration in large or complex systems like grain boundaries or interfaces, where DFT becomes too costly?
A3: Leverage multi-scale simulation strategies.
- Machine-Learned Force Fields (MLFF): Train MLFFs on accurate DFT data. These force fields can approach DFT-level accuracy at a fraction of the computational cost, enabling large-scale MD simulations of complex systems like interfaces [62].
- Classical MD with Reliable Force Fields: Use well-parameterized classical force fields to simulate large systems, such as an electrolyte confined between two electrodes under an applied constant potential [4].
- Topological Analysis (TAPED): This method requires minimal computation and is well-suited for scanning thousands of non-equivalent pathways in large, low-symmetry systems like polycrystals, providing quantitative barrier estimates [60].
Q4: Why does my simulated ionic conductivity not match experimental values?
A4: Discrepancies can arise from several factors related to model realism.
- Charge Carrier Concentration: Simulated conductivity is highly sensitive to the concentration of vacancies or interstitials. Ensure your model's defect concentration reflects the experimental material [60] [61].
- The Role of Interfaces: Bulk simulations may not account for the significant resistance to ion transport at grain boundaries or electrode-electrolyte interfaces. Simulations that explicitly include these interfaces are often necessary [60] [4].
- Applied Electric Field: In devices, ions move under an applied potential. Simulations without this field may overestimate diffusion coefficients. Classical MD with a constant potential method (CPM) can better mimic working conditions [4].
Troubleshooting Guides
Issue: Unphysical Ion Trajectories in MD Simulations
Problem: Ions in your MD simulation are moving erratically or the system energy is exploding.
Check 1: Energy Minimization
- Description: The initial structure may have steric clashes or high-energy configurations.
- Solution: Always perform a thorough energy minimization before starting the production MD run. This relaxes the structure and removes any high-energy contacts [63].
- Protocol: Use a steepest descent algorithm with an energy tolerance of 1000 kJ/(mol·nm) and a step size of 0.01 nm for 50,000 steps or until convergence [63].
Check 2: Equilibration Phases
- Description: Jumping directly from minimization to production MD can shock the system.
- Solution: Implement a two-step equilibration [63].
- NVT Ensemble: Equilibrate the system at constant Number of particles, Volume, and Temperature (e.g., 300 K) for ~100 ps. This allows the solvent and ions to reach the target temperature while the solute (e.g., protein) is restrained.
- NPT Ensemble: Further equilibrate at constant Number of particles, Pressure (e.g., 1 bar), and Temperature for ~100 ps. This allows the system density to adjust to the correct value.
Check 3: Force Field Parameters
- Description: Incorrect or missing parameters for ions or ligands can cause instability.
- Solution: Use a well-established force field (e.g., OPLS/AA, AMBER) and ensure all parameters, especially for non-standard residues or ions, are correctly assigned [63].
Issue: Low Accuracy in MLFF-based MD Simulations
Problem: Your Machine-Learned Force Field is not reproducing DFT-level accuracy for migration barriers.
Check 1: Training Data Diversity
- Description: The MLFF was trained on a dataset that does not represent the configurational space explored during ion migration.
- Solution: Ensure the training data includes structures from short MD runs at various relevant temperatures and, crucially, structures near the transition states of migration paths [62].
- Protocol: Validate the MLFF by comparing its predictions for a set of migration barriers against DFT-NEB calculations. The mean absolute error should be on the scale of 0.1 eV or less [62].
Check 2: Defect Charge States
- Description: The charge state of a migrating ion (e.g., a negative iodide interstitial vs. a neutral one) dramatically impacts its mobility and energy landscape [62].
- Solution: Train separate MLFFs for different charge states of the defect system to accurately capture their distinct migration behaviors [62].
Workflow: Choosing a Method for Predicting Migration Barriers
The following diagram outlines a logical workflow for selecting the appropriate computational method based on your system and research question.
Quantitative Data Tables
Table 1: Comparison of Computational Methods for Predicting Migration Barriers
| Method | Typical System Size | Time Scale | Key Outputs | Advantages | Limitations |
|---|---|---|---|---|---|
| DFT-NEB [60] [61] | ~100 atoms | N/A | Migration Barrier (eV), Minimum Energy Path (MEP) | High accuracy; no empirical parameters; reveals atomic-scale mechanism | Computationally expensive; requires initial path guess; limited to simple systems |
| Ab Initio MD (AIMD) [4] | ~100 atoms | ~100 ps | Diffusion Coefficient, Conductivity, Activation Energy | No force field needed; captures anharmonicity & complex mechanisms | Very high computational cost; limited spatiotemporal scale |
| Classical MD (CMD) [4] [63] | 10,000+ atoms | ~100 ns | Diffusion Coefficient, Conductivity, Radial Distribution Functions | Fast; enables large systems & long times; good for screening | Accuracy depends on force field; cannot model bond breaking/forming |
| MLFF-MD [62] | ~1,000 atoms | ~1-10 ns | Diffusion Coefficient, Complex Mechanisms, Activation Energy | Near-DFT accuracy; faster than AIMD; good for charged defects | Requires training data & validation; risk of poor transferability |
| Topological (TAPED) [60] | 1,000+ atoms | N/A | Migration Pathway, Relative Barriers | Extremely fast; automated for complex systems; excellent for initial NEB guess | Neglects structural relaxation; barriers are relative, need calibration |
Table 2: Exemplary Ion Migration Barriers from Different Materials
| Material | Migrating Ion | Method | Activation Barrier (eV) | Notes | Source |
|---|---|---|---|---|---|
| Li₃OCl (Anti-perovskite) | Li⺠| DFT-CI-NEB | Varies (e.g., 0.2 - 0.6) | Barriers for different pathways in bulk | [60] |
| Li₆PS₅Cl (LPSCl) | Li⺠| Classical MD | 0.20 (no field), 0.32 (with field) | Shows effect of applied electric potential | [4] |
| NaGaPO₄F (KTP-type) | Na⺠| AIMD | 0.11 - 0.22 (vacancy), 0.48 - 0.63 (Frenkel) | Ultra-low barrier for vacancy mechanism | [61] |
| CsPbI₃ (Perovskite) | I⻠(II⻠defect) | MLFF-MD / DFT-NEB | ~0.45 | Migration barrier for a charged iodide interstitial | [62] |
| CsPbI₃ (Perovskite) | I⻠(Vᵢ⺠defect) | MLFF-MD / DFT-NEB | ~0.40 | Migration barrier for a charged iodide vacancy | [62] |
The Scientist's Toolkit: Essential Research Reagents & Software
| Item Name | Type | Primary Function | Relevance to Ion Migration Studies |
|---|---|---|---|
| VASP [60] [62] | Software Package | First-Principles DFT Calculations | The benchmark for performing NEB and AIMD calculations to determine precise energy barriers and mechanisms. |
| GROMACS [64] [63] [65] | Software Package | Molecular Dynamics Simulation | A high-performance, open-source MD engine for simulating ion transport in complex systems (proteins, electrolytes) using classical force fields. |
| LAMMPS [61] | Software Package | Molecular Dynamics Simulator | A versatile MD code that can be used with classical potentials, reactive force fields, and MLFFs for materials science applications. |
| Python Materials Genomics (pymatgen) [61] | Python Library | Materials Analysis | Provides robust tools for analyzing crystal structures, manipulating molecules, and processing computational materials data. |
| RDKit [66] | Cheminformatics Library | Molecular Informatics | Handles chemical information, molecular representation, and fingerprinting, useful for preparing ligands or small molecules in a simulation. |
| AutoDock Suite [67] | Software Suite | Molecular Docking & Virtual Screening | Used in drug discovery to predict how small molecules (e.g., ions or drugs) bind to a protein target, related to studying binding sites. |
| OPLS/AA & SPC/E Water [63] | Force Field & Model | Classical Interaction Potentials | A widely used combination of a protein/ligand force field and a water model for setting up biologically relevant MD simulations. |
| Machine-Learned Force Fields (MLFF) [62] | Computational Method | Bridge between DFT and MD | Trained on DFT data to enable large-scale MD simulations with quantum mechanical accuracy, crucial for realistic defect migration studies. |
Troubleshooting Guides & FAQs
Frequently Asked Questions (FAQs)
Q1: What are the common symptoms of ion migration in my halide perovskite solar cell? Ion migration in halide perovskites often manifests as current-voltage (J-V) hysteresis, where the power output changes depending on the voltage scan direction [1]. You may also observe transient phenomena, such as a slow recovery of the open-circuit voltage after a light or voltage pulse, and in severe cases, phase segregation or device degradation over time [20] [1].
Q2: How does the primary migrating species differ between these material systems? In halide perovskites, the most mobile species are typically halide anions (e.g., iodide vacancies) and, to a lesser extent, organic cations like MA⺠[1] [68]. In contrast, solid-state batteries are designed for the highly selective and reversible migration of a single working ion, such as the alkali metal cation (e.g., Liâº, Naâº) within the solid electrolyte [69] [70].
Q3: Our team is observing inconsistent results in film quality during perovskite deposition. Could nucleation be a factor? Yes, uncontrolled nucleation is a primary source of inconsistency. Stochastic (random) nucleation leads to non-uniform crystal size and distribution, which creates varied defect densities and ion migration pathways across the film [20] [19]. Implementing controlled nucleation techniques, such as antisolvent dripping or substrate temperature modulation, is crucial for achieving reproducible, high-quality films [20].
Q4: Why is quantifying ion migration so challenging in these materials? The main challenge stems from their nature as Mixed Ionic-Electronic Conductors (MIECs) [70] [68]. The electrical signals from mobile ions are often convoluted with those from electronic charge carriers (electrons and holes). Accurate quantification requires techniques that can effectively separate the ionic and electronic contributions to the total conductivity [68].
Troubleshooting Guide: Common Experimental Issues
| Problem | Possible Cause | Solution / Mitigation Strategy |
|---|---|---|
| Hysteresis in J-V Curves [1] | High ion mobility, especially of halide vacancies, leading to interfacial polarization. | • Reduce halogen vacancy concentration via stoichiometric tuning [1].• Use larger A-site cations (e.g., FAâº, GAâº) to tighten the lattice [1].• Employ interface and grain boundary passivation [20]. |
| Poor Device Stability [1] | Migrating ions reaching and reacting with charge transport layers or electrodes. | • Incorporate low-dimensional perovskite phases (e.g., 2D Ruddlesden-Popper) to suppress ion migration [1].• Optimize extraction layers to create non-reactive interfaces [1].• Control crystallization to form large, uniform grains with fewer grain boundaries [20]. |
| Inconsistent Film Morphology [20] [19] | Uncontrolled, stochastic nucleation during the film formation process. | • Implement controlled nucleation methods like antisolvent treatment [20].• Pre-condition the coating substrate to a specific temperature [20].• Use pressure manipulation techniques (e.g., vacuum) to induce uniform nucleation [19]. |
| Difficulty in Measuring Ionic Conductivity [70] [68] | Overlap of electronic and ionic currents; sensitivity to environmental stimuli. | • Use solid-state ionics techniques like DC polarization (Hebb-Wagner method) to block ionic current [70].• Conduct measurements under controlled, inert atmospheres to avoid interference from moisture/oxygen [68]. |
The table below summarizes key quantitative parameters related to ion migration, enabling a direct comparison between the material systems.
Table 1: Quantitative Comparison of Ion Migration Parameters
| Parameter | Halide Perovskites | Solid-State Batteries (Typical Electrolytes) | Measurement Technique & Notes |
|---|---|---|---|
| Ion Diffusion Coefficient (Dᵢₒₙ) | ~10â»â¸ to 10â»Â¹âµ cm²/s [1]• MAPbI₃: ~10â»â¸ cm²/s• CsPbBr₃: ~10â»Â¹Â² to 10â»Â¹Â³ cm²/s• 2D Perovskites: ~10â»Â¹Â² to 10â»Â¹âµ cm²/s | ~10â»â¶ to 10â»Â¹Â¹ cm²/s (for Li⺠in solid electrolytes like LLZO, LATP) [69] [70] | Calculated from electrical conductivity or measured directly via techniques like Transient Ion Drift (TID) and Electrochemical Impedance Spectroscopy (EIS) [1] [68]. |
| Activation Energy (Eâ‚) | ~0.1 to 0.6 eV for halide vacancies [68] | ~0.2 to 0.8 eV for Li⺠migration [70] | Determined from temperature-dependent conductivity measurements (Arrhenius plot) [70] [68]. The soft lattice of perovskites contributes to low Eâ‚. |
| Ionic Conductivity (σᵢₒₙ) | ~10â»Â³ to 10â»â´ S/cm (estimated for halides) [69] | ~10â»Â² to 10â»â¸ S/cm (for Liâº) [70] | Measured using EIS with ion-blocking electrodes or DC polarization methods [70] [68]. |
| Impact of Nucleation Control | Directly affects defect density and T₈₀ lifetime. Controlled nucleation is key to reducing Dion and improving stability [20] [1]. | Governs interface contact and dendrite suppression. Uniform nucleation of Li plating is critical for cycle life [69] [70]. | A universal principle: controlling the initial nucleation step is foundational for managing long-term ion migration and stability. |
Experimental Protocols
Protocol 1: Controlled Nucleation of Perovskite Films via Antisolvent Engineering
Principle: Induce supersaturation and uniform nucleation by rapidly removing the solvent during spin-coating, thereby controlling the initial crystal formation and subsequent growth [20].
Step-by-Step Methodology:
- Solution Preparation: Prepare your perovskite precursor solution (e.g., MAPbI₃ in DMF/DMSO) and filter it (0.45 µm PTFE filter) to remove particulate contaminants.
- Substrate Pre-treatment: Clean the substrate (e.g., ITO/glass) and treat it with UV-Ozone for 15-20 minutes to ensure a uniform, hydrophilic surface.
- Spin-coating: Dynamically dispense the precursor solution onto the spinning substrate. A typical two-step program is used (e.g., 1000 rpm for 10 s, then 4000 rpm for 30 s).
- Antisolvent Dripping: At a precisely optimized moment (e.g., 5-10 seconds before the end of the second high-speed step), swiftly dispense a pre-determined volume (e.g., 200 µL) of an antisolvent (e.g., chlorobenzene or toluene) onto the center of the spinning substrate.
- Nucleation and Film Formation: The antisolvent rapidly extracts the main solvent, driving the solution into a state of supersaturation and inducing a shower of uniform nucleation sites across the entire substrate. The film should immediately change color (e.g., become dark brown).
- Annealing: Transfer the film immediately to a hotplate and anneal at 100°C for 10-60 minutes to facilitate crystal growth and remove residual solvent.
Troubleshooting Notes:
- Dripping Timing is Critical: A delay of just a few seconds can drastically alter nucleation density, leading to either discontinuous films or large, isolated crystals [20].
- Antisolvent Choice: The chemical potential of the system is tailored by the antisolvent's miscibility with the host solvent. Test different antisolvents for optimal results [20].
Protocol 2: Quantifying Ionic Conductivity using Thermal Admittance Spectroscopy
Principle: Measure the device's capacitance as a function of frequency at different temperatures to deconvolute the ionic and electronic contributions, which have different response times [70] [68].
Step-by-Step Methodology:
- Device Fabrication: Complete the fabrication of your full device (e.g., ITO/HTL/Perovskite/ETL/Metal).
- Temperature-controlled Setup: Place the device in a temperature-controlled stage or cryostat with electrical probes, ensuring a dark and inert (e.g., Nâ‚‚) environment.
- Impedance Measurement: Apply a small AC bias (e.g., 20 mV) around 0 V DC bias. Sweep the frequency (typically from 1 MHz down to 0.1 Hz) at a series of fixed temperatures (e.g., from 250 K to 320 K in 10 K steps).
- Capacitance Analysis: Extract the capacitance (C) from the impedance data at each temperature and plot it as a function of frequency (C-f plot). Identify features such as the geometric capacitance, bulk ionic capacitance, and electrode polarization.
- Ionic Conductivity Calculation:
- The bulk ionic resistance (Rᵢₒₙ) can be determined from the low-frequency response in the Nyquist plot or the plateau in the C-f plot.
- The ionic conductivity is calculated as: σᵢₒₙ = d / (A × Rᵢₒₙ), where d is the perovskite layer thickness and A is the device area.
- Activation Energy: Plot the extracted σᵢₒₙ versus 1/T (Arrhenius plot). The slope of the linear fit gives the activation energy: E₠= -slope × k, where k is Boltzmann's constant.
Troubleshooting Notes:
- Avoid Electronic Overlap: Using a low DC bias (0 V) helps minimize the injection of electronic charge carriers, making the ionic signal more dominant [68].
- Environmental Control: Perovskites are highly sensitive to light, humidity, and oxygen. Any leakage can severely compromise the measurement accuracy [68].
Diagrams & Visualization
Diagram 1: Ion Migration Pathways & Nucleation Control Logic
Diagram Title: Ion Migration and Nucleation Control Logic
Diagram 2: Experimental Workflow for Film Fabrication & Analysis
Diagram Title: Film Fabrication and Analysis Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Ion Migration and Nucleation Control Experiments
| Reagent / Material | Function / Application | Key Consideration |
|---|---|---|
| Antisolvents (Chlorobenzene, Toluene) | Induces supersaturation and controlled nucleation during perovskite film casting by rapidly extracting the host solvent [20]. | Purity is critical. Dripping timing and volume must be rigorously optimized for reproducibility. |
| Precursor Salts (PbI₂, MAI, FAI, CsI) | Forms the perovskite crystal lattice (ABX₃). Used for compositional engineering to tune ion migration rates [69] [1]. | Precise, stoichiometric weighing and mixing in inert atmosphere (glovebox) is essential to control defect chemistry. |
| Large Organic Cations (PEAâº, BAâº) | Used to create low-dimensional (2D/3D) perovskites. These large cations suppress ion migration by acting as structural barriers [1]. | Concentration must be optimized to balance ion migration suppression with charge transport efficiency. |
| Passivation Agents | Molecules (e.g., Lewis bases) that bond to under-coordinated Pb²⺠ions and other defect sites at grain boundaries and surfaces, reducing pathways for ion migration [20]. | Typically added in small quantities (<1 mol%) to the precursor solution or as a post-treatment. |
| Ion-Blocking Electrode Materials | Essential for electrical characterization. Electron-blocking (e.g., Ti) and ion-blocking (e.g., C) contacts are used to isolate and quantify ionic conductivity [70] [68]. | Electrode selection is fundamental to the interpretation of impedance and DC polarization data. |
Troubleshooting Guides
FAQ: Ion Migration in Perovskite Photovoltaics
Q1: My perovskite solar cell shows good initial efficiency but rapid performance degradation. Could ion migration be the cause, and how can I confirm this?
Yes, ion migration is a primary cause of rapid degradation in perovskite photovoltaics. You can confirm this through several characterization techniques:
- Hysteresis Index (HI) Analysis: Measure current-voltage (J-V) curves at different scan rates. A strong dependence of efficiency on scan rate indicates significant ion migration, as ions cannot redistribute fast enough to keep up with the voltage changes [8].
- Bias-Assisted Charge Extraction (BACE) and Fast Hysteresis (FH) J-V: These techniques specifically quantify mobile ion density and its impact on device performance, revealing ionic losses separate from charge carrier losses [71].
- X-ray Photoelectron Spectroscopy (XPS) Under Bias: Applying reverse bias while monitoring interfacial chemistry with XPS can directly detect ion migration across layers, helping quantify the barrier energy needed to suppress it [3].
Q2: I've applied a phenethylammonium iodide (PEAI) passivation layer, which improved my open-circuit voltage (VOC) and fill factor (FF) but caused a significant short-circuit current density (JSC) loss. What is happening?
This is a documented effect of certain 2D passivation layers. The JSC loss is not primarily due to lower conductivity but rather increased mobile ion density induced by the PEAI layer itself. These accumulated ions screen the internal electric field, reducing charge collection efficiency and manifesting as JSC loss [71].
- Solution: Implement a bilayer passivation strategy. Introduce an additional interlayer such as ammonium benzenesulfonate (ABS) or ethylenediammonium diiodide (EDAIâ‚‚) between the perovskite and the PEAI. This bilayer structure stabilizes the 2D perovskite phase and reduces mobile ion density, mitigating JSC losses while preserving VOC and FF benefits [71].
Q3: In all-perovskite tandem solar cells, how do I determine which sub-cell's ion migration is limiting overall stability?
The stability of all-perovskite tandem cells is dominantly controlled by the current-limited sub-cell. The hysteresis index (HI) of the tandem device is more influenced by ion migration in the sub-cell that produces less photocurrent [8].
- Diagnostic Method: Use a dual-light source compensation setup to independently control the current generated in each sub-cell. Monitor the stable power output and hysteresis behavior under different light bias conditions. The sub-cell whose current limitation correlates most strongly with changes in tandem device hysteresis is the dominant source of ion migration instability [8].
- Mitigation: Focus your ion suppression strategies (defect passivation, composition engineering) on the current-limited sub-cell identified through this testing protocol [8].
Key Experimental Protocols
Protocol 1: Quantifying Barrier Energy to Suppress Iodide Migration
This protocol determines the minimum energy barrier required to prevent iodide ion migration from the perovskite layer into adjacent charge transport layers, a critical factor in enhancing device longevity [3].
- Objective: Determine the threshold barrier energy (in eV) to suppress iodide ion migration at the perovskite/HTL interface.
- Materials:
- Completed perovskite solar cells (various compositions)
- Reverse bias source (source measurement unit)
- X-ray Photoelectron Spectroscopy (XPS) system
- Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS) (optional)
- Methodology:
- Age Control Devices: Subject finished devices to light soaking (e.g., 1 sun illumination) for an extended period (e.g., 500 hours) without electrical bias.
- Interface Analysis: Use XPS to detect and quantify the presence of iodine (I 3d peaks) on the surface of the hole transport layer (e.g., PTAA) of the aged control devices. This establishes the baseline level of iodide migration.
- Reverse Bias Application: Apply a reverse electrical bias (e.g., -0.8 V for FAPbI₃-based devices) to fresh devices during a similar light-soaking aging process.
- Equilibrium Confirmation: Perform XPS analysis on the HTL surface of the reverse-biased devices. The specific bias value required to eliminate the I 3d peaks indicates the point where ionic drift and diffusion have reached equilibrium, confining iodide ions.
- Barrier Energy Calculation: Calculate the built-in field potential within the HTL depletion region under the determined reverse bias condition. This potential drop, obtained through electrical simulation matching your device structure, equals the quantitative barrier energy (e.g., 0.911 eV for FAPbI₃) needed to prevent iodide loss [3].
Protocol 2: Implementing a Composite Scattering and Drift Barrier Layer
This method creates a quantified barrier on the perovskite surface that combines physical and electrostatic mechanisms to suppress ion migration by over 99.9%, dramatically improving thermal stability [3].
- Objective: Fabricate a composite blocking layer that meets or exceeds the quantified barrier energy threshold without impeding charge carrier transport.
- Materials:
- Prepared perovskite film
- Atomic Layer Deposition (ALD) system
- Hafnium precursor (e.g., HfClâ‚„) and water (Hâ‚‚O) for ALD
- Dipole molecule solution (e.g., (4-(2-(trifluoromethyl)pyrimidin-5-yl)phenyl) boronic acid (CF3-PBAPy) in isopropanol)
- Poly(N-vinylcarbazole) (PVK) for hole transport layer
- Methodology:
- Scattering Barrier Deposition:
- Use ALD to deposit a thin, continuous HfOâ‚‚ layer (~1.5 nm) directly onto the perovskite film. This layer acts as a physical barrier that scatters migrating ions.
- Characterization: Confirm uniform coverage and thickness via Scanning Electron Microscopy (SEM). Verify that carrier transport is not hindered using Time-Resolved Photoluminescence (TRPL), as carriers can tunnel through this ultra-thin layer [3].
- Drift Barrier Formation:
- Spin-coat the CF3-PBAPy solution onto the HfOâ‚‚ layer. The dense hydroxyl groups on ALD HfOâ‚‚ provide excellent anchoring sites, promoting the formation of a uniform, ordered dipole monolayer.
- This monolayer creates a permanent, directional electric field (drift field) that electrostatically repels negatively charged iodide ions, adding to the total blocking energy [3].
- Band Alignment Correction:
- Deposit a PVK layer as the HTL. Its high work function compensates for any band shifts induced by the dipole monolayer's electric field, ensuring efficient hole extraction is maintained [3].
- Validation:
- Performance: Measure J-V characteristics to ensure the power conversion efficiency (PCE) is maintained or improved.
- Stability: Perform maximum power point (MPP) tracking at elevated temperatures (e.g., 85°C). Devices employing this composite barrier have demonstrated >95% of initial PCE after 1500 hours of testing [3].
- Scattering Barrier Deposition:
Quantitative Data Tables
Table 1: Quantified Barrier Energies for Different Perovskite Compositions [3]
| Perovskite Composition | Required Reverse Bias (V) to Suppress Iâ» Migration | Calculated Barrier Energy (eV) |
|---|---|---|
| FAPbI₃ | -0.80 | 0.911 |
| FAâ‚€.₉MAâ‚€.â‚PbI₃ | -0.75 | 0.854 |
| FAâ‚€.₉Csâ‚€.â‚PbI₃ | -0.70 | 0.797 |
| FA₀.₉MA₀.₀₅Cs₀.₀₅PbI₃ | -0.65 | 0.740 |
Table 2: Performance Comparison of Ion Migration Suppression Strategies
| Strategy | Key Metric(s) | Reported Outcome | Reference |
|---|---|---|---|
| Composite HfO₂/CF3-PBAPy Barrier | Steady-state PCE / Stability (85°C, MPP) | 25.7% certified PCE / >95% retention after 1500 h | [3] |
| Bilayer ABS/PEAI Passivation | Initial JSC / PCE | Reduced JSC loss to ~0.5 mA cmâ»Â² / PCE ≈ 25% | [71] |
| PEAI-only Passivation | Initial JSC / Operational Degradation | JSC loss of 1.3 mA cmâ»Â² / Rapid ionic JSC loss (14 mA cmâ»Â² after 160 h) | [71] |
| Current Matching in Tandems | Hysteresis Index (HI) Control | Tandem HI is dominated by the current-limited sub-cell | [8] |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Ion Migration Suppression Experiments
| Material | Function/Application | Key Consideration |
|---|---|---|
| Phenethylammonium Iodide (PEAI) | Forms a 2D perovskite passivation layer on a 3D perovskite surface, reducing non-radiative recombination and improving VOC. | Can increase mobile ion density, leading to JSC loss. Requires co-passivation for mitigation [71]. |
| Ethylenediammonium Diiodide (EDAIâ‚‚) | Used as an interlayer beneath PEAI to stabilize the 2D phase and reduce the mobile ion density induced by PEAI [71]. | Part of a bilayer passivation strategy to achieve high VOC without JSC penalties. |
| Ammonium Benzenesulfonate (ABS) | Functions similarly to EDAIâ‚‚ as an interlayer to suppress PEAI-induced ionic losses [71]. | Offers an alternative chemical approach for bilayer passivation. |
| HfOâ‚‚ (via ALD) | Creates an ultra-thin, pinhole-free scattering barrier on the perovskite surface to physically impede ion migration [3]. | Thickness is critical (~1.5 nm); must be thin enough to allow carrier tunneling. |
| CF3-PBAPy Dipole Molecule | Forms an ordered monolayer on HfOâ‚‚, generating a permanent drift electric-field that repels iodide ions [3]. | The anchored dipole provides a quantifiable electrostatic barrier energy. |
| Poly(N-vinylcarbazole) (PVK) | High-work-function hole transport material used in conjunction with dipole layers to maintain optimal band alignment for hole extraction [3]. | Corrects for band shifts caused by interfacial dipole fields. |
Supporting Diagrams
Diagram 1: Ion Migration Equilibrium at a Blocked Interface
Diagram 2: Workflow for Composite Barrier Layer Fabrication
Conclusion
The strategic management of ion migration through nucleation and grain boundary control represents a fundamental advance in materials science, moving beyond heuristics to a predictive framework. The key insight is that many crystallization additives function not by altering nucleation but by enhancing ion mobility across grain boundaries during coarsening, directly influencing the final microstructure's resilience to ion migration. This unified principle, validated by advanced characterization and computational modeling, bridges additive engineering with post-processing techniques. Future directions involve the rational design of multi-functional additives that simultaneously passivate defects, guide coarsening, and create intrinsic energy barriers against ion movement. The convergence of high-throughput experimentation, multi-scale modeling, and operando analysis will accelerate the development of next-generation materials where ion migration is not just mitigated, but masterfully controlled for ultimate device stability and performance.
References
- 1. Assessing the Drawbacks and Benefits of Ion Migration in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9578653/]
- 2. Advanced Strategies to Tailor the Nucleation and Crystal ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.842924/full]
- 3. Confining iodide migration with quantified barrier for ... [https://www.nature.com/articles/s41467-025-64390-2]
- 4. Unveiling Li-ions migration mechanism in Li 6 PS 5 Cl ... [https://www.nature.com/articles/s41524-025-01832-x]
- 5. Ion migration and defect effect of electrode materials in ... [https://www.sciencedirect.com/science/article/abs/pii/S0079642521001353]
- 6. Mechanosensitive ion channels in cell migration - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8240554/]
- 7. Methods of suppressing ion migration in n-i-p perovskite ... [https://www.sciopen.com/article/10.23919/IEN.2024.0029]
- 8. Insight into ion-induced stability degradation in all ... [https://www.sciencedirect.com/science/article/abs/pii/S2095927325003548]
- 9. Multi-physics mechanisms and regulation of perovskite ... [https://pubs.rsc.org/en/content/articlelanding/2025/ee/d5ee02240a]
- 10. Underlying factors determining grain morphologies in high ... [https://www.nature.com/articles/s41467-023-38885-9]
- 11. Recent advances in tin halide perovskite solar cells: a critical ... [https://pubs.rsc.org/nb/content/articlehtml/2025/ta/d5ta04568a]
- 12. Controlling grain structure in metallic additive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10281942/]
- 13. Grain Size Analysis in Metals and Alloys [https://evidentscientific.com/en/applications/grain-size-analysis]
- 14. AI-supported material analysis for precision [https://www.zeiss.com/metrology/us/software/zeiss-zen-core/intelligent-material-analysis-with-ki.html]
- 15. Grain Size Measurement | Particle Analysis | Fiber Thickness [https://merrowscientific.com/https-www-mipar-us-applications-materials-htm/]
- 16. Quantifying and Reducing Ion Migration in Metal Halide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10343672/]
- 17. Spectroscopic methods (NMR, IR, UV-Vis) | Intro to Polymer Science Class Notes | Fiveable | Fiveable [https://library.fiveable.me/introduction-polymer-science/unit-7/spectroscopic-methods-nmr-ir-uv-vis/study-guide/njmGbIF5Ry9eVnDq]
- 18. Self-Reducing Copper Precursor Inks and Photonic Additive Yield Conductive Patterns under Intense Pulsed Light - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6641306/]
- 19. A Practical Method for Resolving the Nucleation Problem in ... [https://www.bioprocessintl.com/formulation/a-practical-method-for-resolving-the-nucleation-problem-in-lyophilization]
- 20. Advanced Strategies to Tailor the Nucleation and Crystal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9043105/]
- 21. uv-vis ir nmr: Topics by Science.gov [https://www.science.gov/topicpages/u/uv-vis+ir+nmr]
- 22. How crystallization additives govern halide perovskite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12603034/]
- 23. Cementing the grain boundary defects in the strain relaxed ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725046261]
- 24. Understanding ion migration suppression in all-inorganic ... [https://www.sciopen.com/article/10.26599/NRE.2025.9120166]
- 25. Ion Migration and Interface Engineering in Emerging ... [https://www.sciencedirect.com/science/article/pii/S2468217925001674]
- 26. The role of ion migration, octahedral tilt, and the A-site ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10739958/]
- 27. Recent advances in the stability of Sn–Pb mixed perovskite ... [https://pubs.rsc.org/en/content/articlehtml/2025/tc/d5tc02757h?page=search]
- 28. Recent progress in Pb-, Sn-, and Pb–Sn-based inorganic ... [https://pubs.rsc.org/en/content/articlehtml/2025/el/d5el00065c]
- 29. Formation of lattice vacancies and their effects on lithium-ion ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d4ta05713a]
- 30. A Single-Entity Method for Actively Controlled Nucleation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10308327/]
- 31. SnO2 anode surface passivation by atomic layer deposited ... [https://pubmed.ncbi.nlm.nih.gov/24634208/]
- 32. Threshold Switching and Resistive Switching in SnO 2 [https://www.mdpi.com/2073-4352/14/10/909]
- 33. Tailoring SnO2 Defect States and Structure [https://pmc.ncbi.nlm.nih.gov/articles/PMC10303380/]
- 34. Effect of SnO2 particulate characteristics on mechanical ... [https://www.sciencedirect.com/science/article/abs/pii/S0020768325001246]
- 35. Combustion-assisted low-temperature solution process for ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884222011683]
- 36. Preparation and characterization of HfO2/VO2 ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884221036476]
- 37. First-principles predictions of HfO2-based ferroelectric ... [https://www.nature.com/articles/s41524-024-01344-0]
- 38. Bidirectional passivation of the SnO 2 /perovskite interface ... [https://www.sciencedirect.com/science/article/pii/S2949746923000125]
- 39. Manipulating the crystallization kinetics of halide ... [https://www.nature.com/articles/s43246-024-00566-5]
- 40. Quantitatively analyzing the failure processes of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8580315/]
- 41. Transition metal coordination to degradation products in ... [https://pubs.rsc.org/en/content/articlehtml/2025/ee/d5ee01250c]
- 42. Failure modes, safety concerns, testing protocol, and ... [https://www.sciencedirect.com/science/article/pii/S2950264025000929]
- 43. Enhancing metabolite coverage using dedicated mobile ... [https://link.springer.com/article/10.1007/s00216-025-06189-0]
- 44. Elucidating the charge-transfer and Li-ion-migration ... [https://www.sciopen.com/article/10.26599/NRE.2022.9120031]
- 45. Ionic Liquids in Pharmaceutical and Biomedical Applications [https://www.mdpi.com/1999-4923/16/1/151]
- 46. Grain size control for successfully fabricating stainless and ... [https://www.thefabricator.com/tubepipejournal/article/tubepipeproduction/grain-size-control-for-successfully-fabricating-stainless-and-inconel-alloy-tubing]
- 47. How crystallization additives govern halide perovskite grain growth [https://www.nature.com/articles/s41467-025-65484-7?error=cookies_not_supported&code=33f21bc3-4092-4324-9232-22305026873e]
- 48. Developing low-resistance ion migration pathways using ... [https://pubs.rsc.org/en/content/articlehtml/2025/ee/d5ee00132c]
- 49. sciencedirect.com/topics/engineering/ grain - coarsening [https://www.sciencedirect.com/topics/engineering/grain-coarsening]
- 50. Enhanced charge carrier transport and defects mitigation of ... [https://www.nature.com/articles/s41467-024-52925-y]
- 51. Transient Response and Ionic Dynamics in Organic ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11219702/]
- 52. Elevating Modern Drug-Delivery Devices with Advanced ... [https://www.medicaldesignbriefs.com/component/content/article/53779-elevating-modern-drug-delivery-devices-with-advanced-polymers]
- 53. Understanding Galvanic Corrosion: Concepts, Causes, ... [https://armoloy.com/understanding-galvanic-corrosion-causes-prevention-and-solutions/]
- 54. Current status of electrode corrosion passivation and its ... [https://www.sciencedirect.com/science/article/pii/S025527012500042X]
- 55. Cathodic Corrosion of Metal Electrodes—How to Prevent It in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8431381/]
- 56. Maximizing interface stability in all-solid-state lithium ... [https://www.nature.com/articles/s41467-024-51123-0]
- 57. Applying in Situ Bias During TOF-SIMS Analysis ... [https://www.osti.gov/biblio/2574749]
- 58. Back to the basics of time-of-flight secondary ion mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10063322/]
- 59. Monitoring Ion Track Formation Using In Situ RBS/c, ToF- ... [https://pubmed.ncbi.nlm.nih.gov/28878186/]
- 60. Enhancing first-principles simulations of complex solid ... [https://www.nature.com/articles/s41524-022-00877-6]
- 61. A new class of solid electrolytes with an ultra-low Na-ion ... [https://www.sciencedirect.com/science/article/abs/pii/S0378775325008158]
- 62. Tracing Ion Migration in Halide Perovskites with Machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12105036/]
- 63. Running molecular dynamics simulations using GROMACS [https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/md-simulation-gromacs/tutorial.html]
- 64. Introduction to Molecular Dynamics [https://tutorials.gromacs.org/md-intro-tutorial.html]
- 65. GROMACS Tutorials [http://www.mdtutorials.com/gmx/]
- 66. Top 20+ Essential Resources for AI-Driven Computational ... [https://dev.to/vaib/top-20-essential-resources-for-ai-driven-computational-drug-discovery-faa]
- 67. Resource for Structure-based Computational Drug Discovery ... [https://rsd3.scripps.edu/]
- 68. Toolsets for assessing ionic migration in halide perovskites [https://www.sciencedirect.com/science/article/pii/S2542435124001065]
- 69. Exploring metal halide perovskites as active architectures in ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d5ta04267d]
- 70. Solid-State Ionics of Hybrid Halide Perovskites - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6727625/]
- 71. Mitigating Mobileâ€Ionâ€Induced Instabilities and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12306393/]